Introduction
Hypereosinophilic syndrome (HES) is defined as the
presence of a peripheral blood eosinophil (PBE) count of
≥1.5×109/l for at least 6 months [a shorter duration is
acceptable in the presence of symptoms that require eosinophil
(EOS)-lowering therapy] with clinical end-organ damage (1). The age-adjusted incidence rate of HES
was ~0.36 per 1,000,000 people between 2001 and 2005, according to
the Surveillance, Epidemiology and End Results database (2). HES may be divided into three subtypes,
namely primary (clonal), secondary (reactive) and idiopathic
eosinophilia (IHES), according to its pathogenesis (1). Numerous diseases may cause secondary
eosinophilia, including infection (particularly by tissue-invasive
parasites), allergy/atopy, hypersensitivity, drug reactions and
collagen-vascular disease (3,4). Control
of the primary disease is therefore essential for the treatment of
secondary eosinophilia. Furthermore, the evaluation of a primary
bone marrow disorder should be conducted if secondary causes of
eosinophilia are excluded (5).
Screening for factor interacting with PAPOLA and CPSF1
(FIP1L1)-platelet-derived growth factor receptor (PDGFR) A using
fluorescent in situ hybridization (FISH) or polymerase chain
reaction (PCR) and cytogenetic analysis for reciprocal
translocations involving 4q12 (PDGFRA), 5q31-33 (PDGFRB), 8p11-12
(fibroblast growth factor receptor 1; FGFR1) or 9p24 (janus kinase
2; JAK2) are necessary for the diagnosis of primary eosinophilia
(5). However, IHES is a diagnosis
made when the possibility of secondary and primary eosinophilia are
excluded (5). Lymphocyte-variant HSE
is a subtype of primary eosinophilia characterized by an abnormal
T-cell population (6). Imatinib is a
recommended treatment for patients exhibiting rearrangements of
PDGFRA or PDGFRB (7,8) and corticosteroids are the first-line
therapy for patients with lymphocyte-variant hypereosinophilia and
IHES (5,9). Interferon-α targeted antibodies and
hydroxyurea, as well as other chemotherapies, may clinically
benefit patients with primary eosinophilia that have variable
response durability (10–12).
Immunoglobulin G4 (IgG4)-related disease (IgG4-RD)
is a recently defined clinical illness that is characterized by
tissue infiltration by IgG4-positive plasma cells and/or elevated
serum IgG4 concentration alongside chronic inflammation and
primarily affects middle aged or elderly men (13). Additionally, IgG4-RD encompasses a
variety of conditions, including Mikulicz's syndrome, chronic
sclerosing sialadenitis, hypophysitis, Riedel thyroiditis,
inflammatory artificial tumors, chronic interstitial pneumonitis,
interstitial nephritis, autoimmune pancreatitis, retroperitoneal
fibrosis, sclerosing cholangitis, sclerosing cholecystitis,
prostatitis and lymphadenopathy (13). However, the pathogenesis, diagnostic
criteria and role of increased serum IgG4 remain controversial
(14).
The present study reported the case of a 9 year-old
Chinese boy who presented with eosinophilia and elevated serum
levels of IgG4. Following systematic evaluation, he was diagnosed
with HES and IgG4-associated disease. Corticosteroids were
administered for treatment and were proven to be effective and the
patient remains asymptomatic.
Case report
A 9-year-old Chinese boy was admitted to the
Hangzhou First People's Hospital (Hangzhou, China) due to suspected
leukocytosis and eosinophilia for ~1 week on July 17, 2015. The
appropriate examinations were performed in the hospital and written
informed consent was obtained from the patient's family. The
results of a routine blood test identified an abnormal EOS count of
7.01×109/l (normal range, 0.02–0.52×109/l). A
high erythrocyte sedimentation rate (64 mm/h; normal range, 0–15
mm/h), high IgG (41.8 g/l; normal range, 6.09–12.85 g/l) and high
IgE (1,755.5 kU/l; normal range, <87 kU/l) counts were also
observed. However, the IgA (1.84 g/l; normal range, 0.52–2.16 g/l)
and IgM (2.33 g/l; normal range, 0.67–2.48 g/l) counts were within
the normal ranges. An upper abdominal B ultrasonography identified
multiple peripancreatic hypoechoic nodules. Furthermore, an
ultrasound of the mesenteric lymph nodes revealed multiple
mesenteric lymph nodes, some of which were enlarged. However, the
echocardiography was normal. A bone marrow puncture identified an
elevated EOS rate of 23% (normal range, <5%), which indicated
eosinophilia. The boy was permitted to leave the hospital when the
EOS count was reduced to 2.94×109/l following therapy
with anti-allergic drugs administered orally (4 mg chlorphenamine
maleate three times a day for 1 week and 4 mg singulair per day for
1 month) and oral antiparasitic drugs (zentel, 200 mg per day for 3
days). The boy had no history of allergies, contact with pets or
stay in an epidemic area and he did not have a fever, cough,
expectoration, abdominal pain or diarrhea.
On March 31, 2016, the patient was admitted to the
hospital again due to long-term eosinophilia and right posterior
auricular lymph node swelling. A physical examination identified
enlargement of several peanut-sized lymph nodes in the
retroauricular region and on both sides of the neck. The nodes were
homogeneous, smooth-surfaced, mobile and not tender. A routine
blood examination indicated an increased EOS count of
12.6×109/l compared with that detected previously and
high IgE (875.3 kU/l), IgG (43.9 g/l) and IgG4 (14.2 g/l; normal
range, 0.03–2.01 g/l) counts. Allergic source tests identified an
allergy to house dust mites due to elevated IgE (IgE; 16.26 kU/l;
normal range, <0.35 kU/l). A T-SPOT test for tuberculosis and
the results of autoantibody tests [including anti-laminoid
antibody, anti-nucleosome antibody, anti-Ro52 antibody, anti-PM-Scl
antibody, anti-Scl-70 antibody, antinuclear antibody, anti-Sm
antibody, anti-SSB antibody, anticardiolipin antibody (IgA, IgG and
IgM), anti-actin antibody, anti-PCNA antibody, anti-centromere
antibody, anti-ribosomal P protein antibody, anti-mitochondrial
(M2) antibody, anti-Jo1 antibody, anti-U1-nRNP antibody, anti-SSA
antibody, anti-histone antibody and anti-double stranded DNA
antibody tests] were all negative. Superficial lymphadenopathy B
ultrasonography identified several smooth-surfaced hypoechoic
nodules with irregular shapes in the bilateral neck (the largest
one being 3.2×1.2 cm). A magnetic resonance imaging examination of
the head displayed abnormal signals in the right thalamus, possibly
indicating an eosinophilic granuloma. A lung computed tomography
(CT) scan identified several small nodules in the right lung and an
upper abdominal CT scan detected abnormal lesions in the left liver
and spleen.
The patient subsequently underwent a cervical lymph
node biopsy. All specimens were fixed in 4% buffered formalin at
room temperature (37°C) for 24 h and added to paraffin-embedded
blocks. Sections were cut to a thickness of 4 µm and stained using
hematoxylin and eosin (H&E). Following washing, sections were
soaked in hematoxylin for 5 min at 37°C, washed with water, then
subsequently washed with hydrochloric acid alcohol for 30 sec and
then with water for 5min at 50°C. Finally, sections were stained
with eosin for 2 min at 37°C. EOS, plasma cells and macrophages
were counted in 10 random high power fields of view using a light
microscope at a magnification of ×400). The EOS was identified
following H&E staining. Plasma cells and macrophages were
identified by H&E staining combined with staining for cluster
of differentiation (CD)138 (cat. no. EP201; 1:300) and CD68 (cat.
no. KP1; 1:100), respectively. The number of IgG+ and IgG4+ cells
were counted from 10 different high-power fields of view
(magnification, ×400). The quantification of cells was conducted
manually using 10 high power fields, and 100 cells were counted for
every HPF. The percentage of cells was then calculated.
Histomorphological results were replicated three experienced
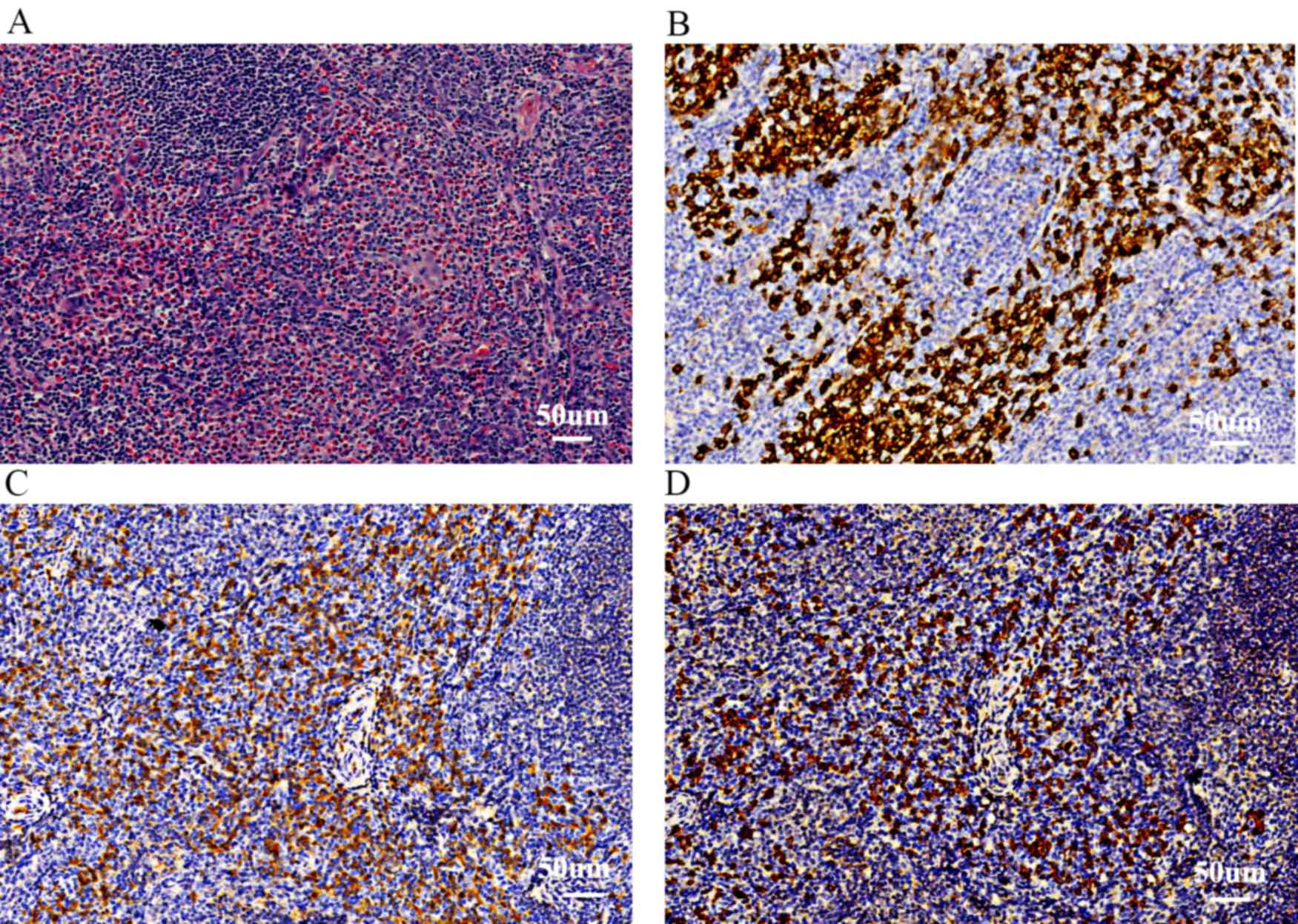
pathologists. The pathological results identified high EOS
infiltration (>80% infiltrating cells) in the lymph nodes
(Fig. 1A). The infiltrate also
contained CD138+ plasma cells (<20% infiltrating cells; Fig. 1B) and a small number of multinucleate
giant cells (<2% infiltrating cells). However, no clear lymph
node fibrosis as detected using H&E, was observed.
Immunohistochemical analysis of the patient's
cervical lymph node was then conducted. Samples were fixed in 4%
paraformaldehyde-0.1 mmol/l phosphate buffer at 4°C for 24h and
then underwent paraffin blocking and were cut to a thickness of 3
µm. Sections were then place in water at 40°C, picked up using
glass slides and dried using an incubator at 37°C for 8 h). Glass
slides were then washed with 100% xylene and rehydrated in
descending alcohol series (100, 95, 90, 80 and 70%) for 10 min
each. Samples were washed using water and 3%
H2O2 for 10 min. Following this, citrate
buffer was added, heated for 3 min until boiled and cooled to room
temperature. This temperature protocol was then repeated. The
citrate buffer was removed and the samples were washed with water
twice and subsequently washed with PBS for 5 min. Samples were then
added to 10% goat serum (cat. no. C-0005; Jiangxi Haoran Bio-Pharma
Co., Ltd., Jiangxi, China; dilution by PBS) for 10 min at 37°C.
Primary antibodies were subsequently added and incubated for 1 h at
37°C. Samples were then washed three times with PBS for 5 min.
Horseradish peroxidase conjugated secondary antibodies were added
(1% BSA-PBS dilution) and incubated for 30 min at 37°C. Samples
were then washed three times with PBS for 5 min. DAB was then
applied for 1 min and observed using a light microscope
(magnification, ×400). Immunohistochemical analysis was performed
using an automated Benchmark XT slide stainer (Ventana Medical
Systems Inc., Tucson, AZ, USA). The following antibodies were used
in this analysis: CD2 (cat. no. UMAB6; 1:100), CD3 (cat. no. SP(F);
1:100), CD5 (cat. no. UMAB9; 1:100), CD20 (cat. no. L26; 1:200),
CD79a (cat. no. SP18; 1:200), CD138 (cat. no. EP201; 1:300), IgG
(cat. no. RA; 1:3,000), IgG4 (cat. no. RA; 1:3,000), Ki-67 (cat.
no. UMAB107; 1:100). All antibodies were purchased from Dako;
Agilent Technologies, Inc. (Santa Clara, CA, USA). Expression
intensity was determined with a scoring system based on the
immunoreactive score (IRS) system proposed by Kay et al
(15) using 5 random fields of view
(magnification, ×200). The point system was based on staining
strength and was as follows: i) 0 points for no staining; ii) 1
point for a light yellow stain; iii) 2 points for medium yellow or
brown appearance without background staining or a deep brown stain
with light brown background and iv) 3 points for a deep brown
appearance without background staining. The percentage of positive
cells was then calculated using 5 random fields of view with a
light microscope (magnification, ×40) and points were allocated
based on their values: i) 0 points for 0% positive cells; ii) 1
point for 0–25% positive cells; iii) 2 points for 25–50% positive
cells; iv) 3 points for 50–75% positive cells; and v) 4 points for
>75% positive cells. The points allocated from staining strength
and the percentage of positive cells were added together. Final
scores of 0, 1–3, 4–5 and 6–7 were deemed negative (−), weak (+),
focal (++) and strong (+++), respectively. Thus, the results of
immunohistochemistry were as follows: CD2 focal (++); CD3 focal
(++); CD5 focal (++); CD20 focal (++); CD79α focal (++); S100 focal
(++); CD1α focal (++); CD68 focal (++); CD13 weak (+); CD34 (−);
myeloperoxidase weak (+); CD138 focal (++); IgG focal (++); IgG4
focal (++); IgG4/IgG=50% (Fig. 1C and
D).
Peripheral blood flow cytometric analysis was then
performed. All analyses were performed according to standard
techniques using a FACS CantoTM II (BD Biosciences, Franklin Lakes,
NJ, USA). The panel of fluorochrome conjugated monoclonal
antibodies presented in Table I
(mAbs; dilution: 1:2) were utilized to diagnose and classify each
cell group, including lymphocytes. A total of 100 µl peripheral
blood was obtained from the patient. The sample was stained with 1
test mAb for each marker (20 µl mAbs conjugated to FITC/PE; 5 µl,
all other mAbs) and then incubated for 20 min at room temperature
in dark. A total of 1 ml of 1×BD FACS Lysing solution (cat. no.
349202; BD Biosciences) was added and cell suspensions were washed
once with PBS. Cells were vortexed at a speed of 850 × g for 5 min
at 37°C. Cells were then washed once with PBS and pellets were
resuspended in 300 µl PBS. A total of 500 µl 1X Permabilizing
Solution 2 (cat. no. GAS002S100; Invitrogen; Thermo fisher
Scientific, Inc.) were added to the cell pellet for the staining of
intracellular antibodies and then incubated for 10 min at room
temperature in the dark. Following incubation, cells were
centrifuged at a speed of 850 × g for 5 min at 37°C. MPO and cCD79a
were subsequently added to the cell pellet and incubated for 20 min
at room temperature in the dark. Samples were then analyzed using a
flow cytometer (FACS CantoTM II; BD Biosciences) with Cell Quest
soft (BD Biosciences; version 5.1) software. The results confirmed
that the rate of EOS was increased (48.9%; normal range,
0.4–8.0%).
 | Table I.Monoclonal antibodies utilized for
the identification of each cell group. |
Table I.
Monoclonal antibodies utilized for
the identification of each cell group.
| Cell | Panel | Conjugate | Catalogue
number |
|---|
| Lymphocytes | HLA-DR | APC-CY7 | 335796 |
|
| CD2 | FITC | 347593 |
|
| CD3 | PE | 347347 |
|
| CD4 | PE-CY7 | 348789 |
|
| CD5 | APC | 340583 |
|
| CD7 | PE | 340581 |
|
| CD8 | APC-CY7 | 348793 |
|
| CD10 | PE-CY7 | 341092 |
|
| CD11b | APC | 301310 |
|
| CD16 | APC-CY7 | 302018 |
|
| CD19 | APC | 652804 |
|
| CD20 | APC-CY7 | 335829 |
|
| CD22 | PE | 347577 |
|
| CD34 | FITC | 348053 |
|
| CD38 | PE-CY7 | 303516 |
|
| CD56 | PE-CY7 | 335791 |
|
| cCD79a | PE | 340579 |
|
| cCD3 | PE | 347347 |
|
| TdT | FITC | 347194 |
|
| sKappa | FITC | 349516 |
|
| sLambda | PE | 349516 |
|
| CD45 | Percp | 347464 |
| Monocyte | HLA-DR | APC-CY7 | 335796 |
|
| CD11b | APC | 301310 |
|
| CD13 | PE | 347837 |
|
| CD14 | APC-CY7 | 333951 |
|
| CD15 | FITC | 332778 |
|
| CD16 | APC-CY7 | 302018 |
|
| CD33 | APC | 340474 |
|
| CD34 | FITC | 348053 |
|
| CD38 | PE-CY7 | 303516 |
|
| CD64 | FITC | 555527 |
|
| CD117 | PE | 340529 |
|
| MPO | FITC | 340580 |
|
| CD45 | Percp | 347464 |
| Granulocyte | HLA-DR | APC-CY7 | 335796 |
|
| CD11b | APC | 301310 |
|
| CD13 | PE | 347837 |
|
| CD15 | FITC | 332778 |
|
| CD16 | APC-CY7 | 302018 |
|
| CD33 | APC | 340474 |
|
| CD34 | FITC | 348053 |
|
| CD38 | PE-CY7 | 303516 |
|
| CD64 | FITC | 555527 |
|
| CD117 | PE | 340529 |
|
| MPO | FITC | 340580 |
|
| CD45 | Percp | 347464 |
An analysis of the patient's genes was then
conducted. For all genes analyzed. RNA was extracted from patient
heparinized blood using an OMEGA Blood RNA Maxi kit (Omega Bio-Tek,
Inc., Norcross, GA, USA). cDNA was constructed via reverse
transcription using random ligomer Primers (6 mer; Takara
Biotechnology Co., Ltd., Dalian, China) and cDNA synthesis was
performed using a PrimeScript™ RT reagent kit (Takara Biotechnology
Co., Ltd.) following the manufacturer's protocol.
Quantitative-polymerase chain reaction (qPCR) was then performed
using a 20 µl system, containing 16 µl pre-mixed solution [10 µl
Promega GoTaq Green Mix (Promega Corporation, Madison, WI, USA) and
6 µl double distilled (dd)H2O], 2 µl of specific primers
(all, Invitrogen; Thermo Fisher Scientific, Inc.) and 2 µl cDNA,
positive control samples, negative control samples or
ddH2O. The primers utilized for each gene and the
thermocycling conditions of PCR are presented in Tables II and III respectively. PCR products were
separated by electrophoresis using 2% agarose gel. Products were
analyzed using the Bio-Rad ChemiDoc XRS+ Gene Genius Bio-imaging
system (Bio-Rad Laboratories, Inc., Hercules, CA, USA). PCR
products were subsequently separated by electrophoresis using 2%
agarose gel. Products were analyzed using the Bio-Rad ChemiDoc XRS+
Gene Genius Bio-imaging system (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). The appearance of specific amplification strips
relevant to each gene indicated a positive result. These were as
follows: i) BCR/ABL: b3a2, 360 bp, b2a2 and 285 bp; ii) JAK2-V617F:
203 bp and 364 bp; iii) FIP1L1/PDGFRA: 700 bp; iv) ETV6/PDGFRB: 700
bp.
| Table II.Forward and reverse primers of each
gene utilized in PCR. |
Table II.
Forward and reverse primers of each
gene utilized in PCR.
| Gene | First primer | Reverse primer |
|---|
| BCR/ABL |
5′-TCCGCTGACCATCAATAAGGA-3′ |
5′-CACTCAGACCCTGAGGCTCAA-3′ |
| JAK2-V617F |
5′-TGCTGAAAGTAGGAGAAAGTGCAT-3′ |
5′-TCCTACAGTGTTTTCAGTTTCAA-3′ |
| FIP1L1/PDGFRA |
|
|
| First
PCR | FIP1L1-F1,
5′-ACCTGGTGCTGATCTTTCTGAT-3′ | PDGFRA-R1,
5′-TGAGAGCTTGTTTTTCACTGGA-3′ |
| Second
PCR | FIP1L1-F2,
5′-AAAGAGGATACGAATGGGACTTG-3′ | PDGFRA-R2,
5′-GGGACCGGCTTAATCCATAG-3′ |
| ETV6/PDGFRB |
|
|
| First
PCR | ETV6-3F,
5′-CTGCTGACCAAAGAGGACTT-3′ |
PD-C,5′-TGGCTTCTTCTGCCAAAGCA-3′ |
| Second
PCR | ETV6-J,
5′-TTCACCATTCTTCCACCCTGGA-3′ | PD-J,
5′-GGAGATGATGGTGAGCACCA-3′ |
| Table III.Thermocycling conditions for each
gene utilized in PCR. |
Table III.
Thermocycling conditions for each
gene utilized in PCR.
| Gene | Condition |
|---|
| BCR/ABL | 10 min at 94°C,
followed by 33 cycles of 30 sec at 94°C, 30 sec at 57°C, 45 sec at
72°C and 10 min at 72°C |
| JAK2-V617F | 5 min at 95°C for 1
cycle, followed by 35 cycles of 30 sec at 95°C, 30 sec at 57°C, 1
min at 72°C and 7 min at 72°C for 1 cycle |
| FIP1L1/PDGFRA |
|
| First
PCR | 10 min at 95°C,
followed by 33 cycles of 45 sec at 95°C, 45 sec at 57°C, 75 sec at
72°C and 5 min at 72°C |
| Second
PCR | 10 min at 95°C,
followed by 25 cycles of 30 sec at 94°C, 30 sec at 60°C, 60 sec at
72°C and 5 min at 72°C |
| ETV6/PDGFRB |
|
| First
PCR | 10 min at 95°C,
followed by 28 cycles of 30 sec at 94°C, 45 sec at 60°C, 75 sec at
72°C and 5 min at 72°C |
| Second
PCR | 10 min at 95°C,
followed by 25 cycles of 30 sec at 94°C, 45 sec at 64°C, 60 sec at
72°C and 5 min at 72°C. |
The results of the gene analysis were negative for
janus kinase-V617F, breakpoint cluster region protein/abelson
murine leukemia viral oncogene homolog 1 fusion,
PDGFRA/retinoblastoma protein and ETV6/PDGFRB.
A two-color fluorescence in situ
hybridization (FISH) was then performed for the detection of a
CHIC2 deletion, acting as a surrogate marker for the FIP1L1-PDGFRA
fusion gene at 4q12. Patient peripheral blood was sampled and 24010
(telomeric of PDGFRA), 3H20 (between FIP1L1 and PDGFRA) and
Bacterial artificial chromosome (BAC) clones 120K16 (centromeric of
FIP1L1) were used as probes (Abbott, 05N52-020). For PDGFRB and
FGFR1, the probe was from Abbott (06N24-010) and CYTOTEST
(CT-PACO56-10-GO) respectively. FISH data were collected using an
Olympus fluorescence microscope (Provis, Olympus Corporation,
Tokyo, Japan) and analyzed using ASI-fishview expo 6.0 analyze
system software (Applied Spectral Imaging, Inc., Carlsbad, CA,
USA). The results demonstrated that there was a negative expression
of PDGFRA/RB- and fibroblast growth factor receptor 1.
Cytokine measurements were then obtained from the
patient. IL-1β (cat. no. KT98060), IL-6 (cat. no. kt34251), IL-8
(cat. no. KT98090), IL-10 (cat no. KT98105), TNF-α (cat. no.
KT98069) were measured (all kits were sourced from Bender
MedSystems, Burlingame, CA, USA) using a Siemens
Immulite® 1000 (Siemens Healthineers, Erlangen,
Germany). The results identified increased levels of interleukin
(IL)-6 (38.3 pg/ml; normal range 0–3.4 pg/ml), tumor necrosis
factor-α (TNF-α; 26.3 pg/ml; normal range 0–8.1 pg/ml), IL-1β (27.7
pg/ml; normal range 0–5 pg/ml) and IL-8 (620 pg/ml; normal range
0–62 pg/ml). IL-10 levels were <5 pg/dl (normal range 0–9.1
pg/dl). The peripheral blood flow cytometric analysis also detected
CD19 low CD38+CD20-CD27+ plasmablasts at 4,110/ml (positive
predictive value, >2,000/ml). Following therapy with 20 mg
prednisone per day for one week, blood tests were performed twice a
week at the hospital until the patient's EOS count was reduced to
5.3×109/l. Following this, the patient was discharged.
There after, prednisone treatment was gradually decreased and
stopped after 6 months. This consisted of 20 mg for first 2 weeks,
15 mg for the following month, 10 mg for the months after that and
2.5 mg per month for the remainder of the treatment period. The
clinical course and concomitant EOS count, IgG4 levels and response
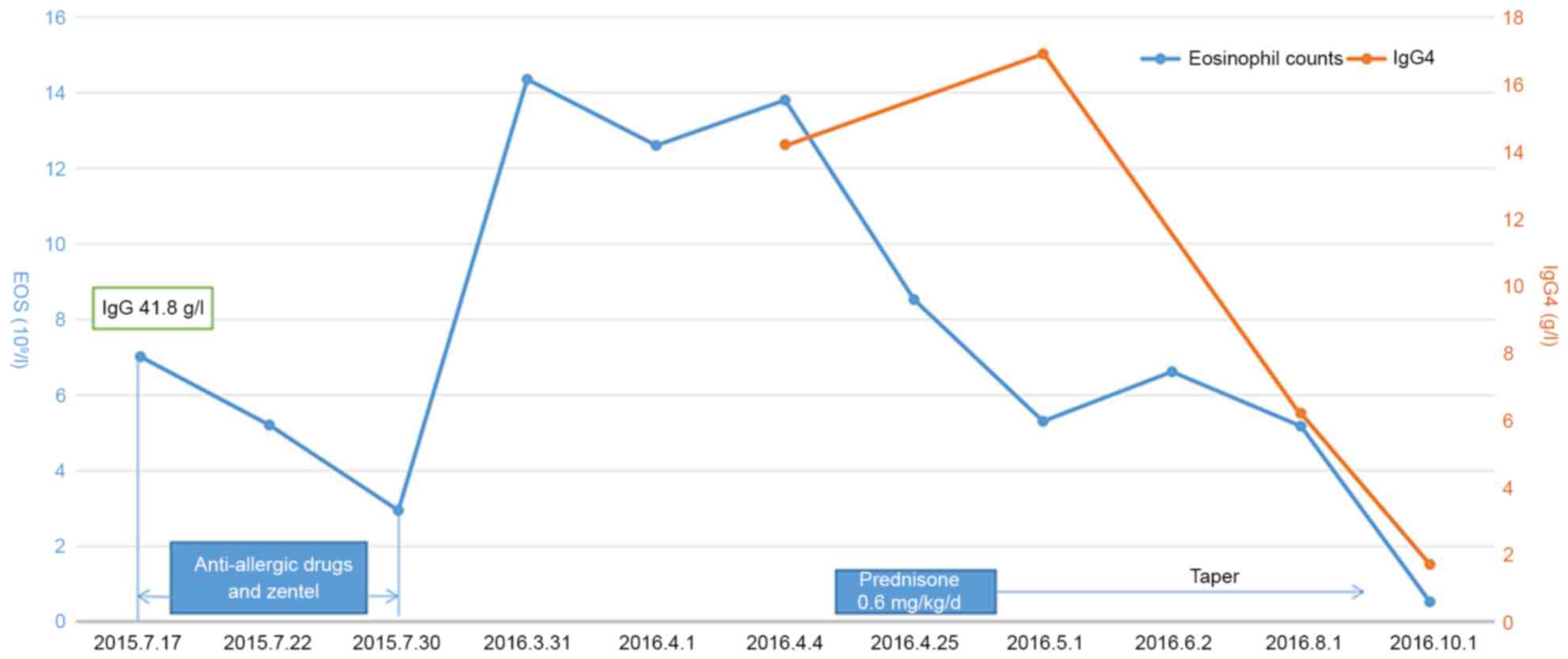
to prednisone therapy are demonstrated in Fig. 2. Additionally, a comparison of
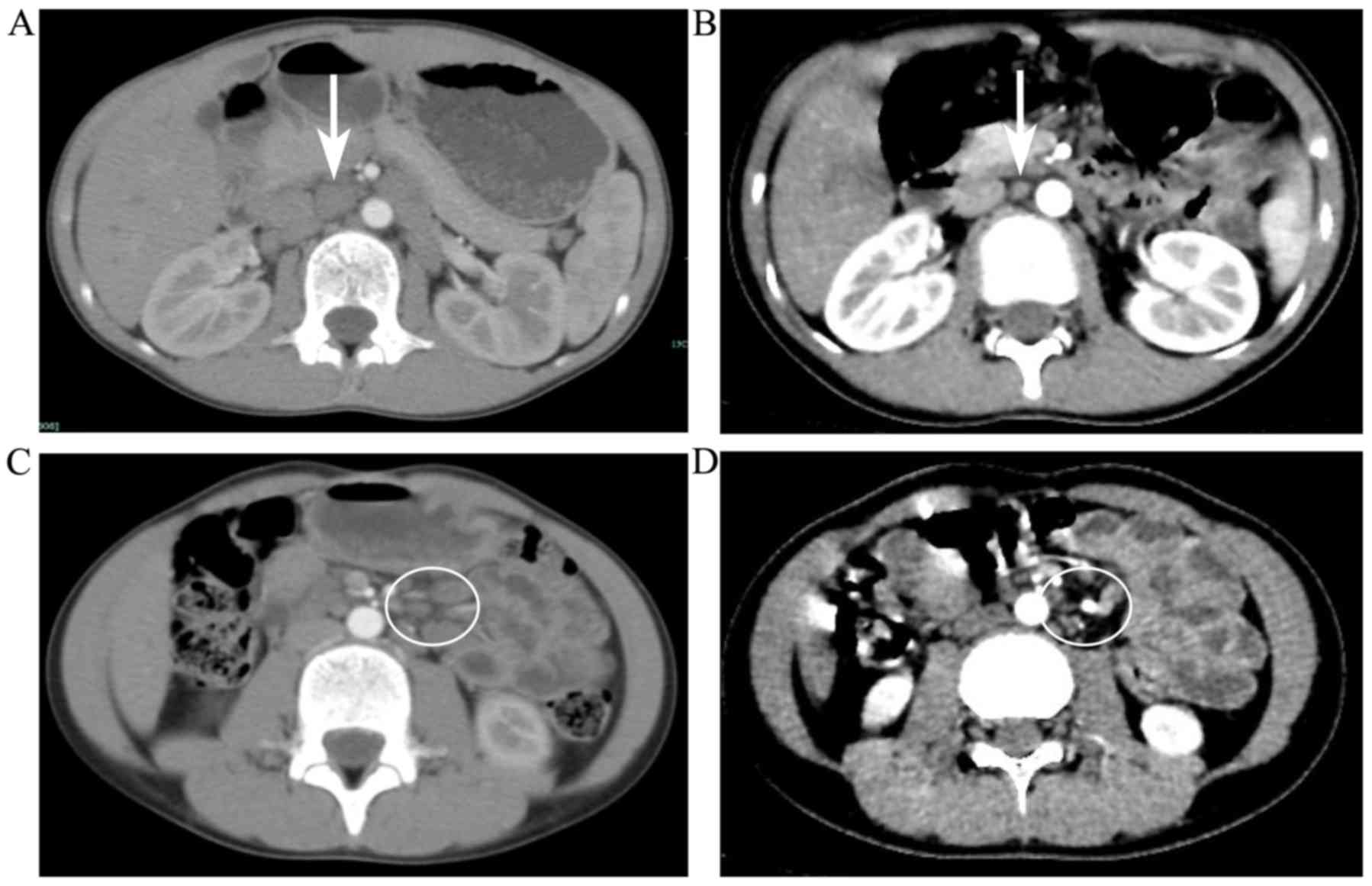
abdominal CT scans obtained pre- and post-treatment is depicted in
Fig. 3. A clinical follow-up was
conducted every week during the first 2 months, which included
routine blood analysis, routine blood biochemical tests, IgG and
IgG4 tests and an abdominal and superficial lymph node ultrasound.
An additional follow-up was conducted twice a month during the
period of prednisone treatment, which consisted of the same tests
conducted in the first 2 months, with the addition of an abdominal
CT scan. The patient then received a follow-up every 3 months,
which included a systemic examination, blood tests and
imageological examination if necessary. The last follow-up was
conducted in July 2017 and the boy remains asymptomatic.
Discussion
The patient initially visited Hangzhou First
People's Hospital due to increased levels of EOS and leukocytosis.
An allergen test identified an allergy to dust mites. Despite the
decrease in EOS count following desensitization therapy, it is
unlikely that the patient's condition was solely due to a dust mite
allergy that lacked systemic symptoms such as a cough, skin rash or
other related reactions. Recurrence of increased EOS count was
observed, along with multiple organ infiltration of EOS', which
could not be explained by an allergic reaction. The possible
diagnoses of clonal eosinophilia and lymphatic variation
eosinophilia were excluded according to the results of peripheral
blood flow cytometry, gene analysis and FISH. The patient was also
treated with antiparasitics but treatment was not effective. Thus,
secondary eosinophilia caused by parasites or drugs was excluded. A
lymph node biopsy identified infiltration of EOS, which indicated a
diagnosis of HES. However, the patient exhibited high levels of
IgG4 and imaging examinations, including those of the
retroperitoneal and whole body lymph nodes and thalamic region,
indicated a diagnosis of IgG4-RD, which required further
differentiation from HES secondary to IgG4-RD.
According to the diagnostic criteria for IgG4-RD
proposed by the Japanese G4 team in 2012 (16) (Fig.
4), a consensus has been reached on two essential diagnostic
criteria: i) serum IgG4 concentration >135 mg/dl and ii)
IgG4+/IgG+ plasma cells >40% and >10 IgG4+ cells per
high-power field of biopsy samples. Storiform fibrosis and
obliterative phlebitis may be clearly identified in pancreatic,
biliary tract and retroperitoneal lesions, but are rarely observed
in the salivary glands or lymph nodes. The patient was highly
suspected to have IgG4-related lymphadenopathy, due to the presence
of enlarged lymph nodes and results of biopsy.
IgG4-related lymphadenopathy is characterized by
diffuse lymphoplasmacytic infiltration, an increased percentage of
IgG4 (+) plasma cells and fibrosis and should be tested for
reactive lymphoid hyperplasia, Castleman's disease, inflammatory
pseudotumor and angioimmunoblastic T cell lymphoma (17). Furthermore, IgG4-related
lymphadenopathy may be characterized into five histological
subtypes: Multicentric Castleman's disease-like, reactive
follicular hyperplasia-like, reactive follicular hyperplasia,
progressively transformed germinal center-type and inflammatory
pseudotumor-like IgG4-related lymphadenopathy (18). The first two subtypes are
characterized by systemic lymphadenopathy, while the last three
subtypes primarily involve local lymph nodes (19). Based on the infiltrative patterns of
IgG4-positive cells, IgG4-related lymphadenopathy can also be
divided into interfollicular plasmacytosis and germinal center
types (20). Additionally, the
diagnosis of IgG4-related lymphadenopathy requires awareness and a
high index of suspicion regarding unexplained lymphadenopathy with
scattered EOSs and numerous plasma cells (20). In the present case, histological
results were not specific to IgG4-related lymphadenopathy due to a
lack of fibrosis. However, the abundant infiltration of plasma
cells and EOS prompted a careful diagnosis. Immunohistochemical
studies are also important in the pathological diagnosis of IgG4-RD
(21). In the present case, the
ratio of IgG4/IgG was increased to 50%, which supports the
diagnosis of IgG4-RD. Blood plasmablast concentrations may be more
meaningful than serum IgG4 levels for the diagnosis of IgG4-RD
(14). Additionally, a study by
Wallace et al (22) observed
that patients with IgG4-RD exhibited markedly increased total
plasmablast counts (median: 4,698/ml; range: 610–79,524/ml), which
was also evaluated in the current patient (4,110/ml, positive
predictive value: >2,000/ml), thus verifying that plasmablast
counts are a potentially useful biomarker for the diagnosis of
IgG4-RD. Considering these previous findings and the present
clinical data, it is probable that the patient of the present study
had IgG4-RD.
The high level of IL-6 also caused difficulty in the
diagnosis of the patient, as this is an indicator of Castleman's
disease. However, he did not present other associated symptoms,
including hypoproteinemia, hypocholesterolemia, anemia and
increased C-reactive protein (CRP) levels (23), which did not support the diagnosis of
Castleman's disease. IL-6 is a type of a multifunctional cytokine
that may regulate the immune and stress responses, haematopoietic
function and bone metabolism (23).
Masaki et al (24) reported
that serum IL-6 was an important tool for differential diagnosis
between Castleman's disease and IgG4-RD. However, in a study by
Sato et al (25) of 9
patients with IgG4-RD, two patients exhibited slightly increased
levels IL-6; however lacked increased CRP levels and were
responsive to hormone therapy. Another study by Yokoyama (26) documented that increased IL-6 levels
(>25 pg/ml) were present in 7% of healthy subjects, which
indicated that levels of IL-6 may vary among individuals (25). Furthermore, it is difficult to
identify IgG4-RD and Castleman's disease when the IL-6 titer may
undergo random fluctuations (27).
Nakamura et al (27) reported a patient with increased IL-6
levels of up to 83.5 pg/ml and complications of IgG4-RD. They also
proposed an improved diagnostic criteria for IgG4-RD (Fig. 5), which was based on a study
performed by Umehara et al (16). The present case was highly suspected
to be IgG4-RD according to the modified criteria.
IgG4-RD is typically observed in older patients,
with a median age of onset of 50 years old (28), though a number of studies have
reported cases in children (29–51).
While no retrospective study has been reported to date, Karim et
al (52) recently reviewed
published data and performed a systematic literature search of
IgG4-RD in pediatrics. The median age of onset was 13 years (3 to
17 years old) and 64% of patients were girls. IgG4-RD in children
is potentially the same as in adults, primarily affecting the
orbit, lymph nodes, salivary tract and pancreas (52,53). The
percentage of pediatric patients with elevated serum IgG4 levels
was 70%, which is similar to that in adult patients (30–50%)
(54). However, the positive
predictive value of serum IgG4 is limited. The present diagnosis
also required histological observations, assessment of clinical
symptoms and serological and radiological observations. The first
choice therapy for pediatric IgG4-RD is prednisone, however, no
consensus on the dosage has been reached. For cases not responding
to prednisone, disease-modifying antirheumatic drugs, including
mycophenolate mofetil, azathioprine and methotrexate have
demonstrated efficacy in some patients (38,45,47).
Idiopathic HES is rare in children and a limited
number of cases have been reported (55–57). The
true incidence is unknown and data on the efficacy of treatment
regarding the response and survival of patients is also
insufficient (55). Pediatric HES
has been reported to have a slight male prevalence (56), though a recent study by Williams
et al (57) observed that the
female/male occurrence ratio was increased to 23/9 (57). Additionally, they documented that
children with HES typically exhibit higher peak EOS counts than
adults. Presentation of HES is variable, though fever, arthralgias
and rash were common presentation symptoms (57). In pediatric idiopathic HES, pulmonary
and cardiac involvement may be the most common clinical
manifestations (55). However, in
the current patient, there was no specific clinical presentation
except painless and enlarged lymphoma.
Treatment strategies are based on the disease
severity. Prevention of organ dysfunction and reductions in EOS
count are priority and corticosteroids are recommended as the
first-line therapy for HES (5). To
the best of our knowledge, no published data has demonstrated a
relationship between HES and IgG4-RD in children, whereas several
studies (58–61) have described a potential correlation
between these two illnesses in adults.
Previous studies have reported that an immune
reaction mediated by T helper (Th) 2 cells is predominant in
IgG4-RD (62), which may contribute
to disease pathogenesis (63,64).
However, Th2 memory cells have not been detected in a large number
of individuals with severe IgG4-RD, despite the patients didn't
having undergone immunosuppressive therapy (62). Therefore, a Th2 immune response in
IgG4-RD may not represent a fundamental feature of IgG4-RD but
rather a potential phenomenon or process (62). Additionally, cytokines IL-4, IL-5 and
IL-13 that are secreted by Th2 cells may induce the transition
between IgG and IgE and promote the release of PBEs (65). A recent study evaluated a large
cohort of IgG4-RD patients exhibiting multiple organ involvement
and indicated that processes inherent to IgG4-RD itself rather than
atopy may contribute to eosinophilia and IgE elevation (66). The median count of EOS in IgG4-RD was
641 cells/µl (ranging between 40 to 1,500 cells/µl) (66), which differs to that of the current
patient (2,940–12,600 cells/µl). Thus, combined with the
pathological observations and clinical features, a diagnosis of
IHES was more probable.
Hypereosinophilia and high levels of IgE may cause
T-reg cells to secrete IL-10 and tumor growth factor β and increase
the serum titer of IgG4 (67). IL-10
is often overexpressed in IgG4-RD (68). Furthermore, hypereosinophilia and
IgG4-RD can occur at the same time (58). In the current patient, increased
levels of EOS and serum IgG4 were concurrent with a low titer of
IL-10, which indicated that elevated IgG4 was not simply due to
HES. To date, the correlation between HES and IgG4-RD remains
unclear (14). A study by Tabata
et al (69) reviewed 554
IgG4-RD patients who had their IgG4 levels measured and 2/16
patients with HES exhibited an increased serum titer of IgG4.
Although the author did not confirm the diagnosis of IgG4-RD in the
two patients, one of them was sensitive to hormone therapy.
Existing data on the concurrence of HES with IgG4-RD
is rare. Nagao et al (59)
reported a patient with lymphoproliferative variant HES who was
highly suspected to have IgG4-RD, which was combined with an
increased titer of TNF-α and IL-10. Aoyama et al (60) also reported a case of HES-related
acute hepatitis caused by HES and associated with elevated serum
IgG4 levels, which is similar to the results reported by Nagamura
et al (58). They concluded
that HES associated with an elevated concentration of serum IgG4 is
difficult to differentiate from IgG4-RD and a careful diagnosis
based on clinical symptoms and pathological observations is
required which is also agreed by Aoyama et al (60). Clayton et al (61) hypothesized that in adults,
eosinophilic esophagitis may be mediated by IgG4 and not an
IgE-induced allergy. Existing data on concurrent HES and IgG4-RD
lacks full support of the pathology. These data indicate that the
association between HES and IgG4-RD remains unclear and further
studies are required to focus on the mechanism between these two
illnesses.
In conclusion, the present study described a case of
HES with highly suspected IgG4-RD. However, due to insufficient
pathological observations, a definitive pathological diagnosis of
IgG4-RD could not be made. Nevertheless, the clinical presentation,
immunohistochemical observations, abundant peripheral blood
plasmablast and response to prednisone therapy confirmed the
diagnosis. As IgG4-RD typically affects middle-aged and elderly
males, published cases of IgG4-RD in children are extremely rare.
Therefore, the present case serves as a reminder that IgG4-RD may
occur in children and clinical doctors should not neglect the
possibility of IgG4-RD in children.
Glossary
Abbreviations
Abbreviations:
|
IgG4
|
immunoglobulin G4
|
|
IgG4-RD
|
immunoglobulin G4-related disease
|
|
HES
|
hypereosinophilic syndrome
|
|
IHES
|
idiopathic hypereosinophilic
syndrome
|
|
EOS
|
eosinophils
|
|
IL
|
interleukin
|
|
TNF
|
tumor necrosis factor
|
|
CRP
|
C-reactive protein
|
|
FISH
|
fluorescent in situ
hybridization
|
|
CT
|
computed tomography
|
|
MRI
|
magnetic resonance imaging
|
|
AIP
|
autoimmune pancreatitis
|
|
MCD
|
multiple Castleman's disease
|
References
|
1
|
Tefferi A, Gotlib J and Pardanani A:
Hypereosinophilic syndrome and clonal eosinophilia: Point-of-care
diagnostic algorithm and treatment update. Mayo Clinic Proc. 85:pp.
158–164. 2010; View Article : Google Scholar
|
|
2
|
Crane MM, Chang CM, Kobayashi MG and
Weller PF: Incidence of myeloproliferative hypereosinophilic
syndrome in the United States and an estimate of all
hypereosinophilic syndrome incidence. J Allergy Clin Immunol.
126:179–181. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tefferi A, Patnaik MM and Pardanani A:
Eosinophilia: Secondary, clonal and idiopathic. Br J Haematol.
133:468–492. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ganeva M, Gancheva T, Lazarova R, Troeva
J, Baldaranov I, Vassilev I, Hristakieva E and Tzaneva V:
Carbamazepine-induced drug reaction with eosinophilia and systemic
symptoms (DRESS) syndrome: Report of four cases and brief review.
Int J Dermatol. 47:853–860. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gotlib J: World Health
Organization-defined eosinophilic disorders: 2014 update on
diagnosis, risk stratification, and management. Am J Hematol.
89:325–337. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Vardiman JW: The World Health Organization
(WHO) classification of tumors of the hematopoietic and lymphoid
tissues: An overview with emphasis on the myeloid neoplasms. Chem
Biol Interact. 184:16–20. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Schaller JL and Burkland GA: Case report:
Rapid and complete control of idiopathic hypereosinophilia with
imatinib mesylate. MedGenMed. 3:92001.PubMed/NCBI
|
|
8
|
Cheah CY, Burbury K, Apperley JF, Huguet
F, Pitini V, Gardembas M, Ross DM, Forrest D, Genet P, Rousselot P,
et al: Patients with myeloid malignancies bearing PDGFRB fusion
genes achieve durable long-term remissions with imatinib. Blood.
123:3574–3577. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ogbogu PU, Bochner BS, Butterfield JH,
Gleich GJ, Huss-Marp J, Kahn JE, Leiferman KM, Nutman TB, Pfab F,
Ring J, et al: Hypereosinophilic syndromes: A multicenter,
retrospective analysis of clinical characteristics and response to
therapy. J Allergy Clin Immunol. 124:1319–1325.e3. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fauci AS, Harley JB, Roberts WC, Ferrans
VJ, Gralnick HR and Bjornson BH: NIH conference. The idiopathic
hypereosinophilic syndrome. Clinical, pathophysiologic, and
therapeutic considerations. Ann Intern Med. 97:78–92. 1982.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Quiquandon I, Claisse JF, Capiod JC,
Delobel J and Prin L: alpha-Interferon and hypereosinophilic
syndrome with trisomy 8: Karyotypic remission. Blood. 85:2284–2285.
1995.PubMed/NCBI
|
|
12
|
Cofrancesco E, Cortellaro M, Pogliani E,
Boschetti C, Salvatore M and Polli EE: Response to vincristine
treatment in a case of idiopathic hypereosinophilic syndrome with
multiple clinical manifestations. Acta Haematol. 72:21–25. 1984.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Umehara H, Okazaki K, Masaki Y, Kawano M,
Yamamoto M, Saeki T, Matsui S, Sumida T, Mimori T, Tanaka Y, et al:
A novel clinical entity, IgG4-related disease (IgG4RD): General
concept and details. Mod Rheumatol. 22:1–14. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Stone JH, Brito-Zerón P, Bosch X and
Ramos-Casals M: Diagnostic approach to the complexity of
IgG4-related disease. Mayo Clin Proc. 90:pp. 927–939. 2015;
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Friedrichs K, Gluba S, Eidtmann H and
Jonat W: Overexpression of p53 and prognosis in breast cancer.
Cancer. 72:3641–3647. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Umehara H, Okazaki K, Masaki Y, Kawano M,
Yamamoto M, Saeki T, Matsui S, Yoshino T, Nakamura S, Kawa S, et
al: Comprehensive diagnostic criteria for IgG4-related disease
(IgG4-RD), 2011. Mod Rheumatol. 22:21–30. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jingzhi S, Lifang Z and Meiyun F:
IgG4-related lymphadenopathy: A case report. Zhong hua shi yong nei
ke za zhi. 08:721–723. 2015.
|
|
18
|
Sato Y and Yoshino T: IgG4-related
lymphadenopathy. Int J Rheumatol. 2012:5725392012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sato Y, Kojima M, Takata K, Morito T,
Mizobuchi K, Tanaka T, Inoue D, Shiomi H, Iwao H and Yoshino T:
Multicentric Castleman's disease with abundant IgG4-positive cells:
A clinical and pathological analysis of six cases. J Clin Pathol.
63:1084–1089. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sato Y, Notohara K, Kojima M, Takata K,
Masaki Y and Yoshino T: IgG4-related disease: Historical overview
and pathology of hematological disorders. Pathol Int. 60:247–258.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Stone JH, Zen Y and Deshpande V:
IgG4-related disease. N Engl J Med. 366:539–551. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wallace ZS, Mattoo H, Carruthers M,
Mahajan VS, Della Torre E, Lee H, Kulikova M, Deshpande V, Pillai S
and Stone JH: Plasmablasts as a biomarker for IgG4-related disease,
independent of serum IgG4 concentrations. Ann Rheum Dis.
74:190–195. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Frizzera G, Peterson BA, Bayrd ED and
Goldman A: A systemic lymphoproliferative disorder with morphologic
features of Castleman's disease: Clinical findings and
clinicopathologic correlations in 15 patients. J Clin Oncol.
3:1202–1216. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Masaki Y, Dong L, Kurose N, Kitagawa K,
Morikawa Y, Yamamoto M, Takahashi H, Shinomura Y, Imai K, Saeki T,
et al: Proposal for a new clinical entity, IgG4-positive multiorgan
lymphoproliferative syndrome: Analysis of 64 cases of IgG4-related
disorders. Ann Rheum Dis. 68:1310–1315. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sato Y, Kojima MK, Takata K, Morito T,
Asaoku H, Takeuchi T, Mizobuchi K, Fujihara M, Kuraoka K, Nakai T,
et al: Systemic IgG4-related lymphadenopathy: A clinical and
pathologic comparison to multicentric Castleman's disease. Mod
Pathol. 22:589–599. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yokoyama A: Interleukin-6 (IL-6)/soluble
IL-6 receptor. Nippon Rinsho Japanese J Clin Med. 63 Suppl
8:S72–S74. 2005.
|
|
27
|
Nakamura M, Iwamoto O, Chino T, Todoroki K
and Kusukawa J: Diagnostic dilemma of IgG4-related primary
localized cervical lymphadenopathy associated with aberrant IL-6
expression level. Diagn Pathol. 11:432016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kamisawa T, Zen Y, Pillai S and Stone JH:
IgG4-related disease. Lancet. 385:1460–1471. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bain BJ, Gilliland DG, Vardiman JW and
Horny HP: Chronic eosinophilic leukaemia, not otherwise specified.
2008.
|
|
30
|
Batu ED, Arici ZS, Orhan D, Kiratli H and
Özen S: Immunoglobulin G4-related orbital disease: Report of two
pediatric cases. Clin Exp Rheum. 33:409–410. 2015.
|
|
31
|
Caso F, Fiocco U, Costa L, Sfriso P, Punzi
L and Doria A: Successful use of rituximab in a young patient with
immunoglobulin G4-related disease and refractory scleritis. Joint
Bone Spine. 81:190–192. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Corujeira S, Ferraz C, Nunes T, Fonseca E
and Vaz LG: Severe IgG4-related disease in a young child: A
diagnosis challenge. Case Rep Pediatr. 2015:1407532015.PubMed/NCBI
|
|
33
|
Gillispie MC, Thomas RD and Hennon TR:
Successful treatment of IgG-4 related sclerosing disease with
rituximab: A novel case report. Clin Exp Rheumatol. 33:549–550.
2015.PubMed/NCBI
|
|
34
|
Griepentrog GJ, Vickers RW, Karesh JW,
Azari AA, Albert DM and Bukat CN: A clinicopathologic case study of
two patients with pediatric orbital IgG4-related disease. Orbit.
32:389–391. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hasosah MY, Satti MB, Yousef YA, Alzahrani
DM, Almutairi SA, Alsahafi AF, Sukkar GA and Alzaben AA:
IgG4-related sclerosing mesenteritis in a 7-year-old Saudi girl.
Saudi J Gastroenterol. 20:385–388. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Jariwala MP, Agarwal M, Mulay K and
Sawhney S: IgG4-related orbital inflammation presenting as
unilateral pseudotumor. Indian J Pediatr. 81:1108–1110. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kalapesi FB, Garrott HM, Moldovan C,
Williams M, Ramanan A and Herbert HM: IgG4 orbital inflammation in
a 5-year-old child presenting as an orbital mass. Orbit.
32:137–140. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Mannion M and Cron RQ: Successful
treatment of pediatric IgG4 related systemic disease with
mycophenolate mofetil: Case report and a review of the pediatric
autoimmune pancreatitis literature. Pediatr Rheum Online J.
9:12011. View Article : Google Scholar
|
|
39
|
Miglani RK, Murthy D, Bhat R and Kumar AK:
Immunoglobulin G4-associated cholangitis mimicking
cholangiocarcinoma in a young boy. J Postgrad Med. 56:140–142.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Mittal R, Ganguly A, Rath S, Das B and
Mishra A: IgG4-related orbital inflammation presenting as bilateral
proptosis in a child. Eye (Lond). 28:1264–1266. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Nada R, Gupta A, Kang M, Rawat A, Sood A,
Ahluwalia J and Singh S: Hepatic mass and coagulopathy in a
ten-year-old boy with fever. Arthritis Rheumatol. 67:19772015.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Naghibi M, Ahmed Aal Badri AM, Bateman AC,
Shepherd HA and Gordon JN: The successful treatment of
IgG4-positive colitis with adalimumab in a patient with
IgG4-related sclerosing disease-a new subtype of aggressive
colitis? J Crohns Colitis. 7:e81–e84. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Pasic S, Ristic G and Djuricic S:
PReS-FINAL-2276: IgG4 related disease in a 10-year-old girl.
Pediatr Rheumatol Online J. 11 Suppl 2:P2662013. View Article : Google Scholar
|
|
44
|
Prabhu SM, Yadav V, Irodi A, Mani S and
Varghese AM: IgG4-related disease with sinonasal involvement: A
case series. Indian J Radiol Imaging. 24:117–120. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Sane M, Chelnis J, Kozielski R and
Fasiuddin A: Immunoglobulin G4-related sclerosing disease with
orbital inflammation in a 12-year-old girl. J AAPOS. 17:548–550.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zakeri H and Kashi Z: Variable clinical
presentations of Riedel's thyroiditis: Report of two cases. Case
Rep Med. 2011:7092642011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ibrahim SH, Zhang L and Freese DK: A
3-year-old with immunoglobulin G4-associated cholangitis. J Pediatr
Gastroenterol Nutr. 53:109–111. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Melo JC, Kitsko D and Reyes-Múgica M:
Pediatric chronic sclerosing sialadenitis: Küttner tumor. Pediatr
Dev Pathol. 15:165–169. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Notz G, Intili A and Bilyk JR:
IgG4-related dacryoadenitis in a 13-year-old girl. Ophthal Plast
Reconstr Surg. 30:e161–e163. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Pifferi M, Di Cicco M, Bush A, Caramella
D, Chilosi M and Boner AL: Uncommon pulmonary presentation of
IgG4-related disease in a 15-year-old boy. Chest. 144:669–671.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Rosen D, Thung S, Sheflin-Findling S, Lai
J, Rosen A, Arnon R and Chu J: IgG4-sclerosing cholangitis in a
pediatric patient. Semin Liver Dis. 35:89–94. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Karim F, Loeffen J, Bramer W, Westenberg
L, Verdijk R, van Hagen M and van Laar J: IgG4-related disease: A
systematic review of this unrecognized disease in pediatrics.
Pediatr Rheumatol Online J. 14:182016. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Islam AD, Selmi C, Datta-Mitra A, Sonu R,
Chen M, Gershwin ME and Raychaudhuri SP: The changing faces of
IgG4-related disease: Clinical manifestations and pathogenesis.
Autoimmun Rev. 14:914–922. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Pieringer H, Parzer I, Wöhrer A, Reis P,
Oppl B and Zwerina J: IgG4-related disease: An orphan disease with
many faces. Orphanet J Rare Dis. 9:1102014. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Tavil B, Aytaç S, Unal S, Kuskonmaz B,
Gumruk F and Cetin M: Hypereosinophilic syndrome: Hacettepe
experience. J Pediatr Hematol Oncol. 38:539–543. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Alfaham MA, Ferguson SD, Sihra B, et al:
The idiopathic hypereosinophilic syndrome. Arch Dermatol.
132:583–585. 2001.
|
|
57
|
Williams K, Ware JA, Abiodun A,
Holland-Thomas NC, Khoury P and Klion AD: Hypereosinophilia in
children and adults: A retrospective comparison. J Allergy Clin
Immunol Pract. 4:941–947.e1. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Nagamura N, Ueno S, Fujishiro H and Oonuma
H: Hepatitis associated with hypereosinophilia suspected to be
caused by HES that also presented with the pathological features of
IgG4-related disease. Intern Med. 53:145–149. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Nagao Y, Yamanaka H and Harada H: A
patient with hypereosinophilic syndrome that manifested with
acquired hemophilia and elevated IgG4: A case report. J Med Case
Rep. 6:632012. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Aoyama T, Matsumoto T, Uchiyama A, Kon K,
Yamashina S, Suzuki S, Ikejima K, Yao T, Kuwatsuru R and Watanabe
S: Recurrent severe acute hepatitis caused by hypereosinophilic
syndrome associated with elevated serum immunoglobulin G4 levels.
Clin J Gastroenterol. 7:516–522. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Clayton F, Fang JC, Gleich GJ, Lucendo AJ,
Olalla JM, Vinson LA, Lowichik A, Chen X, Emerson L, Cox K, et al:
Eosinophilic esophagitis in adults is associated with IgG4 and not
mediated by IgE. Gastroenterology. 147:602–609. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Mattoo H, Della-Torre E, Mahajan VS, Stone
JH and Pillai S: Circulating Th2 memory cells in IgG4-related
disease are restricted to a defined subset of subjects with atopy.
Allergy. 69:399–402. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Zen Y, Fujii T, Harada K, Kawano M, Yamada
K, Takahira M and Nakanuma Y: Th2 and regulatory immune reactions
are increased in immunoglobin G4-related sclerosing pancreatitis
and cholangitis. Hepatology. 45:1538–1546. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Tanaka A, Moriyama M, Nakashima H, Miyake
K, Hayashida JN, Maehara T, Shinozaki S, Kubo Y and Nakamura S: Th2
and regulatory immune reactions contribute to IgG4 production and
the initiation of Mikulicz disease. Arthr Rheum. 64:254–263. 2012.
View Article : Google Scholar
|
|
65
|
Okazaki K, Uchida K, Ohana M, Nakase H,
Uose S, Inai M, Matsushima Y, Katamura K, Ohmori K and Chiba T:
Autoimmune-related pancreatitis is associated with autoantibodies
and a Th1/Th2-type cellular immune response. Gastroenterology.
118:573–581. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Della Torre E, Mattoo H, Mahajan VS,
Carruthers M, Pillai S and Stone JH: Prevalence of atopy,
eosinophilia, and IgE elevation in IgG4-related disease. Allergy.
69:269–272. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Tsuboi H, Matsuo N, Iizuka M, Tsuzuki S,
Kondo Y, Tanaka A, Moriyama M, Matsumoto I, Nakamura S and Sumida
T: Analysis of IgG4 class switch-related molecules in IgG4-related
disease. Arthr Res Ther. 14:R1712012. View
Article : Google Scholar
|
|
68
|
Nakashima H, Miyake K, Moriyama M, Tanaka
A, Watanabe M, Abe Y, Sato H, Nakamura S and Saito T: An
amplification of IL-10 and TGF-beta in patients with IgG4-related
tubulointerstitial nephritis. Clin Nephrol. 73:385–391. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Tabata T, Kamisawa T, Takuma K, Egawa N,
Setoguchi K, Tsuruta K, Obayashi T and Sasaki T: Serial changes of
elevated serum IgG4 levels in IgG4-related systemic disease. Intern
Med. 50:69–75. 2011. View Article : Google Scholar : PubMed/NCBI
|