Introduction
Myocardial ischemia-reperfusion (I/R) injury is
considered to have a detrimental role in coronary heart disease
(CHD), which is considered to be the leading cause of death
worldwide (1–3). Myocardial infarction is caused by
coronary occlusion and the subsequent insufficient supply of blood
to the myocardium, which may then cause irreversible necrosis to
occur (4,5). Restoring blood flow to the ischemic
myocardium is the most effective treatment to rescue ischemic
myocardium cells and to save the life of the patient (6). However, this treatment can cause an
abrupt restoration of the oxygen supply and the reperfusion of
myocardium may aggravate myocardial injury, leading to a
reperfusion injury (7,8). Therefore, it is necessary to explore
and to better understand the potential molecular mechanism of
myocardial I/R injury.
MicroRNAs (miRNAs or miRs) are small,
single-stranded, non-coding RNAs (20–22 nucleotides) that are
involved in numerous biological processes (9). miRNAs participate in many biological
processes, such as cell apoptosis, cell differentiation and cell
development (10). More and more
evidence has indicated that several miRNAs are expressed abnormally
during myocardial I/R injury, suggesting the involvement of miRNAs
in myocardial I/R injury development (11–13).
Furthermore, the inhibition of mitochondria-mediated apoptosis to
reduce cardiomyocyte apoptosis is considered to be an important
mechanism (14,15). However, little evidence is available
regarding the role of miRNAs and mitochondria-mediated apoptosis in
myocardial I/R injury.
Hypoxia-inducible factor (HIF) 1 is known as a
heterodimeric transcription factor composed of an oxygen-labile α
subunit (HIF1-α) and a constitutive β subunit (HIF1-β), and HIF1
can bind to the hypoxia response element to regulate gene
expression (16,17). Furthermore, HIF1-α is the regulatory
subunit that senses tissue oxygen level, responds to various types
of cellular stimulation and exerts a vast array of physiological
functions, enabling cells to adapt to temporary hypoxia (18). Some studies found that HIF1-α
activity showed some effects in preventing diabetic cardiomyopathy
and cardiac remodeling (19,20). However, the role of HIF1-α in
myocardial I/R injury is incompletely understood.
The aim of the current study was to investigate the
effect of miRNAs and mitochondria-mediated apoptosis in a
myocardial I/R injury model. In the current study, the authors
uncovered the pivotal role of miR-138 in the myocardial I/R injury
model, which may be connected with the inhibition of myocardial
I/R-induced mitochondrial apoptosis.
Materials and methods
Ethical statement
The experiments involving animals conform to local
and national Guide for the Care and Use of Laboratory Animal
guidelines. Furthermore, these experimental methods are also
approved by the Clinical Ethics Committee of Affiliated Hospital of
Weifang Medical University (Shandong, China).
Mouse model of myocardial I/R
Injury
A total of 60 mice (30 males and 30 females; age,
8–10 weeks; weight, 18–25 g) were obtained from the Animal Center
of Weifang Medical University (Weifang, China). Animals were housed
at a temperature of 23–25°C and a relative humditity of 40–60%,
under a 14/10 h light/dark cycle and free access to water and food.
Housing conditions also included a light intensity of ~40 lux at
the position of the animal in cage. Mice were divided into five
groups (each, n=12): A sham group (mice without injury), a control
group (I/R injury with miR-138 negative control injection), an I/R
injury group, a miR-138 mimic group (I/R injury with miR-138 mimic
injection) and a miR-138 inhibitor group (I/R injury with miR-138
inhibitor injection). The mouse model of myocardial I/R injury was
established as previously described (21). In short, pentobarbital (50 mg/kg) was
used to anesthetize the mice. Then, a left horizontal incision was
made at the third intercostal space. Subsequently, a silk suture we
used to tie around the left anterior descending artery and a
silicon tube (1 mm outside diameter). In the sham group, the mouse
only received the identical surgical procedure without ligature.
The silicon tube was then removed to achieve reperfusion for 6 h
after 30 min of ischemia. After reperfusion, the heart samples were
collected as quickly as possible. For some experiments, miR-138
mimic (20 nmol/l), miR-138 inhibitor (20 nmol/l), HIF1-α siRNA (100
nmol/l) or miR-138 negative control (20 nmol/l) were administered
intraperitoneally for seven days before modeling. miR-138 mimic,
miR-138 inhibitor, HIF1-α siRNA were all purchased from Genscript
(Piscataway, NJ, USA). The miR-138 mimic sequence was
5-AGCUGGUGUUGUGAAUCAGGCCG-3, the miR-138 inhibitor sequence was
5-CGGCCUGAUUCACAACACCAGCU-3, the HIF1-α siRNA sequence was
5-AUCCAGAGUCACUGGAACU-3′ and the NC sequence was
5-UGAAUCCUUGAAUAGGUGUGUU-3. Carbon dioxide was used to euthanize
the mice with a fill rate of 10–30% of the volume of the cage per
minute. The authors of the current study followed the American
Veterinary Medical Association guidelines' recommendations to
confirm the death of the mice, including heartbeat, breathing,
corneal reflex and responses to firm toe pinch, graying of the
mucous membranes and rigor mortis.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
RNA from heart samples from each group was extracted
using the TRIzol® reagent (Invitrogen). Subsequently,
the RNA was reverse transcribed into cDNA using the cDNA Reverse
Transcription kit (Applied Biosystems; Thermo Fisher Scientific,
Inc., Waltham, MA, USA), and we perform RT-qPCR with SYBR Premix Ex
Taq (Takara Biotechnology Co., Ltd.). GAPDH was used as the
internal normalized reference. The relative levels of miR-138 and
HIF1-α were calculated using the 2−∆∆Cq method (22). The thermocycing conditions were as
follows: 45°C for 10 min and one cycle of 95°C for 10 min, followed
by 40 cycles at 95°C for 15 sec and 60°C for 45 sec. The primers
sequences were as follows: miR-138: Forward,
5-TCCGAGCCTGACTAAGTGTTGTGGTCGA-3 and reverse, 5-GTGCAGGGTCCGAGGT-3;
GAPDH: Forward, 5-TGGTATCGTGGAAGGACTC-3 and reverse
5-AGTAGAGGCAGGGATGATG-3; HIF1-α: Forward, 5-CTCAGCCCCAGTGCATTGTA-3
and reverse 5-GAACCTCCTATAGCCACCGC-3.
Western blot analysis
Myocardial tissues from each group were lysed using
radioimmunoprecipitation assay buffer. Subsequently, a
bicinchoninic acid protein assay to detect the concentration of
protein. Protein lysates (20 µl) were then separated using 10%
SDS-PAGE and transferred to polyvinylidene fluoride membranes (EMD
Millipore, Billerica, MA, USA). Membranes were then blocked with 5%
skim milk in TBST for 1 h at room temperature. The membranes were
incubated with the following antibodies obtained from Abcam
(Cambridge, UK): HIF1-α (1:2,000; cat. no. ab51608), cleaved
caspase-9 (1:2,000; cat. no. ab2324), cleaved caspase-3 (1:2,000;
cat. no. ab2302), Drp1 (1:2,000; cat. no. ab184247), Fis1 (1:2,000;
cat. no. ab71498) and GAPDH (1:5,000; Santa Cruz Biotechnology,
Inc.) at 4°C overnight. Then, they were washed using TBST and
incubated with secondary antibodies (horseradish peroxidase
conjugated Goat Anti-Rabbit IgG; 1:5,000; cat. no. ab205718, Abcam)
at 4°C for 1 h. ECL Western blotting substrate (Pierce; Thermo
Fisher Scientific, Inc.) was used to visualize and detect the
results. ImageJ software 1.43v (National Institutes of Health,
Bethesda, MD, USA) was used to quantify protein levels.
Assessment of myocardial infarct
size
After sacrificing the mice in each group, evans blue
dye was used to demarcate the ischemic area-at-risk. Subsequently,
heart tissues were excised and sliced (50 µm). These samples were
stained with 1% triphenyltetrazolium chloride at 37°C for 20 min
and then fixed using 4% paraformaldehyde for 8 h at 4°C. Infarcted
myocardium was separated from the non-infarcted myocardium and
weighed carefully. Heart tissue from each group were dissected and
weighed. The infarct size was presented as a percentage of the
total ischemic area.
Measurement of serum myocardial
enzymes
After 6 h of reperfusion, blood samples from each
group were collected, centrifuged at 2,000 × g for 30 min at 4°C
and transferred to Eppendorf tubes. Subsequently, ELISA was
performed to measure the serum levels of troponin I (cat. no.
BEK-2212-1P/2P; Biosensis), cardiac muscle (cTn I; cat. no.
EKC40439, R&D Systems Inc.) and creatine kinase M-type/B-type
(CK-MB; cat. no. ABIN415661; R&D Systems Inc.) according to the
manufacturer's protocol.
Electron microscopy analysis
The method of electron microscopy analysis was
described as previously (23). The
electron microscopy images of mitochondria were analyzed using
PhotoshopCS5.0 software (Adobe, Inc.). For the analysis, the number
of the myocardial mitochondria in each group were measured.
Luciferase reporter assay
TargetScan (www.targetscan.org/) was used to predict the target
gene of miR-138. Following, a wild-type (WT) 3-untranslated region
(UTR) fragment of HIF1-α containing the putative miR-138 binding
sequence was inserted into a pmirGlO Dual-luciferase miRNA Target
Expression Vector (Promega Corporation, Madison, WI, USA), while
mutant (MUT) 3′-UTR was also cloned into the vector to generate a
mutated binding site. Subsequently, the cells obtained from the
American Type Culture Collection were co-transfected with HIF1-α-WT
or HIF1-α-MUT and miR-138 mimics (20 nmol/l) using Lipofectamine™
2000 (Thermo Fisher Scientific, Inc.). Dual Luciferase reporter
assay system (DLR® Assay, Promega Corporation) was used
to evaluate the luciferase activity after 48 h. Renilla Luciferase
was used as a normalizing transfection control.
Statistical analysis
GraphPad Prism 4 software was used to analyze the
experimental data. The data were presented as mean ± standard
deviation. Statistical analyses were performed using Student's
t-test and one-way analysis of variance, followed by a Tukey's
post-hoc test. P<0.05 indicated that the difference between
groups was statistically significant.
Results
miR-138 is downregulated and HIF1-α is
upregulated after myocardial I/R injury
To identify whether miR-138 performed important
functions in myocardial I/R injury, its expression level in adult
mouse myocardium with and without I/R injury was examined. The
results indicated that the miR-138 expression level was
significantly downregulated in the myocardium of the myocardial I/R
injury model compared with the control myocardium (Fig. 1A). Moreover, the expression level of
HIF1-α was significantly upregulated in the myocardium of the
myocardial I/R injury model compared with the control myocardium
(Fig. 1B). The western blotting
results also confirmed the alteration of HIF1-α expression level as
HIF1-α protein expression significantly upregulated in the
myocardium of the I/R injury group compared with the control group
(Fig. 1C and D).
Overexpression of miR-138 reduces the
myocardial I/R-induced increase in infarct size and myocardial
enzymes
As mentioned previously, compared with the control
group, the miR-138 expression level was significantly decreased in
the myocardial I/R injury group. Subsequently, to further explore
the role of miR-138 in the myocardial I/R injury model, miR-138
mimic, miR-138 inhibitor or a negative control were administered
intraperitoneally before modeling. Compared with the I/R injury
group, the infarct size of, and serum CTn I and CK-MB levels in the
myocardium with I/R injury were significantly decreased in the
miR-138 mimic group and increased in the miR-138 inhibitor group
(Fig. 2).
miR-138 inhibits the expression of
cleaved caspase-9 and −3 in myocardial I/R injury
The number of the mitochondria in the myocardial I/R
injury group per area was significantly decreased compared with the
sham group. However, in the miR-138 mimic group, the number of
mitochondria per area were increased significantly, while in the
miR-138 inhibitor group, the number of mitochondria was
significantly reduced compared with the I/R injury group (Fig. 3A). Notably, the results of the
western blot analysis indicated that the expression levels of
cleaved caspase-9 and −3 were significantly increased in the
myocardial I/R injury group compared with sham mice, and after the
injection of the miR-138 mimic, expression levels were decreased
compared with the I/R injury group (Fig.
3B-D). In the miR-138 inhibitor group, cleaved caspase-9 and −3
expression levels were significantly increased reduced compared
with the I/R injury group.
miR-138 inhibits the expression of
proteins related to mitochondrial morphology
To explore the mechanism of miR-138 in relation to
morphological alterations of mitochondria in myocardial I/R injury,
the expression levels of the mitochondrial fission-related
proteins, Drp1 and Fis1 (24,25),
were examined using western blot analysis. The results indicated
that the expression levels of Drp1 and Fis1 in the myocardial I/R
injury group were both increased compared with the sham group
(Fig. 4). When treated with miR-138
mimic, however, Drp1 and Fis1 expression levels were significantly
decreased, while their expression levels were both increased in the
miR-138 inhibitor group compared with the I/R injury group.
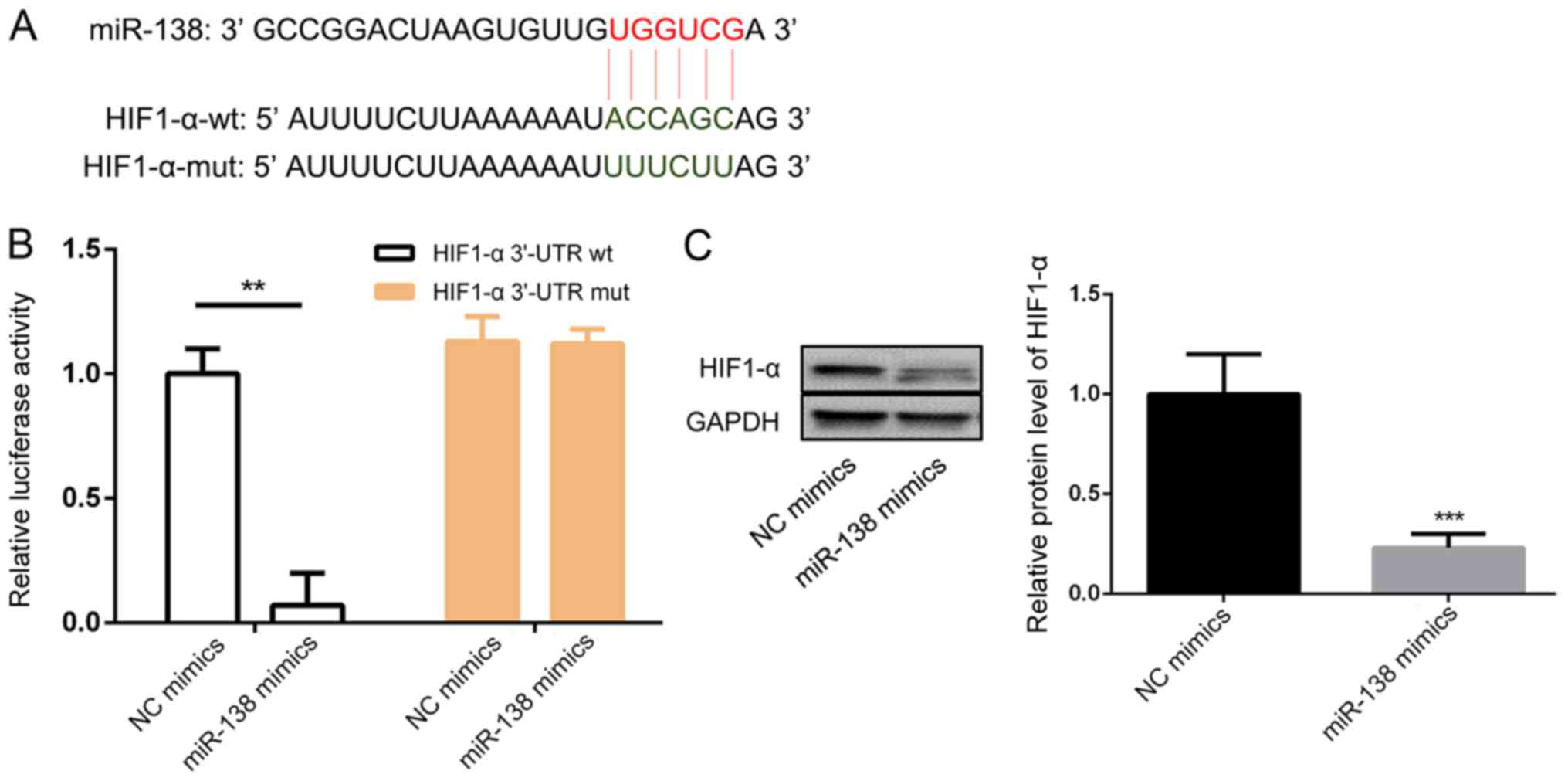
HIF1-α is the target of miR-138
The bioinformatic results were confirmed by a
luciferase reporter assay. Fig. 5A
shows the predicted miR-138 binding sequence in HIF1-α. The
luciferase activity of the construct with the WT 3′-UTR was
significantly inhibited after transfection with the miR-138 mimic
(Fig. 5B). Western blotting results
indicated that miR-138 mimic transfection markedly decreased the
expression of HIF1-α compared with NC mimic transfection (Fig. 5C).
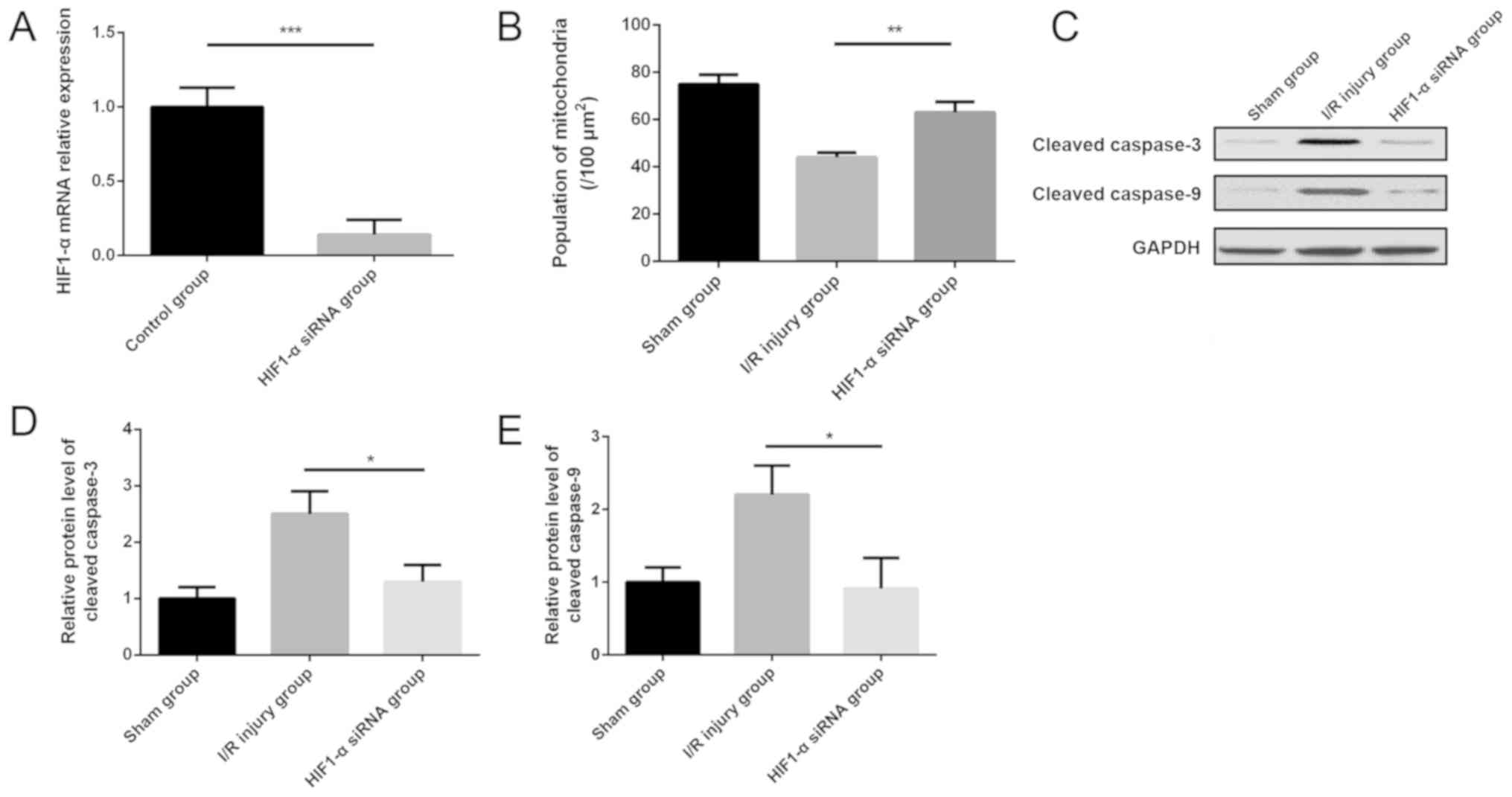
Downregulation of HIF1-α inhibits the
expression of cleaved caspase-9 and caspase-3 in myocardial I/R
injury
To explore the role of HIF1-α in the model of
myocardial I/R injury, siRNA of HIF1-α were administered
intraperitoneally before modeling. HIF1-α siRNA was shown to
significantly decrease the expression of HIF1-α in the myocardium
compared with the control group (Fig.
6A). The number of mitochondria per area was increased
significantly in the mouse myocardium of the HIF1-α siRNA group
compared with the I/R injury group (Fig.
6B). Notably, western blotting results indicated that the
protein expression levels of cleaved caspase-9 and −3 were
decreased significantly in the myocardium after HIF1-α siRNA
administration compared with the I/R injury group (Fig. 6C-E).
Discussion
CHD is considered to be the leading cause of death
worldwide, with approximately 17.5 million people dying because of
cardiovascular disease according to the estimates from the World
Health Organization (26–28). Acute myocardial I/R usually causes
the detrimental effects of CHD (29). Therefore, to explore the molecular
mechanism involved in myocardial I/R injury, the authors of the
current study conducted functional experiments and identified the
critical role of miR-138 and mitochondria-mediated apoptosis in the
myocardial I/R injury model. The results indicated that low
expression levels of miR-138 were found in the myocardium with I/R
injury compared with that of control myocardium. Furthermore, the
level of HIF1-α was significantly upregulated in the myocardium of
the myocardial I/R injury model compared with control myocardium.
Overexpression of miR-138 reduced the myocardial I/R-induced
increase in infarct size and myocardial enzymes by targeting HIF1-α
to inhibit myocardial I/R-induced mitochondrial apoptosis. These
findings suggest that miR-138 may prevent damage of the myocardium
after I/R injury.
miRNAs are a type of small non-coding RNAs that bind
to their target mRNAs specifically, and subsequently cause the
downregulation of the target gene by repressing degradation or
translation (30). In recent years,
miRNAs have been implicated in many processes related to the heart,
such as cardiac hypertrophy, cardiac development and heart failure
(31). miRNAs function as endogenous
intracellular regulators of mRNA translation, but the significance
of miR-138 in the process of myocardial I/R injury has not been
reported previously, particularly its role in myocardial
I/R-induced mitochondrial apoptosis. In the current study, the
results indicated that miR-138 was downregulated in myocardium with
I/R injury compared with control myocardium. The results also
showed that the infarct size and serum CTn I and CK-MB levels of
the myocardium with I/R injury were significantly decreased in the
miR-138 mimic group, but were significantly increased in the
miR-138 inhibitor group compared with the control group.
Apoptosis is known as a critical pathological
process in the course of myocardial I/R injury and the amount of
apoptosis determines the severity of myocardial I/R injury
(32,33). Therefore, understanding the mechanism
involved in cardiomyocyte apoptosis in myocardial I/R injury is
critically important in the development of effective treatment
methods. Prior research has shown that myocardial I/R-induced
mitochondrial dysfunction and apoptosis are responsible for the
exacerbation of cardiac ischemic injury in diabetic patients
(15). In the present study, the
authors found that in the miR-138 mimic group, the mitochondria per
area were significantly increased, while the number of mitochondria
was significantly decreased in the miR-138 inhibitor group.
Furthermore, the expression levels of cleaved caspase-9 and −3 were
both significantly increased in the myocardial I/R injury model
compared with control mice. Additionally, after injection of the
miR-138 mimic, their expression levels were decreased. After
treatment with the miR-138 mimic, protein expression levels of Drp1
and Fis1, proteins related to mitochondrial morphology, were both
significantly decreased, while their expression levels were
increased in the miR-138 inhibitor group. Moreover, HIF1-α was
confirmed as the target of miR-138. Therefore, the aforementioned
results showed that miR-138 might lessen myocardial ischemia
reperfusion injury by inhibiting mitochondria-mediated apoptosis,
due to its associated targeting of HIF1-α.
There are some limitations in the current study. The
mitochondrial membrane potential generated by proton pumps is an
essential component in the process of energy storage during
oxidative phosphorylation, and it is associated with cells'
capacity to generate ATP by oxidative phosphorylation (34,35).
Several fluorescent lipophilic cationic dyes (including,
tetramethylrhodamine methyl ester and tetramethylrhodamine ethyl
ester, Rhodamine 123, 3,3′-dihexyloxacarbocyanine iodide and
5,5′,6,6′-tetrachloro-
1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide) have become
important tools for directly measuring the mitochondrial membrane
potential (36). Furthermore,
apoptosis inducing factor (AIF), such as Cyt-C, Smac and Apaf-1,
are proteins that trigger chromatin condensation and DNA
fragmentation in a cell in order to induce programmed cell death
(37,38). The mitochondrial AIF protein was
found to be a caspase-independent death effector that can allow
independent nuclei to undergo apoptotic changes (39). Therefore, future studies should
detect the change of mitochondrial membrane potential and apoptosis
factors to detect mitochondrial apoptosis.
This study indicated that overexpression of miR-138
reduces the myocardial I/R-induced increase in infarct size and
myocardial enzymes by targeting HIF1-α to inhibit myocardial
I/R-induced mitochondrial apoptosis. These results demonstrated a
cardioprotective effect of miR-138 and suggested the potential to
become a promising target to alleviate myocardial I/R injury.
Acknowledgements
Not applicable.
Funding
The present study was supported by Shandong Province
Natural Science Foundation (grant no. ZR2015HL011) and Shandong
Medical and Health Technology development Project (grant no.
2016WS0687).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
QZ conceptualized the idea; YL and JFZ performed the
experiments; YL and XYL searched the literature; YL, XYL and JFZ
analyzed the data; YL and JFZ designed and made the figures; YL
created the tables; YL and QZ wrote the manuscript. QZ reviewed the
paper.
Ethics approval and consent to
participate
This current study was approved by the Clinical
Ethics Committee of Affiliated Hospital of Weifang Medical
University (Shandong, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Nichols M, Townsend N, Scarborough P and
Rayner M: Cardiovascular disease in europe 2014: Epidemiological
update. Eur Heart J. 35:2950–2959. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cuevas P, Carceller F and Gimenez-Gallego
G: Fibroblast growth factors in myocardial ischemia/reperfusion
injury and ischemic preconditioning. J Cell Mol Med. 5:132–142.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Song CL, Liu B, Wang JP, Zhang BL, Zhang
JC, Zhao LY, Shi YF, Li YX, Wang G, Diao HY, et al: Anti-apoptotic
effect of microRNA-30b in early phase of rat myocardial
ischemia-reperfusion injury model. J Cell Biochem. 116:2610–2619.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jennings RB and Reimer KA: The cell
biology of acute myocardial ischemia. Annu Rev Med. 42:225–246.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mu F, Duan J, Bian H, Yin Y, Zhu Y, Wei G,
Guan Y, Wang Y, Guo C, Wen A, et al: Cardioprotective effects and
mechanism of radix salviae miltiorrhizae and lignum dalbergiae
odoriferae on rat myocardial ischemia/reperfusion injury. Mol Med
Rep. 16:1759–1770. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yellon DM and Hausenloy DJ: Myocardial
reperfusion injury. New Engl J Med. 357:1121–1135. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Prompunt E, Sanit J, Barrere-Lemaire S,
Nargeot J, Noordali H, Madhani M and Kumphune S: The
cardioprotective effects of secretory leukocyte protease inhibitor
against myocardial ischemia/reperfusion injury. Exp Ther Med.
15:5231–5242. 2018.PubMed/NCBI
|
|
8
|
Li X, Liu M, Sun R, Zeng Y, Chen S and
Zhang P: Protective approaches against myocardial ischemia
reperfusion injury. Experimental and therapeutic medicine 12. Exp
Ther Med. 12:3823–3829. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shi Z, Zhou H, Lu L, Li X, Fu Z, Liu J,
Kang Y, Wei Z, Pan B, Liu L, et al: The roles of microRNAs in
spinal cord injury. Int J Neurosci. 127:1104–1115. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen C, Zhou Y, Wang J, Yan Y, Peng L and
Qiu W: Dysregulated microRNA involvement in multiple sclerosis by
induction of T helper 17 cell differentiation. Front Immunol.
9:12562018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yang J, Chen L, Yang J, Ding J, Li S, Wu
H, Zhang J, Fan Z, Dong W and Li X: MicroRNA-22 targeting CBP
protects against myocardial ischemia-reperfusion injury through
anti-apoptosis in rats. Mol Biol Rep. 41:555–561. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang SB, Liu TJ, Pu GH, Li BY, Gao XZ and
Han XL: MicroRNA-374 exerts protective effects by inhibiting SP1
through activating the PI3K/Akt pathway in rat models of myocardial
ischemia-reperfusion after sevoflurane preconditioning. Cell
Physiol Biochem. 46:1455–1470. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhu ZD, Ye JY, Niu H, Ma YM, Fu XM, Xia ZH
and Zhang X: Effects of microRNA-292-5p on myocardial
ischemia-reperfusion injury through the peroxisome
proliferator-activated receptor-alpha/-gamma signaling pathway.
Gene Ther. 25:234–248. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Huang LH, Li J, Gu JP, Qu MX, Yu J and
Wang ZY: Butorphanol attenuates myocardial ischemia reperfusion
injury through inhibiting mitochondria-mediated apoptosis in mice.
Eur Rev Med Pharmacol Sci. 22:1819–1824. 2018.PubMed/NCBI
|
|
15
|
Wu Y, Leng Y, Meng Q, Xue R, Zhao B, Zhan
L and Xia Z: Suppression of excessive histone deacetylases activity
in diabetic hearts attenuates myocardial ischemia/reperfusion
injury via mitochondria apoptosis pathway. J Diabetes Res.
2017:82080652017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu J, Zhang C, Zhao Y, Yue X, Wu H, Huang
S, Chen J, Tomsky K, Xie H, Khella CA, et al: Parkin targets
HIF-1alpha for ubiquitination and degradation to inhibit breast
tumor progression. Nat Commun. 8:18232017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Semenza GL, Shimoda LA and Prabhakar NR:
Regulation of gene expression by HIF-1. Novartis Found Symp.
272:2–8. 2006.PubMed/NCBI
|
|
18
|
Semenza GL, Jiang BH, Leung SW, Passantino
R, Concordet JP, Maire P and Giallongo A: Hypoxia response elements
in the aldolase A, enolase 1, and lactate dehydrogenase A gene
promoters contain essential binding sites for hypoxia-inducible
factor 1. J Biol Chem. 271:32529–32537. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen JX and Stinnett A: Ang-1 gene therapy
inhibits hypoxia- inducible factor-1alpha
(HIF-1alpha)-prolyl-4-hydroxylase-2, stabilizes HIF-1alpha
expression, and normalizes immature vasculature in db/db mice.
Diabetes. 57:3335–3343. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xue W, Cai L, Tan Y, Thistlethwaite P,
Kang YJ, Li X and Feng W: Cardiac-specific overexpression of
HIF-1{alpha} prevents deterioration of glycolytic pathway and
cardiac remodeling in streptozotocin-induced diabetic mice. Am J
Pathol. 177:97–105. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Elrod JW, Calvert JW, Morrison J, Doeller
JE, Kraus DW, Tao L, Jiao X, Scalia R, Kiss L, Szabo C, et al:
Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury
by preservation of mitochondrial function. Proc Natl Acad Sci USA.
104:15560–15565. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gupta S and Knowlton AA: HSP60, Bax,
apoptosis and the heart. J Cell Mol Med. 9:51–58. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Frank S, Gaume B, Bergmann-Leitner ES,
Leitner WW, Robert EG, Catez F, Smith CL and Youle RJ: The role of
dynamin-related protein 1, a mediator of mitochondrial fission, in
apoptosis. Dev Cell. 1:515–525. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Perrelli MG, Pagliaro P and Penna C:
Ischemia/reperfusion injury and cardioprotective mechanisms: Role
of mitochondria and reactive oxygen species. World J Cardiol.
3:186–200. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang W, Li Y and Wang P: Long non-coding
RNA-ROR aggravates myocardial ischemia/reperfusion injury. Braz J
Med Biol Res. 51:e65552018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Arora M, Kaul D and Sharma YP: Human
coronary heart disease: Importance of blood cellular miR-2909
RNomics. Mol Cell Biochem. 392:49–63. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Venardos KM, Zatta AJ, Marshall T, Ritchie
R and Kaye DM: Reduced L-arginine transport contributes to the
pathogenesis of myocardial ischemia-reperfusion injury. J Cell
Biochem. 108:156–168. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hausenloy DJ and Yellon DM: Myocardial
ischemia-reperfusion injury: A neglected therapeutic target. J Clin
Invest. 123:92–100. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Esteller M: Non-coding RNAs in human
disease. Nat Rev Genet. 12:861–874. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cheng J, Wu Q, Lv R, Huang L, Xu B, Wang
X, Chen A and He F: MicroRNA-449a inhibition protects H9C2 cells
against hypoxia/reoxygenation-induced injury by targeting the
notch-1 signaling pathway. Cell Physiol Biochem. 46:2587–2600.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Konstantinidis K, Whelan RS and Kitsis RN:
Mechanisms of cell death in heart disease. Arterioscler Thromb Vasc
Biol. 32:1552–1562. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Geng YJ: Molecular mechanisms for
cardiovascular stem cell apoptosis and growth in the hearts with
atherosclerotic coronary disease and ischemic heart failure. Ann N
Y Acad Sci. 1010:687–697. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Basheer WA, Fu Y, Shimura D, Xiao S,
Agvanian S, Hernandez DM, Hitzeman TC, Hong T and Shaw RM: Stress
response protein GJA1-20k promotes mitochondrial biogenesis,
metabolic quiescence, and cardioprotection against
ischemia/reperfusion injury. JCI Insight. 3:2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kadenbach B, Ramzan R, Moosdorf R and Vogt
S: The role of mitochondrial membrane potential in ischemic heart
failure. Mitochondrion. 11:700–706. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Solaini G, Sgarbi G, Lenaz G and Baracca
A: Evaluating mitochondrial membrane potential in cells. Biosci
Rep. 27:11–21. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Muzaffar S and Chattoo BB:
Apoptosis-inducing factor (Aif1) mediates anacardic acid-induced
apoptosis in Saccharomyces cerevisiae. Apoptosis. 22:463–474. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhang X, Chen J, Graham SH, Du L, Kochanek
PM, Draviam R, Guo F, Nathaniel PD, Szabó C, Watkins SC and Clark
RS: Intranuclear localization of apoptosis-inducing factor (AIF)
and large scale DNA fragmentation after traumatic brain injury in
rats and in neuronal cultures exposed to peroxynitrite. J
Neurochem. 82:181–191. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Joza N, Susin SA, Daugas E, Stanford WL,
Cho SK, Li CY, Sasaki T, Elia AJ, Cheng HY, Ravagnan L, et al:
Essential role of the mitochondrial apoptosis-inducing factor in
programmed cell death. Nature. 410:549–554. 2001. View Article : Google Scholar : PubMed/NCBI
|