Introduction
Hypophosphatemic osteomalacia (HO) is an uncommon
metabolic disease characterized by low concentrations of serum
phosphate levels, which leads to reduced mineralization of the bone
matrix (1). It may affect
individuals of all ages and either gender. Typically, HO is either
inherited (2), a result of
tumor-induced osteomalacia (TIO) (3), drug-induced (4) or a symptom of chronic kidney disease
(5). Patients with inherited or TIO
have been widely reported on by endocrinology or oncology
specialists, however HO is often misdiagnosed in clinical practice
as ankylosing spondylitis (AS), chronic arthritis, lumbar disc
disease, osteoporosis and somatoform disorder, as it typically
presents with the same signs and symptoms of these rheumatologic
diseases, including bone pains, thoracic or back pain, muscle
weakness, proximal myopathy and arthralgia (6). Diagnosis of HO remains a challenge to
rheumatologists and physicians due to its low prevalence and
nonspecific manifestations. Screening blood tests for electrolytes,
particularly serum phosphate and bone mineral density (BMD) is
basic clue for diagnosis. Screening for hidden tumors, which are
classified as phosphaturic mesenchymal or phosphaturic mesenchymal
tumor mixed connective tissue variants is important for patients
with HO without an obvious etiology or history. The majority of the
tumors are of bone or soft tissue origin and positron emission
tomography (PET)/computed tomography (CT) scans may be used to
identify them (3). The prognosis of
the disease depends on the etiology. The treatment or removal of
secondary etiologies, including drugs and tumors has been reported
to be particularly effective at improving the condition. It is
necessary to supply basic supplementation to all patients with
sufficient oral phosphate, elemental calcium and active vitamin D
(7). The present study reported 9
misdiagnosed cases of HO in order to improve the recognition of
this disease amongst rheumatologists and physicians. Adefovir
dipivoxil-induced Fanconi syndrome was present in 6 of the cases, 2
were caused by tumors and 1 case was due to chronic
nephropathy.
Case report
Patients
A total of 9 patients with terminal diagnoses of HO
were diagnosed and treated in the Department of Rheumatology at
Guangdong General Hospital (Guangzhou, China) between January 2011
to August 2015. All patients had clear etiologies and no family
history of the disease. The length of disease history ranged from
10 months to 5 years, and the male-to-female ratio was 7:2. Patient
ages ranged from 22 to 55 years (mean, 40.9 years). Patients with
endocrine or metabolic diseases were excluded. All patients were
negative for autoantibodies and human leukocyte antigen-B27.
Methods
The whole diagnosis and treatment information of the
9 cases were retrospectively analyzed, including etiology, patient
complaints, clinical manifestations, physical examinations,
laboratory and radiology examinations, bone mineral density
examinations, and all treatments and prognoses. A literature review
was also conducted.
Etiology
As demonstrated in Table
I, all patients had acquired HO, and 6 patients with chronic
hepatitis B had adefovir dipivoxil-induced Fanconi syndrome. The
duration of drug treatment was 4–6 years (mean, 4.8 years) and the
duration of symptoms was 2–4 years (mean, 3.5 years). A total of 2
cases presented with tumors (one giant cell tumor in the forearm
tendon sheath and one sub-skull tumor). Furthermore, 1 case
presented with chronic nephropathy with insufficient function and
tubule acidosis.
 | Table I.General characteristics of patients
with hypophosphatemic osteomalacia enrolled in the present
study. |
Table I.
General characteristics of patients
with hypophosphatemic osteomalacia enrolled in the present
study.
| Case | Sex | Age, years | Duration of
complaint | Etiology | Misdiagnosis | Total drug history,
years | Drug exposure to
symptom, years |
|---|
| 1 | F | 39 | 3 years | Tumor (giant cell
tumor) | Lumbar disc
disease, | – | – |
|
|
|
|
|
| osteoporosis |
|
|
| 2 | M | 47 | 5 years | Tumor
(sub-skull) | AS, osteoporosis | – | – |
| 3 | M | 43 | 2 years | Drug-induced | Lumbar disc
disease, | 6 | 4 |
|
|
|
|
| Fanconi syndrome | somatoform disorder,
AS |
|
|
| 4 | M | 43 | 2 years | Drug-induced fanconi
syndrome | Chronic
arthritis | 4 | 2 |
| 5 | M | 34 | 17 months | Chronic nephropathy
with acidosis | AS | – | – |
| 6 | M | 22 | 10 months | Drug-induced fanconi
syndrome | AS, chronic
arthritis | 4 | 3 |
| 7 | M | 50 | 1 year | Drug-induced fanconi
syndrome | Hematological
disease | 6 | 5 |
| 8 | F | 35 | 2 years | Drug-induced fanconi
syndrome | AS, osteoporosis | 4 | 2 |
| 9 | M | 55 | 1 year | Drug-induced fanconi
syndrome | Lumbar disc
disease, | 5 | 4 |
|
|
|
|
|
| osteoporosis |
|
|
Clinical manifestations
All cases developed gradually and presented with
thoracic and back pain and arthralgia of the hips, feet or
shoulders, which was accompanied by gradually aggravated muscle
weakness and severe limitation of movement. In 1 case, foot
numbness developed with muscle spasms, and 1 case presented with
dramatically decreased height, weakness in chewing and worn
teeth.
Course of treatment and
misdiagnosis
Of the patients, 5 were misdiagnosed with AS, 3 of
which accepted etanercept treatment. Furthermore, 2 cases had
chronic arthritis, 3 had lumbar vertebral disc disease, 4 had
primary osteoporosis and 1 had somatoform disorder (Table I). These patients were transferred
between 3–6 hospitals and departments, including orthopedics, the
Traditional Chinese Medicine Department, psychology, nephrology and
rheumatology. This is a common practice in China, as due to the
organization of the medical system the patients are able to freely
select their hospital. The longest period of misdiagnosis was 5
years, with no effective treatment.
Laboratory examination
As demonstrated in Table
II, basic laboratory examinations were conducted on all
patients and the results were as follows: Serum phophatase (P),
0.37–0.72 mmol/l (mean, 0.55 mmol/l); serum Ca, 2.01–2.28 mmol/l
(mean, 2.16 mmol/l); parathyroid hormone, 17.8–108.7 pg/ml (mean,
43.1 pg/ml; normal range, 15–65 pg/ml); and alkaline phosphatase,
155–492 U/l (mean, 273.6 U/l;. In 2 patients, an insufficiency of
25-OH vitamin D (VitD) was observed. Results of 24 h urine P were
within 21.8–60.04 mmol (normal, 3–42 mmol/24-h urine) in 6 cases
(mean, 43. mmol). Serum fibroblast growth factor-23 (FGF-23) levels
were not monitored.
| Table II.Biochemical test results for patients
with hypophosphatemic osteomalacia enrolled in the present
study. |
Table II.
Biochemical test results for patients
with hypophosphatemic osteomalacia enrolled in the present
study.
|
|
Biochemical
parameter |
|---|
|
|
|
|---|
| Case | Serum P, mmol/l | Serum Ca, mmol/l | PTH, pg/ml | ALP, U/l | Cl−,
mmol/l | Plasma pH | Plasma BE,
mmol/l | Serum
HCO3−, mmol/l | 24 h U P, mmol | U glucose,
mmol/l | U protein, g/l | U pH |
|---|
| 1 | 0.40 | 2.18 | 95.0 | 155 | – | – | – | – | 60.0 | 56 | – | – |
| 2 | 0.40 | 2.28 | 108.7 | 230 | 106.6 | 7.45 |
1.7 | – | 24.7 | – | – | – |
| 3 | 0.72 | 2.01 |
19.2 | 271 | 110.8 | 7.30 |
−4.3 | 19.7 | 23.3 | 3 | 0.75 | 8.0 |
| 4 | 0.54 | 2.05 |
21.6 | 343 | 108.6 | – | – | – | – | – | – | 7.5 |
| 5 | 0.57 | 2.13 |
17.8 | 185 | 120.1 | 7.24 | −13.9 | 12.0 | – | – | 0.25 | 9.0 |
| 6 | 0.37 | 2.14 |
50.9 | 492 | 113.6 | 7.31 |
−9.1 | 17.1 | – | 56 | 0.75 | 7.0 |
| 7 | 0.65 | 2.23 |
28.8 | 352 | 109.2 | 7.26 |
−3.2 | 21.1 | 22.5 | 3 | 0.25 | 7.0 |
| 8 | 0.60 | 2.20 |
21.4 | 259 | 112.3 | 7.30 |
−4.2 | 20.5 | 24.4 | 5 | 0.75 | 7.0 |
Fanconi syndrome-related
examination
A total of 6 cases with hepatitis B had drug-induced
Fanconi syndrome (7), induced by
adefovir dipivoxil. Blood gas analyses revealed that blood pH was
7.24–7.45 (mean, 7.28; normal, 7.35–7.45), Cl− was
106.6–120.1 mmol/l (mean, 110.8 mmol/l; normal, 95–105 mmol/l),
bases excess was −13.9 to 1.7 mmol/l (mean, −4.9 mmol/l; normal,
−3-3) and HCO3− was 12–21.1 mmol/l (mean, 19.7 mmol/l;
normal, 22–27 mmol/l. Urine pH was 7–9 (mean, 7.25; normal, 5–8),
urine protein was 0.25–0.75 g/l (normal, negative) and urine
glucose was 3–56 mmol/l (normal, negative; Table II).
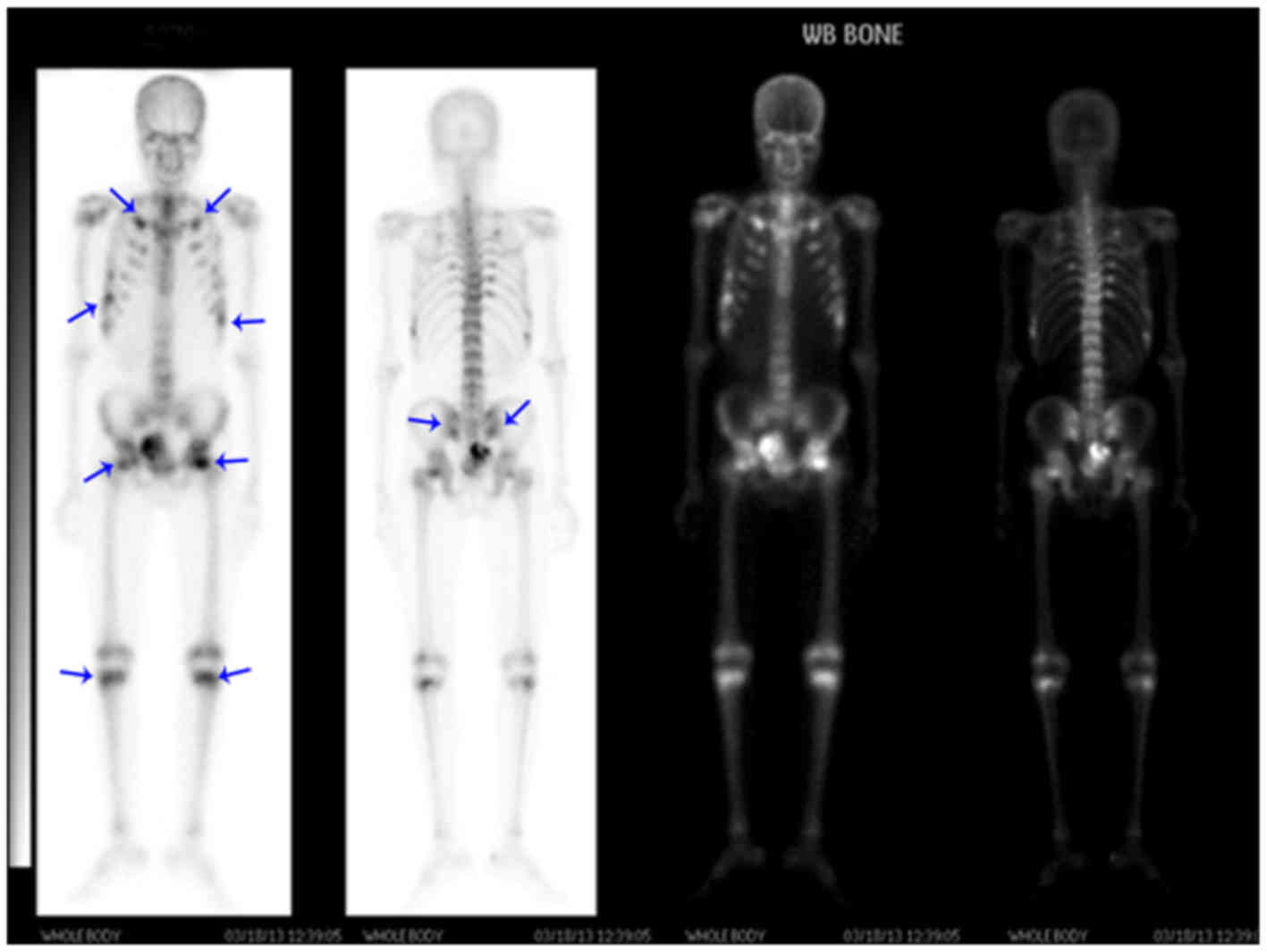
Bone scan
Bone scans revealed decreased methylene
diphosphonate uptake in all bones, and multiple hot spots of
fractured ribs and involved joints, consistent with metabolic bone
disease (Fig. 1).
Bone densitometry
All patients were demonstrated to have markedly low
bone densities, with a Z score of <-2.5 (normal, >-1).
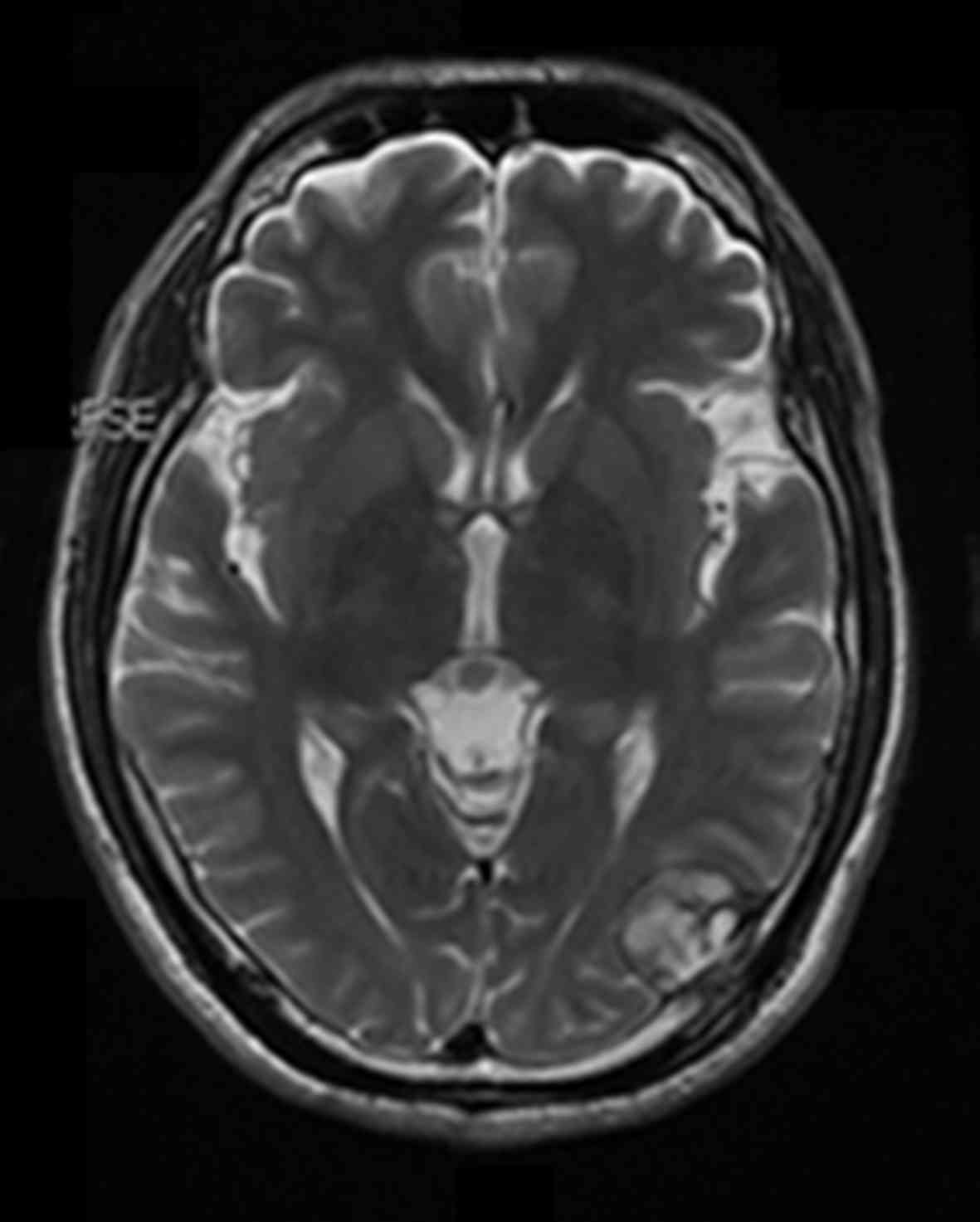
Imageology
X-ray and CT scans were performed and these revealed
multiple fractures located in the ilium, pubis, lumbar and ribs in
5 cases. Magnetic resonance imaging of the head revealed a 25×18 mm
tumor under the skull plate and outside the brain in 1 patient
(Fig. 2).
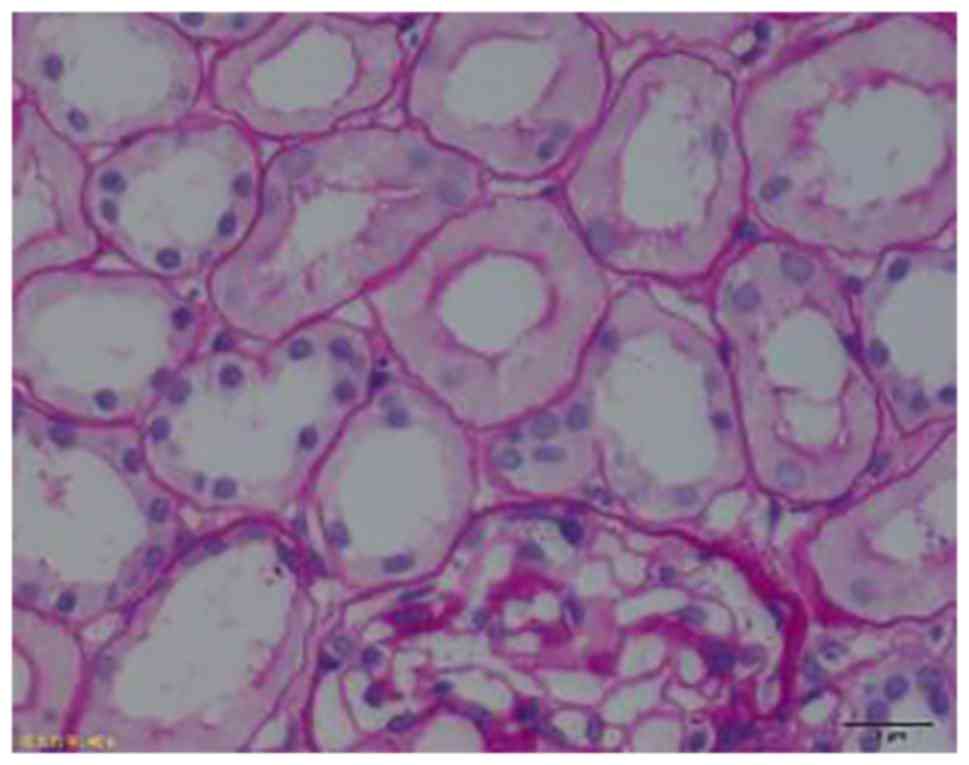
Biopsy
Two patients accepted kidney biopsies and the
results revealed proximal renal tubule lesions associated with the
Fanconi syndrome (Fig. 3) as
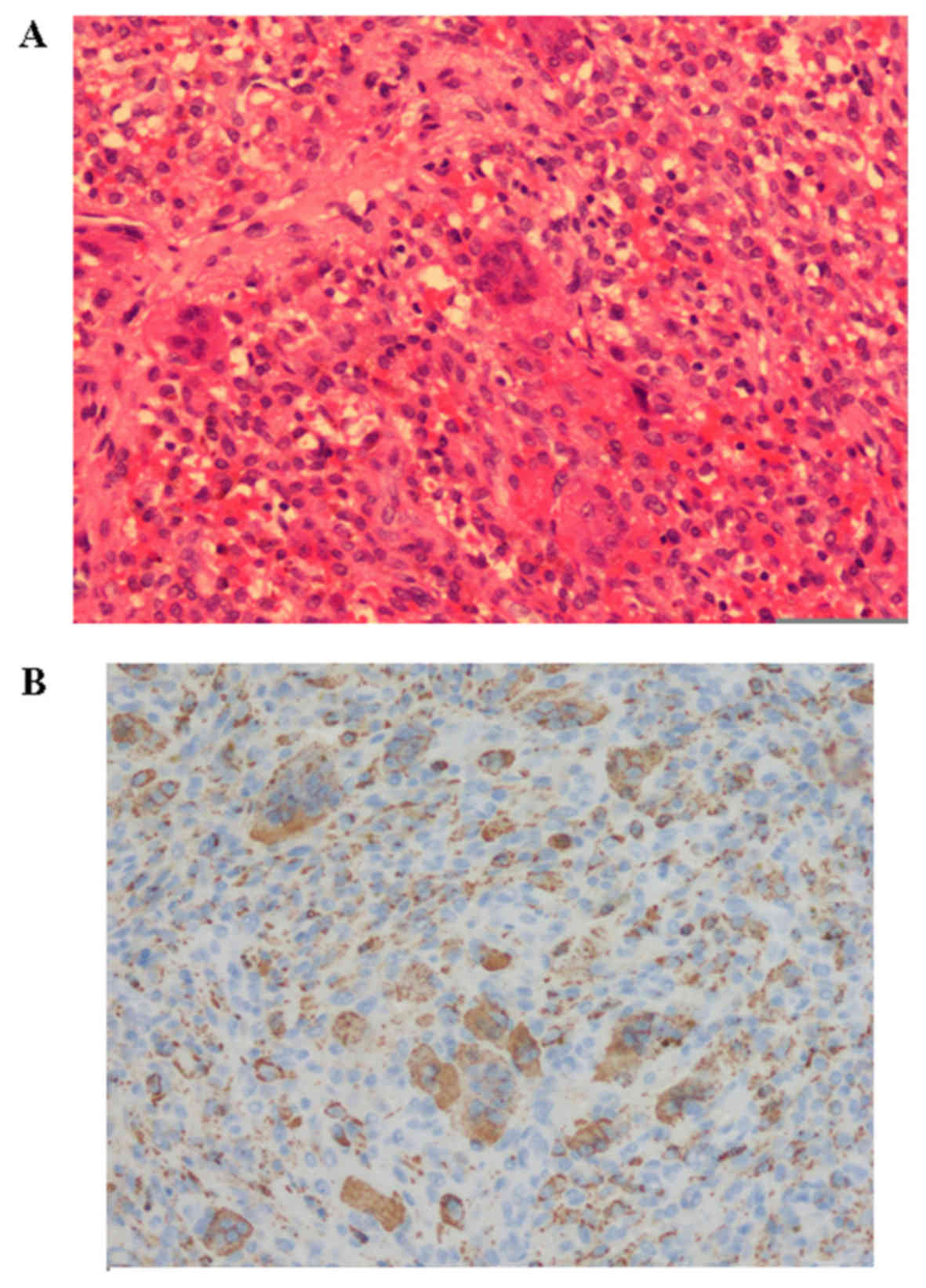
confirmed by histopathology. The tumor identified in the left
forearm tendon sheath of 1 patient was identified as a tenosynovial
giant cell tumor (Fig. 4) as
confirmed by histopatology.
Treatment and prognosis
All patients accepted basic supplementation with
oral phosphate supplements (30 doses, 50 mg/kg/day in divided
doses), elemental calcium 1 g/day and active vitamin D supplements
(calcitriol 0.5 µg/day). The 6 patients with hepatitis B ceased
treatment with adefovir dipivoxil and recovered within 6 months. In
1 patient, hypophosphatemia was completely eradicated 3 days
following tumor resection (patient was discharged after 1 week and
no further follow up was performed). The patient that presented
with a sub-skull tumor refused surgery, and at the 6 month
follow-up his height had decreased by 10 cm and he reported
prominent pain across the whole body. The patient with chronic
kidney disease and tubule acidosis had a poor prognosis at the
6-month follow-up due to insufficient kidney function.
Discussion
Hypophosphatemia is defined as a serum phosphate
level of <0.8 mmol/l (2.5 mg/dl); <0.32 mmol/l (1 mg/dl) is
regarded as severe (8). It is caused
by decreased intake and absorption of, and the increased loss or
transcellular shift of phosphorus (9). The most common causes of HO are
inherited or tumor-induced(TIO), and have been widely studied by
endocrinology or oncology specialists (2). Reports from rheumatologists of clinical
musculoskeletal manifestations of HO are rare, with the exception
of a report by Reginato et al (10). The authors of the present study
reported a case of adefovir dipivoxil-induced Fanconi syndrome and
HO in 2011 (7). In our previous
report, the literature was reviewed and it was reported that <10
similar cases of this disease had been reported in clinical
practice (7). The cases presented
with clinical manifestations that mimicked primary musculoskeletal
disease, and the course of treatment was typically difficult, which
is indicative of the lack of recognition of HO by physicians.
The renal tubule regulates the excretion and
absorption of calcium and phosphorus, which in turn affects bone
tissues (8). In clinical practice,
20–25% of males and >5% of females with osteoporosis exhibit
renal tubule disorders including phosphate wasting, hypercalciuria
and tubular acidosis (11).
Adefovir dipivoxil is commonly used for the
treatment of chronic hepatitis B (12). Its renal toxicity is dose- and
time-related, and often occurs in patients with a daily dose of
>30 mg and those with impaired renal function (13). The mechanism by which adefovir
dipivoxil causes kidney damage is that its product is mainly
excreted by the kidneys and influences the reabsorption of renal
tubule cells (13).
Hyperphosphaturia is typically accompanied by hypophosphatemia
(13).
The 6 patients with hepatitis B in the present study
were treated with normal doses (10 mg/day) of adefovir dipivoxil
for 2–4 years. This indicates that clinical attention should be
given to adefovir dipivoxil-induced kidney damage in patients with
a drug exposure >2–3 years (14),
even if the drug dosage was within the normal range. TIO typically
occurs in the bone and soft tissues of the upper or lower limbs and
skull, and rarely in the trunk and axial bone (15). A study by Jiang et al
(3) reported that the majority of
tumors (85%) in TIO were classified as phosphaturic mesenchymal
tumors or phosphaturic mesenchymal tumor mixed connective tissue
variants. These tumors are of bone (40%) or soft tissue (55%)
origin, and 42% are located in the lower extremities (3). TIO is a common cause of adult-onset
hypophosphatemia in China (15). It
was reported that 68Ga DOTA-octreotate PET/CT performed better than
18F-fluorodeoxyglucose PET/CT, and is useful for the
detection of tumors that cause oncogenic osteomalacia (15). In the majority of cases, successful
removal of tumors leads to recovery; however, long term follow-up
should be performed in case of recurrence (16).
Of the phosphate excreted by the kidney, ~80% is
reabsorbed by the proximal renal tubule through the
natrium-potassium co-transfer protein IIa (NaP IIa) (17). These TIO associated tumors excrete
FGF-23, and suppress the recruitment, and expression of NaP IIa and
the reabsorption of phosphorus (17), increasing phosphorus drainage.
Furthermore, FGF-23 influences the activity of 1-α hydroxylase in
kidneys to decrease the formation of 1.25(OH)2 D3 and
the intake of phosphate in the intestine (18). Bone mineralization is hindered by
hypophosphatemia and the insufficiency of serum
1.25(OH)2 D3 (9).
Fracture lines in osteomalacia are very common
(19), and may consist of pseudo
fractures, true fractures or insufficiency fractures. Pseudo
fractures are characteristic of HO (20). The diagnostic criteria of a pseudo
fracture are as follows: i) Imaging manifestations of osteomalacia;
and ii) the predilection site of the pseudo fracture is the pubic
branch, medial of the femur neck, medial of the femur shaft, the
femur lesser trochanter, lateral of the scapula, rib, or proximal
and posterior of the ulna. Thin slice CT scans and
three-dimensional reconstruction techniques are able to accurately
display the pseudo fracture line (20). The pseudo fracture may be used as an
imaging index to evaluate patient condition (20).
The differential diagnoses of HO include primary
osteoporosis, multiple myeloma and hyperparathyroidism with
consensus (21). However, it must be
noted that the clinical presentation of HO typically mimics
rheumatologic diseases, in particular AS, chronic arthritis or
myopathy, and primary hospitals often misdiagnose HO due to
insufficient examination. Treatment methods and prognoses depend on
the etiology of the disease, and the most effective treatments
target the underlying cause of HO (22). Clinically, physicians should perform
basic electrolyte examinations when presented with young patients
with unexplained back pain and muscle weakness. Screening tumors in
soft tissues and bones is also important to achieve early
diagnosis, effective treatment and a positive long-term prognosis
(23).
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Carpenter TO: The expanding family of
hypophosphatemic syndromes. J Bone Miner Metab. 30:1–9. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Che H, Roux C, Etcheto A, Rothenbuhler A,
Kamenicky P, Linglart A and Briot K: Impaired quality of life in
adults with X-linked hypophosphatemia and skeletal symptoms. Eur J
Endocrinol. 174:325–333. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jiang Y, Xia WB, Xing XP, Silva BC, Li M,
Wang O, Zhang HB, Li F, Jing HL, Zhong DR, et al: Tumor-induced
osteomalacia: An important cause of adult-onset hypophosphatemic
osteomalacia in China: Report of 39 cases and review of the
literature. J Bone Miner Res. 27:1967–1975. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang XB, Zhu XC, Huang XY, Ye WJ and Wang
LX: Fanconi syndrome due to prolonged use of low-dose adefovir. J
Res Med Sci. 20:416–419. 2015.PubMed/NCBI
|
|
5
|
Kazama JJ, Matsuo K, Iwasaki Y and
Fukagawa M: Chronic kidney disease and bone metabolism. J Bone
Miner Metab. 33:245–52. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yuan T, Shi L, Xia WB, Xing XP and Meng
XW: Fanconi syndrome misdiagnosed as ankylosing spondylitis for
several years. Chin J Osteoporosis and Bone Mineral Res. 4:281–284.
2012.
|
|
7
|
Li L, Dong GF, Zhang X and Xie YS:
Adefovir dipivoxil-induced Fanconi syndrome and hypophosphatemic
osteomalacia associated with muscular weakness in a patient with
chronic hepatitis B. Nan Fang Yi Ke Da Xue Xue Bao. 31:1956–1957.
2011.PubMed/NCBI
|
|
8
|
Berkelhannner C and Bear RA: A clinical
approach to common electrolyte problems: 3. Hypophosphatemia. Can
Med Assoc J. 130:17–23. 1994.
|
|
9
|
Munoz J, Ortega Michel R, Celzo F and
Donthireddy V: Tumour-induced osteomalacia. BMJ Case Rep.
25:bcr.03201259752012.
|
|
10
|
Reginato AJ, Falasca GF, Pappu R, McKnight
B and Agha A: Musculoskeletal manifestations of osteomalacia:
Report of 26 cases and literature review. Semin Arthritis Rheum.
28:287–304. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Laroche M, Cesini J and Tack I:
Osteoporosis and renal tubular Dysfunction. Joint Bone Spine.
79:S96–S98. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gara N, Zhao X, Collins MT, Chong WH,
Kleiner DE, Liang Jake T, Ghany MG and Hoofnagle JH: Renal tubular
dysfunction during long-term adefovir or tenofovir therapy in
chronic hepatitis B. Aliment Pharmacol Ther. 35:1317–1325. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Izzedine H, Hulot JS, Launay-Vacher V,
Marcellini P, Hadziyannis SJ, Currie G, Brosgart CL, Westland C,
Arterbrun S, Deray G, et al: Renal safety of asdefovir dipivoxil in
patients with chronic hepatitis B: Two double-blind, randomized,
placebo-controlled studies. Kidney Int. 66:1153–1158. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Law ST, Li KK and Ho YY: Nephrotoxicity,
including acquired fanconi's syndrome, caused by adefovir dipivoxil
is there a safe dose? J Clin Pharm Ther. 37:128–131. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Agrawal K, Bhadada S, Mittal BR, Shukla J,
Sood A, Bhattacharya A and Bhansali A: Comparison of 18F-FDG and
68Ga DOTATATE PET/CT in localization of tumor causing oncogenic
osteomalacia. Clin Nucl Med. 40:e6–e10. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ray S, Chakraborty PP, Biswas K, Beatrice
AM, Ghosh S, Mukhopadhyay S and Chowdhury S: Oncogenic osteomalacia
caused by occult nasal mesenchymal tumor: A monster in the cave.
Oxf Med Case Reports. 265–268. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jonsson KB, Zahradnik R, Larsson T, White
KE, Sugimoto T, Imanishi Y, Yamamoto T, Hampson G, Koshiyama H,
Ljunggren O, et al: Fibroblast growth factor 23 in oncogenic
osteomalacia and X-linked hypophosphatemia. N Engl J Med.
348:1656–1663. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gattineni J and Baum M: Regulation of
phosphate transport by fibroblast growth factor 23 (FGF23):
Implications for disorders of phosphate metabolism. Pediatr
Nephrol. 25:591–601. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lee YS, Kim BK, Lee HJ and Dan J:
Pathologic femoral neck fracture due to fanconi syndrome induced by
adefovir dipivoxil therapy for hepatitis B. Clin Orthop Surg.
8:232–236. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lee C and Lashari S: Pseudofracture of the
neck of femur secondary to osteomalacia. J Bone Joint Surg Br.
89:956–958. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Reilly BM, Hart PD, Mascarell S and
Chatrath H: Clinical problem-solving. A question well put. N Engl J
Med. 360:1446–1451. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
de Beur Jan SM: Tumor-induced
osteomalacia. JAMA. 294:1260–1267. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yu WJ, He JW, Fu WZ, Wang C and Zhang ZL:
Reports of 17 Chinese patients with tumor-induced osteomalacia. J
Bone Miner Metab. 35:298–307. 2017. View Article : Google Scholar : PubMed/NCBI
|