Introduction
Increasing evidence suggests that immune and
inflammatory responses serve pivotal roles in obesity, diabetes and
cardiovascular diseases (1,2). Metabolic signals emerging from
metabolic cells initiate inflammatory responses and damage
metabolic homeostasis, thus leading to infiltration of macrophages
into metabolic tissues, inflammation and insulin resistance
(1). Macrophages are essential
components of the innate immunity system, and serve a dynamic role
in host defense, inflammation and tissue homeostasis (3). Monocyte/macrophages are the major
source of inflammatory cytokines, which participate in the
pathogenesis of diabetes, obesity and insulin resistance through
paracrine and endocrine mechanisms (4–6).
The properties and activation state of macrophages
change in different local environments, thus exhibiting an
important heterogeneity (6). Under
inflammatory and noninflammatory stimuli environments, macrophages
display various status (3). M1 and
M2 are two separate polarization activation states of macrophages,
which are usually defined in vitro, while tissue macrophages
may be activated at intermediate states between M1 and M2 (7–9).
Proinflammatory mediators such as lipopolysaccharides (LPS) and
interferon-γ can induce M1 macrophages, which are also named
‘classically activated’ macrophages (7). M1 macrophages produce proinflammatory
cytokines, including tumor necrosis factor-α (TNF-α), interleukin
(IL)-6 and IL-12, and generate reactive oxygen species such as
nitric oxide (NO) via activation of inducible nitric oxide synthase
(iNOS) (7). M2, or ‘alternatively
activated’ macrophages, are generated in vitro by exposure
to IL-4 and IL-13 (4). M2
macrophages have low proinflammatory cytokine expression and
generate high levels of the anti-inflammatory cytokine IL-10
(4). Additionally, M2-polarized
macrophages can enhance arginase production. This enzyme blocks
iNOS activity by competing for the arginine substrate that is
required for NO production (9). M2
macrophages are considered to block inflammatory responses and
repair tissue during inflammatory responses as well as being
involved in the promotion of tissue repair (7–9). Upon
induction, macrophage state can switch from activated M1 state to
M2 and vice versa (3,7–9).
Glucagon-like peptide-1 (GLP-1) is secreted from
intestinal L-cells and functions in nutrient ingestion. It is
considered to have numerous glucose-lowering actions, including
potentiating glucose-dependent insulin secretion, inhibiting
glucagon secretion, enhancing β cell growth, suppressing appetite
and delaying gastric emptying (10–12).
Additionally, GLP-1 appears to improve insulin sensitivity in
patients with type 2 diabetes and animal models (13). Previous studies have demonstrated
that GLP-1 reduces the accumulation of monocytes/macrophages and
the expression of inflammatory mediators such as TNF-α and monocyte
chemotactic protein in activated macrophages (14). A previous study demonstrated that
GLP-1 reduces the numbers of M1 macrophages and the mRNA expression
levels of M1 marker genes, and reduces the expression levels of
inflammatory factors in adipose tissue and peritoneal macrophages
(15). Considering that GLP-1
receptors are abundantly expressed in the surface of numerous cell
types besides pancreatic islet cells, gastrointestinal cells,
neural cells and mononuclear macrophages, GLP-1 may serve more
important roles than expected. An in vitro study has
elucidated that GLP-1 and GLP-1 agonists increase M2
macrophage-related markers and the secretion of the
anti-inflammatory cytokine IL-10 when acting on human mononuclear
macrophages, and has observed that GLP-1 induces macrophages into
the M2 phenotype by signal transduction and transcriptional
activation factor 3 (STAT3) activation (16). It is well known that STAT3 serves a
key role in macrophage activation towards the M2 phenotype
(17). As a repressor protein of the
inflammatory response, STAT3 in resident macrophages acts as a
transcription factor mediating the anti-inflammatory effects of
IL-10 (18). STAT3 is the dominant
mediator of the anti-inflammatory effects exhibited by IL-10, which
acts to inhibit LPS-mediated TNF-α and IL-6 generation in
macrophages (19). The effects of
intracellular cAMP elevation on the production of inflammatory
mediators in macrophages were originally reported to be mediated by
protein kinase A (PKA) (20).
Furthermore, cyclic adenosine monophosphate (cAMP) is a paramount
factor for macrophage activation towards the M2 phenotype (20–22).
While the roles of STAT3 in macrophages are well
supported, little is known about how GLP-1/GLP-1 receptor (GLP-1R)
activates STAT3 signaling and the underlying mechanisms. With
regard to macrophage polarization, the effects of GLP-1 on signal
transduction have scarcely been documented to date, to the best of
our knowledge. The present study elucidated that GLP-1R signaling
contributes to the inhibition of JNK activation through the
cAMP/PKA pathway, resulting in the activation of STAT3, which
inhibits inflammation and M1 activation and promotes M2 activation.
These findings suggest that modulations of signaling pathways are
essential underlying mechanisms of GLP-1 on a broad spectrum of
metabolic diseases.
Materials and methods
Reagents
Recombinant human GLP-1 (cat. no. 130–08) and murine
IL-4 (cat. no. 214-14) were purchased from PeproTech EC Ltd.
(London, UK). LPS was purchased from Sigma-Aldrich (Merck KGaA,
Darmstadt, Germany). Enhanced BCA Protein Assay kit (cat. no.
P0010S) was purchased from Beyotime Institute of Biotechnology
(Haimen, China). Forskolin and H89/2HCl were purchased from Selleck
Chemicals (Houston, TX, USA). The anti-c-Jun N-terminal kinase
(JNK; cat. no. 9252), anti-phosphorylated JNK (cat. no. 4668),
anti-phosphorylated STAT3 (cat. no. 9145), anti-STAT3 (cat. no.
4904) and anti-GAPDH antibodies (cat. no. 2118) were all obtained
from Cell Signaling Technology, Inc. (Danvers, MA, USA). The Cyclic
AMP EIA kit (cat. no. 581001) was purchased from Cayman Chemical
Company (Ann Arbor, MI, USA).
Cells and cell culture conditions
RAW264.7 cells were provided by Professor Zheng
(Wuhan Union Hospital, Wuhan, China), and were cultured in
Dulbecco's modified Eagle's medium (DMEM)-high glucose (HG) medium
(Hyclone; GE Healthcare Life Sciences, Logan, UT, USA) supplemented
with 10% fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) and antibiotics (100 U/ml penicillin and 100 U/ml
streptomycin). Cells were maintained in an incubator at 37°C under
a humidified atmosphere of 5% CO2. RAW264.7 cells were
seeded in 6-well plates at a density of 5×105 cells/well
and the adherent cells were grown in serum-free DMEM-HG overnight
for cell growth synchronization. Then, RAW264.7 cells were cultured
with or without GLP-1 (10 nM) for 24 h at 37°C, followed by absence
or addition of LPS (100 ng/ml) or IL-4 (10 ng/ml) for inducing
polarization, and were preincubated at 37°C with the corresponding
adenylyl cyclase activator Forskolin (10 µM) or PKA inhibitor
H89/2HCl (10 µM) for 30 min prior to GLP-1 intervention.
Enzyme immunoassay (EIA) method to
measure intracellular cAMP levels
RAW264.7 cells were treated at 37°C without or with
GLP-1 (0.5, 1, 5, 10 or 30 nM) for 8 h, followed by incubation and
solubilization with 0.1 M HCl at room temperature for 20 min to
avoid degradation of cAMP at 37°C. Upon centrifugation at 1,000 × g
for 10 min at 4°C, the cAMP content of the supernatant was
quantified using the aforementioned Cyclic AMP EIA kit according to
the manufacturer's instructions. Additionally, the Forskolin (10
µM) group was used as a positive control. The optical density of
the plate was read at a wavelength of 412 nm using a
Spectrophotometer and an Absorbance Reader (each, Bio-Rad
Laboratories, Inc.). cAMP concentration was calculated using a
computer spreadsheet named EIADouble (2011) available for data
analysis (Cayman Chemical Company).
Western blot analysis
Briefly, macrophages were solubilized and total
protein was extracted with radioimmunoprecipitation assay lysis
buffer including phosphorylated protease inhibitors A and B and
phenylmethanesulfonyl fluoride (Beyotime Institute of
Biotechnology). Upon centrifugation at 13,275 × g at 4°C for 10
min, the supernatant was collected and the protein content was
quantified by employing a bicinchoninic acid protein assay kit. A
total of 25 µg protein per lane was separated by 8–10% SDS-PAGE and
transferred onto a polyvinylidene difluoride transfer membrane (EMD
Millipore, Billerica, MA, USA) by applying an electro-blotting
apparatus. Then, the membranes were blocked with 5% (w/v) skimmed
milk in TBS with Tween-20 (TBST) for 1–2 h at 37°C, followed by
incubation overnight at 4°C with the following primary antibodies:
Anti-STAT3 (1:2,000), anti-phosphorylated STAT3 (Tyr705; 1:2,000),
anti-phosphorylated JNK (Thr183/Tyr185; 1:1,000) and anti-JNK
(1:1,000). The membranes were also blotted with an anti-GAPDH
antibody (1:1,000) serving as an internal calibration control.
Subsequently, the membranes were washed with TBST and incubated
with a horseradish peroxidase-labeled goat anti-rabbit
immunoglobulin G (cat. no. GGHL-90P; 1:5,000; Immunology
Consultants Laboratory, Inc., Portland, OR, USA) at room
temperature for 1 h. The protein bands were visualized by an
enhanced chemiluminescence kit (Thermo Fisher Scientific, Inc.).
The images were captured using the GelDoc XR System (Bio-Rad
Laboratories, Inc.). Quantitative analysis of western blots was
performed using ImageJ 1.4 software (National Institutes of Health,
Bethesda, MD, USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from RAW264.7 cells with
RNAiso Plus (Takara Bio, Inc., Otsu, Japan). RT was carried out
using a PrimeScript RT Reagent kit (Perfect Real Time; Takara Bio,
Inc.) following the manufacturer's protocol. A total of 2 µg total
RNA was reverse-transcribed in a volume of 10 µl for cDNA
synthesis. The resulting cDNA was amplified using a
SYBR® Premix Ex Taq (Tli RNaseH Plus) kit (Takara Bio,
Inc.). qPCR was performed in the CFX96™ Real-Time PCR system
(Bio-Rad Laboratories, Inc.,). The PCR primers used are presented
in Table I. The reaction system
consisted of 2 µl sense primer, 2 µl anti-sense primer, 12.5 µl
SYBR® PCR Master mix, 2 µl template cDNA (25 ng/µl) and
double-distilled water to a final volume of 25 µl. PCR was
performed for 40 cycles, and each cycle included denaturation at
95°C for 5 sec, annealing and extension at 60°C for 30 sec, which
followed pre-denaturation at 95°C for 30 sec. The relative copy
number was analyzed by using the threshold crossing point (Cq),
which was calculated by the system's software, combining with the
2−ΔΔCq calculations (23).
 | Table I.Gene and primer sequences. |
Table I.
Gene and primer sequences.
| Gene | Primer
sequences |
|---|
| ARG-1 | Forward,
5′-CTCCAAGCCAAAGTCCTTAGAG-3′ |
|
| Reverse,
5′-AGGAGCTGTCATTAGGGACATC-3′ |
| MGL-1 | Forward,
5′-TGAGAAAGGCTTTAAGAACTGGG-3′ |
|
| Reverse,
5′-GACCACCTGTAGTGATGTGGG-3′ |
| MRC-1 | Forward,
5′-TGGGCTACAGGAGAACCCAACTTT-3′ |
|
| Reverse,
5′-GCAGTGGCATTGATGCTGCTGTTA-3′ |
| IL-10 | Forward,
5′-GCTCTTACTGACTGGCATGAG-3′ |
|
| Reverse,
5′-CGCAGCTCTAGGAGCATGTG-3′ |
| IL-6 | Forward,
5′-ACAAAGCCAGAGTCCTTCAGAGAG-3′ |
|
| Reverse,
5′-TTGGATGGTCTTGGTCCTTAGCCA-3′ |
| iNOS | Forward,
5′-AATCTTGGAGCGAGTTGTGG-3′ |
|
| Reverse,
5′-CAGGAAGTAGGTGAGGGCTTG-3′ |
| TNF-α | Forward,
5′-TCTCAGCCTCTTCTCATTCCTGCT-3′ |
|
| Reverse,
5′-AGAACTGATGAGAGGGAGGCCATT-3′ |
| GAPDH | Forward,
5′-TGAAGCAGGCATCTGAGGG-3′ |
|
| Reverse,
5′-CGAAGGTGGAAGAGTGGGAG-3′ |
Statistical analysis
Statistical analysis was conducted with one-way
analysis of variance followed by a Tukey's post-hoc test using SPSS
v.22 (IBM Corp., Armonk, NY, USA). P<0.05 was considered to
indicate a statistically significant difference.
Results
GLP-1 induces M2 polarization and
inhibits M1 polarization in RAW264.7 cells
In an attempt to elucidate the effects of GLP-1 on
M1/M2 polarization in RAW264.7 cells with RT-qPCR analysis,
macrophage-specific markers, including proinflammatory factors for
M1 (iNOS, IL-6 and TNF-α) and M2-specific genes IL-10, mannose
receptor-1 (MRC-1), macrophage galectin-1 (MGL-1) and arginine-1
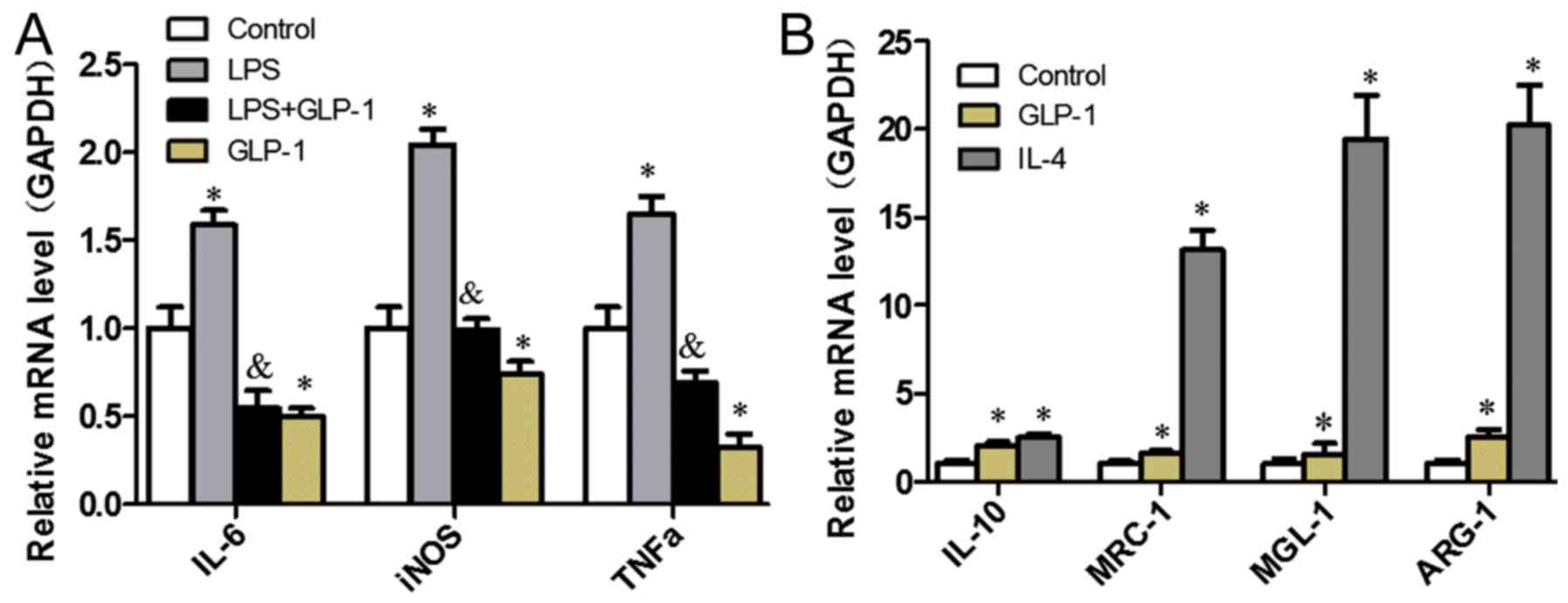
(ARG-1), were profiled. As presented in Fig. 1A, GLP-1 led to a decrease in the
expression levels of M1-specific genes when compared with the
control group (P<0.05), which were similarly reduced in cells
pretreated with GLP-1 compared with those treated with LPS
(P<0.05). GLP-1 augmented the expression levels of M2-specific
genes (Fig. 1B). These results
indicate that GLP-1 induces macrophage polarization toward M2 and
inhibits M1 polarization.
GLP-1 induces STAT3 signal activation
in RAW264.7 cells
In order to elucidate the molecular mechanisms
underlying the effect of GLP-1 on macrophage polarization, the
effect of STAT3 signaling on macrophage polarization toward M2
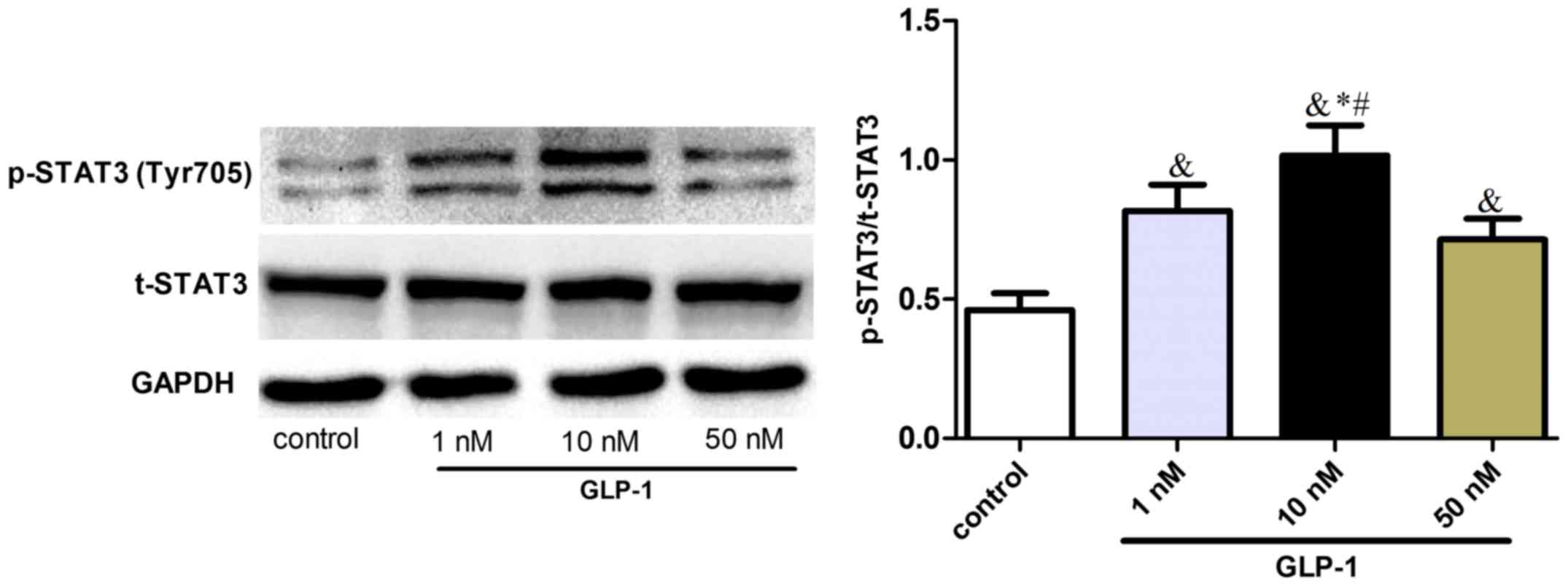
status was investigated in RAW264.7 cells. It was observed that
GLP-1 upregulated the phosphorylation of STAT3 (Tyr705) (Fig. 2), suggesting that GLP-1 could induce
STAT3 signaling activation and promote M2 activation of
macrophages.
GLP-1 suppresses JNK phosphorylation
activation and increases STAT3 signal activation in RAW264.7
cells
To elucidate the molecular mechanisms underlying
STAT3 signaling activation by GLP-1, its inhibition effects on JNK
were investigated. GLP-1 and IL-4 treatment increased STAT3
phosphorylation (P<0.05). However, JNK (Thr183/Tyr185)
phosphorylation of GLP-1 was downregulated (P<0.05), while the
total JNK protein levels were not significantly different (Fig. 3). Suppression of JNK phosphorylation
accordingly upregulated the intracellular levels of phosphorylated
STAT3 in RAW264.7 cells, suggesting that the JNK-STAT3 signaling
pathway could regulate macrophage polarization, resulting in
counteraction between M1 and M2.
 | Figure 3.GLP-1 reduces the phosphorylation of
JNK and increases the phosphorylation of STAT3 in RAW264.7 cells.
RAW264.7 cells were incubated with or without 10 nM GLP-1 for 24 h,
followed by IL-4 (10 ng/ml) for 24 h. Cell extracts were prepared
and analyzed by western blotting using anti-STAT3, anti-p-STAT3,
anti-JNK, anti-p-JNK and anti-GAPDH antibodies. To determine the
expression of phosphorylated JNK, JNK was used as the loading
control, while the expression of phosphorylated STAT3 was
determined using STAT3 as the loading control. The results are
representative of three independent experiments.
&P<0.05 vs. control group. GLP-1, glucagon-like
peptide-1; STAT3, signal transduction and transcriptional
activation factor 3; JNK, c-Jun N-terminal kinase; IL, interleukin;
p, phosphorylated; t, total. |
GLP-1/GLP-1R signaling inhibits M1
activation and induces M2 activation by cAMP/PKA-mediated JNK
downregulation in RAW264.7 cells
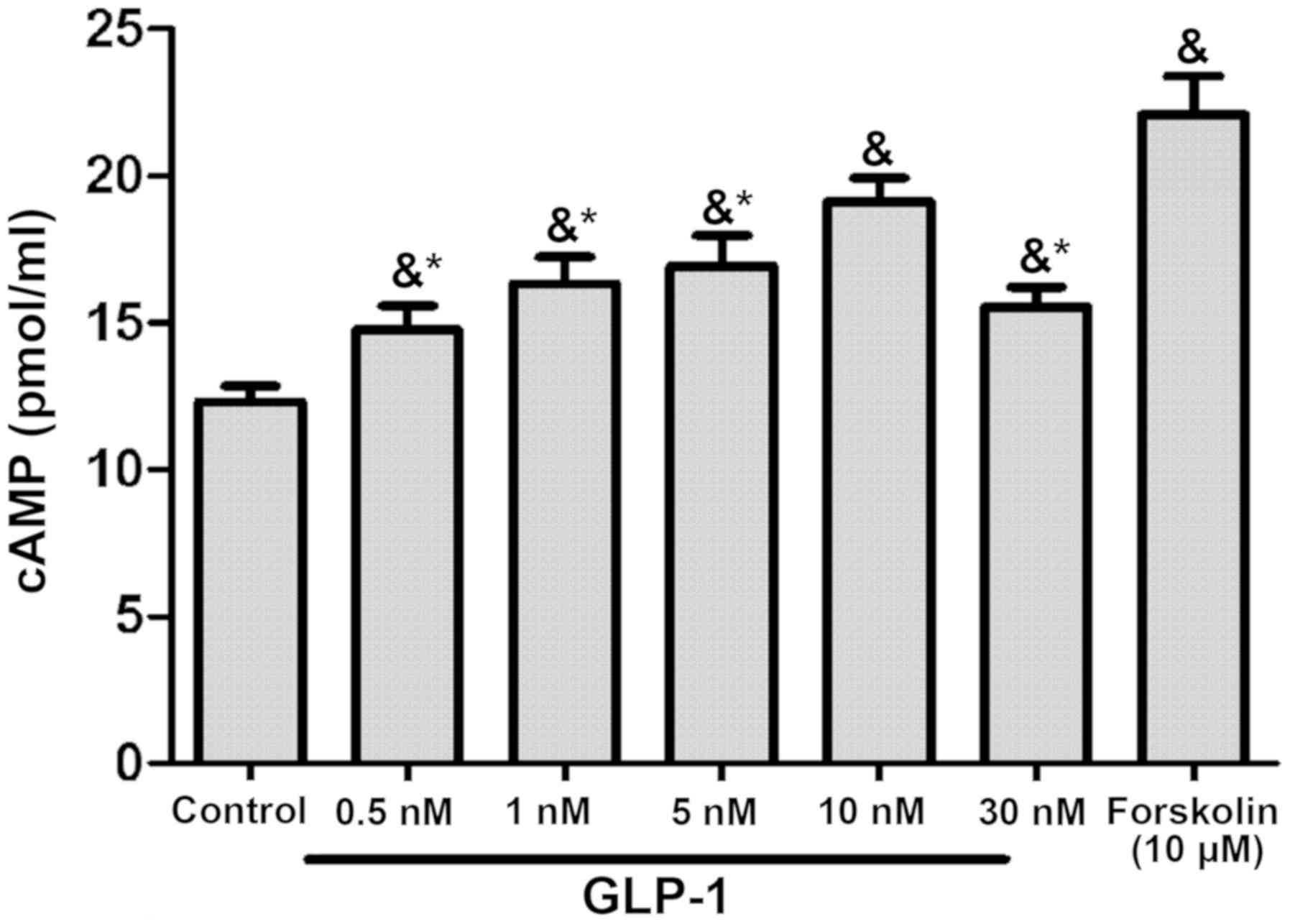
GLP-1 could increase cAMP levels in RAW264.7 cells
in a concentration-dependent manner, with the most marked effect
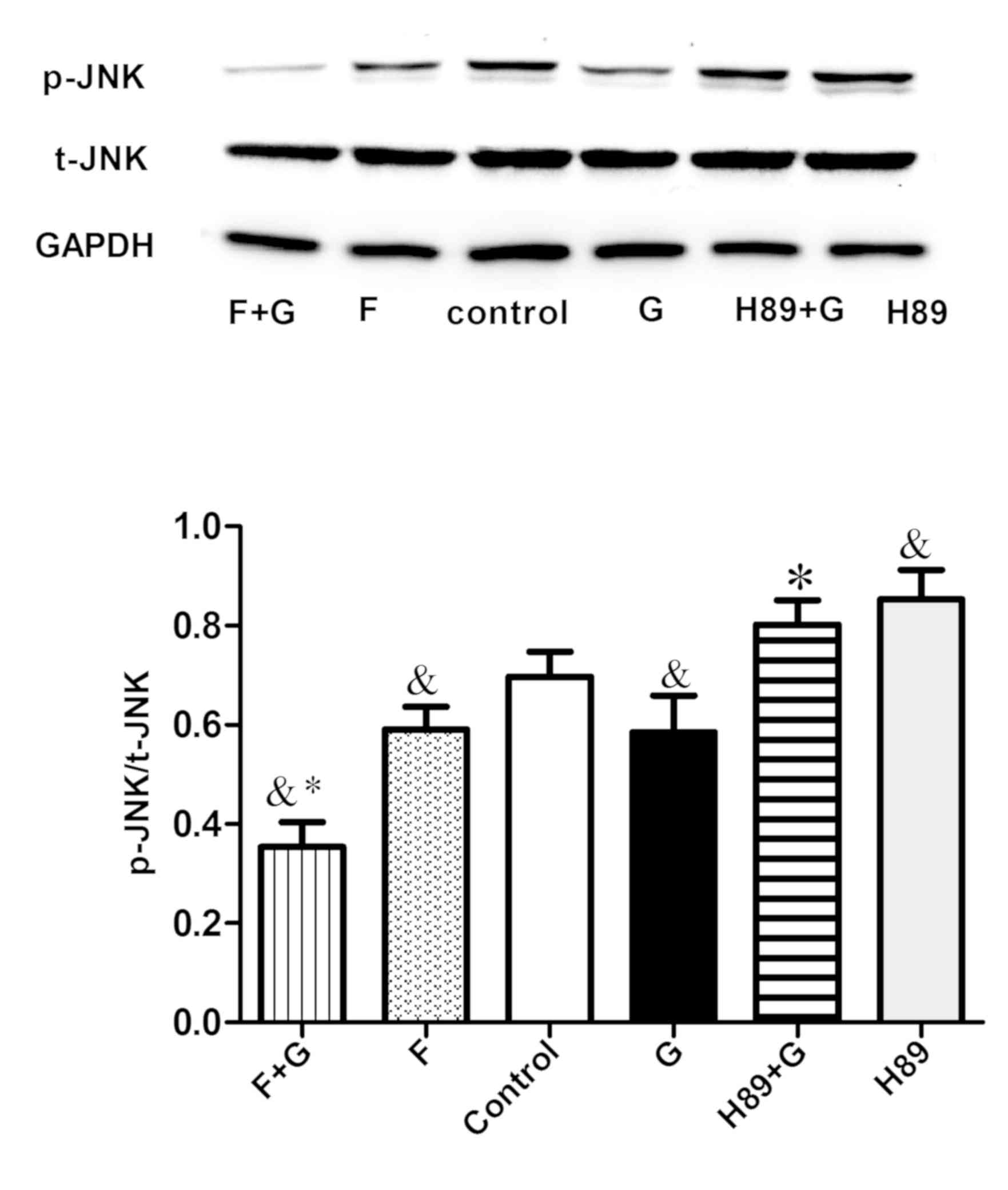
observed in the 10 nM group (P<0.05; Fig. 4). To identify the target molecule,
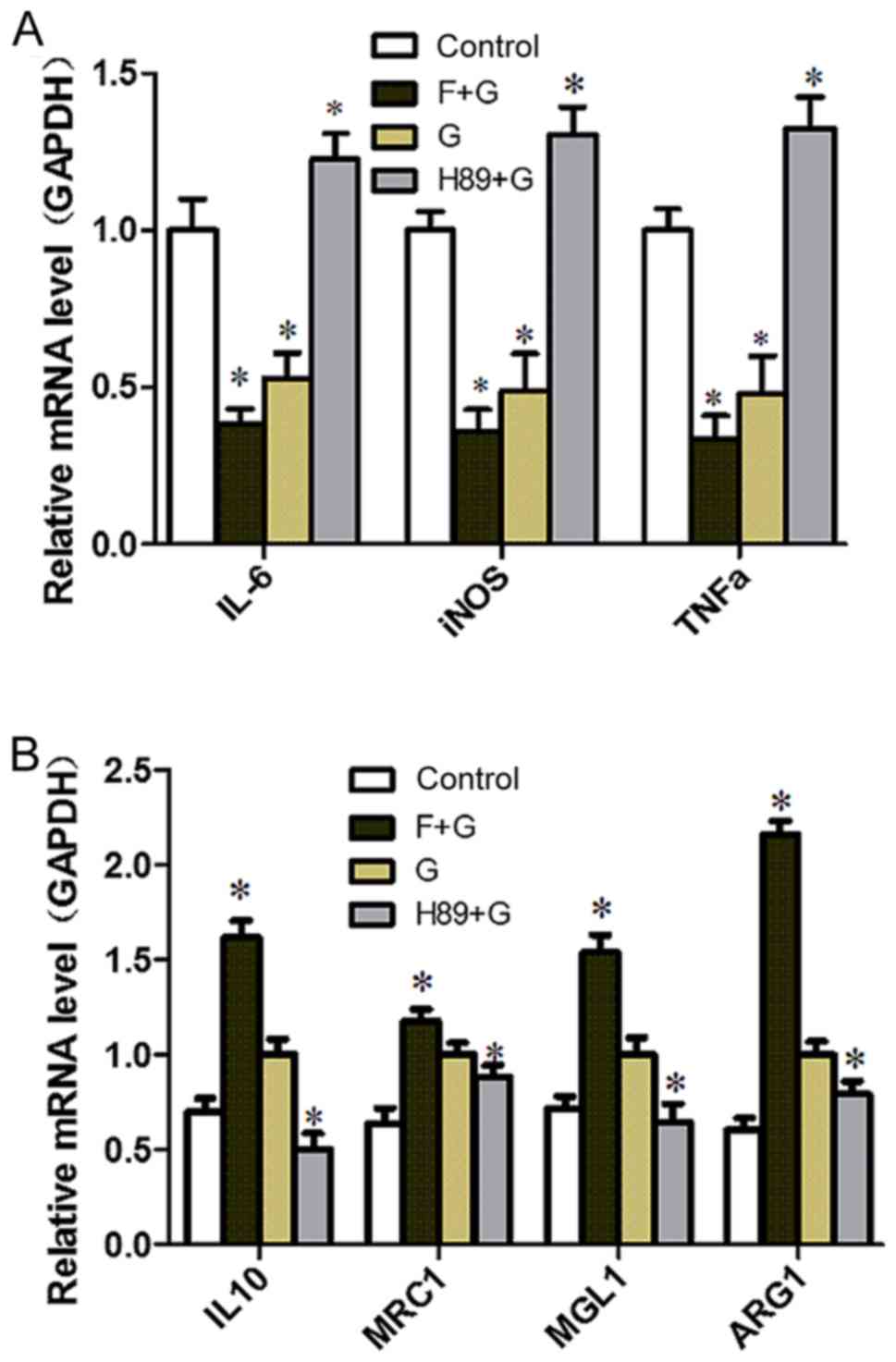
the adenylate cyclase inhibitor Forskolin and the PKA inhibitor H89
were used. H89 pretreatment augmented the expression of M1-related
inflammation genes (P<0.05) and reversed the inhibitory effect
of GLP-1 on inflammatory factors (P<0.05) (Fig. 5A). Conversely, the expression of
M2-specific genes was significantly increased with the addition of
Forskolin, and was reduced with the addition of H89 (P<0.05;
Fig. 5B); GLP-1 and Forskolin
pretreatment reduced the expression of M1-related inflammation
genes (P<0.05). These results suggest that GLP-1R signaling can
promote M2 polarization and reduce M1 polarization through the
cAMP/PKA signaling pathway, and JNK downregulation may serve a
paramount role. Western blot analysis revealed that GLP-1 and
Forskolin treatment suppressed the phosphorylation of JNK
(P<0.05), while H89 treatment facilitated JNK phosphorylation
(P<0.05) and blocked the inhibitory effect of GLP-1 on JNK
phosphorylation (P<0.05) (Fig.
6). These results suggest that GLP-1/GLP-1R signaling inhibited
JNK activation via the cAMP/PKA signaling pathway in RAW264.7
cells.
Discussion
The fact that macrophage activation acts on the
development of inflammatory metabolic disorders such as insulin
resistance and atherosclerosis is useful to investigate the
mechanisms underlying insulin resistance. In the present study,
GLP-1 was demonstrated to induce macrophage polarization toward M2
and inhibit M1 polarization. The present study elucidated that
GLP-1R signaling contributes to the inhibition of JNK activation
and leads to the activation of STAT3, which inhibits inflammation
and M1 activation and promotes M2 activation. It was also
demonstrated that GLP-1R signaling could promote M2 polarization
and reduce M1 polarization through the cAMP/PKA-mediated JNK
downregulation pathway. These findings suggest that modulation of
signaling pathways may serve as pharmacological targets of
macrophage activation and essential underlying mechanisms of GLP-1
on a broad spectrum of metabolic diseases.
Obesity and metabolic syndrome are accompanied by a
phenotypic switch in the activation state of macrophages from an
anti-inflammatory M2 polarization state to a proinflammatory M1
polarization state (9,24,25).
GLP-1 is known to improve insulin sensitivity, and may decrease
macrophage infiltration and suppress the inflammatory response
(13,15). Consistent with the study of Shiraishi
et al (16), it was confirmed
that GLP-1 could increase the expression of M2-specific genes and
induce the activation of STAT3, which is of paramount importance in
macrophage differentiation toward the M2 phenotype. In addition, it
was observed that GLP-1 could decrease M1-specific gene expression
and inhibit M1 phenotype polarization, which were broadly congruent
with the results of Arakawa et al and Chang et al
(13,22). Accordingly, prompting M2 polarization
and suppressing M1 polarization may be effective treatments of
insulin resistance and other inflammation-related diseases.
However, little is known about how GLP-1/GLP-1R activates STAT3
signaling and the underlying mechanisms.
Activation of the stress-activated protein
kinase/JNK serves an important role in macrophage polarization and
insulin resistance. The JNK/mitogen-activated protein kinase
pathways, located upstream of STAT3 signaling, also regulate STAT3
activity. In previous studies, JNK activation in Kupffer cells,
peritoneal macrophages and adipose tissue macrophages led to M1
polarization, tissue inflammation and systemic insulin resistance
(26–28). These data suggest that JNK expression
in macrophages has a paramount importance on macrophage
accumulation, M1 polarization and obesity-induced insulin
resistance. The present study observed that GLP-1 inhibited JNK
activation, which led to the reduction of M1 polarization and
inflammatory factors, and accordingly ameliorated insulin
sensitivity. Lim and Cao (29)
proposed that dual regulation of STAT3 by JNK could upregulate
serine phosphorylation and negatively regulate tyrosine
phosphorylation. Overall, STAT3 serves a crucial role in M2
polarization, which mediates the anti-inflammation response
(30–32). It was hypothesized that
GLP-1/JNK/STAT3 signaling may induce macrophage polarization, which
is in accordance with the inhibition of GLP-1 on JNK activation
observed in the present study. The present results revealed that
the effect of GLP-1 on STAT3 in RAW264.7 cells suppressed the
effect of activated JNK on phosphorylated STAT3. Therefore, GLP-1
may suppress JNK phosphorylation and accordingly upregulate the
intracellular levels of phosphorylated STAT3 in RAW264.7 cells,
which promotes M2 polarization and is associated with
anti-inflammatory effects. These results indicated that the
JNK-STAT3 signaling pathway could regulate macrophage polarization,
resulting in counteraction between M1 and M2.
Previous studies demonstrated that the main effects
of GLP-1 are mediated through the activation of adenylate cyclase
and the generation of cAMP, which subsequently activates PKA signal
transduction (22,33). The effects of increased intracellular
cAMP levels in macrophages on inflammatory mediator generation were
originally reported to be mediated by PKA, being cAMP a key event
for macrophage activation toward the M2 phenotype (13,20–22).
Regarding the cytoprotective action of GLP-1 on pancreatic β-cells,
Ferdaoussi et al (34)
proposed the potent inhibitory effect of GLP-1R activation via the
cAMP/PKA signaling pathway on JNK activity as a major mechanism for
preventing β-cells apoptosis. Thus, the present study revealed that
GLP-1 increased intracellular cAMP levels in RAW264.7 cells within
a certain range, in a concentration-dependent manner. The present
study also demonstrated that GLP-1-cAMP/PKA was located upstream of
JNK signaling and was involved in restraining JNK activity. This
data suggested that GLP-1 promoted M2 polarization and inhibited M1
polarization through the cAMP/PKA pathway, whereby JNK activity
served a vital role. As a result of JNK activity, M1 is inhibited,
while STAT3 activity leads to M2 growth. M2 activation has been
largely defined by its ability to antagonize and counteract M1
polarization and the inflammatory response, including insulin
resistance (4,35). The present findings suggest that
there is crosstalk between M1 and M2, where the JNK-STAT3 signaling
pathway is a major contributor and regulates the counterpoise
between M1 and M2. The findings also suggest that the effects of
GLP-1 in endocrine and metabolic diseases are possibly mediated by
the modulatory association of the signaling pathways, which
provides insight into the molecular mechanisms behind the
progression of metabolic diseases and may help to identify better
drug targets. The therapeutic implications of this conclusion are
notable as they suggest that pharmacological targeting of
macrophage activation, rather than purely inflammation, may be
efficacious in treating this global epidemic.
Due to the limitations of the present in
vitro experiments, future investigations will focus on
identifying factors that trigger the phenotypic switch of
macrophage polarization using in vivo experiments and
whether these changes in polarization state are generalizable to
macrophages in tissues. Animal models are required to address these
challenges, since these approaches allow specific deletion and cell
type-specific strategies involved in macrophage polarization.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Natural
Science Foundation of Hubei Province, China (grant no.
ZRY1073).
Availability of data and materials
The datasets used or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HS conceived and designed the study. SW performed
the experiments and wrote the paper. HS reviewed and edited the
manuscript. All authors read and approved the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no conflict of
interest.
References
|
1
|
Gregor MF and Hotamisligil GS:
Inflammatory mechanisms in obesity. Annu Rev Immunol. 29:415–445.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Aroor AR, McKarns S, Demarco VG, Jia G and
Sowers JR: Maladaptive immune and inflammatory pathways lead to
cardiovascular insulin resistance. Metabolism. 62:1543–1552. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sica A and Mantovani A: Macrophage
plasticity and polarization: In vivo veritas. J Clin Invest.
122:787–795. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Goerdt S and Orfanos CE: Other functions,
other genes: Alternative activation of antigen-presenting cells.
Immunity. 10:137–142. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nathan C and Ding A: Nonresolving
inflammation. Cell. 140:871–882. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gordon S and Taylor PR: Monocyte and
macrophage heterogeneity. Nat Rev Immunol. 5:953–964. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mantovani A, Sica A, Sozzani S, Allavena
P, Vecchi A and Locati M: The chemokine system in diverse forms of
macrophage activation and polarization. Trends Immunol. 25:677–686.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dall'Asta M, Derlindati E, Ardigò D,
Zavaroni I, Brighenti F and Del Rio D: Macrophage polarization: The
answer to the diet/inflammation conundrum? Nutr Metab Cardiovasc
Dis. 22:387–392. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Odegaard JI and Chawla A: Alternative
macrophage activation and metabolism. Annu Rev Pathol. 6:275–297.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yabe D and Seino Y: Two incretin hormones
GLP-1 and GIP: Comparison of their actions in insulin secretion and
beta cell preservation. Prog Biophys Mol Biol. 107:248–256. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Friedrichsen BN, Neubauer N, Lee YC, Gram
VK, Blume N, Petersen JS, Nielsen JH and Møldrup A: Stimulation of
pancreatic beta-cell replication by incretins involves
transcriptional induction of cyclin D1 via multiple signalling
pathways. J Endocrinol. 188:481–492. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Drucker DJ: The biology of incretin
hormones. Cell Metab. 3:153–165. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Arakawa M, Mita T, Azuma K, Ebato C, Goto
H, Nomiyama T, Fujitani Y, Hirose T, Kawamori R and Watada H:
Inhibition of monocyte adhesion to endothelial cells and
attenuation of atherosclerotic lesion by a glucagon-like peptide-1
receptor agonist, exendin-4. Diabetes. 59:1030–1037. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li L, Yang G, Li Q, Tan X, Liu H, Tang Y
and Boden G: Exenatide prevents fat-induced insulin resistance and
raises adiponectin expression and plasma levels. Diabetes Obes
Metab. 10:921–930. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lee YS, Park MS, Choung JS, Kim SS, Oh HH,
Choi CS, Ha SY, Kang Y, Kim Y and Jun HS: Glucagon-like peptide-1
inhibits adipose tissue macrophage infiltration and inflammation in
an obese mouse model of diabetes. Diabetologia. 55:2456–2468. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shiraishi D, Fujiwara Y, Komohara Y,
Mizuta H and Takeya M: Glucagon-like peptide-1 (GLP-1) induces M2
polarization of human macrophages via STAT3 activation. Biochem
Biophys Res Commun. 425:304–308. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Takaishi K, Komohara Y, Tashiro H, Ohtake
H, Nakagawa T, Katabuchi H and Takeya M: Involvement of
M2-polarized macrophages in the ascites from advanced epithelial
ovarian carcinoma in tumor progression via Stat3 activation. Cancer
Sci. 101:2128–2136. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yu H, Pardoll D and Jove R: STATs in
cancer inflammation and immunity: A leading role for STAT3. Nat Rev
Cancer. 9:798–809. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Williams L, Bradley L, Smith A and Foxwell
B: Signal transducer and activator of transcription 3 is the
dominant mediator of the anti-inflammatory effects of IL-10 in
human macrophages. J Immunol. 172:567–576. 2003. View Article : Google Scholar
|
|
20
|
Serezani CH, Ballinger MN, Aronoff DM and
Peters-Golden M: Cyclic AMP: Master regulator of innate immune cell
function. Am J Respir Cell Mol Biol. 39:127–132. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mustafa SB and Olson MS: Expression of
nitric-oxide synthase in rat Kupffer cells is regulated by cAMP. J
Biol Chem. 273:5073–5080. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chang SY, Kim DB, Ryu GR, Ko SH, Jeong IK,
Ahn YB, Jo YH and Kim MJ: Exendin-4 inhibits iNOS expression at the
protein level in LPS-stimulated Raw264.7 macrophage by the
activation of cAMP/PKA pathway. J Cell Biochem. 114:844–853. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Weisberg SP, McCann D, Desai M, Rosenbaum
M, Leibel RL and Ferrante AW: Obesity is associated with macrophage
accumulation in adipose tissue. J Clin Invest. 112:1796–1808. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lumeng CN, Bodzin JL and Saltiel AR:
Obesity induces a phenotypic switch in adipose tissue macrophage
polarization. J Clin Invest. 117:175–184. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Odegaard JI and Chawla A: Mechanisms of
macrophage activation in obesity-induced insulin resistance. Nat
Clin Pract Endocrinol Metab. 4:619–626. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang X, Xu A, Chung SK, Cresser JH,
Sweeney G, Wong RL, Lin A and Lam KS: Selective inactivation of
c-Jun NH2-terminal kinase in adipose tissue protects against
diet-induced obesity and improves insulin sensitivity in both liver
and skeletal muscle in mice. Diabetes. 60:486–495. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Han MS, Jung DY, Morel C, Lakhani SA, Kim
JK, Flavell RA and Davis RJ: JNK expression by macrophages promotes
obesity-induced insulin resistance and inflammation. Science.
339:218–222. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lim CP and Cao X: Serine phosphorylation
and negative regulation of Stat3 by JNK. J Biol Chem.
274:31055–31061. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li SN, Wang W, Fu SP, Wang JF, Liu HM, Xie
SS, Liu BR, Li Y, Lv QK, Li ZQ, et al: IL-21 Modulates release of
proinflammatory cytokines in LPS-stimulated macrophages through
distinct signaling pathways. Mediators Inflamm. 2013:5480732013.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Matsukawa A, Kudo S, Maeda T, Numata K,
Watanabe H, Takeda K, Akira S and Ito T: Stat3 in resident
macrophages as a repressor protein of inflammatory response. J
Immunol. 175:3354–3359. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gaba A, Grivennikov SI, Do MV, Stumpo DJ,
Blackshear PJ and Karin M: IL-10-mediated tristetraprolin induction
is part of a feedback loop that controls Macrophage STAT3
activation and cytokine production. J Immunol. 189:2089–2093. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Makranz C, Cohen G, Reichert F, Kodama T
and Rotshenker S: cAMP cascade (PKA, Epac, adenylyl cyclase, Gi,
and phosphodiesterases) regulates myelin phagocytosis mediated by
complement receptor-3 and scavenger receptor-AI/II in microglia and
macrophages. Glia. 53:441–448. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ferdaoussi M, Abdelli S, Yang JY, Cornu M,
Niederhauser G, Favre D, Widmann C, Regazzi R, Thorens B, Waeber G
and Abderrahmani A: Exendin-4 protects beta-cells from
interleukin-1 beta-induced apoptosis by interfering with the c-Jun
NH2-terminal kinase pathway. Diabetes. 57:1205–1215. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gordon S: Alternative activation of
macrophages. Nat Rev Immunol. 3:23–35. 2003. View Article : Google Scholar : PubMed/NCBI
|