Introduction
Intrahepatic cholangiocarcinoma (ICC) represents the
second most commonly diagnosed primary liver malignant tumor,
accounting for ~3% of all gastrointestinal malignancies globally,
and both incidence and mortality rates are increasing (1). Only small proportion of patients with
early-stage ICC are likely to survive in the long-term, even
following radical hepatectomy; furthermore, the majority of
patients are diagnosed at a later stage and have poor prognosis
(2). Furthermore, all available
treatment strategies for ICC, including chemotherapy and
immunotherapy, induce a poor response from patients. Thereby,
developing novel therapeutic agents against ICC remains highly
important.
The epidermal growth factor receptor (EGFR) belongs
to the ErbB receptor tyrosine kinase family. Through the binding of
ligands to its extracellular ligand-binding domain, EGFR can be
phosphorylated to form homodimers or heterodimers, and consequently
initiates extensive intracellular signaling cascades (3,4). As one
of the most important downstream effectors, signal transducer and
activator of transcription 3 (STAT3) can be further phosphorylated
at the sites of Tyr-705 or Ser-727 by activated EGFR, and can then
translocate into the nucleus to exert transcriptional regulations,
primarily contributing to cell proliferation, resistance to
apoptosis and angiogenesis (5,6). Thus
far, aberrant EGFR-STAT3 flux has been widely implicated to various
neoplasms, such as colorectal, breast and lung cancer (4,5,7). All these features indicate that EGFR
may be an intriguing target for developing novel antitumor agents.
Numerous small-molecule EGFR inhibitors known as tyrosine kinase
inhibitors (TKIs) have exhibited varying anticancer effects to date
(3,8,9).
However, due to the secondary resistance primarily induced by EGFR
mutations, various TKIs have caused unsatisfactory therapeutic
effects in previous clinic studies (8,10,11).
A novel irreversible TKI agent known as afatinib has
been identified (10,11). Due to its increased inhibiting
potency and multitarget suppressing ability on ErbB receptor
tyrosine kinases, afatinib exhibits relative superiority over other
conventional first-generation EGFR inhibitors (8,12). To
date, studies have demonstrated that afatinib has extensive
antitumor efficacies on various neoplasms, particularly on
metastatic or refractory tumors (10,11).
These findings further establish afatinib among the first-line
options of EGFR-targeted therapeutic strategies, whether as
monotherapy or an important compound in combined treatment. More
recently, phase III clinic studies of afatinib on non-small cell
lung cancer have also been conducted (8). However, the potential carcinogenic
impact of EGFR-STAT3 signaling axis on ICC has not been fully
validated. In addition, to the best of our knowledge, the effect of
afatinib treatment on ICC has not been investigated previously.
Therefore, the present study was conducted with the aim of
identifying the effect of afatinib treatment on ICC.
Materials and methods
Cells and reagents
Human ICC cell lines OZ and JCK were purchased from
the Chinese Academy of Sciences (Shanghai, China). Afatinib (cat.
no. SC-364398) was obtained from Santa Cruz Biotechnology, Inc.
(Santa Cruz, CA, USA). Dulbecco's modified Eagle's medium, fetal
bovine serum, MTT cell proliferation and cytotoxicity assay (cat.
no. C0009) and BCA protein assay (cat. no. P0012S) kits were all
obtained from Beyotime Institute of Biotechnology (Nanjing, China).
The Apo-BrdU-Red In Situ DNA Fragmentation Assay kit (cat.
no. K404-60) was from BioVision, Inc. (Tucson, AZ, USA). Primary
rabbit monoclonal antibodies against phosphorylated EGFR (pEGFR;
cat. no. ab40815), pSTAT3 (cat. no. ab76315) and GAPDH (cat. no.
8227), as well as the horseradish peroxidase-conjugated goat
anti-rabbit IgG secondary antibody (cat. no. 97080) were all
purchased from Abcam (Cambridge, MA, USA).
Immunohistochemical analysis
In order to determine the protein levels of
phosphorylated EGFR and STAT3, 15 ICC specimens (T group) and
matched adjacent normal tissues (N group) were retrieved from the
Department of Pathology for immunohistochemistry assessment.
Written informed consent was obtained from all participants. The
participants constituted of 9 male and 6 female patients with a
mean age of 53.5 years old. No patients received chemotherapy prior
to anatomic heptectomy, which was performed between July 2013 and
October 2014. ICC specimens which, were 20 µm thick were then fixed
in 10% formaldehyde at 4°C for 24 h prior to paraffin embedding.
All cases were approved by the Ethical Review Committee
(Institutional Ethical Board of Taizhou People's Hospital, Taizhou,
China) and conformed closely to the Declaration of Helsinki.
Consecutive sections (4 µm) of the paraffin-embedded
normal and tumor specimens were prepared. The protein levels of
phosphorylated EGFR and STAT3 in these sections were detected using
antibodies against pEGFR (1:100) and pSTAT3 (1:150), respectively.
Two individual pathologists blinded to the groups scored these
sections using an Olympus CX32 microscope (Olympus Corp., Tokyo,
Japan). The protein expression extent was scored into four
gradients according to the staining intensity, as follows: 0,
absence of staining (<9% staining); 1, mild expression (10–19%
staining); 2, moderate expression (20–49% staining); and 3, high
expression (>49% staining). Subsequently, the mean percentage of
positive tumor cells from at least five different fields-of-view at
magnification of ×400 was calculated for each sample. The
percentages of positive tumor cells and the staining intensity were
multiplied to produce a weighted score for each case, with the
total scores ranging between 0 (<10%) to 3 (>49%).
Cell preparation and viability
measurement
To determine the inhibitory effect of afatinib on
ICC, the human ICC cell lines JCK and OZ were incubated in 96-well
plates (Nunc; Thermo Fisher Scientific, Inc., Waltham, MA, USA) at
a density of 5×103 cells/well and cultured in Dulbecco's
modified Eagle's medium supplemented with 10% fetal bovine serum at
37°C for 4 h. Subsequently, cells were exposed to afatinib at
concentrations of 1, 10, 50, 100 and 150 nM, respectively. Cells
without afatinib treatment were used as the control (ctrl) group.
When cells in the ctrl group had almost reached confluence, 20 µl
MTT (5 mg/ml) was added to each well. The ICC cells were then
incubated at 37°C for a further 4 h. Subsequently, the supernatant
was removed and 150 µl 0.1% dimethyl sulfoxide (vehicle) was added
to every well to resolve the MTT. The absorbance of each well was
ultimately measured at 450 nm on a Multiskan photometer (Thermo
Fisher Scientific, Inc.). Subsequently, the half maximal inhibitory
concentration (IC50) values of afatinib in the JCK and
OZ cell lines were calculated by statistical analysis, and found to
be 54.6 and 35.2 nM, respectively. Furthermore, the growth
responses of the two cell lines to an intermediate concentration of
afatinib (30 nM) at different time points (0, 24, 48 and 72 h) were
determined in order to determine the optimum incubation time.
For subsequent apoptosis measurement and protein
detection experiments, the ICC cell lines were cultured in 6-well
plates (Nunc; Thermo Fisher Scientific, Inc.) with 30 nM afatinib
and harvested after 48 h incubation.
Apoptosis detection
To evaluate the apoptosis variances in ICC cell
lines following afatinib exposure, the commercial Apo-BrdU-Red
In Situ DNA Fragmentation assay kit (BioVision, Inc.,
Milpitas, CA, USA) was employed. The BrdUTP in this TUNEL-staining
kit was able to actively bind DNA strand breaks, which were then
identified by a red fluorescence labeled anti-BrdU monoclonal
antibody, and read by flow cytometry (Excitation/Emittance
wavelengths, 488/576 nm). For sample preparation, ICC cells were
collected following afatinib treatment, resuspended into
phosphate-buffered saline containing 1% (w/v) formaldehyde and
stored at 4°C. Fixed cells were washed with precooling wash buffer
twice at 4°C. Cells were incubated in the DNA Labeling Solution for
60 min at 37°C, then Rinse Buffer were added and the sample was
centrifuged for 5 min. Cells were subsequently incubated with
antibody solution in the dark for 30 min at room temperature. A
total of 0.5 ml of propidium iodide/RNase A solution was added and
the sample was incubated in the dark for 30 min at room
temperature. Flow cytometry was performed using a Beckman Coulter
Epics XL instrument (Beckman Coulter, Inc., Brea, CA, USA) and
analyzed using EXPO32TM ADC software (Beckman Coulter, Inc.). All
above mentioned buffers were part of the Apo-BrdU-Red in
situ DNA Fragmentation assay kit (BioVision, Inc.). Further
procedures were performed according to the manufacturer's
instructions.
Western blotting
Cell samples grown in 6-well plates were incubated
with ice-cold lysis buffer containing 0.1% Triton X-100, 50 mM
HEPES (pH 7.5), 150 mM NaCl, 10% (v/v) glycerol, 1.5 mM
MgCl2, 1 mM dithiothreitol, 1 mM sodium fluoride, 0.1 mM
sodium orthovanadate, 1 mM phenylmethylsulfonyl fluoride, 2 mg/ml
leupeptin and 2 mg/ml aprotin in. Total protein was centrifuged at
12,000 × g at 4°C for 10 min, and the supernatants were
subjected to bolting at 100°C by iron heating for a further 10 min
to degrade protein. The protein concentration of the sample was
determined by a BCA protein assay kit, and equivalent protein
samples (30 mg) were then subjected to 12% SDS-PAGE. Subsequently,
the proteins were transferred to nitrocellulose membranes and
probed with specific primary antibodies against pEGFR (1:1,500),
pSTAT3 (1:2,000) and GAPDH (1:2,000) at 4°C for 12 h. Next, the
samples were incubated with the secondary antibody (1:2,000) at
room temperature for 1 h. The molecular sizes of the targeted
proteins were determined by comparison with prestained Precision
Plus Protein™ Dual Xtra protein markers (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). The protein bands were then
scanned using the Western Lightning Chemiluminescent Reagent Plus
(PerkinElmer, Inc., Boston, MA, USA) detection system. Protein
immunoreactivity was subsequently detected by the Gel Doc XR system
(170–8170) and analyzed using Image J software (https://imagej.nih.gov/ij/docs/index.html). GAPDH
served as the loading control in the western blotting
experiments.
Statistical analysis
All parametric data are expressed as the mean ±
standard error, and were analyzed by Student's t-test.
Non-parametric data analysis was performed by Mann-Whitney test.
For all tests, analyses were conducted using the SPSS version 19.0
statistical software (IBM SPSS, Armonk, NY, USA) and a two-sided
P<0.05 was considered to indicate statistically significant
differences.
Results
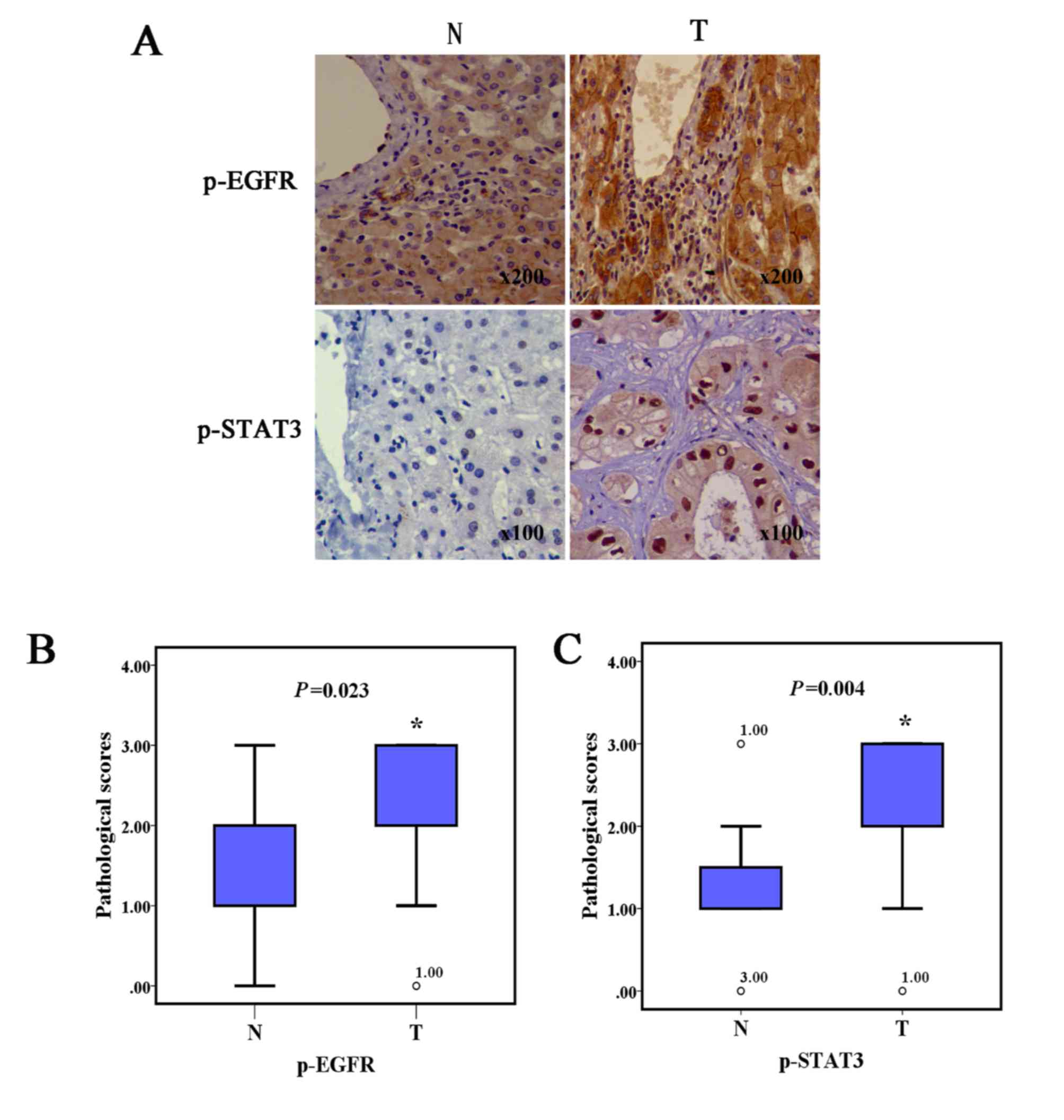
ICC specimens demonstrate increased
pEGFR and pSTAT3 protein expression levels compared with normal
tissues
In order to investigate the pEGFR and downstream
pSTAT3 protein variance between human ICC and normal liver tissues,
immunohistochemical assays were performed. As shown in Fig. 1, ICC specimens both presented notably
increased pEGFR and pSTAT3 protein expression levels representing a
higher pathological score, compared with the adjacent noncancerous
tissues.
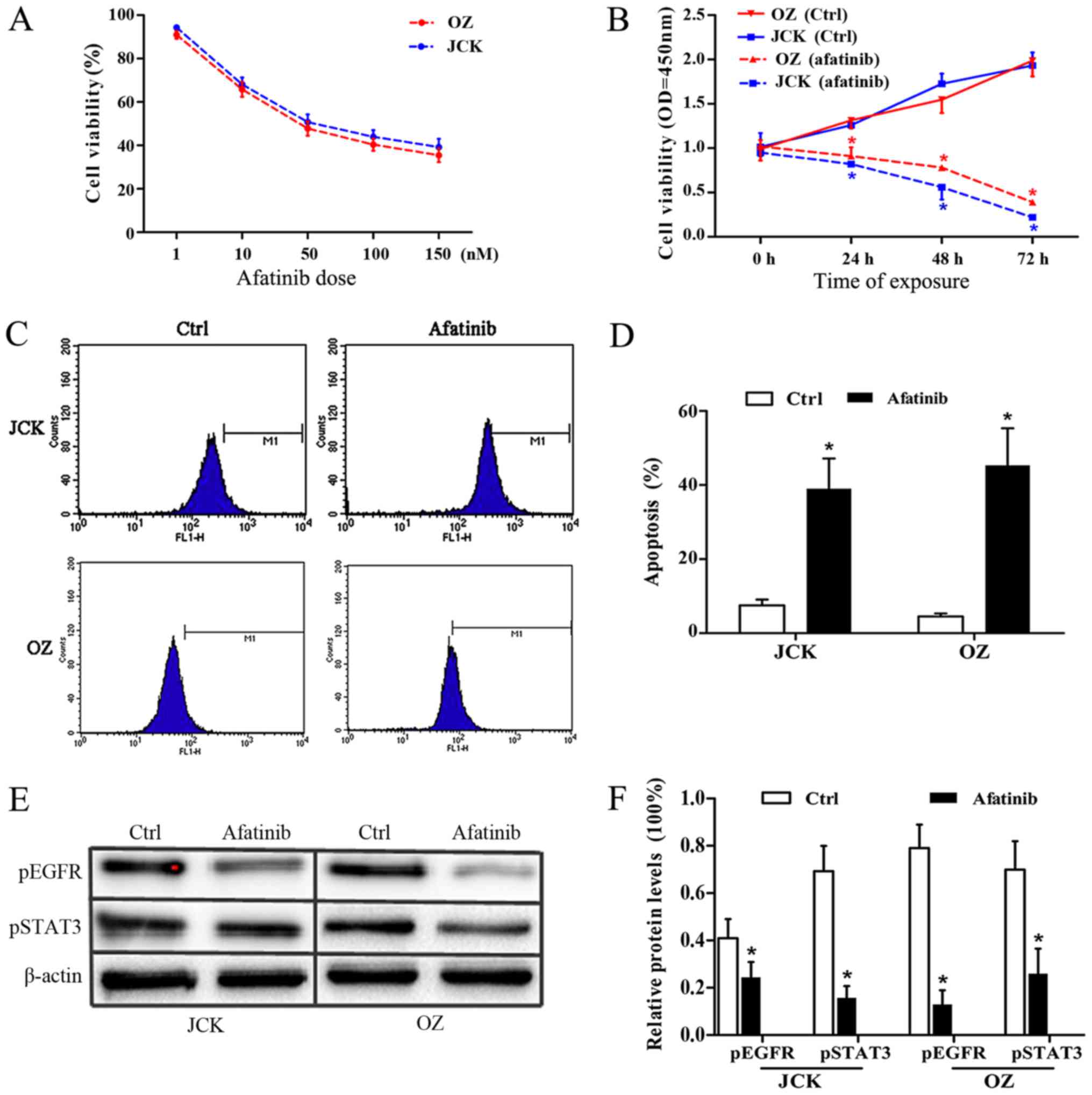
Afatinib evidently induces growth
arrest in ICC cell lines
To verify the antitumor activity of afatinib, the
IC50 values of afatinib treatment on the ICC cell lines
JCK and OZ were determined by MTT assay. Following
afatinib-pretreatment at concentrations of 1, 10, 50, 100 and 150
nM, the JCK cell viability rates were 95±8, 68±9, 51±6, 44±8 and
39±5%, respectively. Similarly, the corresponding viability values
in the OZ cell lines were 91±10, 65±8, 40±7 and 35±6% respectively
(Fig. 2A).
Following statistical analysis, the IC50
values of afatinib in control JCK and OZ cells were found to be
54.6 and 35.2 nM, respectively. Meanwhile, the JCK cell viability
OD values at the time points of 24, 48 and 72 h after 30 nM
afatinib exposure were 0.81±0.02, 0.54±0.11 and 0.23±0.04,
respectively, which were significantly lower than the corresponding
values in the ctrl group (1.01±0.04, 1.73±0.11 and 1.93±0.14;
Fig. 2B). Consistently, the OZ cell
survival ratios at 24, 48 and 72 h in the afatinib group were
0.91±0.10, 0.77±0.04 and 0.36±0.06, respectively, which were all
lower compared with the associated rates in the control group
(1.31±0.09, 1.55±0.15 and 1.99±0.18; Fig. 2B). These results demonstrated that
afatinib treatment had a strong antiproliferative impact on the ICC
cell lines.
Afatinib evidently promotes ICC cell
apoptosis
The apoptosis response of ICC cells to afatinib
treatment was detected by flow cytometry assay. After 48 h of
exposure to 30 nM afatinib, the cell apoptosis ratios in the JCK
and OZ cell lines were 0.39±0.08 and 0.46±0.11 respectively, which
were both markedly higher compared with the rates observed in the
control groups (0.07±0.02 and 0.05±0.01, respectively; Fig. 2C and D). These results indicated a
marked pro-apoptotic effect of afatinib on ICC cells.
Afatinib significantly inhibits
protein levels of pEGFR and pSTAT3
To determine the variation in the protein expression
levels of pEGFR and pSTAT3 in the two ICC cell lines following
afatinib pretreatment, immunoblotting assays were performed
(Fig. 2E and F). At the protein
level, the relative pEGFR contents in afatinib-treated JCK and OZ
cell were 0.24±0.07 and 0.13±0.06, which were both significantly
reduced (P<0.05) compared with those in the corresponding
control groups (0.41±0.08 and 0.79±0.10, respectively). Relative
pSTAT3 protein levels in the JCK and OZ cell lines following
afatinib exposure were 0.16±0.05 and 0.26±0.11, respectively, which
were significantly lower compared with the corresponding control
group levels (0.69±0.11 and 0.70±0.12, respectively; all P<0.05;
Fig. 2F). These data revealed the
evident suppressing impact of afatinib on ICC cell lines.
Discussion
ICC remains one of the most challenging tumors for
clinicians to deal with, due to inadequate therapeutic strategies.
For the successful treatment of patients, appropriate hepatectomy
plus systematic lymphadenectomy is the gold standard; however, the
majority of patients cannot receive these treatments due to delayed
initial presentation (2,13). In addition, patients receiving
radical resection may still encounter unclear distal outcomes, and
the 5-year survival rate ranges between 11.2 and 23.6% (2,14).
Therefore, an insight into the oncogenesis of ICC is required to
facilitate the development of suitable diagnostic biomarkers, as
well as antitumor drugs, and is essential for a more effective
therapeutic protocol of ICC. In present study, the levels of
phosphorylated EGFR were initially investigated. Despite the small
samples size involved in the present study, the
immunohistochemistry assay conducted demonstrated that EGFR is
commonly overexpressed in ICC. This result reinforced previous
suggestions that EGFR participates in ICC tumorigenesis (13,15,16).
Furthermore, the presence of overactivated EGFR has been further
associated with poor long-term survival, implying the significance
of conventional postoperative immunohistochemistry for ICC patients
in selecting appropriate therapeutic schedules subsequent to
radical resection, through the assessment of the pEGFR levels
(15–17). Since EGFR can phosphorylate STAT3 at
multiple sites, such as Tyr-705 and Ser-727, to form dimers, it is
not surprising that enhanced pSTAT3 expression was observed in the
present ICC specimens (3). Notably,
the immunohistochemical assay demonstrated the expression of STAT3
in nucleus was higher than in the cytoplasm. This finding was
consistent with conventional concepts that STAT3 mainly exerts
transcriptional regulation by translocating into the nucleus and
binding to specific promoter sequences (18,19). To
a certain extent, it may also support the novel viewpoint that
STAT3 can also be translocated into the mitochondria and
participate in the regulation of aerobic respiration and apoptosis
(20).
Cancer essentially represents a consequence of
imbalance between cell death and proliferation. In this sense,
apoptosis is a natural defense mechanism to eliminate unhealthy
cells and the loss of apoptosis has thereby been a widely accepted
hallmark of cancer (21). To date,
accumulating evidence has revealed that aberrant STAT3 expression
can contribute to the formation of various tumors via multiple
mechanisms, such as promotion of proliferation, suppression of
apoptosis and induction of cancer stem cell renewal (5,6). In the
current study, silencing EGFR via afatinib treatment significantly
promoted cell apoptosis and induced proliferation arrest in the ICC
cell lines, suggesting the important effect of EGFR-STAT3 signaling
on ICC formation. This result coincided closely with the findings
of previous studies on other tumors, and also indicated that TKIs
may be promising anticancer agents against ICC (8,12,22).
Besides downstream STAT3, EGFR itself can also regulate various
genes that operate in tumor cells through multiple interlinked
signaling pathways, such as RAS-ERK-MAPK and PI3K-AKT-mTOR
(3,4). The current study primarily focused on
investigating the STAT3 variance. However, according to the
conclusions of previous studies, it can be speculated that
downstream oncogenic regulators other than STAT3 may also
contribute to the blocking effects of afatinib on ICC cell lines
(3,6).
The definite tumorigenesis role of EGFR resulted in
endeavors to develop numerous EGFR targeting agents consisting of
TKIs and monoclonal antibodies. However, acquired resistance
remains an important challenge for clinicians when providing early
EGFR targeting therapies (8,10,11). To
date, EGFR mutations in 790M and/or S492R sites are known as the
most important contributors accounting for TKI resistance (3). To combat this problem, the novel
quinazoline derivative afatinib has been proven to inhibit EGFR via
irreversible covalent modification of Cys797 in the ATP binding
cleft of EGFR and has displayed considerable superiority to
conventional TKIs (3). Besides EGFR
mutation, the upregulation of human epidermal growth factor
receptor 2 (HER2), a member of the ErbB receptor tyrosine kinase
family, represents another key cause of TKI resistance (3,15). To
date, increasing evidence has generally validated the oncogenic
impact of overactivated HER2 on a variety of tumors, including
breast cancer and non-small cell lung cancer (23,24).
Therefore, afatinib functions as a multitarget TKI by
simultaneously suppressing EGFR and HER2. According to previous
studies, ICC tissues are less likely than EGFR to overexpress HER2
(13,15,16).
However, this conclusion is fundamentally based on
immunohistochemical arrays on postoperative ICC specimens without
using EGFR-targeted therapies. Thus far, to the best of our
knowledge, no studies have been conducted to examine the anticancer
effect of TKIs on ICC. The current study was therefore the first to
investigate the antitumor effect of afatinib.
In conclusion, immunohistochemical assays of human
ICC specimens were performed in the present study in order to
assess the protein expression levels of pEGFR and pSTAT3.
Subsequently, the ICC cell growth variances and protein levels of
pEGFR and pSTAT3 were also detected in the present study. The
findings revealed that overactivated EGFR-STAT3 signaling was
closely associated to ICC formation, while silencing this pathway
through afatinib treatment significantly induced ICC cell apoptosis
and growth arrest. These results suggest that EGFR-targeting
therapies may be promising against ICC and require further
investigation.
Acknowledgements
Not applicable.
Funding
The present research was supported by a grant from
the High Level Talent Project of ‘Top Six Talents’ in Jiangsu
Province (grant no. 2016-WSN-291).
Authors' contributions
CHZ and HX contributed to the study design,
intellectual content, literature research, experimental studies,
data acquisition, data analysis, statistical analysis and
manuscript preparation. ZPZ, YT, XFC, GCC and QHL also contributed
to the literature research, study design and data analysis. QHL
contributed to grant acquisition for this study.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of the Fifth Affiliated Hospital of Nantong University
and experiments were performed in accordance with the approved
guidelines and regulations. Preoperative informed consent was
obtained from all patients.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Consent for publication
Not applicable.
Conflicts of interests
The authors declare that they have no competing
interests.
References
|
1
|
Shishir KM, Gamblin TC, Kamel I,
Corona-Villalobos CP, Thomas M and Pawlik TM: Multidisciplinary
approaches to intrahepatic cholangiocarcinoma. Cancer.
119:3929–3942. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sriputtha S, Khuntikeo N, Promthet S and
Kamsa-Ard S: Survival rate of intrahepatic cholangiocarcinoma
patients after surgical treatment in Thailand. Asian Pacific J
Cancer Prev. 14:1107–1110. 2013. View Article : Google Scholar
|
|
3
|
Robert R Jr: ErbB/HER protein-tyrosine
kinases: Structures and small molecule inhibitors. Pharmacol Res.
87:42–59. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bi WW, Zhang WH, Yin GH, Luo H, Wang SQ,
Wang H, Li C, Yan WQ and Nie DZ: Analysis of indoleamine 2–3
dioxygenase (IDO) and EGFR co-expression in breast cancer tissue by
immunohistochemistry. Asian Pac J Cancer Prev. 15:5535–5538. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhao X, Sun X and Li XL: Expression and
clinical significance of STAT3, p-STAT3, and VEGF-C in small cell
lung cancer. Asian Pac J Cancer Prev. 13:2873–2877. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fang B: Genetic interactions of STAT3 and
anticancer drug development. Cancers (Basel). 6:494–525. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ung N, Putoczki TL, Stylli SS, Ng I,
Mariadason JM, Chan TA, Zhu HJ and Luwor RB: Anti-EGFR therapeutic
efficacy correlates directly with inhibition of STAT3 activity.
Cancer Biol Ther. 15:623–632. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chong CR and Jänne PA: The quest to
overcome resistance to EGFR-targeted therapies in cancer. Nat Med.
19:1389–1400. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ioannou N, Seddon AM, Dalgleish A,
Mackintosh D and Modjtahedi H: Treatment with a combination of the
ErbB (HER) family blocker afatinib and the IGF-IR inhibitor,
NVP-AEW541 induces synergistic growth inhibition of human
pancreatic cancer cells. BMC Cancer. 13:412013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Seiwert TY, Fayette J, Cupissol D, Del
Campo JM, Clement PM, Hitt R, Degardin M, Zhang W, Blackman A,
Ehrnrooth E and Cohen EE: A randomized, phase II study of afatinib
versus cetuximab in metastatic or recurrent squamous cell carcinoma
of the head and neck. Ann Oncol. 25:1813–1820. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lai WY, Chen CY, Yang SC, Wu JY, Chang CJ,
Yang PC and Peck K: Overcoming EGFR T790M-based tyrosine kinase
inhibitor resistance with an allele-specific DNAzyme. Mol Ther
Nucleic Acids. 3:e1502014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Machiels JP, Licitra LF, Haddad RI, Tahara
M and Cohen EE: Rationale and design of LUX-Head & Neck 1: A
randomised, Phase III trial of afatinib versus methotrexate in
patients with recurrent and/or metastatic head and neck squamous
cell carcinoma who progressed after platinum-based therapy. BMC
Cancer. 14:4732014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Harder J, Waiz O, Otto F, Geissler M,
Olschewski M, Weinhold B, Blum HE, Schmitt-Graeff A and Opitz OG:
EGFR and HER2 expression in advanced biliary tract cancer. World J
Gastroenterol. 15:4511–4517. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shen WF, Zhong W, Xu F, Kan T, Geng L, Xie
F, Sui CJ and Yang JM: Clinicopathological and prognostic analysis
of 429 patients with intrahepatic cholangiocarcinoma. World J
Gastroenterol. 15:5976–5982. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yoshikawa D, Ojima H, Iwasaki M, Hiraoka
N, Kosuge T, Kasai S, Hirohashi S and Shibata T:
Clinicopathological and prognostic significance of EGFR, VEGF, and
HER2 expression in cholangiocarcinoma. Br J Cancer. 98:418–425.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Clapéron A, Mergey M, Aoudjehane L,
Ho-Bouldoires TH, Wendum D, Prignon A, Merabtene F, Firrincieli D,
Desbois-Mouthon C, Scatton O, et al: Hepatic myofibroblasts promote
the progression of human cholangiocarcinoma through activation of
epidermal growth factor receptor. Hepatology. 58:2001–2011. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nanda S: Cancer: A limited role for dual
EGFR and ErbB2 inhibition in cholangiocarcinoma? Nat Rev
Gastroenterol Hepatol. 7:5912010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wu J, Patmore DM, Jousma E, Eaves DW,
Breving K, Patel AV, Schwartz EB, Fuchs JR, Cripe TP,
Stemmer-Rachamimov AO and Ratner N: EGFR-STAT3 signaling promotes
formation of malignant peripheral nerve sheath tumors. Oncogene.
33:173–180. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wendt MK, Balanis N, Carlin CR and
Schiemann WP: STAT3 and epithelial-mesenchymal transitions in
carcinomas. JAKSTAT. 3:e289752014.PubMed/NCBI
|
|
20
|
Qi QR and Yang ZM: Regulation and function
of signal transducer and activator of transcription 3. World J Biol
Chem. 5:231–239. 2014.PubMed/NCBI
|
|
21
|
Mobahat M, Narendran A and Riabowol K:
Survivin as a preferential target for cancer therapy. Int J Mol
Sci. 15:2495–2516. 2014. View Article : Google Scholar
|
|
22
|
Concha-Benavente F, Srivastava RM, Ferrone
S and Ferris RL: EGFR-mediated tumor immunoescape: The imbalance
between phosphorylated STAT1 and phosphorylated STAT3.
Oncoimmunology. 2:e272152013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhou J, Liu Y, Wang T, Zhang H, Du M,
Zhang S, Wu S, Song S, Liu B, Zhang H and Jiang Z: Serum HER2 ECD
level and its clinical significance in advanced breast cancer
patients with different molecular subtypes. Zhonghua Yi Xue Za Zhi.
94:1384–1387. 2014.(In Chinese). PubMed/NCBI
|
|
24
|
Parums DV: Current status of targeted
therapy in non-small cell lung cancer. Drugs Today (Barc).
50:503–525. 2014. View Article : Google Scholar : PubMed/NCBI
|