Introduction
Traumatic brain injury (TBI) can be induced by
injuries ranging from simple head impacts to penetrating wounds.
TBI is a severe trauma in neurosurgery, which is associated with
high disability and mortality rates (1). In the USA, TBI has resulted in >5.3
million disabilities, with ~1.7 million people developing TBI per
year (2). Of these, 80% of TBI cases
are considered mild brain injuries, while moderate and severe TBI
is the primary cause of death and disability (3). Severe TBI, combined with pathological
changes, including cephaledema, cerebral ischemia and intracranial
hypertension, may lead to cranial nerve injury and limb dysfunction
(3).
It is well known that necrosis and apoptosis are two
methods of cell Death (4). Necrosis
is a passive and abnormal death process, which is induced by damage
factors or other unfavorable environmental pressures (5). Apoptosis, an active and programmed
process of cell death, is autonomously regulated by genes and
functions to maintain homeostasis (5). A previous study indicated that
autophagy serves as the third method of cell death (5). Autophagy is the process where
intracellular proteins and organelles are degraded by the lysosome,
which serves a vital role in the maintenance of homeostasis in
eukaryotes (5). However, under
pathological conditions, persistent and excessive autophagy
activation results in cell death, which is called autophagic cell
death (5). It is of great importance
to assess the role of autophagy in brain injury as: i) an
interaction exists between autophagy-induced cell death and
apoptotic signaling (6); and ii)
autophagy may activate certain apoptotic promoters in damaged nerve
cells, thus initiating the cell apoptosis program (6).
A previous study has demonstrated that polypeptide
rich arginine provides neuroprotection (7). Furthermore, it has been revealed that
the poly-l-arginine R18 polypeptide and NA-1 also exhibit
neuroprotective effects, with R18 being more protective than NA-1
(7). Cui et al (8) assessed the protective effects of R18,
COG1410 and APP6–110 on neurons following TBI. It was revealed that
R18 could reduce calcium influx and exhibit strong neuroprotection,
while COG1410 and APP6-110 exhibited weak neuroprotection. However,
the mechanism by which the R18 polypeptide exerts neuronal
protective effects has not yet been fully elucidated. We examined
the effect of poly-arginine R18 and its underlying mechanism in
promoting neurocyte cell growth in TBI via autophagy.
Materials and methods
Animals and the controlled cortical
impact (CCI) model
All experimental procedures in the current study
were approved by the Animal Ethics Committee of First Affiliated
Hospital of Xinjiang Medical University (Xinjiang, China). Adult
male Sprague-Dawley rats (n=42; age, 8–10 weeks; weight, 200–230 g)
were obtained from Animal experiment center of Xinjiang medical
university (Xinjiang, China). Animals were maintained under a
controlled temperature (22-23°C) and humidity (55-60%), with a 12 h
light/dark cycle and free access to food and water. All rats were
randomly assigned into the control, model or poly-arginine R18
groups (each, n=6). All rats in the model and poly-arginine R18
groups were anesthetized with 35 mg/kg pentobarbital and a
craniotomy was performed at the bregma and lambda on the right
frontoparietal cortex. In the R18 group, CCI was performed using a
PinPoint™ Precision Cortical Impactor (Hatteras Instruments, Inc.,
Cary, NC, USA) perpendicular to the brain surface and all rats
received a single injection of 300 nmol/kg poly-arginine R18
(synthesized by Sangon Biotech Co., Ltd., Shanghai, China) at 48 h
following surgery, as previously described (9). Rats in the control group were
anesthetized with 35 mg/kg pentobarbital and the same surgical
procedure was performed without CCI.
Brain water content measurement
Following treatment with poly-arginine R18, rats
(n=3/group) were sacrificed, brain samples were collected and
tissue was washed with PBS. The wet weight of brain tissue was
recorded and samples were then dried at 68°C for 48 h to obtain the
dry weight. Brain water content was calculated as follows: wet
weight-dry weight/wet weight ×100%.
Hematoxylin and Eosin (H&E)
staining
Following treatment with poly-arginine R18, rats
(n=3/group) were sacrificed and the hippocampus tissue were fixed
with 4% paraformaldehyde for 24 h at room temperature and embedded
in paraffin. Paraffin-embedded tissue samples were cut into 10
µm-thick sections. Tissue samples were stained with hematoxylin and
eosin (H&E) for 5 min at room temperature and examined using a
confocal microscope (magnification, ×50; Olympus BX51; Olympus
Corp., Tokyo, Japan).
Cell culture
The neuroblastoma neuro-2A (N2A) cell line was
purchased from the Type Culture Collection of the Chinese Academy
of Sciences (Shanghai, China) and cultured in Dulbecco's modified
Eagle's medium (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) supplemented with 10% heat-inactivated fetal bovine serum
(FBS; Gibco; Thermo Fisher Scientific, Inc.), 100 U/ml penicillin
and 100 µg/ml streptomycin in a humidified atmosphere of 5%
CO2 at 37°C. N2A cells in the model group were then
stimulated with 500 ng/ml lipopolysaccharide (LPS; Sigma-Aldrich;
Merck KGaA, Darmstadt, Germany). In the R18 group, N2A cells were
treated with 500 ng/ml LPS and 0.5 µM poly-arginine R18 as
previously described (9). In the
3-MA group, N2A cells were treated with an autophagy inhibitor 3-MA
(5 µM; MedChemExpress, Shanghai, China), 500 ng/ml LPS and 0.5 µM
R18.
MTT and lactate dehydrogenase (LDH)
activity
A total of 10 µl MTT (5 mg/ml; cat. no. C0009;
Beyotime Institute of Biotechnology, Haimen, China) was added to
N2A cells for 4 h at 37°C. DMEM was subsequently removed and
dimethyl sulfoxide was added to cells and incubated for 20 min at
37°C. The optical density in each well was measured at a wavelength
of 492 nm using a microplate reader (Molecular Devices, Sunnyvale,
CA, USA). LDH activity was measured using an LDH Cytotoxicity
Detection kit (cat. no. C0017; Beyotime Institute of
Biotechnology), according to the manufacturer's protocol. The
optical density was measured at a wavelength of 450 nm using a
microplate reader to determine the LDH activity.
Caspase-3/8/9 activity
N2A cells were lysed using radioimmunoprecipitation
assay (RIPA) buffer for 15–30 min at 4°C. The supernatant was then
centrifuged at 2,000 × g for 10 min at 4°C and protein content was
measured using a bicinchoninic acid (BCA) assay. The activity of
caspase-3/8/9 was assessed in total protein (10 µg) using a
caspase-3 (cat. no. C1115), caspase-8 (cat. no. C1151) and
caspase-9 (cat. no. C1158) activity kits (all Beyotime Institute of
Biotechnology). Optical density values in each well were determined
using an enzyme immunoassay analyzer at 405 nm.
Western blot analysis
Tissue samples were homogenized or N2A cells were
lysed using RIPA buffer for 15–30 min at 4°C. The supernatant was
then centrifuged at 2,000 × g for 10 min at 4°C and the protein
content was measured using a BCA assay. Total protein (50 µg) was
separated via SDS-PAGE on a 10% gel. Separated proteins were
transferred onto polyvinylidene fluoride membranes and then blocked
with 5% non-fat milk in TBST for 1 h at 37°C. Membranes were
incubated overnight at 4°C with primary antibodies against Bcl-2
associated X (Bax; 1:1,000; cat. no. 5023), LC3 (1:1,000; cat. no.
4599), Beclin-1 (1:1,000; cat. no. 3495), p62 (1:1,000; cat. no.
23214) and GAPDH (1:1,000; cat. no. 5174; all Cell Signaling
Technology, Inc., Danvers, MA, USA). Subsequently, membranes were
washed with Tris-buffered saline containing 0.1% Tween®
20 (TBST) and membranes were incubated with horseradish
peroxidase-labeled goat anti-rabbit IgG secondary antibody
(1:5,000; cat. no. 7074; Cell Signaling Technology, Inc.) for 1 h
at room temperature. Samples were visualized using an enhanced
chemiluminescence kit (GE Healthcare, Chicago, IL, USA) and exposed
to Image Lab 3.0 software (Bio-Rad Laboratories, Inc., Hercules,
CA, USA).
Immunofluorescence
N2A cells were washed with PBS and fixed using 4%
paraformaldehyde for 20 min at room temperature. Following further
washes with PBS, cells were stained with DAPI (Sigma-Aldrich; Merck
KGaA) for 20 min in darkness at room temperature. Stained cells
were observed using an inverted confocal laser scanning microscope
(magnification, 50×; TCS SP5; Leica Microsystems GmbH, Wetzlar,
Germany).
Statistical analysis
Data are expressed as the mean ± standard deviation.
All statistical analyses were performed using SPSS software
(version 20.0; IBM Corp., Armonk, NY, USA). All data were analyzed
using one-way analysis of variance followed by a Tukey's multiple
comparison test. P<0.05 were considered to indicate a
statistically significant difference.
Results
Poly-arginine R18 promotes neurocyte
cell growth in TBI rats
TBI rats were treated with poly-arginine R18 and the
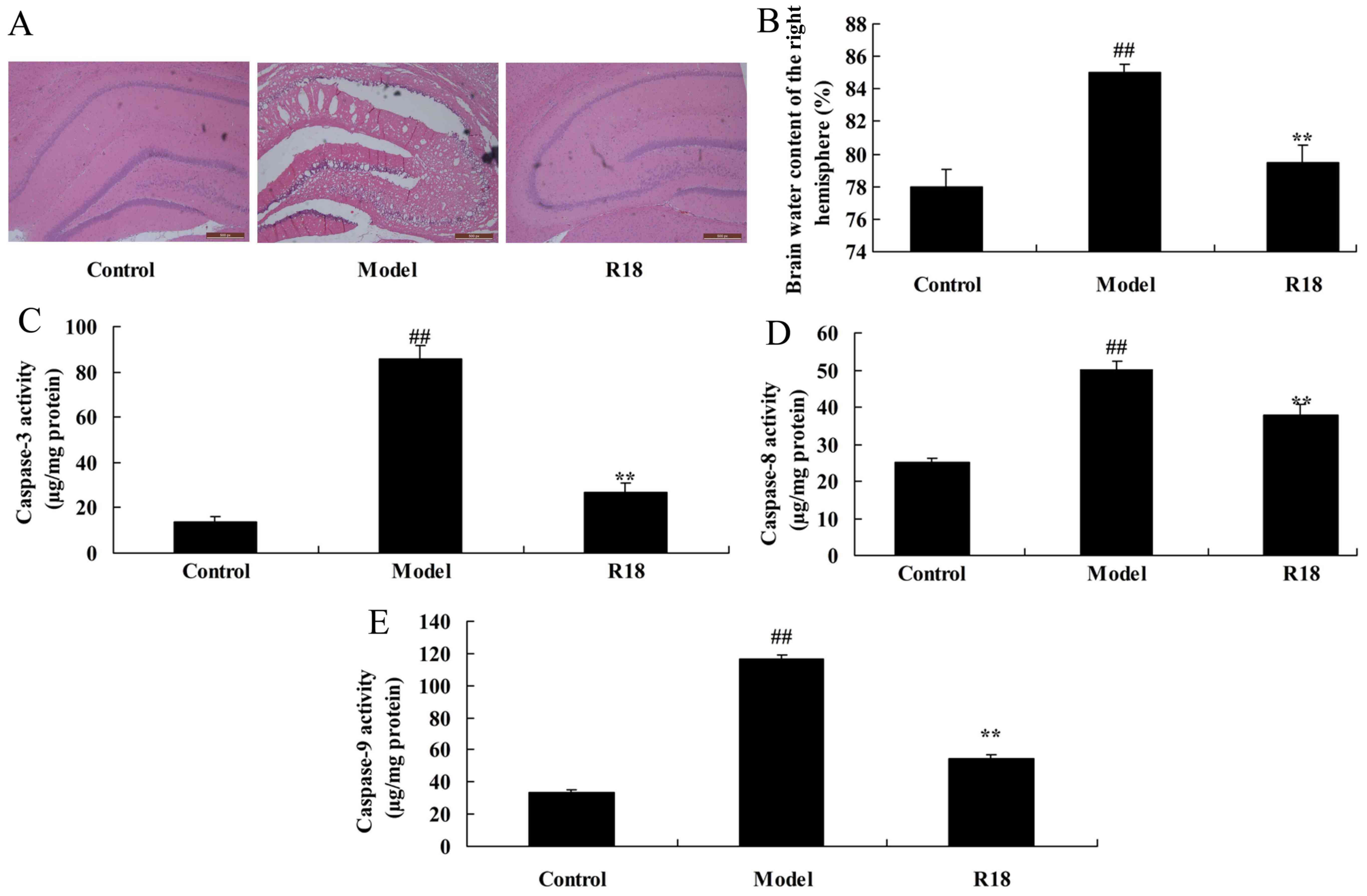
effect on neurocyte cell growth was assessed. As presented in
Fig. 1A and B, neurocyte cell
apoptosis and brain water content was markedly increased in the TBI
model group compared with the control group. Furthermore, the
activity levels of caspase-3/8/9 were increased in the TBI model
group compared with those in the control group (Fig. 1C-E). Treatment with poly-arginine R18
significantly reduced neurocyte cell apoptosis, brain water content
and Bax protein expression whilst also inhibiting caspase-3/8/9
activity in the R18 group compared with the model group (Fig. 1). These results indicate that
poly-arginine R18 promotes neurocyte cell growth in TBI rats.
Poly-arginine R18 induces neurocyte
cell autophagy in TBI rats
The mechanism of poly-arginine R18 in neurocyte cell
growth was assessed in TBI rats by analyzing changes in neurocyte
cell autophagy. As presented in Fig.
2, LC3 and Beclin-1 protein expression was significantly
reduced, whilst p62 and Bax expression was significantly increased
in the TBI model group compared with the control group. Subsequent
treatment with poly-arginine R18 significantly increased LC3 and
Beclin-1 protein expression and reduced p62 and Bax expression in
the R18 group compared with the TBI model group (Fig. 2). Thus, the results indicate that
poly-arginine R18 induces neurocyte cell autophagy in TBI rats.
Poly-arginine R18 induces neurocyte
cell autophagy in vitro
The autophagy of neurocyte cells was assessed
following poly-arginine R18 treatment in an in vitro model
of TBI. As presented in Fig. 3, the
expression levels of LC3 and Beclin-1 was significantly reduced,
whilst p62 and Bax expression was significantly increased in the
in vitro TBI model group compared with the control group.
Following treatment with poly-arginine R18, the expression levels
of LC3 and Beclin-1 were significantly increased and p62 and Bax
protein expression were significantly decreased in the R18 group
compared with the in vitro TBI model group (Fig. 3A-E). Taken together, the results
demonstrated that poly-arginine R18 induces neurocyte cell
autophagy in vitro.
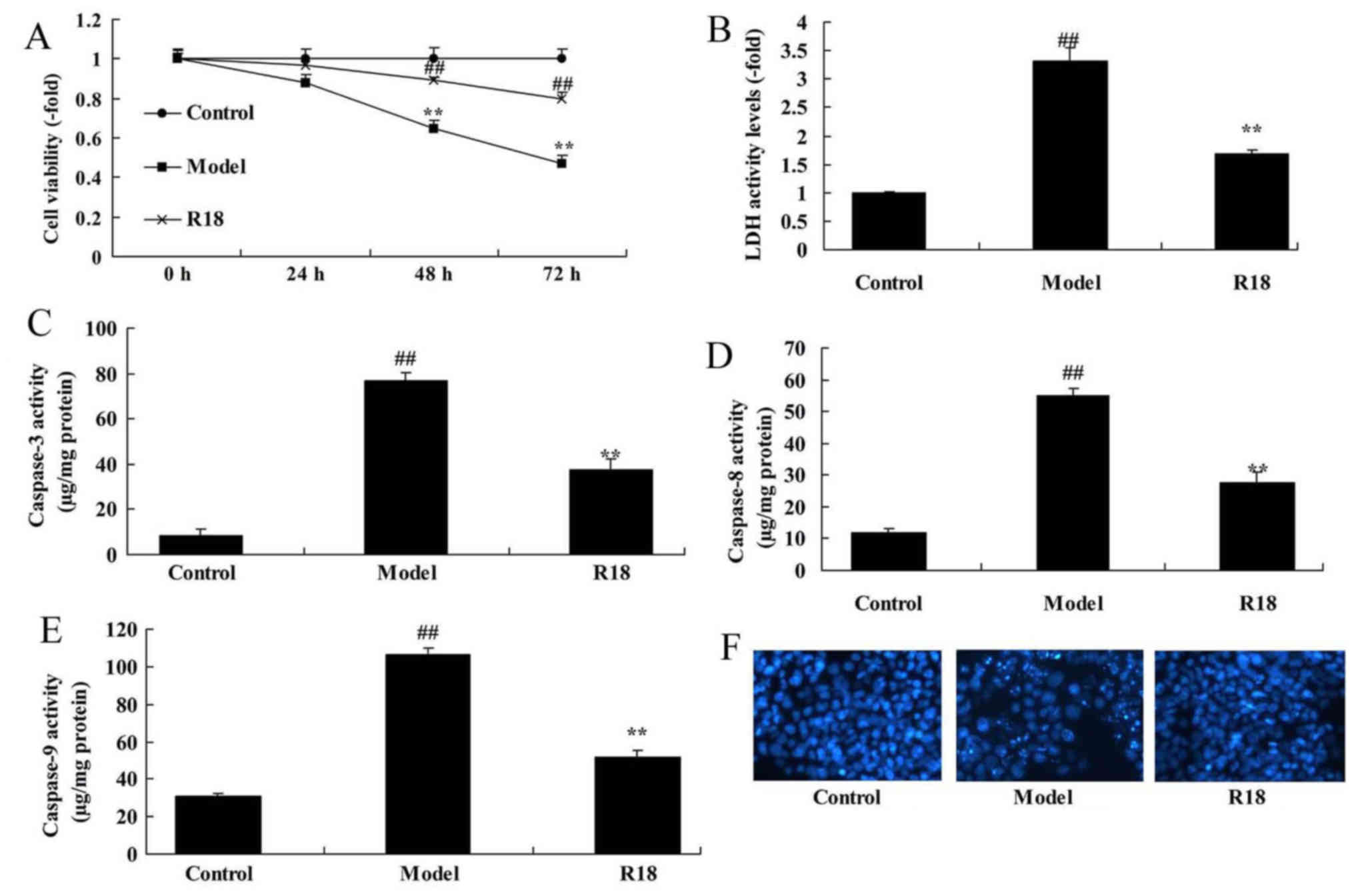
Poly-arginine R18 promotes neurocyte
proliferation and reduces LDH activity in vitro
Neurocyte cell proliferation was significantly
reduced, whilst LDH activity was increased in the in vitro
TBI model group compared with the control group (Fig. 4A and B). In addition, the activity
levels of caspase-3/8/9 and cell nucleus apoptosis were
significantly increased in the in vitro TBI model group
compared with the control group (Fig.
4C-F). Following treatment with poly-arginine R18, neurocyte
proliferation was significantly increased, whilst LDH and
caspase-3/8/9 activity levels were significantly decreased in the
R18 group compared with the in vitro TBI model group
(Fig 4).
Autophagy inhibition attenuates the
effects of poly-arginine R18 on cell proliferation in vitro
An autophagy inhibitor (3-MA) was used to assess the
mechanism of poly-arginine R18 on cell growth in an in vitro
model of TBI. The expression levels of LC3 and Beclin-1 were
significantly reduced, whilst p62 and Bax expression was
significantly increased in the R18 group following treatment with
3-MA (3-MA group) compared with the R18 group (Fig. 5A-E). Furthermore, treatment with 3-MA
attenuated the effects of poly-arginine R18 on neurocyte
proliferation, LDH activity, caspase-3/8/9 activity levels and cell
nucleus apoptosis compared with the R18 group (Fig. 5F-K).
 | Figure 5.Inhibition of autophagy reduces the
effect of poly-arginine R18 on cell growth in an in vitro
model. Quantification of (A) LC3, (B) Beclin-1 and (C) p62 protein
expression and (D) western blotting. (E) Bax expression was also
assessed. (F) Cell proliferation, (G) LDH activity and (H)
caspase-3, (I) caspase-8 and (J) caspase-9 activity was assessed in
N2A cells. (K) DAPI staining. ##P<0.01 vs. the
control group; **P<0.01 vs. the model group;
###P<0.01 vs. the R18 group. Bax, Bcl-2 associated X;
LDH, lactate dehydrogenase; N2A, neruo-2A; model, N2A cells treated
with 500 ng/ml LPS; R18, N2A cells treated with 500 ng/ml LPS and
0.5 µM poly-arginine R18; 3-MA, N2A cells treated with 500 ng/ml
LPS, 0.5 µM poly-arginine R18 and 5 µM 3-MA. LPS,
lipopolysaccharide. |
Discussion
Brain injury is a common trauma that has a growing
incidence rate in China (10). Brain
injury can induce neurological damage through two mechanisms,
including primary injury and secondary injury (10). Secondary brain injury refers to a
series of pathophysiological changes that occur following acute
injury (10). This comprises local
microvascular injury, vascular rupture hemorrhage, spasm or
thrombosis (11). Injuries such as
these subsequently induce tissue ischemia, hypoxia, the
inflammatory response and tissue edema, thus activating
intracellular zymogen (11). Zymogen
destroys various membrane structures within the tissue and
eventually leads to apoptosis and necrosis (12). Therefore, it is important to
understand the changes and repair mechanisms that occur following
brain injury and to effectively suppress cell death, which may
protect neurons and promote the repair and reconstruction of
neurological functional injury (13). In the current study, it was revealed
that poly-arginine R18 promotes neurocyte proliferation in TBI
rats. Chiu et al (14)
demonstrated that poly-arginine R18 reduces axonal injury following
TBI (14). However, the current
study only performed H&E staining to detect brain injury, which
is insufficient for a full assessment. Therefore, future studies
should perform scanning tunneling microscopy to investigate
microstructural organelle damage following injury.
Autophagy is an organelle and protein cycling
process that is controlled by the lysosome degradation pathway
(6). The primary feature of
autophagy activation is the excessive accumulation of
autophagosomes encapsulated by a double-membraned structure or by
autophagic vacuoles (8). It has been
demonstrated that cell autophagy is associated with aging,
immunity, cell death and differentiation (15). Autophagy has also been associated
with the pathogenesis of several types of neurodegenerative disease
and malignant tumors (8). Autophagy
is an early self-adaptive mechanism that eliminates damaged
organelles and protein, and provides nutrients and energy via the
lysosome degradation pathway to restore tissue homeostasis
(8). Excessively activated or
impaired autophagy leads to autophagic cell death, which
accelerates disease development and deterioration (15). A previous study has indicated that
neurological damage is closely associated with cell autophagy
(15), such that the autophagic
marker protein, LC3, is markedly upregulated in brain tissue
subjected to traumatic injury (15).
The present study determined that poly-arginine R18 treatment
induced neurocyte cell autophagy in TBI rats. The N2A neuroblastoma
cell line of mouse origin was utilized in the present study.
However, neuroblastoma cells are different from neurocytes, which
may affect the results of the present study. Future studies should
therefore utilize different cell lines for the construction of an
in vitro model.
Autophagy is involved in the pathological and
physiological process of numerous diseases. However, no consensus
has been reached on the effect of autophagy on organ function
(16). A previous study has
indicated that autophagy may lead to autophagic cell death or
apoptosis, thus aggravating organ dysfunction (17). A previous study demonstrated that
autophagy eliminates damaged mitochondria, blocks the apoptotic
factor from being released into plasma and exhibits protection
(18). The role of autophagy in the
protection or aggravation of injury depends on the precise location
of autophagy and the stage of autophagy (18). Typically, excessive autophagy
following brain injury leads to poor neurological function. In the
present study, the inhibition of autophagy was revealed to reduce
the effects of poly-arginine R18 on cell proliferation in an in
vitro model.
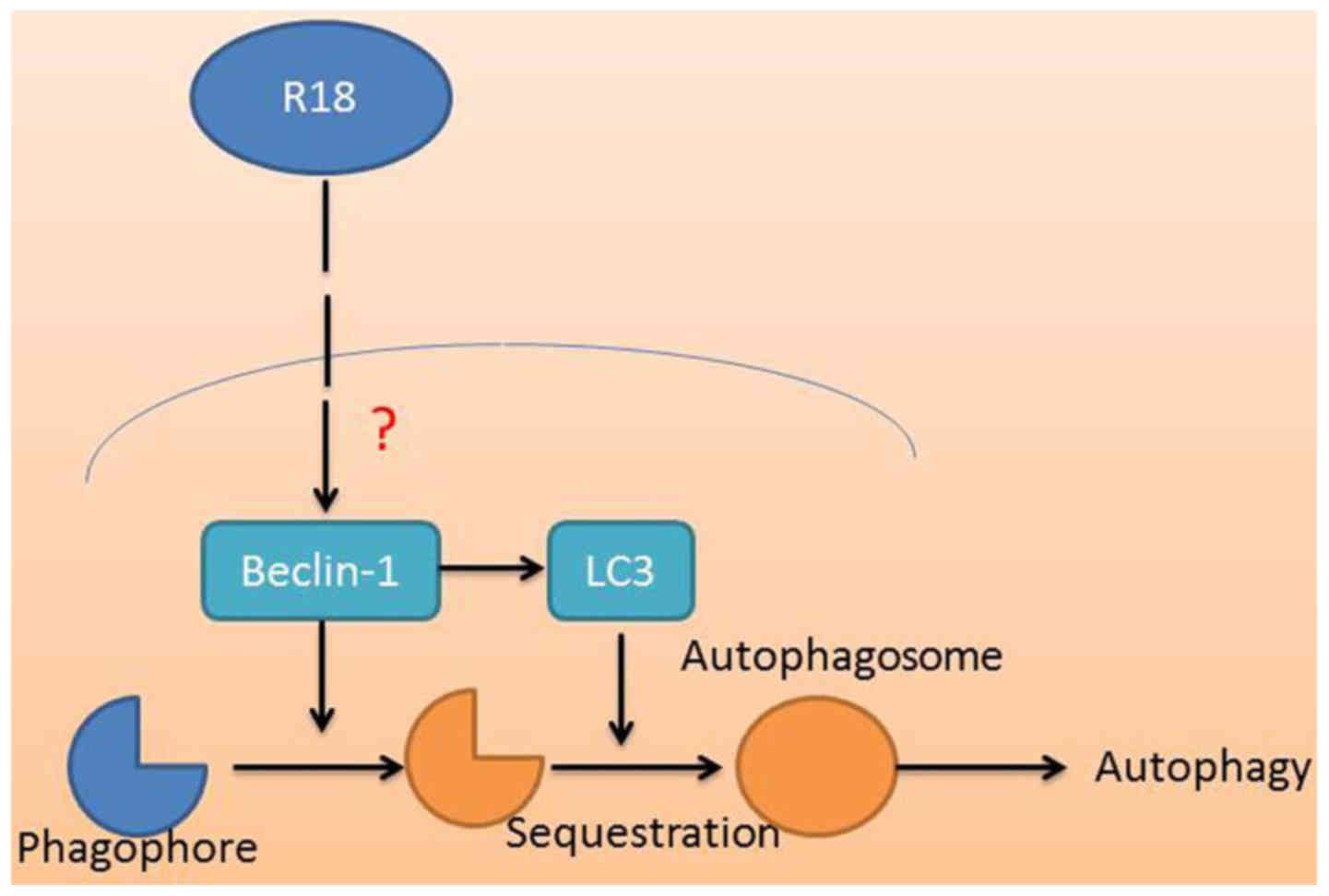
In conclusion, the current study demonstrated that
poly-arginine R18 treatment reduces neurocyte cell apoptosis and
brain water content, and reduces caspase-3/8/9 activity levels in
TBI rats. Therefore, treatment with poly-arginine R18 may induce
neurocyte cell autophagy and promote neurocyte cell growth in TBI
rats (Fig. 6). These findings may
have clinical significance and may therefore justify the further
assessment of poly-arginine R18 as a novel class of neuroprotective
agent in the regulation of autophagy in TBI.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
DG designed the experiment; HB, GD, DL, DS and YF
performed the experiment. DG and HB analyzed the data; DG prepared
the manuscript.
Ethics approval and consent to
participate
All experimental procedures in the current study
were approved by the Animal Ethics Committee of the First
Affiliated Hospital of Xinjiang Medical University (Xinjiang,
China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Yang Z, Lin F, Robertson CS and Wang KK:
Dual vulnerability of TDP-43 to calpain and caspase-3 proteolysis
after neurotoxic conditions and traumatic brain injury. J Cereb
Blood Flow Metab. 34:1444–1452. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Okonkwo DO, Shutter LA, Moore C, Temkin
NR, Puccio AM, Madden CJ, Andaluz N, Chesnut RM, Bullock MR, Grant
GA, et al: Brain oxygen optimization in severe traumatic brain
injury Phase-II: A Phase II randomized trial. Crit Care Med.
45:1907–1914. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Grima NA, Rajaratnam SMW, Mansfield D,
Sletten TL, Spitz G and Ponsford JL: Efficacy of melatonin for
sleep disturbance following traumatic brain injury: A randomised
controlled trial. BMC Med. 16:82018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sun L, Zhao M, Wang Y, Liu A, Lv M, Li Y,
Yang X and Wu Z: Neuroprotective effects of miR-27a against
traumatic brain injury via suppressing FoxO3a-mediated neuronal
autophagy. Biochem Biophys Res Commun. 482:1141–1147. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zeng XJ, Li P, Ning YL, Zhao Y, Peng Y,
Yang N, Zhao ZA, Chen JF and Zhou YG: Impaired autophagic flux is
associated with the severity of trauma and the role of
A2AR in brain cells after traumatic brain injury. Cell
Death Dis. 9:2522018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen X, Wang H, Zhou M, Li X, Fang Z, Gao
H, Li Y and Hu W: Valproic acid attenuates traumatic brain
injury-induced inflammation in vivo: Involvement of autophagy and
the Nrf2/ARE signaling pathway. Front Mol Neurosci. 11:1172018.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Milani D, Knuckey NW, Anderton RS, Cross
JL and Meloni BP: The R18 polyarginine peptide is more effective
than the TAT-NR2B9c (NA-1) peptide when administered 60 minutes
after permanent middle cerebral artery occlusion in the rat. Stroke
Res Treat. 2016:23727102016.PubMed/NCBI
|
|
8
|
Cui C, Cui J, Jin F, Cui Y, Li R, Jiang X,
Tian Y, Wang K, Jiang P and Gao J: Induction of the Vitamin D
receptor attenuates autophagy dysfunction-mediated cell death
following traumatic brain injury. Cell Physiol Biochem.
42:1888–1896. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Milani D, Bakeberg MC, Cross JL, Clark VW,
Anderton RS, Blacker DJ, Knuckey NW and Meloni BP: Comparison of
neuroprotective efficacy of poly-arginine R18 and R18D
(D-enantiomer) peptides following permanent middle cerebral artery
occlusion in the Wistar rat and in vitro toxicity studies. PLoS
One. 13:e01938842018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Blaha RZ, Arnett AB, Kirkwood MW, Taylor
HG, Stancin T, Brown TM and Wade SL: Factors influencing attrition
in a multisite, randomized, clinical trial following traumatic
brain injury in adolescence. J Head Trauma Rehabil. 30:E33–E40.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hammond FM, Alexander DN, Cutler AJ,
D'Amico S, Doody RS, Sauve W, Zorowitz RD, Davis CS, Shin P, Ledon
F, et al: PRISM II: An open-label study to assess effectiveness of
dextromethorphan/quinidine for pseudobulbar affect in patients with
dementia, stroke or traumatic brain injury. BMC Neurol. 16:892016.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Beca J, McSharry B, Erickson S, Yung M,
Schibler A, Slater A, Wilkins B, Singhal A, Williams G, Sherring C,
et al: Hypothermia for traumatic brain injury in children-a phase
II randomized controlled trial. Crit Care Med. 43:1458–1466. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Renaud MI, Lambregts SA, de Kloet AJ,
Catsman-Berrevoets CE, van de Port IG and van Heugten CM:
Activities and participation of children and adolescents after mild
traumatic brain injury and the effectiveness of an early
intervention (Brains Ahead!): Study protocol for a cohort study
with a nested randomised controlled trial. Trials. 17:2362016.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chiu LS, Anderton RS, Cross JL, Clark VW,
Edwards AB, Knuckey NW and Meloni BP: Assessment of R18, COG1410,
and APP96-110 in excitotoxicity and traumatic brain injury. Transl
Neurosci. 8:147–157. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wolf MS, Bayir H, Kochanek PM and Clark
RSB: The role of autophagy in acute brain injury: A state of flux?
Neurobiol Dis. 122:9–15. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sun LQ, Gao JL, Cui Y, Zhao MM, Jing XB,
Li R, Tian YX, Cui JZ and Wu ZX: Neuronic autophagy contributes to
p-connexin 43 degradation in hippocampal astrocytes following
traumatic brain injury in rats. Mol Med Rep. 11:4419–4423. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jin Y, Lin Y, Feng JF, Jia F, Gao G and
Jiang JY: Attenuation of cell death in injured cortex after
post-traumatic brain injury moderate hypothermia: Possible
involvement of autophagy pathway. World Neurosurg. 84:420–430.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cordaro M, Impellizzeri D, Paterniti I,
Bruschetta G, Siracusa R, De Stefano D, Cuzzocrea S and Esposito E:
Neuroprotective effects of Co-UltraPEALut on secondary inflammatory
process and autophagy involved in traumatic brain injury. J
Neurotrauma. 33:132–146. 2016. View Article : Google Scholar : PubMed/NCBI
|