Introduction
Measles is a highly contagious infectious disease
caused by the measles virus (MV), which spreads easily through
coughs and sneezes from infected individuals. Although the measles
vaccine has made an important contribution to the prevention and
control of measles, there is still a long way to go to eliminate
the disease (1). In 2014, an
estimated 114,900 measles-associated deaths were reported across
the globe, with most of these deaths occurring in children aged
<5 years (2). In developed
countries, deaths caused by measles account for 0.1–0.2% of the
population; however, measles-associated mortality may be as high as
10% in low-income areas with a lack of adequate healthcare
(3–5). According to the Chinese Health
Statistics Yearbook, in 2013, the incidence rate of measles was
0.46 per 100,000 Chinese citizens, with a fatality rate of 0.13%
(6–9). Therefore, a further reduction of the
morbidity and mortality associated with measles is required.
However, the complex pathogenesis of measles and the factors
affecting it at the genetic, biochemical and biomechanical level,
have remained to be fully elucidated.
With the continuous development of genomics
technologies, high-throughput microarray and sequencing technology
has also been applied to study gene expression in measles
infections. Microarray analysis has also been increasingly used to
explore disease-associated genes and pathways that may uncover the
molecular mechanisms of measles infection (10). The GSE5808 microarray dataset was
used to analyze the association between differentially expressed
genes (DEGs) in different phases of measles infection and
MV-induced immunological changes in a previous study, which
suggested that certain genes may exhibit an extensive
downregulation of signaling and transcriptional pathways associated
with immunoregulation during the rash and convalescent phases of
measles (10). These studies
suggested that genes involved in immune activation and cytokine
responses may have key roles in measles infection. However, the
exact biological functions of these genes in the development of
measles and whether these genes may be used as candidates for
measles prevention and treatment, requires more detailed analysis.
However, measles is the result of interactions between genes and
environmental factors. The knowledge on the interaction between
environmental factors and DEGs obtained from genome-wide gene
expression profiling studies is currently limited.
The present study used microarray data and
comprehensive bioinformatics analyses to identify DEGs between
healthy controls and measles patient samples at hospital entry.
Furthermore, a functional enrichment analysis for DEGs, a
functional module analysis of the protein-protein interaction (PPI)
network and a change trend profile analysis of the DEGs in
different stages of measles infection was performed. With these
comprehensive analyses, the present study aimed to identify key
genes that may have roles in the development of measles virus
infection and to clarify the interaction networks between
environmental factors and DEGs, which may provide further
information to better elucidate the mechanisms of measles and
potential molecular targets for measles diagnosis and
treatment.
Materials and methods
Data sources
The gene expression profile dataset GSE5808
deposited by Zilliox et al (10) was downloaded from the Gene Expression
Omnibus (GEO) database of the National Center of Biotechnology
Information (NCBI) (http://www.ncbi.nlm.nih.gov/geo/). The data were
produced by an Affymetrix Human Genome U133A Array (HG-U133A;
Affymetrix; Thermo Fisher Scientific, Inc., Waltham, MA, USA) based
on the GPL96 platform, which is an efficient and robust tool for
assessing gene expression changes across the human genome. This
dataset was subjected to a Bioinformatics analysis in the present
study. The microarray included >1,000,000 unique oligonucleotide
features covering >39,000 transcript variants, which in turn
represent >33,000 of the best characterized human genes. The
GSE5808 dataset included data from 3 healthy pediatric control
subjects and from 5 pediatric patients hospitalized with measles at
the University Teaching Hospital in Lusaka (Lusaka, Zambia) with
samples taken at hospital entry, hospital discharge, and 1-month
follow-up. The demographic characteristics of the subjects are
presented in Table I.
 | Table I.Demographics of the cohort subjected
to microarray analysis. |
Table I.
Demographics of the cohort subjected
to microarray analysis.
| Group/age
(months) | Sex |
|---|
| Measles |
|
| 21 | M |
| 79 | F |
|
9 | M |
| 11 | F |
|
8 | M |
| Control |
|
| 26 | F |
| 15 | M |
|
9 |
|
The mRNA from peripheral blood mononuclear cells
(PBMCs) was used for detection. All samples were normalized with
quantile normalization and summarized using robust multi-array
analysis. The information on the 22,283 genes in GSE5808 was
employed for bioinformatics analysis in the present study.
Data pre-processing and differential
expression analysis
All the raw expression data from GSE5808 was
preprocessed using R software (www.bioconductor.org) and normalization was conducted
using the robust multiarray average algorithm (RMA). RMA is an
algorithm used to create an expression matrix from Affymetrix data.
The raw intensity values are background corrected, log2 transformed
and quantile normalized. A linear model is then fitted to the
normalized data to obtain an expression measure for each probe set
on each array (http://www.molmine.com/magma/loading/rma.htm). To
explore the DEGs between patients with measles upon hospital
admission, at discharge and one month after discharge,
respectively, and the control group, the Student's t-test method
implemented in the Linear Models for Microarray Data package
(http://www.bioconductor.org/packages/release/bioc/html/limma.html)
in R (v.3.4.0) (https://www.r-project.org/) and gene-cloud of
biotechnology information (GCBI; https://www.gcbi.com.cn/gclib/html/index.) were
utilized to calculate the significant P-values of the DEGs. The
cut-off criteria for DEG screening were selected as follows:
P<0.05, false discovery rate (FDR)<0.05 and the absolute
value of fold-change (FC) ≥2. Subsequently, the DEGs were further
identified based on the gene symbols from the original gene
expression profile data.
Identification environmental chemicals
affecting DEGs associated with measles
Environmental chemicals that may interact with
measles-associated DEGs were accessed from the Comparative
Toxicogenomics Database (CTD; http://ctdbase.org/), which provides information about
interactions between chemicals and gene products and their
association with diseases.
On 31 January 2017, the CTD content contained
>30.5 million toxicogenomic connections. First, significant DEGs
between the controls and patients with measles at hospital entry
were defined by P<0.05, FDR<0.05 and the absolute value of
FC≥2. Subsequently, environmental chemicals which interacted with
measles-associated DEGs were identified from the CTD database.
Data analysis
Changes in the peripheral blood transcriptional
profiles of patients with measles at different stages were analyzed
using R and GCBI. To further understand the molecular functions and
biological processes of the DEGs in the blood of patients with
measles, Gene Ontology (GO) enrichment analysis and protein-protein
interaction (PPI) analysis were performed to identify Kyoto
Encyclopedia of Genes and Genomes (KEGG) pathways and an
interaction network for different stages of measles infection. GO
terms included three categories: Biological process, cellular
component and molecular function. Fisher's exact test (two-sided)
or the χ2 test was performed to classify the GO and KEGG
pathway categories. The FDR (indicated by the Q-value) was used for
P-value correction. Significantly enriched GO terms and KEGG
pathways (P≤0.05; number of enriched genes, ≥2) were identified.
STRING 10.5 (http://string-db.org/), a database of
known and predicted protein interactions that covers >9.6
million proteins from ≥2,000 organisms, was also used in the
analyses. The protein interaction reliability score was 0.700 (high
confidence).
Results
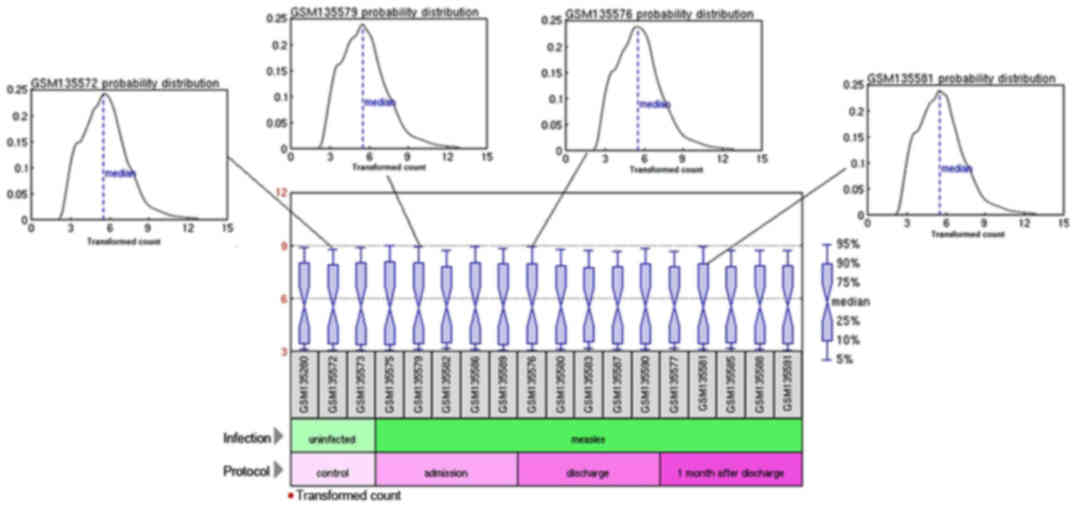
Data pre-processing
The standardized expression profiling data after
pre-processing are presented in Fig.
1. The volatility of data after normalization was greatly
reduced and the medians of the expression values of all genes are
on the same line, which provided more reliable data and information
for the subsequent analyses.
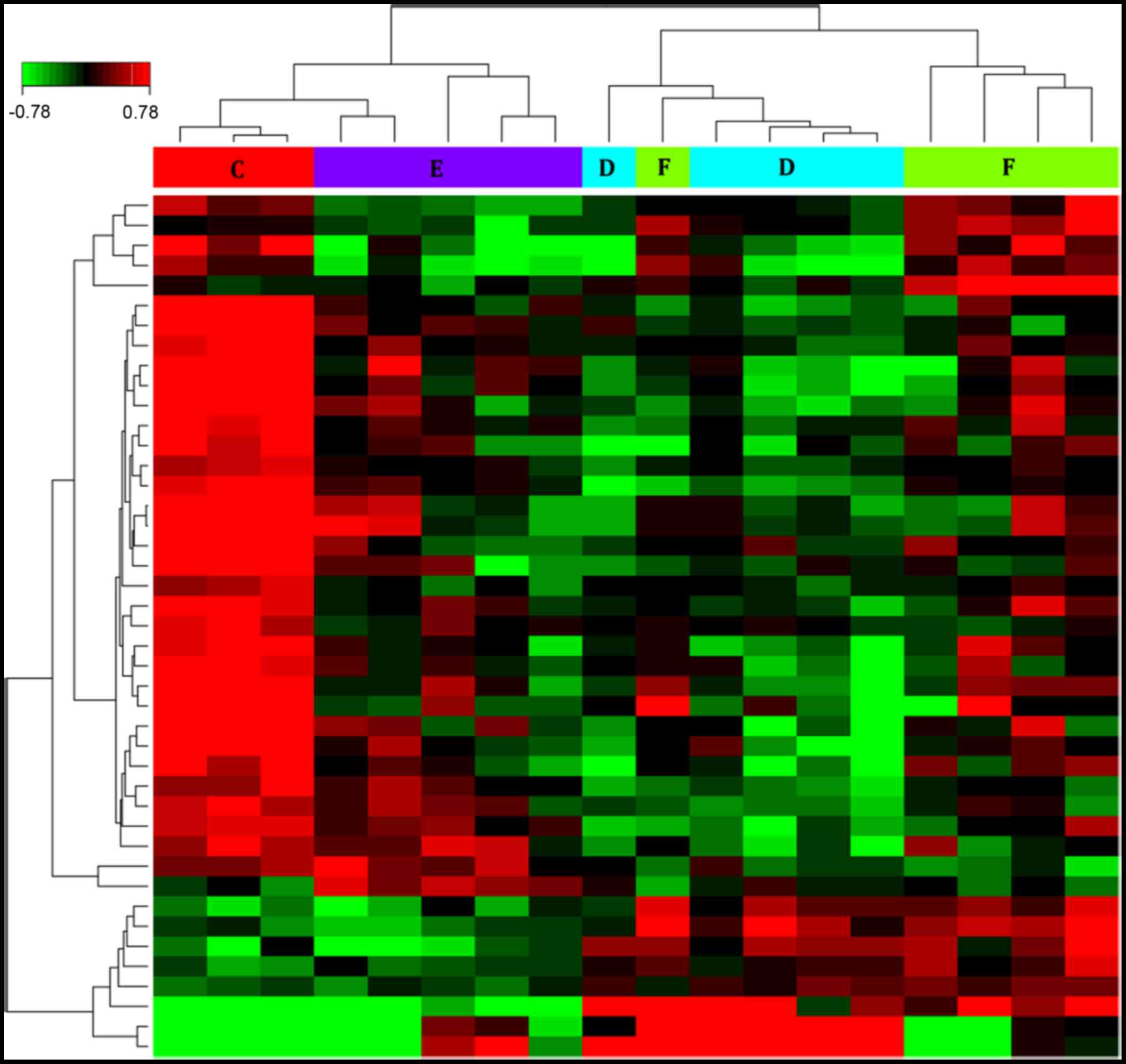
Identification of DEGs in different
stages of measles
A total of 43 DEGs were obtained from the groups
(patients with measles vs. healthy controls), including 10
upregulated genes and 33 downregulated genes. The heat map in
Fig. 2 depicts the DEGs in the
different groups determined by clustering analysis. The heat map of
the DEGs at different stages of measles infection and the healthy
controls indicated that, compared with the controls, 33 genes had a
decreased expression when the patients entered hospital, reached a
minimum value at discharge and then increased, while 10 genes
exhibited the opposite trend.
GCBI was used to explore and examine the temporal
expression pattern of all significant DEGs to identify other trends
in measles-associated gene expression. Each profile contained
clusters of multiple genes that had similar temporal expression
patterns. The analysis identified that the 43 DEGs were involved in
a total of 26 clusters (Fig. 3).
Series test cluster (STC) (11)
indicated that the P-values (The P-value is p(X≥t(mi)),
X~Bin(|G|, Ei/|G|) and is the significant level of
single model profile. t(mi) is the number of genes in
the mith model profile. |G| and Ei/|G| are
parameters of binomial distribution which the number of genes in
the model profile should obey.) (11) of clusters 3, 21 and 5 were all
<0.05, which meant that those clusters exhibited most potential
gene expression trends of patients with measles at different
stages. Clusters 3 and 5 were comprised of genes that were
downregulated in measles. Cluster 21 was comprised of genes that
were clearly upregulated in patients with measles when they were
admitted to hospital and discharged. The gene speckle-type POZ
protein (SPOP) is included in profiles 3 and 5, indicating that
SPOP is co-expressed in these two profiles.
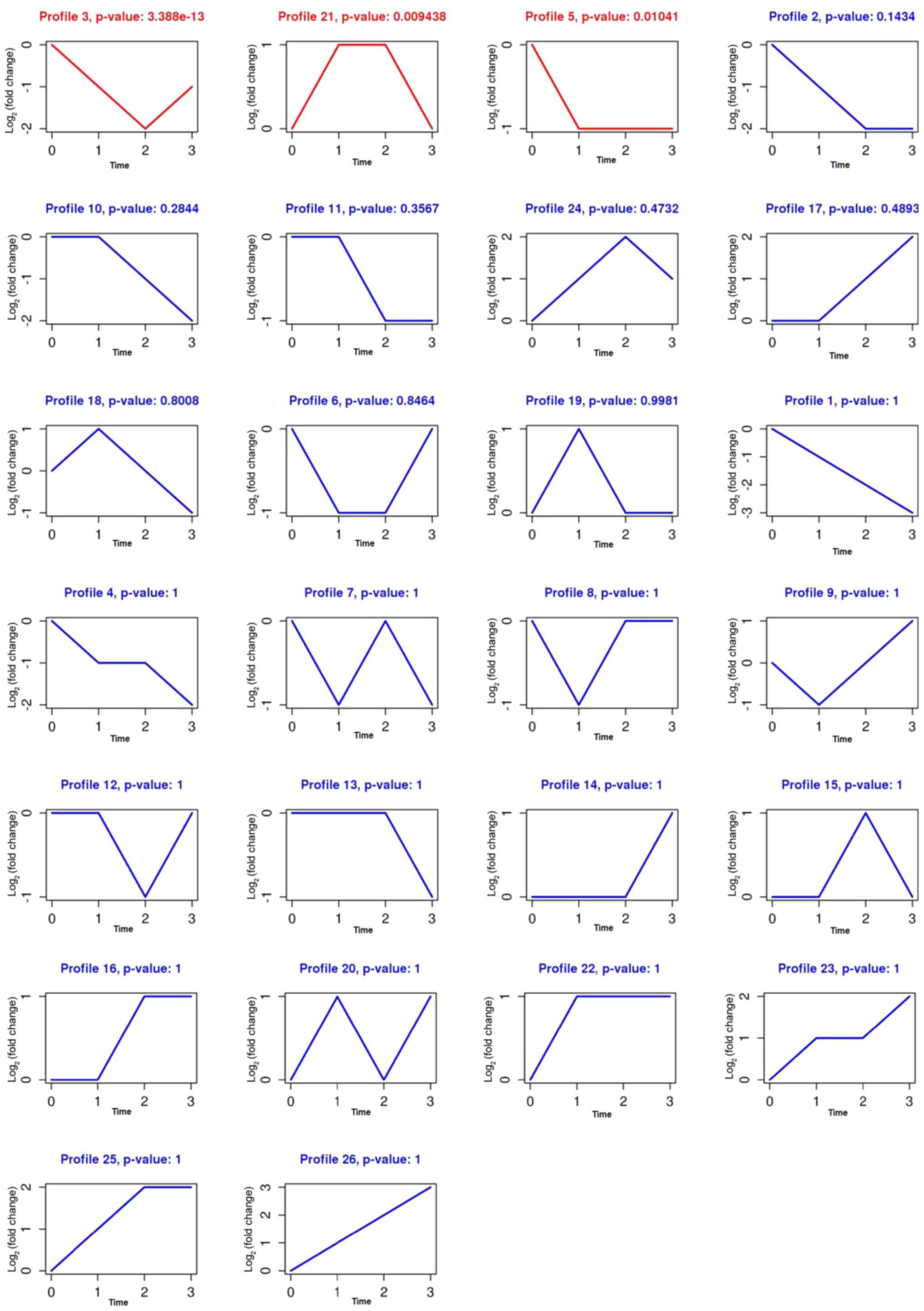 | Figure 3.Profiles containing a cluster of
multiple genes that have similar expression patterns were
identified by gene-cloud of biotechnology information (0, control;
1, admission; 2, discharge; 3, 1 month after discharge). Of these
clusters, profiles 3, 5 and 21 are statistically significant (The
three Ps refer to profiles 3, 5 and 21<0.05). Clusters 3 and 5
were comprised of genes that were downregulated in patients with
measles, but when the patients were discharged, the genes in
cluster 3 had the lowest expression. Cluster 21 was comprised of
genes that were clearly upregulated in patients with measles when
they were admitted to hospital and discharged. The gene SPOP is
included in profiles 3 and 5, indicating that SPOP is co-expressed
in these profiles. SPOP, speckle-type POZ protein. |
GO enrichment analysis
The top five enriched terms in molecular function
and biological process and two enriched terms in cellular component
are listed in Table II.
| Table II.Enrichment terms in molecular
function, biological process and cellular component. |
Table II.
Enrichment terms in molecular
function, biological process and cellular component.
| GO ontologies | GO name | P-value | FDR | Gene number |
|---|
| Molecular
function | Protein binding | 0.0000 | 0.0006 | 16 |
|
| Chemokine
activity | 0.0016 | 0.0331 | 2 |
|
| HMG box domain
binding | 0.0003 | 0.0121 | 2 |
|
| Peptide disulfide
oxidoreductase activity | 0.0018 | 0.0331 | 1 |
|
| Type III
transforming growth factor beta receptor binding | 0.0018 | 0.0331 | 1 |
| Cellular
component | Nucleus | 0.0000 | 0.0014 | 15 |
|
| Cytoplasm | 0.0001 | 0.0030 | 13 |
| Biological
process | Positive regulation
of signal transduction | 0.0001 | 0.0209 | 2 |
|
| Cellular component
movement | 0.0002 | 0.0209 | 3 |
|
| Embryonic
hemopoiesis | 0.0003 | 0.0209 | 2 |
|
| Lens development in
camera-type eye | 0.0005 | 0.0209 | 2 |
|
| Response to
activity | 0.0005 | 0.0209 | 2 |
The results demonstrated that the DEGs (patients
with measles vs. Control) exhibited many functions, including
protein binding, chemokine activity, HMG box domain binding,
peptide disulfide oxidoreductase activity and type III transforming
growth factor beta receptor binding. Biological process were
predominantly invovled in the positive regulation of signal
transduction, cellular component movement, embryonic hemopoiesis,
lens development in camera-type eye and response to activity. DEGs
were primarily enriched in two cellular sites: The nucleus and the
cytoplasm.
KEGG enrichment analysis
The most enriched KEGG pathways were fatty acid
elongation, cytokine-cytokine receptor interaction and RNA
degradation. The three enriched KEGG pathways are listed in
Table III.
| Table III.Enriched KEGG pathways. |
Table III.
Enriched KEGG pathways.
| Pathway ID | Pathway name | Number of DEGs in
pathway | P-value | FDR | Genes |
|---|
| 62 | Fatty acid
elongation | 2 | 0.0003851 | 0.015018644 | ACOT7, HADH |
| 4060 | Cytokine-cytokine
receptor interaction | 3 | 0.0034654 | 0.049204993 | CCL4, TGFBR2,
CCL20 |
| 3018 | RNA
degradation | 2 | 0.003785 | 0.049204993 | TOB2, LSM1 |
PPI network analysis
PPI network analysis was performed using the encoded
proteins from the 43 DEGs based on the STRING 9.05 database of
known and predicted protein interactions. The results are presented
in Fig. 4.
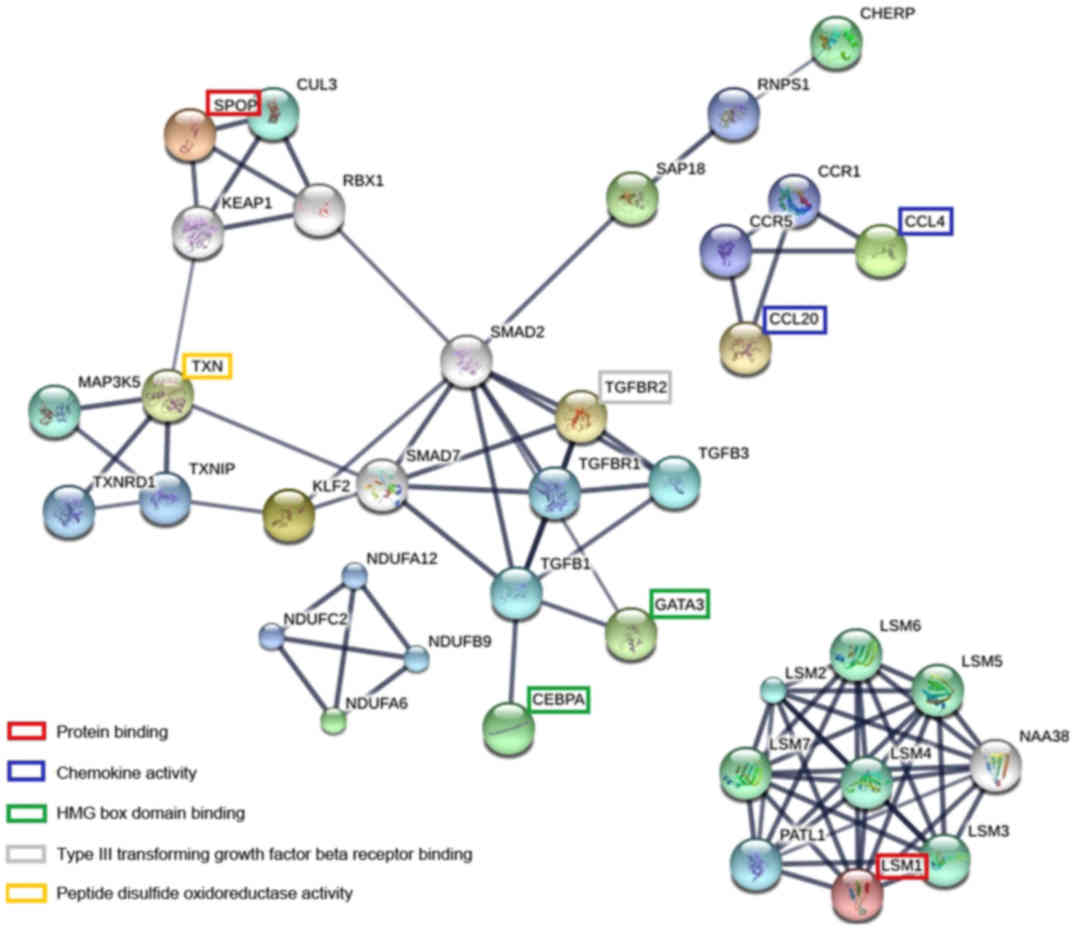 | Figure 4.PPI network (confidence, ≥0.70; PPI
enrichment P=4.28×10−11) of 38 coding proteins of the 43
DEGs generated with STRING. The network was expanded by including
20 additional partner proteins (CCR1 and CCR5, C-C motif chemokine
receptor; CUL3, Cullin 3; LSM2, LSM3, LSM4, LSM5, LSM6 and LSM7,
LSMs homolog, mRNA degradation associated; MAP3K5,
mitogen-activated protein kinase kinase kinase 5; NDUFA12,
Ubiquinone oxidoreductase subunit A12; NDUFB9, Ubiquinone
oxidoreductase subunit B9; NDUFC2, Ubiquinone oxidoreductase
subunit C2; PATL1, PAT1 homolog 1, processing body mRNA decay
factor; RNPS1, RNA binding protein with serine rich domain 1;
TGFB1, transforming growth factor beta 1; TGFB3, transforming
growth factor beta 3; TGFBR1, transforming growth factor beta
receptor 1; TXNIP, thioredoxin interacting protein; TXNRD1,
thioredoxin reductase 1) that share similar physiological functions
to the included DEGs. The genes with names are mainly associated
with protein binding and chemokine activity. PPI, Protein-protein
interaction; DEG, Differentially expressed gene. SPOP, Speckle-type
POZ protein. |
STRING interaction network analysis was performed
for the 43 identified DEGs in the four groups and 28 encoded
proteins were identified. Subsequently, the PPI network was widened
by adding 20 partner proteins (CCR1, CCR5, C-C motif chemokine
receptor; CUL3, Cullin 3; LSM2, LSM3, LSM4, LSM5, LSM6 and LSM7,
LSMs homolog mRNA degradation associated; MAP3K5, mitogen-activated
protein kinase kinase kinase 5; NDUFA12, Ubiquinone oxidoreductase
subunit A12; NDUFB9, Ubiquinone oxidoreductase subunit B9; NDUFC2,
Ubiquinone oxidoreductase subunit C2; PATL1, PAT1 homolog 1,
processing body mRNA decay factor; RNPS1, RNA binding protein with
serine rich domain 1; TGFB1, transforming growth factor beta 1;
TGFB3, transforming growth factor beta 3; TGFBR1, transforming
growth factor beta receptor 1; TXNIP, thioredoxin interacting
protein; TXNRD1, thioredoxin reductase 1) with similar
physiological functions to each other. A PPI with a confidence of
≥0.7 and degree of interaction of nodes of >5 was considered as
the key central node in the network in Fig. 4. The genes in the PPI network were
mainly associated with chemokine activity and protein binding.
Identification of chemicals that
interact with DEGs
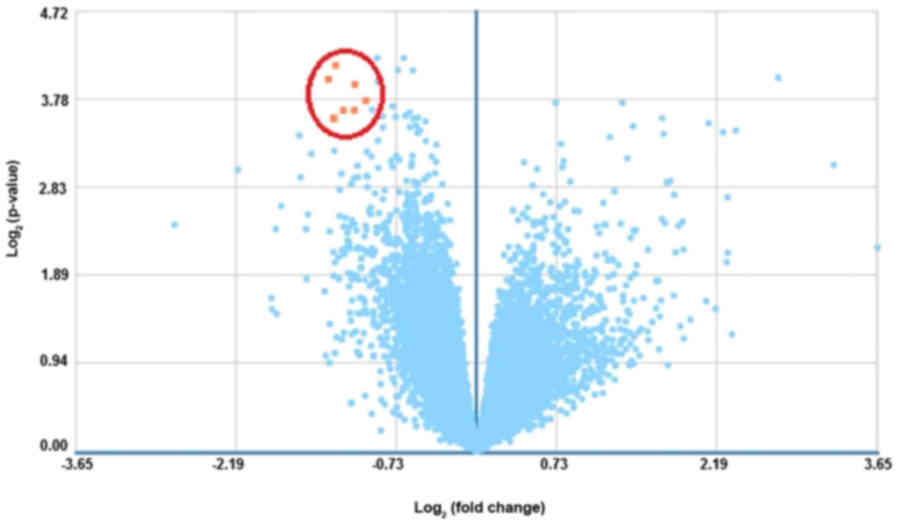
A total of 7 DEGs (Q<0.05; P<0.05; FC>2)
between the control group and patients with measles at the
time-point of hospital entry are displayed in Table IV (as GTPase, IMAP family members
1–5 were detected detected as a whole in a probe, no detailed
analysis of these genes was performed (Six genes in Table IV were analyzed in detail). These 6
DEGs all exhibited reduced expression compared with the control
group, as indicated in Fig. 5.
| Table IV.Six DEGs between control group and
patients with measles when they entered hospital. |
Table IV.
Six DEGs between control group and
patients with measles when they entered hospital.
| Gene
abbreviation | Gene
definition | Fold-change | P-value | FDR |
|---|
| ANKRD46 | Ankyrin repeat
domain 46 | −2.435009 | 0.0001 | 0.0000 |
| HLA-DPB1 | Major
histocompatibility complex, class II, DP β1 | −2.157143 | 0.0001 | 0.0000 |
| SPOP | Speckle-type POZ
protein | −2.013933 | 0.0002 | 0.0411 |
| KLF12 | Kruppel-like factor
12 | −2.164959 | 0.0002 | 0.0411 |
| ZBTB18 | Zinc finger and BTB
domain containing 18 | −2.319983 | 0.0002 | 0.0411 |
| TGFBR2 | Transforming growth
factor, β receptor II | −2.465444 | 0.0003 | 0.0411 |
A total of 219 chemicals were identified that may
either singly or jointly interact with the 6 DEGs between control
group and patients with measles on admission. Chemicals that may
affect the expression of >3 DEGs are listed in Table V. The results indicated that
benzo(a)pyrene (BaP) and tetrachlorodibenzodioxin (TCDD) interact
with the expression of 6 DEGs (Ankyrin repeat domain 46, Major
histocompatibility complex class II, DP β1, Speckle-type POZ
protein, Kruppel-like factor 12, Zinc finger and BTB domain
containing 18, Transforming growth factor and β receptor II), and
these DEGs may have an important role in the development of measles
infection.
| Table V.Identified differently expressed
environmental response genes in the blood samples of patients with
measles (upon hospital entry) and healthy controls. |
Table V.
Identified differently expressed
environmental response genes in the blood samples of patients with
measles (upon hospital entry) and healthy controls.
| Environmental
response DEGs between measles and controls | Chemicals |
|---|
| ANKRD46,
HLA-DPB1, KLF12, SPOP, TGFBR2, ZBTB18 | Benzo(a)pyrene |
|
|
Tetrachlorodibenzodioxin |
| ANKRD46,
HLA-DPB1, KLF12, TGFBR2, ZBTB18 | Valproic acid |
| ANKRD46, KLF12,
SPOP, TGFBR2, ZBTB18 | Cyclosporine |
|
| Pirinixic acid |
|
|
Tetradecanoylphorbol acetate |
|
| Acetaminophen |
|
| Ionomycin |
|
| Bisphenol A |
| ANKRD46,
HLA-DPB1, SPOP, TGFBR2 | Antirheumatic
agents |
| ANKRD46, KLF12,
SPOP, TGFBR2 | Cadmium
chloride |
|
| Diuron |
|
| Copper sulfate |
|
| Nanotubes,
carbon |
|
| Phenobarbital |
|
| Estradiol |
|
|
2,4,5,2′,4′,5′-hexachlorobiphenyl |
| HLA-DPB1, KLF12,
SPOP, ZBTB18 | Methyl
methanesulfonate |
| ANKRD46,
HLA-DPB1, TGFBR2 | Potassium
chromate(VI) |
|
| Isotretinoin |
|
| Dexamethasone |
|
| Carbamazepine |
|
|
N,N,N',N'-tetrakis(2-pyridylmethyl)ethylenediamine |
|
| Methotrexate |
| ANKRD46, SPOP,
TGFBR2 |
N-Methyl-3,4-methylenedioxyamphetamine |
|
| Oxygen |
|
|
7,8-Dihydro-7,8-dihydroxybenzo(a)pyrene
9,10-oxide |
|
|
Dextroamphetamine |
|
| Trichostatin A |
|
| Decitabine |
|
| Cobaltous
chloride |
|
| Dietary fats |
|
|
9,10-Dimethyl-1,2-benzanthracene |
|
| vinclozolin |
|
| Ethinyl
estradiol |
|
| C646 compound |
| ANKRD46, KLF12,
TGFBR2 | Tretinoin |
|
| Arsenic
trioxide |
|
| Propiconazole |
|
| Quercetin |
|
| Vancomycin |
|
|
Diethylnitrosamine |
|
| Carbon
tetrachloride |
|
| Vanadates |
|
| Tobacco smoke
pollution |
|
|
Lipopolysaccharides |
|
| Isoproterenol |
|
| Gentamicins |
| ANKRD46, TGFBR2,
ZBTB18 | Ammonium
chloride |
|
| 8-Bromo cyclic
adenosine monophosphate |
| KLF12, SPOP,
ZBTB18 | Dibutyl
phthalate |
|
| Oxaliplatin |
Discussion
Measles, caused by the MV, is an acute infection
that mainly affects children. Measles is a highly contagious
disease and despite the availability of an effective vaccine,
measles was estimated to be a direct cause of 114,900 mortalities
in 2014 (2). At present, measles has
become a significant public health issue worldwide. Although the
burden of measles has decreased significantly during the last few
decades due to the success of global measles control programs,
there is still a long way to go before the goal of measles
eradication is achieved. At present, a number of studies have
suggested that specific gene expression signatures are produced
during pathogen infection in a variety of tissues (12–14).
Zilliox et al (10) reported
that gene expression patterns during measles infection reflect
immune responses, and that extensive downregulation of signaling
and transcription pathways may be associated with these immune
responses. However, specific gene expression signatures in the
human genome and the mechanism of the immune response in measles
infection have remained to be fully elucidated.
Studies on the molecular regulation of measles
infection have made marked progress in recent years. To better
characterize MV-induced immunological changes at the
transcriptional level, microarrays have been used to survey global
mRNA levels in PBMCs during acute measles infection and
convalescence (11). In order to
obtain more valuable information, state-of-the-art bioinformatics
analysis techniques were employed in the present study to
re-analyze these massive gene expression datasets. A total of 43
DEGs were identified in samples from patients with measles (at
hospital entry and discharge) compared with the control samples,
including 10 upregulated genes and 33 downregulated genes, which
suggests that in the acute phase of measles, the gene expression
profiles of the patients had changed and may have been associated
with the body's immune status.
Previous clinical studies suggest that at different
stages of the disease, the disease characteristics have different
manifestations, including gene transcription (15–17). In
the present study, 26 clusters with different gene expression
patterns at different stages of measles infection were identified
by GCBI and profiles 3, 5 and 21 were statistically significant.
Profiles 3 and 5 comprised genes that were downregulated in
patients with measles infection, but when the patients were
discharged, the genes in profile 3 had the lowest expression
levels. Cluster 21 comprised genes that were clearly upregulated in
patients with measles when they were admitted to hospital and
discharged. The SPOP gene is included in profiles 3 and 5,
indicating that SPOP is involved in the two profiles. SPOP was
first reported as a mutated gene in human prostate cancer and its
substrates are implicated in several essential cellular functions,
including apoptosis, secondary messenger formation and
transcriptional repression (18–20). The
results of the present analysis indicated that SPOP expression in
PBMCs from patients with measles was significantly lower than that
in healthy controls.
Chemokines are a large family of proteins (6–14 kDa)
that mediate a wide range of biological activities, particularly in
the normal immune system (21).
Chemokines are essential for immune system architecture and
development, and immune surveillance, which are mediated by the
chemokine signaling pathway (22).
Chemokines activate leukocytes and stimulate various effector
functions of these cells (23). The
results of the present study indicated that the chemokine signaling
pathway may have an important role in MV-induced immunological
changes, which may help to explain, at least partly, the prolonged
alteration of lymphocyte responses that are characteristic of
measles.
BaP is a member of the benzopyrenes, which are
formed by the fusion of a benzene ring to pyrene. BaP is considered
to be important due to its toxicity and abundance in the
environment, and has been reported to induce immune suppression,
phototoxicity, as well as neurological and reproductive dysfunction
in in vivo experiments (24–26).
However, to the best of our knowledge, no previous study has
reported on the association between BaP and infectious diseases.
However, the immune suppression induced by BaP in animals or humans
may affect the development of infectious diseases. For instance,
BaP may inhibit white blood cells from differentiating into
macrophages, which may affect the first line of defense to fight
infections in the body (27). The
molecular mechanisms of the effects of BaP on the immune system
have remained elusive. The results of the present study indicated
that the 6 DEGs between patients with measles and healthy controls
are all able to interact with BaP, which suggests that upon MV
infection of the body, BaP may serve a role in the occurrence and
development of measles. TCDDs, also called dioxins, are
environmental, highly lipophilic and persistent pollutants usually
emitted from combustion facilities. TCDDs may contaminate the food
chain and accumulate in adipose tissues, and the diet is considered
one of the major sources of exposure to TCDD for the general
population (28). TCDDs are known to
be endocrine-disrupting chemicals and are suspected to increase the
risk of cancer, including breast cancer (29–31). To
the best of our knowledge, no epidemiological or basic research
studies have reported an association between TCDD or dioxin
exposure and measles infection. The present results indicated that
TCDD also interacts with 6 DEGs between patients with measles and
healthy controls, and these interactions may affect the immune
system directly or indirectly (for instance, they may first disrupt
the normal biological activities through the chemokine pathway),
which may increase the risk of measles development.
In conclusion, the present study performed detailed
bioinformatics analyses of a dataset from the NCBI's GEO database
to identify key biomarkers and their interactions with
environmental chemicals in the development of measles. The
genome-wide analysis included differential expression analysis, GO
enrichment analysis, KEGG analysis and PPI analysis, as well as an
analysis of the interaction of the DEGs with environmental
chemicals. The present results may provide insight into the genetic
basis of measles infection and the possible role of environmental
chemicals in the development of measles. Further functional studies
are required to validate the results of the present study and
further elucidate the mechanisms of measles infection.
Acknowledgements
Not applicable.
Funding
This study was funded by Youth Research Project of
Shaanxi University of Chinese Medicine (grant number:
2015NQ05).
Availability of data and materials
The analyzed data sets generated during the study
are available from the corresponding author on reasonable
request.
Authors' contributions
RZ and FL planned the study. RZ, FL, HJ, JW, NS, YD,
XM, and DR performed the data analysis, wrote and modified the
manuscript. RZ and HJ submitted the study.
Ethical approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
World Health Organization, . Measles Fact
Sheets. 2016.http://www.who.int/mediacentre/factsheets/fs286/en/September
29–2017
|
|
2
|
Centers for Disease Control and Prevention
(CDC): Measles-United States, 2011. MMWR Morb Mortal Wkly. Rep.
61:253–257. 2012.
|
|
3
|
World Health Organization: Measles-Rubella
Bulletin. 2017.http://iris.wpro.who.int/bitstream/handle/10665.1/13539/Measles-Rubella-Bulletin-2017-Vol-11-No-01.pdf?ua=1September
29–2017
|
|
4
|
No authors listed: Measles vaccines: WHO
position paper. Wkly Epidemiol Rec. 84:349–360. 2009.(In English,
French). PubMed/NCBI
|
|
5
|
Zheng J, Zhou Y, Wang H and Liang X: The
role of the China experts advisory committee on immunization
program. Vaccine. 28 Suppl 1:A84–A87. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang RQ, Li HB, Li FY, Han LX and Xiong
YM: Epidemiological characteristics of measles from 2000 to2014:
Results of a measles catch-up vaccination campaign in Xianyang,
China. J Infect Public Health. 10:624–629. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li L, Yu WZ, Shui TJ, Ma C, Wen N and
Liang XF: Analysis on epidemiological characteristics of age
distribution of measles in China during 2003–2006. Chin J Vaccines
Immun. 13:101–105. 2007.
|
|
8
|
Wang Z, Yan R, He H, Li Q, Chen G, Yang S
and Chen E: Difficulties in eliminating measles and controlling
rubella and mumps: A Cross-sectional study of a first measles and
rubella vaccination and a second measles, mumps, and rubella
vaccination. PLoS One. 9:e893612014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu Y, Lu P, Hu Y, Wang Z, Deng X, Ma F,
Tao H, Jia C, Ding X, Yang H, et al: Cross-sectional surveys of
measles antibodies in the Jiangsu Province of China from 2008 to
2010: The effect of high coverage with two doses of measles vaccine
among children. PLoS One. 8:e667712013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zilliox MJ, Moss WJ and Griffin DE: Gene
expression Changes in Peripheral blood mononuclear cells during
measles virus infection. Clin Vaccine Immunol. 14:918–923. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xiao S, Mo D, Wang Q, Jia J, Qin L, Yu X,
Niu Y, Zhao X, Liu X and Chen Y: Aberrant host immune response
induced by highly virulent PRRSV identified by digital gene
expression tag profiling. BMC Genomics. 11:5442010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Honda M, Kaneko S, Kawai H, Shirota Y and
Kobayashi K: Differential gene expression between chronic hepatitis
B and C hepatic lesion. Gastroenterology. 120:955–966. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sankaran S, Guadalupe M, Reay E, George
MD, Flamm J, Prindiville T and Dandekar S: Gut mucosal T cell
responses and gene expression correlate with protection against
disease in long-term HIV-1-infected nonprogressors. Proc Natl Acad
Sci USA. 102:9860–9865. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yu SY, Hu YW, Liu XY, Xiong W, Zhou ZT and
Yuan ZH: Gene expression profiles in peripheral blood mononuclear
cells of SARS patients. World J Gastroenterol. 11:5037–5043. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rota PA, Liffick SL, Rota JS, Katz RS,
Redd S, Papania M and Bellini WJ: Molecular epidemiology of measles
viruses in the United States, 1997–2001. Emerg Infect Dis.
8:902–908. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rota PA, Featherstone DA and Bellini WJ:
Molecular epidemiology of measles virus. Curr Top Microbiol
Immunol. 330:129–150. 2009.PubMed/NCBI
|
|
17
|
Riddell MA, Rota JS and Rota PA: Review of
the temporal and geographical distribution of measles virus
genotypes in the prevaccine and postvaccine eras. Virol J.
2:872005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Blattner M, Liu D, Robinson BD, Huang D,
Poliakov A, Gao D, Nataraj S, Deonarine LD, Augello MA, Sailer V,
et al: SPOP mutation drives prostate tumorigenesis in vivo through
coordinate regulation of PI3K/mTOR and AR signaling. Cancer Cell.
3:436–451. 2017. View Article : Google Scholar
|
|
19
|
Zhang L, Peng S, Dai X, Gan W, Nie X, Wei
W, Hu G and Guo J: Tumor suppressor SPOP ubiquitinates and degrades
EglN2 to compromise growth of prostate cancer cells. Cancer Lett.
390:11–20. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhi X, Tao J, Zhang L, Tao R, Ma L and Qin
J: Silencing speckle-type POZ protein by promoter hypermethylation
decreases cell apoptosis through upregulating Hedgehog signaling
pathway in colorectal cancer. Cell Death Dis. 7:e25692016.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rump L, Mattey DL, Kehoe O and Middleton
J: An initial investigation into endothelial CC chemokine
expression in the human rheumatoid synovium. Cytokine. 97:133–140.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zheng Y, Han GW, Abagyan R, Wu B, Stevens
RC, Cherezov V, Kufareva I and Handel TM: Structure of CC Chemokine
receptor 5 with a potent chemokine antagonist reveals mechanisms of
chemokine recognition and molecular mimicry by HIV. Immunity.
46:1005–1017.e5. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Prabhakaran S, Rizk VT, Ma Z, Cheng CH,
Berglund AE, Coppola D, Khalil F, Mulé JJ and Soliman HH:
Evaluation of invasive breast cancer samples using a 12-chemokine
gene expression score: Correlation with clinical outcomes. Breast
Cancer Res. 19:712017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Arfsten DP, Schaeffer DJ and Muneny DC:
The effects of near ultraviolet radiation on the toxic effects of
polycyclic aromatic hydrocarbons in animals and plants: A review.
Ecotoxicol Environ Saf. 33:1–24. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Saunders CR, Ramesh A and Shockley DC:
Modulation of neurotoxic behavior in F-344 rats by temporal
disposition of benzo(a)pyrene. Toxicol Lett. 129:33–45. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yamaguchi K, Near R, Shneider A, Cui H, Ju
ST and Sherr DH: Fluoranthecene-induced apoptosis in murine T cell
hybridomas is independent of the aromatic hydrocarbon receptor.
Toxicol Appl Pharmacol. 139:144–152. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kang HG, Jeong SH, Cho MH and Cho JH:
Changes of biomarkers with oral exposure to benzo(a)pyrene,
phenanthrene and pyrene in rats. J Vet Sci. 8:361–368. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Fries GF: A review of the significance of
animal food products as potential pathways of human exposures to
dioxins. J Anim Sci. 73:1639–1650. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kogevinas M: Human health effects of
dioxins: Cancer, reproductive and endocrine system effects. Hum
Reprod Update. 7:331–339. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Schug TT, Janesick A, Blumberg B and
Heindel JJ: Endocrine disrupting chemicals and disease
susceptibility. J Steroid Biochem Mol Biol. 127:204–215. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liem AK, Fürst P and Rappe C: Exposure of
populations to dioxins and related compounds. Food Addit Contam.
17:241–259. 2000. View Article : Google Scholar : PubMed/NCBI
|