Introduction
Sepsis is a type of systemic inflammatory response
syndrome with high morbidity and mortality due primarily to
multiple organ failure (MOF), including lung injury, renal
dysfunction and skeletal problems, particularly in elderly people
(1–3). Despite recent advances, sepsis remains
an economic and humanistic burden to society that is increasing in
developing and developed countries due to delayed diagnoses
(4–6). Identifying the biomarkers of MOF may
increase our understanding of the underlying mechanisms and allow
for earlier diagnosis of sepsis.
Bacterial culturing of blood samples, which requires
several days to complete, is a traditional method used to diagnose
sepsis (4). Polymerase chain
reaction (PCR)-based molecular quantification has emerged as a
novel diagnostic method for several diseases, such as sepsis
(7). For example, 16S ribosomal RNA
gene amplification by quantitative PCR and sequencing has been
reported to improve the sensitivity, specificity, and positive and
negative predictive value of bacteria detection in neonatal sepsis
(7). Furthermore, the development of
‘omics’ technologies, including high-throughput sequencing and mass
spectrometry, has accelerated the exploration of biomarkers for
early diagnosis and treatment of sepsis (8). A study by Scherag et al
(9) identified vacuolar protein
sorting 13 homolog A, cysteine rich secretory protein LCCL domain
containing 2 and chromosome 13 loci as potential hot points for
sepsis via a genome-wide association study. A study by Gosiewski
et al (10) demonstrated that
bacterial DNA in the blood was a valuable biomarker for sepsis via
high-throughput cationic trypsinogen sequencing. It has also been
reported that elevated levels of serum trypsin and the PRSS1
mutation may also contribute to susceptibility of sepsis (11).
Skeletal muscle dysfunction is a major risk factor
of sepsis, as well as a complication, and contributes to
sepsis-associated mortality (12).
The perturbation of several hormones, proteins and gene expression
in skeletal muscle have been demonstrated to be associated with the
outcome of sepsis, including glucocorticoids (13), lipopolysaccharide (14), regulated in development and DNA
damage response 1 (15) and glucose
transporter type 4 (16) expression.
Gene co-expression and coregulation are important indicators of
their functions in some biological processes or diseases (17). Altered co-expression or coregulation
patterns under different conditions may indicate the role of
different genes in specific diseases. To the best of our knowledge,
there have been no previous studies investigating differential
co-expression (DCE) or differential coregulation (DCR) analysis in
the skeletal muscles of patients with sepsis. In the present study,
DCE and DCR analysis of gene expression was performed using the
skeletal muscles of patients with sepsis. Several genes and
transcription factors (TFs) that may contribute to the progression
of sepsis were identified. These results may be helpful to improve
our understanding of the underlying mechanisms and potential
effective treatments for sepsis.
Materials and methods
Microarray datasets
The skeletal muscle transcriptome-wide expression
profiles of 13 patients with sepsis and 8 controls were downloaded
from the Gene Expression Omnibus (ncbi.nlm.nih.gov/geo/; accession number, GSE13205)
deposited by Fredriksson et al (18). No significant differences were
observed in age (P=0.115; Student's t-test) or sex (P=0.336;
Fisher's exact test) between the groups.
Gene expression profile analysis
The analysis of gene expression profiles consisted
of two steps: Preprocessing and differential expression (DE)
analysis. Briefly, the raw microarray datasets were imported into R
version 3.4.3 (https://www.r-project.org/), a free open-source
statistical software package, and the affy version 1.56.0
(http://bioconductor.org/packages/release/bioc/html/affy.html)
(19) package was used for
background correction and normalization in order to compare
expression profiles of different samples. DE genes (DEGs) with the
thresholds of fold change >2 and Benjamin adjusted P<0.05
were screened out using the limma version 3.34.8 (http://www.bioconductor.org/packages/release/bioc/html/limma.html)
(20) package. Additionally
supervised two-way clustering, including cluster analysis of
samples and DEGs, was conducted through the Euclidean distance
method (21) based on pheatmap 1.0.8
package (https://cran.r-project.org/web/packages/pheatmap/index.html).
Functional enrichment analysis
To explore the functions associated with DEGs,
functional enrichment analysis was performed using the Database for
Annotation, Visualization and Integrated Discovery (DAVID;
david.ncifcrf.gov/) (22). Gene Ontology (GO) terms (http://www.geneontology.org/) and Kyoto Encyclopedia
of Genes and Genomes (KEGG) pathways (http://www.kegg.jp/), which had P<0.05 and a gene
count ≥2, were identified in the present study.
DCE and DCR analysis
As a type of polygenic inheritance, the progression
of sepsis is associated with multiple gene interactions; similarly,
the co-expression of two genes indicates that they may serve
similar functions in sepsis (17).
Differences in co-expression patterns in the control and sepsis
groups may reveal the roles of particular genes in a specific
biological process or disease. This type of link is a DCE link
(DCL). Genes involved in DCLs are considered to be DCE genes if the
links surrounding them are significantly enriched in a binomial
probability model (P<0.05); these genes are differential
co-expression genes (DCGs).
TFs serve important roles in the regulation of gene
expression. Multiple genes are regulated by common TFs, the
differential expression of which may result in DCE among those
genes. This is referred to as DCR (17). In the present study, DCE and DCR
analyses were performed based on the DCGL version 2.1.2 (https://cran.r-project.org/web/packages/DCGL/index.html)
(17) package. DCGs were screened
out when the P-value of the differential co-expression enrichment
(DCe) test and Q-value of the differential co-expression profile
(DCp) test in DCGL were <0.05. Two types of DCL were identified:
TF_bridged_DCLs, in which the two DCGs share ≥1 TF, and
TF2target_DCLs, in which TF-gene regulation relationships were
identified. The DCL network was visualized using Cytoscape 3.6.0
software (http://www.cytoscape.org/) with nodes
and edges, representing genes and interactions, respectively.
Results
Differential expression genes
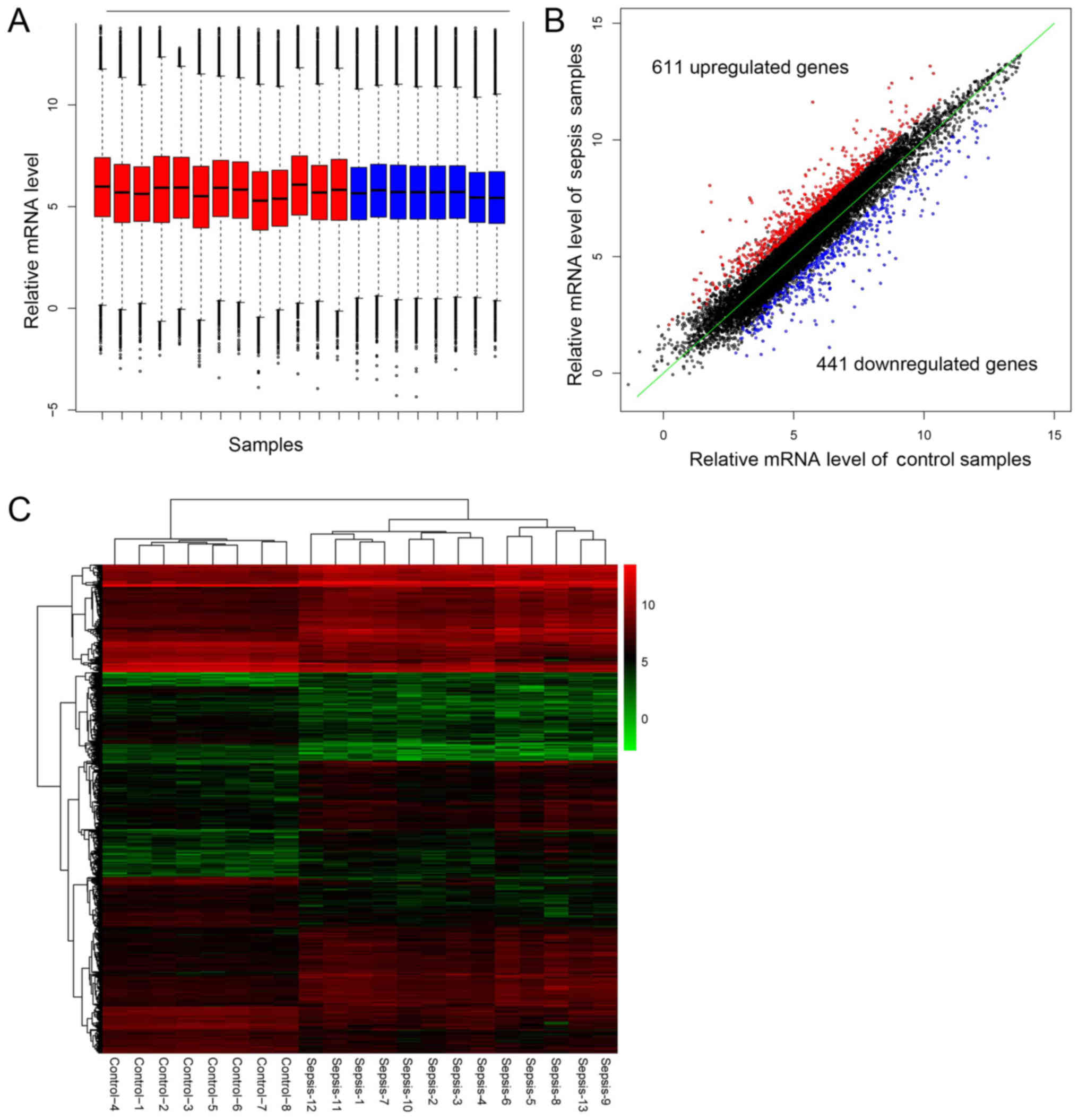
The preprocessing step produced comparable
expression profiles among all the samples (Fig. 1A), which were subsequently used for
DE analysis. A total of 1,052 DEGs were screened out in sepsis
samples, including 441 downregulated and 611 upregulated genes
(Fig. 1B). Supervised two-way
hierarchical clustering of DEGs and samples was performed based on
the pheatmap package (Fig. 1C).
Enriched functions of DEGs
A total of 129 significantly enriched GO terms were
identified using DAVID according to the thresholds of P<0.05 and
gene count ≥2. The 10 most enriched significant GO terms are
presented in Table I. The majority
of GO terms were associated with biological processes in the
skeletal muscle, RNA binding and cell responses to stimuli. A total
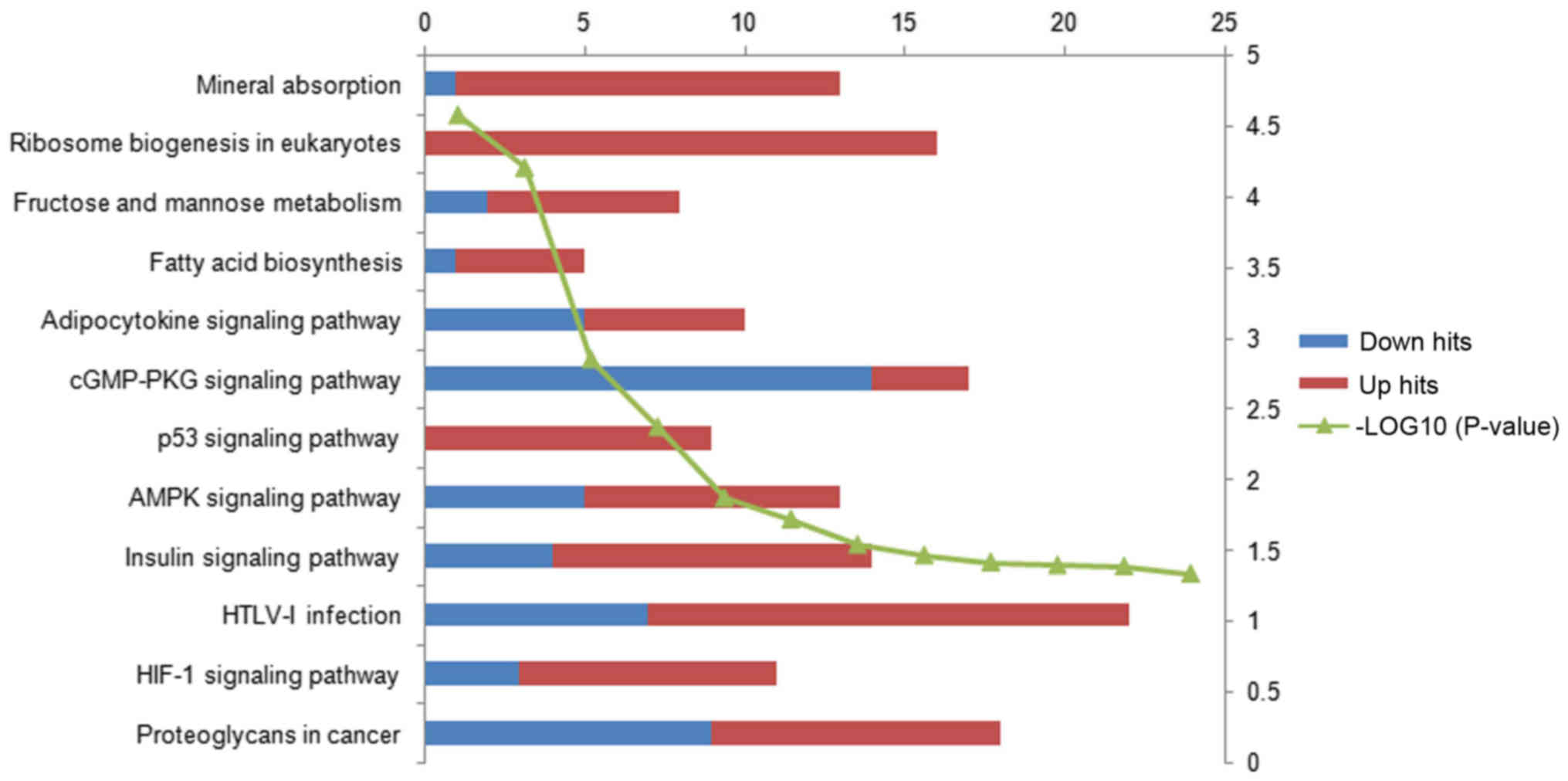
of 12 KEGG pathways were identified and these were primarily
associated with substance metabolism and synthesis, and
inflammatory and cancer-associated processes (Fig. 2). The number of upregulated genes was
markedly greater than the number of downregulated genes in the four
most enriched pathways, suggesting that upregulated genes serve an
important role in these pathways.
 | Table I.Top 10 most significantly enriched GO
terms of differentially expressed genes. |
Table I.
Top 10 most significantly enriched GO
terms of differentially expressed genes.
| Category | GO name | Hits | P-value |
|---|
| CC | Z disc | 19 | <0.001 |
| BP | Skeletal muscle
tissue development | 12 | <0.001 |
| CC | Cytoplasm | 318 | <0.001 |
| CC | T-tubule | 10 | <0.001 |
| BP | Muscle
contraction | 17 | <0.001 |
| BP | Cellular response to
cadmium ion |
7 | <0.001 |
| BP | Skeletal muscle
contraction |
8 | <0.001 |
| CC | Cytosol | 212 | <0.001 |
| CC | Nucleolus | 69 | <0.001 |
| BP | Response to
denervation involved in regulation of muscle adaptation |
5 | <0.001 |
DCLs and DCRs
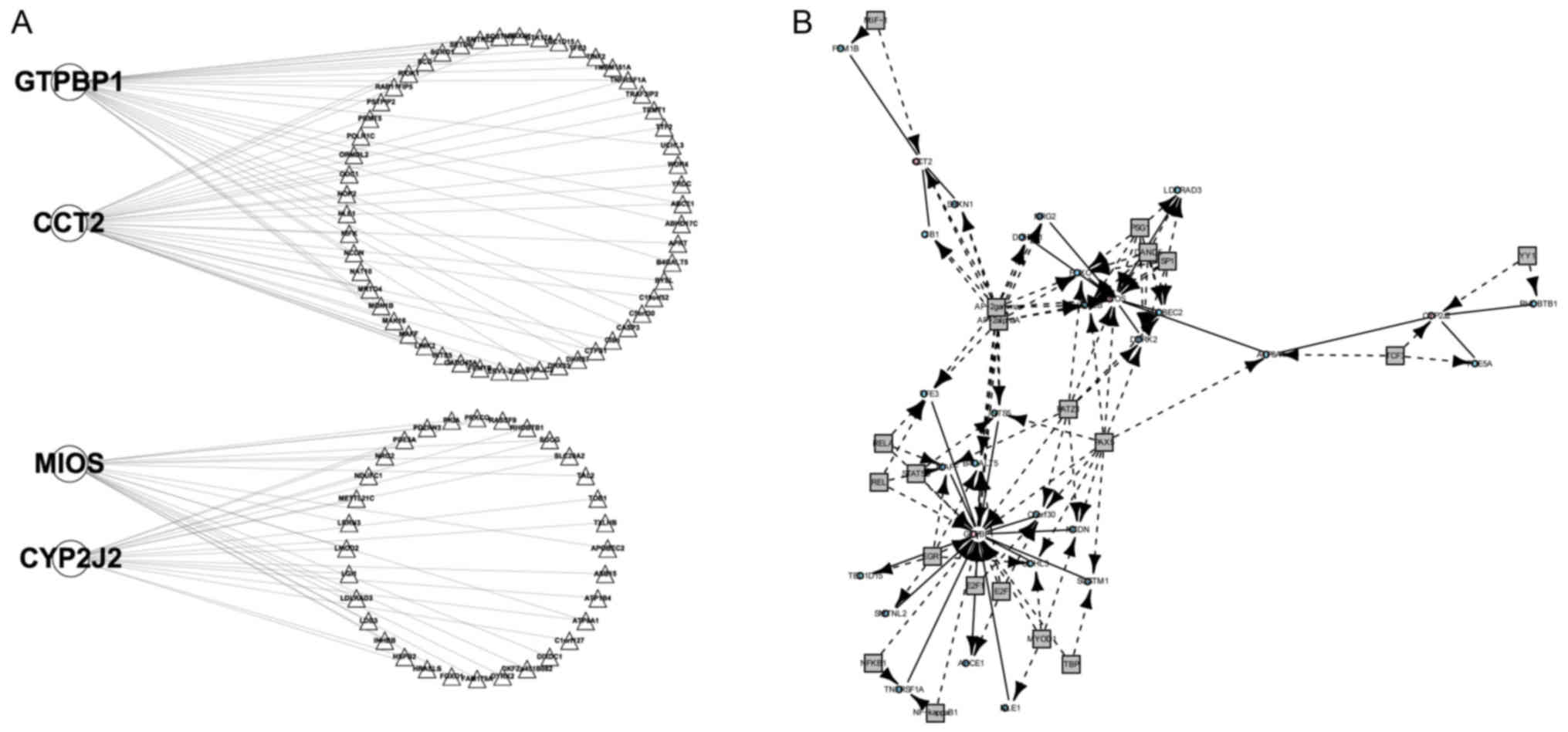
A total of four DCGs were identified using the DCGL
package, including GTP binding protein 1 (GTPBP1), chaperonin
containing T-complex 1 complex (CCT2), meiosis regulator of oocyte
development (MIOS) and cytochrome P450 family 2 subfamily J member
2 (CYP2J2), and 93 DCLs. The network composed by the four DCGs and
90 non-DCGs was visualized using Cytoscape software (Fig. 3A) (23). No TF2target_DCLs were identified;
however, 73 TF_bridged_DCLs were revealed using DCR analysis. The
DCR network is presented in Fig. 3B.
A total of eight TFs, including TATA box binding protein,
melanocyte inhibiting factor-1 (MIF-1), signal transducer and
activator of transcription 5B (STAT5B), POZ-, AT hook-, and zinc
finger-containing protein 1, LIM homeobox 3 (LHX3)a, LHX3b, AP-2γ
and AP-2αA, were considered to be associated with the development
of sepsis due to their target enrichment in the DCGs.
Discussion
Severe sepsis induces skeletal muscle dysfunction
(24,25) and, in turn, skeletal muscle
dysfunction is associated with the outcome of sepsis (12). Identifying biomarkers associated with
this process may increase our understanding of the underlying
mechanisms and allow for the development of therapeutic targets and
novel treatment methods. In the present study, DE, DCE and DCR
analysis were performed for this purpose. Functional enrichment
analysis identified several inflammatory and skeletal muscle
development-associated GO terms and KEGG pathways, which was
consistent with previous studies (26,27).
Several skeletal muscle development-associated
biological processes were identified in the present study,
including skeletal muscle tissue development, muscle contraction
and skeletal muscle atrophy. Several KEGG pathways associated with
substance biosynthesis and metabolism were identified as
significantly enriched in skeletal muscle samples from patients
with sepsis compared with control samples. The majority of genes in
those pathways were upregulated in patients with sepsis and all of
the genes associated with ribosome biogenesis in eukaryotes were
upregulated. Ribosome biogenesis dysfunction is associated with
skeletal muscle inflammation and results in a decrease in muscle
mass, which accounts for ~60% of sepsis-associated mortality
(28,29). It may be a previously unidentified
pathway that is associated with the dysfunction of skeletal muscle
in sepsis. Several cancer-associated pathways were also identified
in the present study, which may be because cancer and sepsis are
inflammation-inducing diseases.
The regulation of gene expression serves a key role
in several diseases (17). In the
present study, 73 TF_bridged_DCLs, which contained four DCGs (CCT2,
CYP2J2, GTPBP1 and MIOS) were identified. A total of 29, 17, 28 and
19 co-expression genes were identified for CCT2, CYP2J2, GTPBP1 and
MIOS, respectively, in the DCE network. The direct association
between these genes and sepsis progression or skeletal muscle
dysfunction was not investigated in the present study. Previous
studies have focused on their association with inflammation and
cancer; for instance, CYP2J2 has been reported to regulate
metabolic dysfunction via peroxisome proliferator-activated
receptor-γ by reducing hepatic inflammation (30) and to protect against lung
ischemia/reperfusion injury based on its anti-inflammatory effects
(31). CCT2 and TCP1 have also been
demonstrated to be valuable potential biomarkers for the prognosis
of breast cancer (32). Such studies
support the diverse functions of these genes and their role as
potential therapeutic targets for sepsis or skeletal muscle
dysfunction. The differential regulated rank (DRrank)
method, which is based on DCGs and their co-expressed genes
surrounding a specific TF, identified AP-2α and AP-2γ as the top
two highest ranked TFs significantly associated with the
development of sepsis, and they are considered to be associated
with the progression of inflammatory diseases, such as sepsis
(33–35). DNA binding of STAT5B, another
potential target of sepsis, was also identified to serve a role in
sepsis in a study by Chen et al (36), along with some novel potential
sepsis-associated TFs, including MIF-1, ETS domain-containing
protein Elk-1.
In conclusion, the present study identified aspects
of the mechanisms of sepsis progression and skeletal muscle
dysfunction using systemic bioinformatics analysis. DCE and DCR
analysis provide an insight into gene regulation loops in sepsis
and may be utilized to develop therapeutic targets and treatment
methods for patients with sepsis.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets generated and/or analyzed during the
current study are available in the GEO repository, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE13205.
Authors' contributions
FY conducted data analysis and wrote the manuscript.
YW designed the study, and corrected spelling and grammar
errors.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Evans CE and Zhao YY: Impact of thrombosis
on pulmonary endothelial injury and repair following sepsis. Am J
Physiol Lung Cell Mol Physiol. 312:L441–L451. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Eisen DP, Moore EM, Leder K, Lockery J,
McBryde ES, McNeil JJ, Pilcher D, Wolfe R and Woods RL: AspiriN To
Inhibit SEPSIS (ANTISEPSIS) randomised controlled trial protocol.
BMJ Open. 7:e0136362017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sehgal V, Bajwa SJ, Consalvo JA and Bajaj
A: Clinical conundrums in management of sepsis in the elderly. J
Transl Int Med. 3:106–112. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Graziani AC, Stets MI, Lopes AL, Schluga
PHC, Marton S, Mendes IF, Andrade ASR, Krieger MA and Cardoso J:
High efficiency binding aptamers for a wide range of bacterial
sepsis agents. J Microbiol Biotechnol. 27:838–843. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tiru B, DiNino EK, Orenstein A, Mailloux
PT, Pesaturo A, Gupta A and McGee WT: The economic and humanistic
burden of severe sepsis. Pharmacoeconomics. 33:925–937. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Perner A, Rhodes A, Venkatesh B, Angus DC,
Martin-Loeches I, Preiser JC, Vincent JL, Marshall J, Reinhart K,
Joannidis M and Opal SM: Sepsis: Frontiers in supportive care,
organisation and research. Intensive Care Med. 43:496–508. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Midan DA, Abo El Fotoh WMM and El
Shalakany AH: The potential role of incorporating real-time PCR and
DNA sequencing for amplification and detection of 16S rRNA gene
signatures in neonatal sepsis. J Matern Fetal Neonatal Med.
30:1476–1483. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ludwig KR and Hummon AB: Mass spectrometry
for the discovery of biomarkers of sepsis. Mol Biosyst. 13:648–664.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Scherag A, Schöneweck F, Kesselmeier M,
Taudien S, Platzer M, Felder M, Sponholz C, Rautanen A, Hill AVS,
Hinds CJ, et al: Genetic factors of the disease course after
sepsis: A genome-wide study for 28 day mortality. EBioMedicine.
12:239–246. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gosiewski T, Ludwig-Galezowska AH,
Huminska K, Sroka-Oleksiak A, Radkowski P, Salamon D, Wojciechowicz
J, Kus-Slowinska M, Bulanda M and Wolkow PP: Comprehensive
detection and identification of bacterial DNA in the blood of
patients with sepsis and healthy volunteers using next-generation
sequencing method-the observation of DNAemia. Eur J Clin Microbiol
Infect Dis. 36:329–336. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen Q, Xue H, Chen M, Gao F, Xu J, Liu Q,
Yang X, Zheng L and Chen H: High serum trypsin levels and the −409
T/T genotype of PRSS1 gene are susceptible to neonatal sepsis.
Inflammation. 37:1751–1756. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shibahashi K, Sugiyama K, Kashiura M and
Hamabe Y: Decreasing skeletal muscle as a risk factor for mortality
in elderly patients with sepsis: A retrospective cohort study. J
Intensive Care. 5:82017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bodine SC and Furlow JD: Glucocorticoids
and skeletal muscle. Adv Exp Med Biol. 872:145–176. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hansen ME, Simmons KJ, Tippetts TS,
Thatcher MO, Saito RR, Hubbard ST, Trumbull AM, Parker BA, Taylor
OJ and Bikman BT: Lipopolysaccharide disrupts mitochondrial
physiology in skeletal muscle via disparate effects on sphingolipid
metabolism. Shock. 44:585–592. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Steiner JL, Crowell KT, Kimball SR and
Lang CH: Disruption of REDD1 gene ameliorates sepsis-induced
decrease in mTORC1 signaling but has divergent effects on
proteolytic signaling in skeletal muscle. Am J Physiol Endocrinol
Metab. 309:E981–E994. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lu GP, Cui P, Cheng Y, Lu ZJ, Zhang LE and
Kissoon N: Insulin control of blood glucose and GLUT4 expression in
the skeletal muscle of septic rats. West Indian Med J. 64:62–70.
2015.PubMed/NCBI
|
|
17
|
Yang J, Yu H, Liu BH, Zhao Z, Liu L, Ma
LX, Li YX and Li YY: DCGL v2.0: An R package for unveiling
differential regulation from differential co-expression. PLoS One.
8:e797292013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fredriksson K, Tjäder I, Keller P,
Petrovic N, Ahlman B, Schéele C, Wernerman J, Timmons JA and
Rooyackers O: Dysregulation of mitochondrial dynamics and the
muscle transcriptome in ICU patients suffering from sepsis induced
multiple organ failure. PLoS One. 3:e36862008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gautier L, Cope L, Bolstad BM and Irizarry
RA: affy-analysis of Affymetrix GeneChip data at the probe level.
Bioinformatics. 20:307–315. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Diboun I, Wernisch L, Orengo CA and
Koltzenburg M: Microarray analysis after RNA amplification can
detect pronounced differences in gene expression using limma. BMC
Genomics. 7:2522006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Greenacre M: Ordination with any
dissimilarity measure: A weighted euclidean solution. Ecology.
98:2293–2300. 2017. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dennis G Jr, Sherman BT, Hosack DA, Yan J,
Gao W, Lane HC and Lempicki RA: DAVID: Database for annotation,
visualization, and integrated discovery. Genome Biol. 4:P32003.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zolfaghari PS, Carré JE, Parker N, Curtin
NA, Duchen MR and Singer M: Skeletal muscle dysfunction is
associated with derangements in mitochondrial bioenergetics (but
not UCP3) in a rodent model of sepsis. Am J Physiol Endocrinol
Metab. 308:E713–E725. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Crowell KT, Soybel DI and Lang CH:
Restorative mechanisms regulating protein balance in skeletal
muscle during recovery from sepsis. Shock. 47:463–473. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang J, Zhao L, Li Y, Feng S and Lv G: HuR
induces inflammatory responses in HUVECs and murine sepsis via
binding to HMGB1. Mol Med Rep. 17:1049–1056. 2018.PubMed/NCBI
|
|
27
|
Rontoyanni VG, Malagaris I, Herndon DN,
Rivas E, Capek KD, Delgadillo AD, Bhattarai N, Elizondo A, Voigt
CD, Finnerty CC, et al: Skeletal muscle mitochondrial function is
determined by burn severity, sex and sepsis and is associated with
glucose metabolism and functional capacity in burned children.
Shock. Dec 4–2017.(Epub ahead of print). View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rocheteau P, Chatre L, Briand D, Mebarki
M, Jouvion G, Bardon J, Crochemore C, Serrani P, Lecci PP, Latil M,
et al: Sepsis induces long-term metabolic and mitochondrial muscle
stem cell dysfunction amenable by mesenchymal stem cell therapy.
Nat Commun. 6:101452015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Figueiredo VC, Markworth JF, Durainayagam
BR, Pileggi CA, Roy NC, Barnett MP and Cameron-Smith D: Impaired
ribosome biogenesis and skeletal muscle growth in a murine model of
inflammatory bowel disease. Inflamm Bowel Dis. 22:268–278. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li R, Xu X, Chen C, Wang Y, Gruzdev A,
Zeldin DC and Wang DW: CYP2J2 attenuates metabolic dysfunction in
diabetic mice by reducing hepatic inflammation via the PPARγ. Am J
Physiol Endocrinol Metab. 308:E270–E282. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen W, Yang S, Ping W, Fu X, Xu Q and
Wang J: CYP2J2 and EETs protect against lung ischemia/reperfusion
injury via anti-inflammatory effects in vivo and in vitro. Cell
Physiol Biochem. 35:2043–2054. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Guest ST, Kratche ZR, Bollig-Fischer A,
Haddad R and Ethier SP: Two members of the TRiC chaperonin complex,
CCT2 and TCP1 are essential for survival of breast cancer cells and
are linked to driving oncogenes. Exp Cell Res. 332:223–235. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sung SJ, Walters JA, Hudson J and Gimble
JM: Tumor necrosis factor-alpha mRNA accumulation in human
myelomonocytic cell lines. Role of transcriptional regulation by
DNA sequence motifs and mRNA stabilization. J Immunol.
147:2047–2054. 1991.PubMed/NCBI
|
|
34
|
Ye X and Liu SF: Lipopolysaccharide
regulates constitutive and inducible transcription factor
activities differentially in vivo in the rat. Biochem Biophys Res
Commun. 288:927–932. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang HJ, Hinney A, Song JY, Scherag A,
Meng XR, Grallert H, Illig T, Hebebrand J, Wang Y and Ma J:
Association of common variants identified by recent genome-wide
association studies with obesity in Chinese children: A
case-control study. BMC Med Genet. 17:72016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chen Y, Sun D, Krishnamurthy VM and Rabkin
R: Endotoxin attenuates growth hormone-induced hepatic insulin-like
growth factor I expression by inhibiting JAK2/STAT5 signal
transduction and STAT5b DNA binding. Am J Physiol Endocrinol Metab.
292:E1856–E1862. 2007. View Article : Google Scholar : PubMed/NCBI
|