Introduction
Pneumonia refers to inflammation in the lower
airways, the alveolar and pulmonary interstitium, which is caused
by micro-organisms (bacteria, viruses or fungi) (1,2),
physical and chemical factors, immune damage, allergies or drugs.
Bacterial pneumonia is one of the most common forms of pneumonia,
as well as one of the most common infectious diseases. Numerous
types of bacteria may cause bacterial pneumonia, including
Streptococcus (S.) pneumoniae, Klebsiella pneumoniae,
Haemophilus influenza and Pseudomonas aeruginosa
(3,4).
Gram-positive pathogens are particularly responsible
for the increasing frequency of pneumonia (5). Bacteria are the most common cause of
pneumonia in adults, while this pathology tends to be more severe
in patients below the age of 5 and above the age of 65 years.
Furthermore, patients with heart failure, diabetes, chronic
obstructive pulmonary disease a or weak immune system due to human
immunodeficiency virus infection/acquired immune deficiency
syndrome or cancer chemotherapy also have a high risk of
contracting bacterial pneumonia (6).
At present, the majority of adult patients with bacterial pneumonia
are successfully cured. In 1955, the mortality of this disease was
as low as <10% (7) in patients of
all ages. However, in infants and elderly people, bacterial
pneumonia remains a lethal lung disease. The mortality rate
increases from 1.3% (in patients <45 years) to 26.1% (in
patients aged ≥85 years).
As the efficiency of the treatment of bacterial
pneumonia is still dependent on the proper use of antibiotic drugs,
an accurate diagnosis to distinguish between Gram-positive and the
Gram-negative pathogens appears to be vital for the success of
pneumonia treatment. Approaches including restriction of
antibacterial drugs and appropriate medicinal therapy have
important roles in improving the survival or cure rate of bacterial
pneumonia. All of these rely on an accurate diagnosis and
evaluation of the prognosis. Thus, it is vital to gain more insight
into the pathogenesis of pneumonia, which may improve the
identification of the pathogen and evaluation of the stage of this
disease.
In addition, the incidence of bacterial pneumonia
has markedly increased and the prognosis remains poor due to the
increasing resistance of several bacterial strains to antimicrobial
agents (8–11). The Tracking Resistance in the United
States Today (TRUST) study revealed that >18% of S.
pneumoniae isolates were penicillin-resistant in 2001 (12). Therefore, the rapid development of
novel types of medicine for bacterial pneumonia is in demand.
With the increasing elucidation of the mechanisms of
the recognition and clearance of bacteria by the immune system, it
has become apparent that pneumonia may alter certain dysregulated
genes and bio-functional pathways in the lungs or in organs next to
the site of the primary infection. A study on Gram-negative
pneumonia identified an increased expression Toll-like receptor 2
and 4, as well as MD2, the determination of which contributed to
the accurate diagnosis of pneumonia patients with sepsis (13). Analytic methods and strategies for
generating gene expression profiles represent popular and feasible
means of biomarker exploration in numerous cancer types. However,
few studies were performed to screen the differentially expressed
genes (DEGs) in bacterial pneumonia and the bio-functional pathways
they participated in.
In the present study, these strategies were applied
to Gram-positive pneumonia by using two independent gene expression
datasets. The DEGs in peripheral blood mononuclear cells between
pneumonia patients and healthy samples were identified. The
Database for Annotation, Visualization and Integrated Discovery
(DAVID) was used to annotate and analyze the DEGs and identify
those enriched in the Gene Ontology (GO) terms and Kyoto
Encyclopedia of Genes and Genomes (KEGG) pathways. Finally, a
protein-protein interaction (PPI) network was mapped to identify
key genes and/or pathways. All of these results provided primary
information and basic knowledge to understand the mechanism of the
pathogenesis. Furthermore, elucidation of the pathogenesis of
bacterial pneumonia may contribute to the development of novel
treatments.
Materials and methods
Data sources
Expression profiles of gene arrays were downloaded
from the Gene Expression Omnibus database (https://www.ncbi.nlm.nih.gov/geo/). Two independent
datasets, GSE6269 (14) and GSE35716
(15), were selected to analyze the
DEGs. The dataset GSE6269 consisted of 44 samples from the blood
leukocytes of pediatric patients with Streptococcus
pneumonia infection and 7 unrelated healthy controls based on
the platform Affymetrix Human Genome U133 Array (Affymetrix; Thermo
Fisher Scientific, Inc, Waltham, MA, USA). A total of 10 pneumonia
samples from peripheral blood mononuclear cells stimulated with
plasma from patients with bacterial pneumonia (Gram-positive) in
vitro and 18 healthy control samples from the dataset GSE35716
were processed on the platform Affymetrix Human Genome U133 Plus
2.0 Array (Affymetrix; Thermo Fisher Scientific, Inc.).
Quality assessment
Quality assessment of the gene array datasets was
performed with the affyPLM package (16) by using the linear modeling procedures
at probe level. AffyRNAdeg was then used for the degradation of
RNA. Relative log expression (RLE) and normalized unscaled standard
errors were determined to assess the consistency of the data
trends. Finally, only the data with a consistent trend as well as
high RNA quality were included in the analysis.
Data preprocessing
In order to maintain the integrity and comparability
of the data, the gcrma package (17)
was used for normalization and adjustment to eliminate system
errors in and between chips. As an important indicator to evaluate
the reliability of experiments and sample selection, the
correlation between gene expression levels was analyzed. Based on
the Pearson correlation coefficient, a correlation chart for all
the samples within one dataset was obtained.
Screening of DEGs
DEGs between patient and healthy samples were
identified by using the Limma package (18). Gene expression was presented as
logarithmic values. The threshold was log2 (fold change) >1 and
P<0.05. Subsequently, the differences were visualized in a
volcano plot, Venn diagram and Heat map by using ggplot2 (19), Venn diagram (20) and pheatmap (21) in R language.
Functional analysis of DEGs
Functional annotation tools in DAVID were used to
annotate and analyze the associated pathways and functions of the
DEGs (22). Furthermore, GO terms
and KEGG pathways in which the key genes were enriched were
determined. P-values were adjusted by using the Benjamini method
(23) or the false discovery rate in
multiple testing calibrations. The threshold was P<0.05.
PPI analysis
STRING (http://string-db.org/), the functional protein
association networks, was used to construct and analyze the
interactions between the proteins encoded by these DEGs. Hereinto,
multiple proteins were applied to map the PPI network (24).
Results
Data source and quality
assessment
Regression analysis of the raw data from the two
databases was performed to control the data quality. Corresponding
boxplots of the RLE were generated to verify the homogeneity
between chips by using affyPLM in R. The majority of the
data-points representing samples from the GSE6269 and GSE35716
datasets centered around 0, having approximately the same
dispersion. The quality of each dataset was appropriate for the
subsequent analysis.
Data preprocessing
The gcrma package was then applied to normalize the
original data of the samples from the two datasets. Based on the
density histograms and boxplots of log-intensities of normalized
data from GSE35716 and GSE6269, the relative expression of samples
from the two databases ranged from 0 to 15, revealing a reasonably
small extent. The general expression of the DEGs in the GSE6269 and
GSE35716 dataset concentrated at around 2, indicating a similar
expression trend.
After normalization, logarithmic expression values
were subjected to Pearson correlation analysis of samples with cor
functions in R. The graphs of the correlation clustering (between
different genes in each sample) indicated that the expression of
each sample in the GSE6269 as well as the GSE35716 dataset was
highly correlated (Fig. 1). The
minimum correlation coefficients of samples in GSE6269 and GSE35716
were 0.901 and 0.953, respectively. However, samples in the control
group and treat group could not be clustered significantly for each
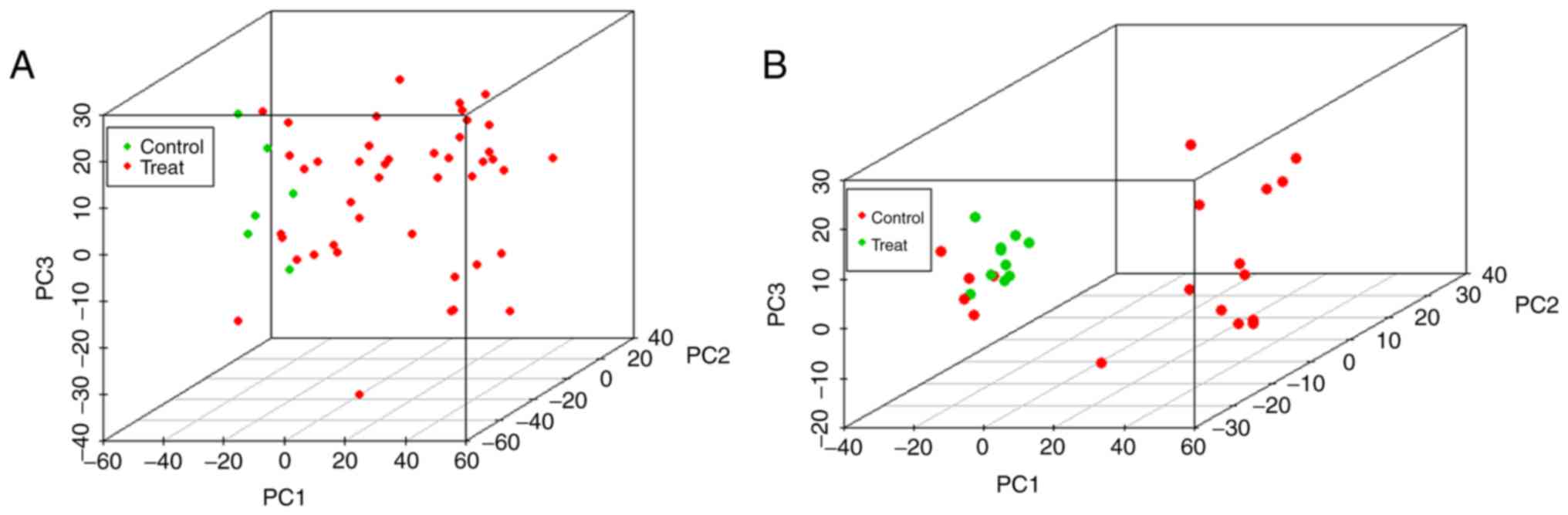
of the two databases. However, when using the first three principal
components for principal component analysis (Fig. 2), the clustering of samples in the
two databases was consistent with the correlation analysis.
DEGs
In the dataset GSE6269, a total of 624 DEGs were
identified between pneumonia samples and healthy samples, including
323 upregulated and 301 downregulated genes. Hereinto, 621 genes
were annotated (Fig. 3A). By
comparing 10 pneumonia samples with 18 healthy samples from the
dataset GSE35716, 398 DEGs and annotation information for 295 of
them were obtained (Fig. 3B). These
DEGS were comprised of 289 significantly upregulated and 109
downregulated genes. Among these the two different databases, 40
common genes were identified. Except for the four genes [adenosine
deaminase, C-C motif chemokine ligand 4 (CCL4), chloride
intracellular channel 3 and inhibitor of DNA binding 3] that
exhibited different expression patterns in the two datasets, a
total of 4 common downregulated and 32 common upregulated DEGs were
identified (Table I).
 | Table I.Common genes from the two
platforms. |
Table I.
Common genes from the two
platforms.
| Regulation | Differentially
expressed genes |
|---|
| Upregulated | PLAUR, ADAP2,
TREM1, TGFBI, CAPG, PPIF, NPL, EREG, LGALS1, TIMP1, PILRA, CTSB,
THBD, PKM, SIRPA, TNFAIP2, ICAM1, FCAR, LILRB1, CD14, S100A12,
MAPKAPK3, PLEC, CES1, IER3, BSG, ANPEP, CORO1C, TOM1, MGLL, GRN,
APLP2 |
| Downregulated | ETS1, IPCEF1,
ABHD10, GVINP1 |
Functional enrichment analysis
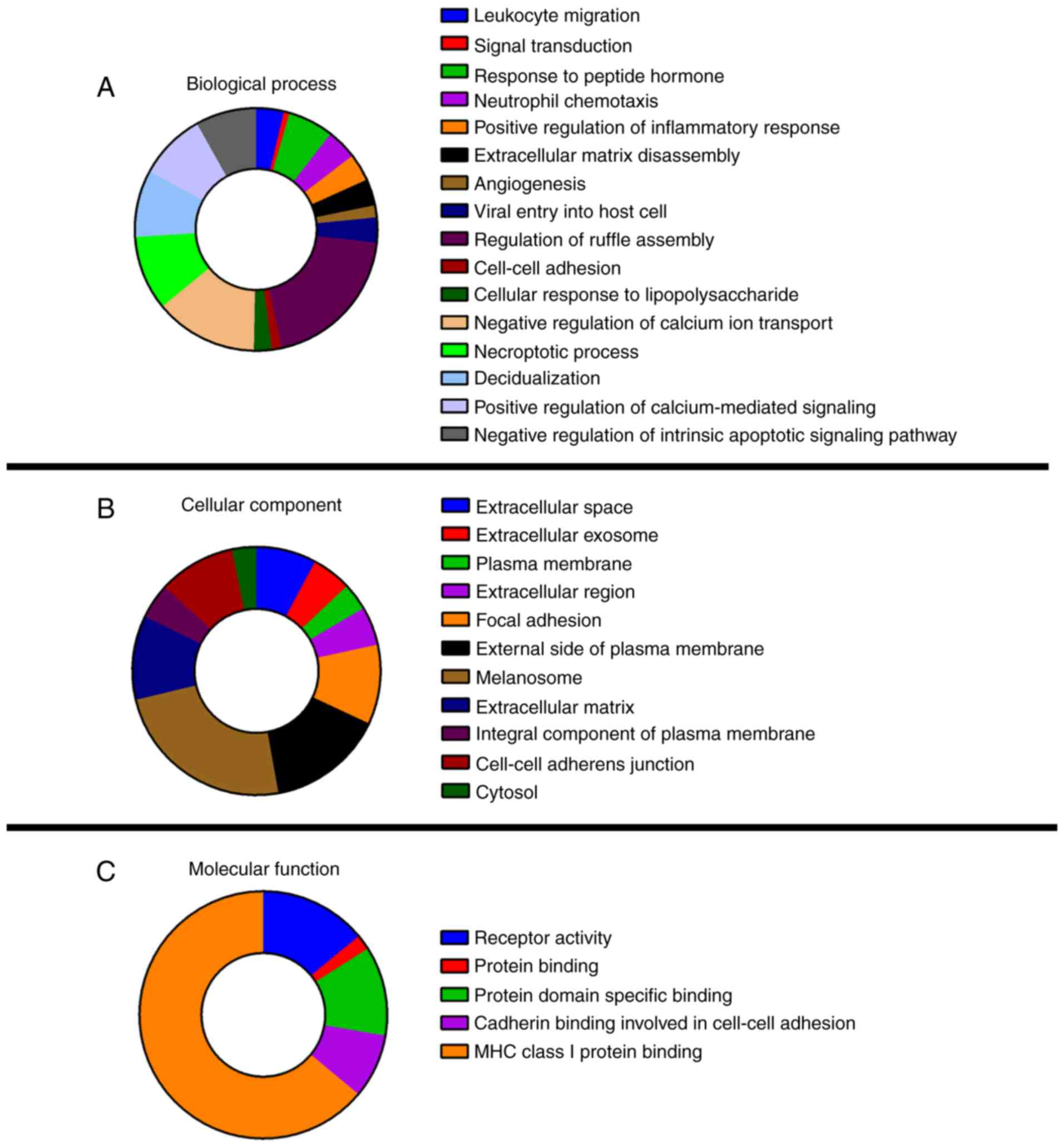
Functional enrichment analysis of 40 common DEGs was
performed using DAVID (Fig. 4). GO
analysis revealed that the DEGs were significantly enriched in 32
GO terms, including 16 terms in the category biological process
(BP), 11 terms in the category cellular component (CC) and 4 terms
in the category molecular function. In the CC category, melanosome
was highly enriched among the 11 terms and in the category BP, the
three most enriched items were regulation of ruffle assembly,
negative regulation of calcium ion transport and necroptotic
process. No DEGs were enriched in the molecular function (MF)
catagory. Among the KEGG terms, only the nuclear factor (NF)-κB
signaling pathway (Homo sapiens 04064) was significantly
enriched.
PPI analysis
To explore the biological and regulating functions
of the common DEGs at the protein level, a PPI network was
constructed identify the key genes associating with pneumonia
caused by Gram-positive bacteria (Fig.
5). Analysis with STRING identified 19 genes that interacted
with each other, generating 19 PPIs. This PPI network was
visualized by using Cytoscape, and hub genes with a degree of
interaction of >3 were selected for further analysis. Five genes
[CCL4, TIMP metallopeptidase inhibitor 1 (TIMP1), intercellular
adhesion molecule 1 (ICAM1), plasminogen activator, urokinase
receptor (PLAUR) and cathepsin B (CTSB)] were identified as hub
genes that strongly interacted with other DEGs (Table II). These five genes may represent
key genes associated with pneumonia caused by Gram-positive
bacteria.
| Table II.Annotation information of the five
key differentially expressed genes. |
Table II.
Annotation information of the five
key differentially expressed genes.
| Gene symbol | GO terms |
|---|
| CCL4 | Extracellular
space, signal transduct binding, neutrophil chemotaxis, positive
regulation of inflammatory response, positive regulation of
calcium-mediated signaling |
| TIMP1 | Extracellular
space, extracellular region, protein binding, extracellular
exosome, response to peptide hormone, extracellular matrix
disassembly |
| ICAM1 | Extracellular
space, protein binding, extracellular exosome, leukocyte migration,
receptor activity, plasma membrane, focal adhesion, external side
of plasma membrane, viral entry into host cell, regulation of
ruffle assembly, integral component of plasma membrane |
| PLAUR | Protein binding,
extracellular exosome, receptor activity, plasma membrane, focal
adhesion, integral component of plasma membrane, signal
transduction, protein domain specific binding, negative regulation
of intrinsic apoptotic signaling pathway |
| CTSB | Extracellular
space, protein binding, extracellular exosome, viral entry into
host cell, extracellular region, melanosome, decidualization,
receptor activity |
Discussion
According to the statistics from the World Health
Organization, pneumonia causes ~1.6 million deaths annually,
becoming a leading cause of morbidity and mortality throughout the
world (25,26). Among the casualties of pneumonia,
>1 million are children under the age of 5 years. An estimate of
90% of pneumonia-associated deaths occur in developing countries.
Individuals aged >60 years are also a major population affected
by pneumonia. Gram-positive bacteria are accountable for a large
proportion of all severe pneumonia cases, including nosocomial
pneumonia and community-acquired pneumonia (27).
Bacteria are commonly present in parts of the upper
respiratory tract; however, they are able to enter alveolar spaces
between the cells and also travel between adjacent alveoli through
connecting pores (28). This
invasion triggers an immune response, comprising the recruitment of
white blood cells (neutrophils) with the capacity to attack
microorganisms to the lungs. A general activation of the immune
system is then triggered by the neutrophils and cytokines. The
neutrophils, bacteria and fluid leaked from surrounding blood
vessels fill the alveoli and result in impaired oxygen
transportation (29). However,
further details regarding the mechanism of the immune response to
Gram-positive bacterial pneumonia remain to be elucidated.
At present, the diagnostic efficacy of Gram-positive
pneumonia is far from satisfactory, and drug resistance among
Gram-positive organisms is now a serious therapeutic problem
despite the availability of novel antimicrobials. An improved
knowledge of the immune mechanisms associated with pneumonia caused
by Gram-positive organisms may contribute to the effective
treatment and the development of more immunogenic vaccines.
At present, advanced biological techniques,
including gene array and high-throughput sequencing are ideal
approaches to assess the mechanisms of the development and immune
responses to various diseases. In the present study, a
bioinformatics analysis of gene array datasets was applied to
determine DEGs in Gram-positive pneumonia and their associated
pathways. Two independent datasets, GSE6269 and GSE35716, were
selected, which contained gene expression profiles of peripheral
blood samples from healthy controls and patients with bacterial
pneumonia. A total of 40 common DEGs associated with pneumonia were
identified between the two databases. All of these DEGs were
annotated subjected to GO/KEGG functional enrichment analysis by
using DAVID. Key DEGs, including CCL4, TIMP1, ICAM1, PLAUR and
CTSB, were further mapped in a PPI network.
CCL4/macrophage inflammatory protein-1β (MIP-1β) is
a CC chemokine with specificity for C-C chemokine receptor type 5
receptors. As a chemoattractant for a variety of other immune
cells, including natural killer cells and monocytes, CCL4/MIP-1β
has a vital role in inflammation caused by bacteria and viruses
(30). It was also identified to be
induced by Gram-positive bacteria including Lactococcus
lactis (31). As an addition to
a previous study reporting that CCL4 interacted with CCL3 (32), the present results also indicated
that CCL4 interacted with ICAM1 and TIMP1 in Gram-positive
pneumonia. ICAM-1/CD54, a protein encoded by the ICAM1 gene in
humans (33,34), is a cell surface glycoprotein
typically expressed on endothelial cells and cells of the immune
system, binding to integrins of the type CD11a/CD18 or CD11b/CD18.
Thus, it is associated with a series of immune responses in
inflammatory diseases. Studies have identified that ICAM-1 was
significantly differentially expressed in S. pneumoniae
infection and bacterial or viral meningitis (35,36).
Another gene interacting with CCL4 is TIMP1, a tissue inhibitor
that regulates matrix metalloproteinases and
disintegrin-metalloproteinases [a disintegrin and metalloproteinase
(ADAMs) and ADAMs with thrombospondin motifs] (37). Studies have reported that TIMP1 was
dysregulated in numerous types of lung cancer (38,39),
breast cancer (40) and nephritis
(41,42). In accordance with the results of the
present study, TIMP1 was also identified to be associated with
interstitial pneumonia (43,44).
CTSB belongs to a family of lysosomal cysteine
proteases and is encoded by the CTSB gene in humans (45,46). It
is an important endogenous protease in intracellular proteolysis,
regulating cell apoptosis and restricting injury-associated
inflammation (47). This protein was
identified to be upregulated in premalignant lesions and various
pathological conditions, as well as in cancer. PLAUR, also known as
urokinase-type plasminogen activator (uPA) receptor or uPAR/CD87,
is a multi-domain glycoprotein tethered to the cell membrane. It
was reported to have important roles in processes associated with
numerous diseases, including tumor infiltration (48) and inflammation (49).
In conclusion, the present study identified five key
DEGs in Gram-positive pneumonia. The type of bioinformatics
analysis performed in the present study has been rarely applied to
study this disease. The results indicated that these five key genes
had a high degree of interaction. It may be suggested that ICAM1,
TIMP1 and CCL4 co-function in Gram-positive bacterial pneumonia by
participating in the regulation of the NF-κB signaling pathway.
However, further laboratory experiments are still required to
confirm the exact association between two correlating genes to
clearly understand what correlation patterns existing between them.
The present study provided basic information paving the road for
future experimental research to explore the mechanisms of the
development of Gram-positive bacterial pneumonia. The increasing
knowledge regarding the mechanisms of this disease may lead to the
improvement of the diagnostic efficacy, as well as the development
of novel treatments.
Acknowledgements
This study was supported by Wu Jieping Medical
Foundation Clinical Research Funding (grant no.
320.6750.16050).
References
|
1
|
Dickson RP, Erb-Downward JR and Huffnagle
GB: The role of the bacterial microbiome in lung disease. Expert
Rev Respir Med. 7:245–257. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Marik PE: Aspiration pneumonitis and
aspiration pneumonia. N Engl J Med. 344:665–671. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Levy SB and Marshall B: Antibacterial
resistance worldwide: Causes, challenges and responses. Nat Med. 10
Suppl 12:S122–S129. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jones RN: Microbial etiologies of
hospital-acquired bacterial pneumonia and ventilator-associated
bacterial pneumonia. Clin Infect Dis. 51 Suppl 1:S81–S87. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fagon J, Patrick H, Haas DW, Torres A,
Gibert C, Cheadle WG, Falcone RE, Anholm JD, Paganin F, Fabian TC
and Lilienthal F: Treatment of gram-positive nosocomial pneumonia:
Prospective randomized comparison of quinupristin/dalfopristin
versus vancomycin. Nosocomial Pneumonia Group. Am J Respir Crit
Care Med. 161:753–762. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Testa A, Giannuzzi R, Daini S, Bernardini
L, Petrongolo L and Gentiloni Silveri N: Psychiatric emergencies
(part III): Psychiatric symptoms resulting from organic diseases.
Eur Rev Med Pharmacol Sci. 17 Suppl 1:S86–S99. 2013.
|
|
7
|
Kirby WM: Treatment of bacterial
pneumonia. AMA Arch Intern Med. 96:809–817. 1955. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Burwen DR, Banerjee SN and Gaynes RP:
Ceftazidime resistance among selected nosocomial gram-negative
bacilli in the United States. National nosocomial infections
surveillance system. J Infect Dis. 170:1622–1625. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
El Moujaber G, Osman M, Rafei R, Dabboussi
F and Hamze M: Molecular mechanisms and epidemiology of resistance
in Streptococcus pneumoniae in the middle east region. J Med
Microbiol. 66:847–858. 2017. View Article : Google Scholar
|
|
10
|
Lee HY, Wu TL, Su LH, Li HC, Janapatla RP,
Chen CL and Chiu CH: Invasive pneumococcal disease caused by
ceftriaxone-resistant Streptococcus pneumoniae in Taiwan. J
Microbiol Immunol Infect. Jun–26;2017.(Epub ahead of print).
View Article : Google Scholar
|
|
11
|
Yusef D, Shalakhti T, Awad S, Algharaibeh
H and Khasawneh W: Clinical characteristics and epidemiology of
sepsis in the neonatal intensive care unit in the era of multi-drug
resistant organisms: A retrospective review. Pediatr Neonatol. Jun
9–2017.(Epub ahead of print).
|
|
12
|
Karlowsky JA, Thornsberry C, Jones ME,
Evangelista AT, Critchley IA and Sahm DF: TRUST Surveillance
Program: Factors associated with relative rates of antimicrobial
resistance among Streptococcus pneumoniae in the United
States: Results from the TRUST surveillance program (1998–2002).
Clin Infect Dis. 36:963–970. 2003. View
Article : Google Scholar
|
|
13
|
Kajikawa O, Frevert CW, Lin SM, Goodman
RB, Mongovin SM, Wong V, Ballman K, Daubeuf B, Elson G and Martin
TR: Gene expression of Toll-like receptor-2, Toll-like receptor-4,
and MD2 is differentially regulated in rabbits with Escherichia
coli pneumonia. Gene. 344:193–202. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ramilo O, Allman W, Chung W, Mejias A,
Ardura M, Glaser C, Wittkowski KM, Piqueras B, Banchereau J,
Palucka AK and Chaussabel D: Gene expression patterns in blood
leukocytes discriminate patients with acute infections. Blood.
109:2066–2077. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Levy H, Wang X, Kaldunski M, Jia S, Kramer
J, Pavletich SJ, Reske M, Gessel T, Yassai M, Quasney MW, et al:
Transcriptional signatures as a disease-specific and predictive
inflammatory biomarker for type 1 diabetes. Genes Immun.
13:593–604. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Heber S and Sick B: Quality assessment of
Affymetrix GeneChip data. OMICS. 10:358–68. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wu C, Irizarry R and Gentry J: gcrma:
Background adjustment using sequence information. 2005.
|
|
18
|
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW,
Shi W and Smyth GK: limma powers differential expression analyses
for RNA-sequencing and microarray studies. Nucleic Acids Res.
43:e472015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wickham H: ggplot2: Elegant Graphics for
Data Analysis. Springer-Verlag; New York: 2009, View Article : Google Scholar
|
|
20
|
Chen H and Boutros PC: VennDiagram: A
package for the generation of highly-customizable Venn and Euler
diagrams in R. BMC Bioinformatics. 12:352011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kolde R: pheatmap. In: Pretty Heatmaps. R
package version 1.0.2, 2015. https://cran.r-project.org/web/packages/pheatmap/pheatmap.pdfJune
12–2016
|
|
22
|
Huang da W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Alishahi K, Ehyaei AR and Shojaie A: A
generalized benjamini-hochberg procedure for multivariate
hypothesis testing. Methodology. 33:2016.
|
|
24
|
Szklarczyk D, Franceschini A, Wyder S,
Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos
A, Tsafou KP, et al: STRING v10: Protein-protein interaction
networks, integrated over the tree of life. Nucleic Acids Res.
43:(Database issue). D447–D452. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mandourah Y, Al-Radi A, Ocheltree AH,
Ocheltree SR and Fowler RA: Clinical and temporal patterns of
severe pneumonia causing critical illness during Hajj. BMC Infect
Dis. 12:1172012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cilloniz C, Martin-Loeches I, Garcia-Vidal
C, San Jose A and Torres A: Microbial etiology of pneumonia:
Epidemiology, diagnosis and resistance patterns. Int J Mol Sci.
17:E21202016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Osiyemi O and Dickinson G: Gram-positive
pneumonia. Curr Infect Dis Rep. 2:207–214. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Scannapieco FA: Role of oral bacteria in
respiratory infection. J Periodontol. 70:793–802. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Garibaldi RA: Epidemiology of
community-acquired respiratory tract infections in adults:
Incidence, etiology and impact. Am J Med. 78:32–37. 1985.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Guan E, Wang J, Roderiquez G and Norcross
MA: Natural truncation of the chemokine MIP-1 beta/CCL4 affects
receptor specificity but not anti-HIV-1 activity. J Biol Chem.
277:32348–32352. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Luther SA and Cyster JG: Chemokines as
regulators of T cell differentiation. Nat Immunol. 2:102–107. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Guan E, Wang J and Norcross MA:
Identification of human macrophage inflammatory proteins 1alpha and
1beta as a native secreted heterodimer. J Biol Chem.
276:12404–12409. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Carlson M, Nakamura Y, Payson R, O'Connell
P, Leppert M, Lathrop GM, Lalouel JM and White R: Isolation and
mapping of a polymorphic DNA sequence (pMCT108.2) on chromosome 18
[D18S24]. Nucleic Acids Res. 16:41881988. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Katz FE, Parkar M, Stanley K, Murray LJ,
Clark EA and Greaves MF: Chromosome mapping of cell membrane
antigens expressed on activated B cells. Eur J Immunol. 15:103–106.
1985. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Murdoch C, Read RC, Zhang Q and Finn A:
Choline-binding protein A of Streptococcus pneumoniae
elicits chemokine production and expression of intercellular
adhesion molecule 1 (CD54) by human alveolar epithelial cells. J
Infect Dis. 186:1253–1260. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
36
|
Shapiro S, Miller A, Lahat N, Sobel E and
Lerner A: Expression of matrix metalloproteinases, sICAM-1 and IL-8
in CSF from children with meningitis. J Neurol Sci. 206:43–48.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Brew K and Nagase H: The tissue inhibitors
of metalloproteinases (TIMPs): An ancient family with structural
and functional diversity. Biochim Biophys Acta. 1803:55–71. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ylisirniö S, Höyhtyä M and
Turpeenniemi-Hujanen T: Serum matrix metalloproteinases-2, −9 and
tissue inhibitors of metalloproteinases-1, −2 in lung cancer-TIMP-1
as a prognostic marker. Anticancer Res. 20:1311–1316.
2000.PubMed/NCBI
|
|
39
|
Fong KM, Kida Y, Zimmerman PV and Smith
PJ: TIMP1 and adverse prognosis in non-small cell lung cancer. Clin
Cancer Res. 2:1369–1372. 1996.PubMed/NCBI
|
|
40
|
Wu ZS, Wu Q, Yang JH, Wang HQ, Ding XD,
Yang F and Xu XC: Prognostic significance of MMP-9 and TIMP-1 serum
and tissue expression in breast cancer. Int J Cancer.
122:2050–2056. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Jiang Z, Sui T and Wang B: Relationships
between MMP-2, MMP-9, TIMP-1 and TIMP-2 levels and their
pathogenesis in patients with lupus nephritis. Rheumatol Int.
30:1219–1226. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kim H, Oda T, López-Guisa J, Wing D,
Edwards DR, Soloway PD and Eddy AA: TIMP-1 deficiency does not
attenuate interstitial fibrosis in obstructive nephropathy. J Am
Soc Nephrol. 12:736–748. 2001.PubMed/NCBI
|
|
43
|
Kakugawa T, Mukae H, Saito M, Ishii K,
Ishimoto H, Sakamoto N, Takazono T, Fukuda Y, Ooe N and Kohno S:
Rapidly progressive interstitial pneumonia associated with
clinically amyopathic dermatomyositis successfully treated with
polymyxin B-immobilized fiber column hemoperfusion. Intern Med.
47:785–790. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Vissers M, Hartman Y, Groh L, de Jong DJ,
de Jonge MI and Ferwerda G: Recognition of Streptococcus
pneumoniae and muramyl dipeptide by NOD2 results in potent
induction of MMP-9, which can be controlled by lipopolysaccharide
stimulation. Infect Immun. 82:4952–4958. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Chan SJ, San Segundo B, McCormick MB and
Steiner DF: Nucleotide and predicted amino acid sequences of cloned
human and mouse preprocathepsin B cDNAs. Proc Natl Acad Sci USA.
83:7721–7725. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Cao L, Taggart RT, Berquin IM, Moin K,
Fong D and Sloane BF: Human gastric adenocarcinoma cathepsin B:
Isolation and sequencing of full-length cDNAs and polymorphisms of
the gene. Gene. 139:163–169. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Foghsgaard L, Lademann U, Wissing D,
Poulsen B and Jaattela M: Cathepsin B mediates tumor necrosis
factor-induced arachidonic acid release in tumor cells. J Biol
Chem. 277:39499–39506. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Borgfeldt C, Bendahl PO, Gustavsson B,
Långström E, Fernö M, Willén R, Grenman S and Casslén B: High tumor
tissue concentration of urokinase plasminogen activator receptor is
associated with good prognosis in patients with ovarian cancer. Int
J Cancer. 107:658–665. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Chavakis T, Kanse SM, May AE and Preissner
KT: Haemostatic factors occupy new territory: The role of the
urokinase receptor system and kininogen in inflammation. Biochem
Soc Trans. 30:168–173. 2002. View Article : Google Scholar : PubMed/NCBI
|