Introduction
Allergic asthma is a common clinical airway
inflammatory disease triggered by antigens (1). It is mediated by T helper (Th) 2 cells
and involves multiple inflammatory cells and molecules (2). Asthma is characterized by reversible
airflow obstruction, high airway reactivity, increased secretion of
airway mucus and immunoglobulin E, and eosinophil infiltration
(3,4). In recent decades, mortality due to
asthma has increased in a number of countries, including the United
States, Great Britain, Australia and New Zealand, making asthma a
serious threat to public health globally (5).
Numerous studies have shown that oxidative stress
plays an important role in the pathogenesis of asthma (6–8). When
airway inflammation occurs, inflammatory mediators such as
histamine and leukotriene are released. These mediators recruit and
activate a variety of inflammatory cells, including eosinophils,
neutrophils, lymphocytes, macrophages and platelets (9). The activation of inflammatory cells
leads to the release of various free radicals and oxidative stress.
Free radicals include reactive oxygen species (ROS) and reactive
nitrogen species. These free radicals can cause serious damage or
apoptosis of airway epithelial cells (10,11). A
number of studies have shown that oxidative damage by lipids,
nucleic acids and proteins is widespread in the lungs of asthmatic
mice sensitized by ovalbumin (OVA) (12–14).
Oxidative stress indicators, such as nitrates and superoxide
anions, are also increased in asthma patients (15). Multiple studies have detected
elevated levels of hydrogen peroxide and nitric oxide in exhaled
gases from asthma patients (16–18),
which suggests potential oxidative injury.
As a free radical scavenger, Edaravone (Eda)
scavenges free radicals and inhibits lipid peroxidation (19). It is currently used for the treatment
of acute cerebral infarction (19).
In recent years, numerous studies have found that it also has a
protective effect against lung injury, due to its ability to
eliminate ROS (20–22). Wang et al (22) found that Eda ameliorated LPS-induced
pulmonary fibrosis by reducing lipopolysaccharide (LPS)-stimulated
oxidative stress and activating transforming growth
factor-β1/mothers against decapentaplegic homolog 3 signaling. In
another study, it was revealed that Eda attenuated oxidative stress
and subsequent lung injury induced by liver reperfusion (23). Herbicide paraquat toxicity can cause
severe oxidative injury in the lungs (24). In A549 cells treated with paraquat,
administration of Eda decreased the levels of intracellular ROS and
malondialdehyde (MDA) but increased the levels of superoxide
dismutase (SOD) (25). In a canine
lung transplantation model, Eda significantly decreased lung
wet/dry ratios, MDA levels and myeloperoxidase activity (26). However, to the best of our knowledge
the effects of Eda on asthma have not yet been investigated.
The nuclear factor erythroid 2-related factor 2
(Nrf2)/antioxidant response element (ARE) signaling pathway is a
defensive pathway in response to oxidative and chemical stress that
is critical for the regulation of antioxidants and phase II
detoxification enzymes. One example of an ARE is hemeoxygenase-1
(HO-1) (27). Under homeostatic
conditions, Nrf2 binds to kelch-like ECH-associated protein-1
(Keap1) in the cytoplasm, remains inactive and is easily degraded.
Under oxidative or chemical stress, Keap1 modification or Nrf2
phosphorylation results in activation of Nrf2 through its
dissociation from Keap1. Activated Nrf2 can then translocate into
the nucleus and interact with HO-1 and other AREs (27). The expression of HO-1 is
transactivated in response to stress. Several studies have shown
that Eda may exert a protective effect through activation of the
Nrf2/HO-1 pathway. Zhang et al (28) revealed that Eda reduced iron-mediated
hydrocephalus and behavioral disorders in rats through Nrf2/HO-1
activation. In an animal model of cognitive damage induced by
chronic cerebral hypoperfusion (CCH), Eda reduced CCH-induced
cognitive damage and increased SOD activity and HO-1 levels, but
decreased MDA levels in the hippocampus through activation of the
Nrf2 pathway (29). Liu et al
(30) found that Eda increased
neuronal density and decreased neuronal damage induced by kainite,
which was administrated in the right hippocampus CA3 region using
the sereotactic technique and this enhanced the expression of Nrf2
and HO-1. However, all of these studies focused on the nervous
system. Whether Eda can activate the Nrf2/HO-1 pathway in the lung
remains to be elucidated.
In order to test the hypothesis that Eda may exert a
protective effect against asthma, asthma was induced in a mouse
model. The effects of Eda on airway responsiveness, cell and
cytokine counts in bronchoalveolar lavage fluid (BALF), and levels
of oxidative products and antioxidant enzymes in lung tissue were
studied. The protein levels of Keap1/Nrf2 and HO-1 in lung tissue
and their roles in the effects of Eda action were examined to
investigate the underlying mechanisms.
Materials and methods
Animals
A total of 96 normal C57BL/6 male mice, 24 male
Nrf2-deficient mice and 24 male HO-1-deficient mice with a
C57BL/6/SV129 background (age, 6–7 weeks; weight, 20±2 g) were
provided by the Experimental Animal Center of Jing'an District
Centre Hospital of Fudan University and kept in a climate
controlled room. The temperature of the animal room was kept at
24±3°C and the relative humidity was kept at 40–60%. The study was
approved by the Institutional Animal Care and Use Committee of the
Jing'an District Centre Hospital of Fudan University (approval no.
AS-17-3652).
After 1 week of adaptive feeding with access to food
and water ad libitum, normal mice were randomly divided into
six groups: Control group, OVA + saline group, OVA + Eda-low (L)
group, OVA + Eda-moderate (M) group, OVA + Eda-high (H) group, and
Eda-H group, with 12 mice in each group. Mice in the control group
received no treatment; mice in the OVA + saline group received OVA
(cat. no. A5503; Grade V; Sigma-Aldrich; Merck KGaA) and
intraperitoneal (i.p.) saline injection; mice in the OVA + Eda-L
group received OVA and i.p. injection of a total of 14 low doses of
Eda (1 mg/kg; cat. no. 443300; Sigma-Aldrich; Merck KGaA) in an
equal volume of saline; mice in the OVA + Eda-M group received OVA
and i.p. injection of a total of 14 moderate doses of Eda (5
mg/kg); mice in the OVA + Eda-H group received OVA and i.p.
injection of a total of 14 high doses of Eda (10 mg/kg); mice in
the Eda-H group received i.p. injection of a total of fourteen high
doses of Eda (10 mg/kg) but were not treated with OVA. In a
separate study, to explore the role of Nrf2 and HO-1 in response to
Eda, normal and deficient mice were divided into control group
(untreated group), OVA + Eda group, OVA + Eda + Nrf2−/−
group, OVA + Eda + HO-1−/− group, Nrf2−/−
group and HO-1−/− group, with 12 mice in each group. All
Eda group mice received i.p. injection of a single moderate dose of
Eda (5 mg/kg).
Asthma induction
On days 1, 8 and 15, mice in the OVA + saline group,
OVA + Eda-L group, OVA + Eda-M group, OVA + Eda-H group, OVA + Eda
+ Nrf2−/− group, and the OVA + Eda + HO-1−/−
group were injected i.p with 0.2 ml of an antigen mixture
(containing 50 µg OVA, 150 µl 10% aluminum hydroxide (cat. no.
1.01091; Sigma-Aldrich; Merck KGaA) and 50 µl physiological
saline). Beginning on day 22, mice inhaled 1% OVA for 30 min daily
in a plastic box using a nebulizer (emka TECHNOLOGIES) for 14 days
as an asthma challenge. I.p Eda was administered 30 min before each
challenge for 14 days. The study period was 36 days.
BALF collection and cell count
Twenty-four h after the last OVA challenge, mice
were anesthetized with sodium pentobarbital (100 mg/kg, i.p.) and
pancuronium bromide (6 µg/g, i.p.), and the lungs were exposed by
thoracotomy. After these procedures were completed, the mice were
sacrificed by cervical dislocation. The spinal column was quickly
dislocated from the brain by pressing the base of the skull and
pulling backward on the tail to provide a fast and painless death
for the mice. BALF was then collected from the left lung. BALF was
centrifuged at 110 × g for 10 min at 4°C, and the cells were
resuspended in 1 ml Hank's solution (cat. no. C0218; Beyotime
Institute of Biotechnology). An aliquot of the cell suspension (100
µl) was put into a blood cell counter to determine the total number
of cells and a further 200 µl taken for Diff-Quick staining (Baxter
Diagnostics, Inc.) (31). At least
200 cells were counted in each sample and classified as
eosinophils, lymphocytes and neutrophils according to their
morphological characteristics (32).
Measurement of airway responsiveness
to inhaled methacholine (Mch)
The procedure for the measurement of airway
responsiveness to inhaled Mch has been described in a previous
publication (33). Briefly, mice
were anesthetized with sodium pentobarbital (100 mg/kg, i.p.) and
pancuronium bromide (6 µg/g, i.p.) and then intubated via
tracheostomy and mechanically ventilated in a plethysmograph
chamber. Intraesophageal and airway pressures were measured with a
dedicated pressure transducer (Validyne DP45; Buxco®,
DSI™, Harvard Bioscience, Inc.). The signals generated were
preamplified (MaxII, Buxco®, DSI™, Harvard Bioscience,
Inc.) and captured with computer software (Biosystem XA; version
2.7.9; Buxco®; DSI™; Harvard Bioscience, Inc.). The
baseline readings were taken and averaged for 3 min. Respiratory
resistance (RR) was measured in response to increasing doses of
inhaled Mch (1, 2 and 4 mg/ml), and 2 µl of each dose of
methacholine was delivered through an ultrasonic nebulizer over 10
consecutive breaths. Tidal volume was increased to 350 µl, and
respiratory rate was reduced to 100 breaths/min during each
nebulization and then returned to pre-nebulization values. RR to
airflow was determined continuously for 10 min, and the peak
responses following each dose of Mch were obtained. After these
procedures were completed, the mice were sacrificed by cervical
dislocation, as described above.
BALF cytokine measurement
BALF was centrifuged at 110 × g for 10 min at 4°C.
The levels of inflammatory cytokines interleukin (IL)-6 (cat. no.
PI326; Beyotime Institute of Biotechnology), IL-13 (cat. no.
ab219634, Abcam), interferon (IFN)-γ (cat. no. PI508; Beyotime
Institute of Biotechnology) and tumor necrosis factor (TNF)-α (cat.
no. PT512; Beyotime Institute of Biotechnology) were measured in
the BALF supernatants using commercial ELISA kits according to the
manufacturer's instructions. Briefly, the monoclonal capture
antibody was pre-coated on the ELISA plate. When the sample was
added, the target protein therein bound to the capture antibody. A
biotinylated anti-target protein antibody was added, which bound to
the target protein to form a sandwich immune complex. Subsequently,
horseradish peroxidase (HRP)-labeled streptavidin was added, which
bound to the sandwich immune complex. Finally, the developer
3,3′,5,5′-tetramethylbenzidine (TMB) solution was added. HRP then
catalyzed the oxidation of the colorless chromogenic agent TMB to a
blue substance, which became yellow after the addition of the stop
solution. The absorbance at 450 nm was then measured using a
Multi-Detection Microplate Reader (BioTek Instruments, Inc.). A
standard curve was generated using known concentrations of each
substance and used to compare with the absorbance values of the
sample, allowing the concentration of cytokines in the sample to be
interpolated.
Histopathology evaluation
At 24 h after the last OVA challenge, the left lobes
of lung tissue samples were collected and fixed with 10% formalin
for 48 h at room temperature. Lung tissues were cut into 5 µm-thick
sections and mounted onto slides, which were stained with
hematoxylin and eosin for 5 min at room temperature. These slides
were then randomly and blindly examined by a pathologist to
evaluate the histopathological appearance (three slides and four
fields of view per sample; magnification, ×200).
Measurement of ROS, total antioxidant
capacity (T-AOC) and MDA in the lung
Levels of intracellular ROS were measured through
oxidative conversion of 2′,7′-dichlorofluorescein diacetate
(DCFH-DA) to the fluorescent compound dichlorofluorescin (34). In brief, 24 h after the last OVA
challenge, lung tissue samples were taken, weighed (50 mg) and
homogenized on ice with ice-cold RIPA lysis buffer (cat. no.
P0013B; Beyotime Institute of Biotechnology) for 30 min. After
centrifugation (16,000 × g, 10 min at 4°C), lung homogenates were
incubated with PBS containing 15 µM DCFH-DA (Nanjing Jiancheng
Bioengineering Institute) for 30 min at 37°C to label intracellular
ROS. The cellular fluorescence was determined using a microplate
reader (Promega Corporation) at 490 and 520 nm. The total
antioxidant capacity (T-AOC) was determined using a detection kit
(cat. no. A015-2-1; Nanjing Jiancheng Bioengineering Institute) and
measuring the absorbance at 520 nm. The MDA content was measured
using a thiobarbituric acid reactive substances assay in accordance
with the manufacturer's instructions (Nanjing Jiancheng
Bioengineering Institute), and the absorbance was measured at a
wavelength of 532 nm (35).
Measurement of SOD, catalase (CAT) and
glutathione peroxidase (GSH-Px) in the lung
A period of 24 h after the last OVA challenge, lung
tissue samples were taken, weighed (50 mg) and homogenized on ice
with RIPA ice-cold lysis buffer (cat. no. P0013B; Beyotime
Institute of Biotechnology) for 30 min. After centrifugation
(16,000 × g, 10 min at 4°C), the activities of SOD, CAT and GSH-Px
in lung homogenate were measured using commercial kits (Beyotime
Institute of Biotechnology), on 96-well plates, according to the
manufacturer's instructions. SOD activity was detected with a total
SOD assay kit with
2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium,
monosodium salt (cat. no. S0101). The absorbance at 450 nm was then
measured using an HT Multi-Detection Microplate Reader (BioTek
Instruments, Inc.). CAT activity was measured with a catalase assay
kit (cat. no. S0051). The plate was measured at 520 nm. GPx
activity was measured with a GPx assay kit (cat. no. S0056). The
plate was measured at 340 nm. Finally, the activities of SOD, CAT
and GSH-Px were calculated by plotting standard curves and
comparing the absorbance values of the samples and the
standards.
Western blot analysis
Twenty-four h after the last OVA challenge, lung
tissue samples were taken, weighed (50 mg) and homogenized on ice
with ice-cold lysis buffer (cat. no. P0013; Beyotime Institute of
Biotechnology) for 30 min. After centrifugation (16,000 × g, 10 min
at 4°C), the supernatant was collected. Proteins in the supernatant
were measured using the BCA method (cat. no. P0010; Beyotime
Institute of Biotechnology). Proteins were separated using 10%
SDS-PAGE and then transferred to PVDF membranes (EMD Millipore).
The blotted membranes were blocked with 5% non-fat dry milk
(Beyotime Institute of Biotechnology). A period of 2 h later (at
room temperature), the membranes were washed with 0.2% Tween-20 in
TBS (TBST) and incubated with anti-Keap1 (1:1,000; cat. no. 4678),
anti-Nrf2 (1:500; cat. no. 4399), anti-HO-1 (1:1,000; cat. no.
70081) and anti-β-actin (1:1,000; cat. no. 3700) antibodies at 4°C
overnight. The membranes were then washed with TBST and probed with
HRP-conjugated anti-rabbit IgG (1:5,000; cat. no. 7074) for 1 h at
room temperature. All antibodies were supplied by CST Biological
Reagents Co., Ltd. After antibody incubations the blots were washed
with TBST and visualized with a peroxidase/luminol enhanced
chemiluminescence reagent (Thermo Fisher Scientific, Inc.) using
Quantity One software (version 4.6.2; Bio-Rad Laboratories,
Inc.).
Statistical analysis
Data are presented as the mean ± SEM (three
repeats). Statistical tests performed were one-way analysis of
variance with Tukey-Kramer or Tamhane's tests by SPSS 17.0 (SPSS
Inc.). GraphPad Prism 5 (GraphPad Software, Inc.) software was used
for plotting graphs. P<0.05 was considered to indicate a
statistically significant difference.
Results
Eda reduces airway responsiveness to
Mch
To measure the effect of Eda on asthma, mice were
treated with different doses of Eda and the airway responsiveness
of the mice to Mch was measured. As shown in Table I, in the control group, the airway
responsiveness to Mch gradually increased as the concentration of
Mch increased. In the animal model of asthma, the airway
responsiveness to Mch was significantly higher than in the control
group. In the OVA + Eda-L group, the airway responsiveness to Mch
was not significantly different when compared with OVA + saline. In
the OVA + Eda-M and OVA + Eda-H groups, however, the airway
responsiveness to Mch was significantly reduced when compared with
OVA + saline. Treatment with Eda-H alone did not affect airway
responsiveness compared with the control.
 | Table I.Airway responsiveness to Mch. |
Table I.
Airway responsiveness to Mch.
|
| RR, % of
baseline |
|---|
|
|
|
|---|
| Group | NS | Mch (1 mg/ml) | Mch (2 mg/ml) | Mch (4 mg/ml) |
|---|
| Control | 102.6±10.5 | 113.6±15.8 | 122.7±10.4 | 152.4±13.2 |
| OVA + saline | 111.2±19.5 |
143.7±15.1a |
188.5±12.6a |
223.1±15.2a |
| OVA + Eda-L | 105.6±16.2 | 129.5±14.7 | 172.9±16.2 | 221.4±12.9 |
| OVA + Eda-M | 103.7±15.5 |
112.2±11.4b |
123.4±11.2b |
152.9±15.8b |
| OVA + Eda-H | 103.3±15.7 |
108.5±10.4b |
120.3±12.8b |
144.6±20.8b |
| Eda-H | 106.8±16.1 | 114.1±12.2 | 123.2±11.9 | 159.7±18.8 |
Eda reduces cell count in BALF
Cell counts, including total cells, eosinophils,
lymphocytes and neutrophils, were subsequently measured in BALF. As
shown in Table II, all cell counts
were significantly increased by OVA treatment when compared with
control. In the OVA + Eda-M and OVA + Eda-H groups, total cells,
eosinophils, lymphocytes and neutrophils in BALF were significantly
reduced when compared with the OVA + saline group. Treatment with
Eda-H alone did not affect the BALF cell count compared with the
control.
| Table II.Cell count in BALF. |
Table II.
Cell count in BALF.
|
| Cell count
(1×104/ml) |
|---|
|
|
|
|---|
| Group | Total cells | Eosinophils | Lymphocytes | Neutrophils |
|---|
| Control | 7.51±1.61 | 0.17±0.05 | 0.44±0.06 | 0.35±0.05 |
| OVA + saline |
21.82±1.92a |
3.52±0.52a |
6.89±0.84a |
4.45±0.59a |
| OVA + Eda-L | 20.65±1.54 | 3.44±0.62 | 6.54±0.78 | 4.29±0.63 |
| OVA +Eda-M |
14.54±1.35b |
2.41±0.52b |
3.56±0.48b |
2.31±0.41b |
| OVA + Eda-H |
10.62±1.56b |
2.03±0.61b |
3.25±0.49b |
2.01±0.52b |
| Eda-H | 7.44±1.53 | 0.16±0.08 | 0.49±0.08 | 0.39±0.08 |
Eda reduces the levels of cytokines in
BALF
To measure the effect of Eda on lung inflammation,
the levels of IL-6, IL-13, IFN-γ and TNF-α were examined in BALF.
As shown in Table III, the levels
of IL-6, IL-13, IFN-γ and TNF-α in BALF were significantly
increased by OVA treatment compared with the control. In the OVA +
Eda-M and OVA + Eda-H groups, however, these parameters were all
significantly reduced when compared with the OVA + saline group.
Treatment with Eda-H alone did not significantly affect cytokine
levels compared with the control.
| Table III.Levels of cytokines in BALF. |
Table III.
Levels of cytokines in BALF.
|
| Levels of cytokines
(ng/l) |
|---|
|
|
|
|---|
| Group | IL-6 | IL-13 | IFN-γ | TNF-α |
|---|
| Control | 15.8±2.2 | 18.6±2.7 | 26.2±6.3 | 11.3±3.3 |
| OVA + saline |
29.6±3.1a |
36.2±3.4a |
56.3±10.2a |
29.6±3.5a |
| OVA + Eda-L | 28.5±3.9 | 35.1±3.3 | 52.3±8.5 | 28.4±2.8 |
| OVA + Eda-M |
20.3±3.5b |
23.6±3.1b |
32.6±6.9b |
20.6±3.7b |
| OVA + Eda-H |
16.5±2.8b |
20.2±2.5b |
28.5±5.5b |
16.9±3.2b |
| Eda-H | 15.4±2.4 | 19.5±2.1 | 23.6±6.7 | 13.4±3.1 |
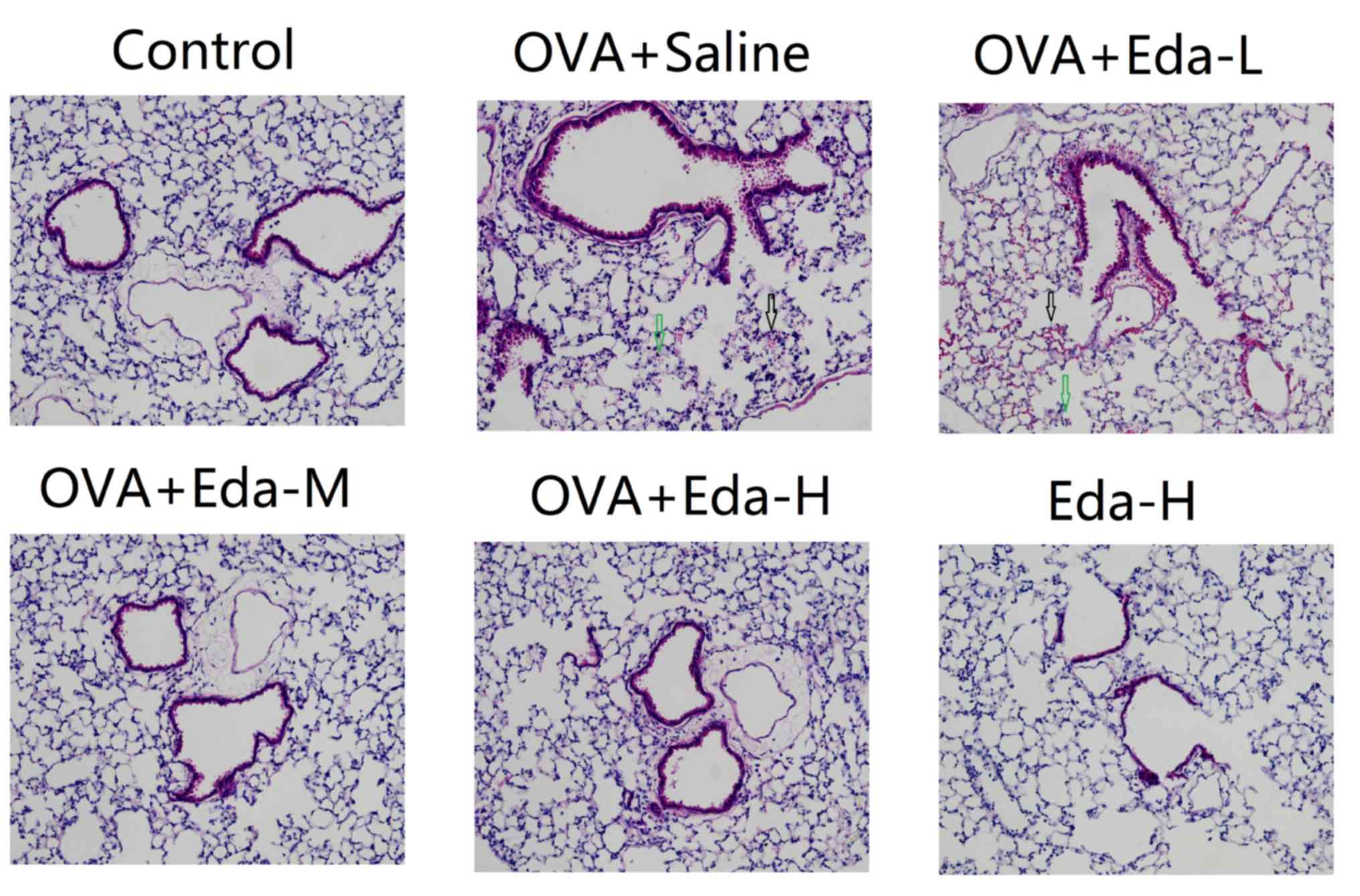
Eda attenuates lung histopathological
changes caused by OVA
Fig. 1 shows
histopathological changes in the mice. The normal control group
showed normal lung architecture. There was no perivascular edema or
peribronchial inflammation. Slides from the OVA + saline and OVA +
Eda-L groups showed perivascular edema and peribronchial
infiltration. The alveolar spaces were filled with alveolar
macrophages and inflammatory cells. In the OVA + Eda-M and OVA +
Eda-H groups, however, there was a marked decrease in perivascular
edema, peribronchial inflammation and macrophage infiltration in
the alveolar space compared to the OVA + saline group. Lung
histopathology was not significantly changed in the Eda-H group
when compared with the control group.
Eda attenuates oxidative stress in the
lung
To measure oxidative stress in the lung, the levels
of ROS, T-AOC and MDA were examined in lung tissue. As shown in
Fig. 2A, ROS level was significantly
increased in the OVA + saline group compared with the control
group. In the OVA + Eda-M and OVA + Eda-H groups, ROS levels were
significantly decreased compared with the OVA + saline group.
Treatment with Eda-H alone did not significantly affect ROS
content. By contrast, T-AOC was significantly decreased in the OVA
+ saline group compared with the control group but greatly
increased in OVA + Eda-M and OVA + Eda-H groups compared with the
OVA + saline group (Fig. 2B).
Treatment with Eda-H alone significantly increased T-AOC level
compared to the control. MDA levels in lung tissues were
significantly enhanced by OVA treatment but greatly decreased by
treatment with moderate and high dosages of Eda when compared with
the OVA + Saline group (Fig. 2C).
Treatment with Eda-H alone did not significantly affect MDA content
compared with the control group.
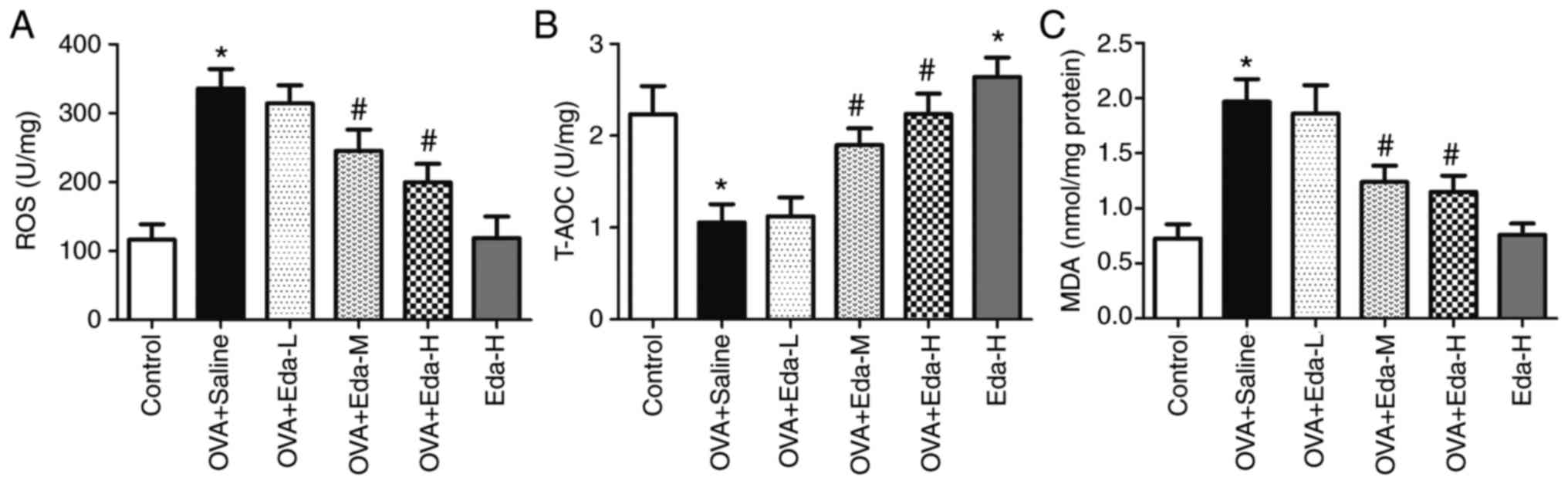 | Figure 2.Effects of Eda on oxidative stress in
the lung. To measure the effects of Eda on oxidative stress in
lungs, the levels of (A) ROS, (B) T-AOC and (C) MDA in lung tissues
were examined. Data are presented as the mean ± SEM. *P<0.05
compared with control; #P<0.05 compared with OVA +
saline. MDA, malondialdehyde; OVA, ovalbumin; ROS, reactive oxygen
species; T-AOC, total antioxidant capacity; Eda, edaravone; L, low
dose; M, moderate dose; H, high dose. |
Eda increases the levels of
antioxidative enzymes in the lung
To measure the effect of Eda on antioxidative
enzymes in the lung, the levels of antioxidative enzymes (SOD, CAT,
GSH-Px) were examined in lung tissues. The results showed that SOD
was greatly decreased in the OVA + saline group but recovered with
treatment with M and H doses of Eda (Fig. 3A). The levels of CAT (Fig. 3B) and GSH-Px (Fig. 3C) were also significantly decreased
in the OVA + saline group when compared with the control but
addition of Eda had no additional effect.
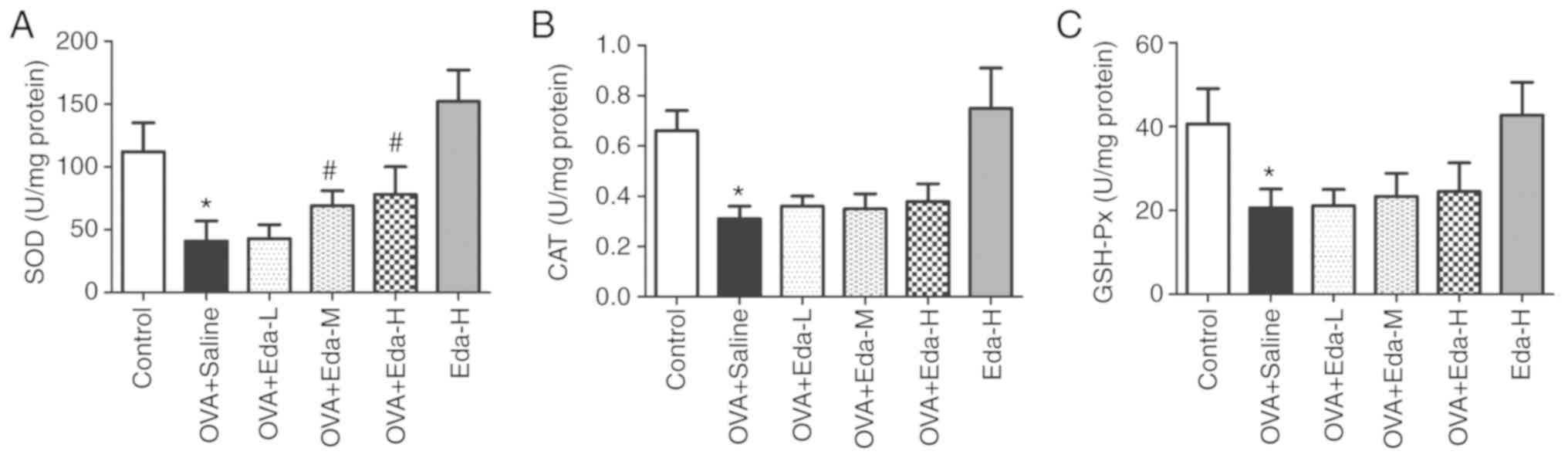 | Figure 3.Effects of Eda on antioxidative
enzymes in the lung. To measure the effects of Eda on antioxidative
enzymes in lungs, the levels of (A) SOD, (B) CAT and (C) GSH-Px in
lung tissues were examined. Data are presented as the mean ± SEM.
*P<0.05 compared with control; #P<0.05 compared
with OVA + saline. CAT, catalase; GSH-Px, glutathione peroxidase;
OVA, ovalbumin; SOD, superoxide dismutase; Eda, edaravone; L, low
dose; M, moderate dose; H, high dose. |
Eda activates the Keap1/Nrf2 pathway
and HO-1 expression in the lung
To explore the effect of Eda on the Keap1/Nrf2
pathway and HO-1 expression in the lung, the expression of Keap1,
Nrf2 and HO-1 was measured in lung tissue by western blotting, as
shown in Fig. 4. The results showed
that after mice were treated with moderate and high doses of Eda,
Keap1/Nrf2 ratio was significantly reduced, and Nrf2 (normalized to
β-actin) and HO-1 (normalized to β-actin) expression was
significantly increased, indicating that the Keap1/Nrf2 pathway was
activated.
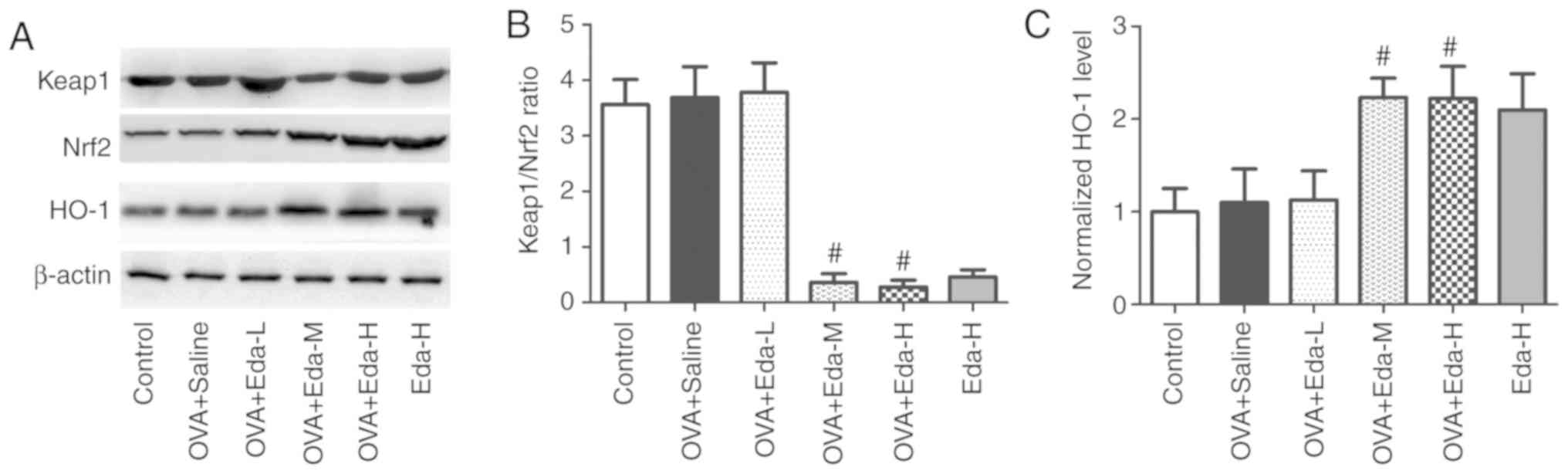 | Figure 4.Effects of Eda on the expression of
Keap1, Nrf2 and HO-1 in lungs. To explore the effect of Eda on the
Keap1/Nrf2 pathway and HO-1 expression in lungs, the expression of
Keap1, Nrf2 and HO-1 was measured in lungs by western blotting. (A)
representative bands of Keap1, Nrf2 and HO-1. (B) Results of
Keap1/Nrf2 ratio. (C) Results of normalized HO-1 levels to β-actin.
Data are presented as the mean ± SEM. #P<0.05
compared with OVA + saline. HO-1, hemeoxygenase-1; Keap1,
kelch-like ECH-associated protein-1; OVA, ovalbumin; Nrf2, nuclear
factor erythroid 2-related factor 2; Eda, edaravone; L, low dose;
M, moderate dose; H, high dose. |
Nrf2 or HO-1 deficiency abolishes the
effect of Eda on airway responsiveness to Mch
To explore the role of the Keap1/Nrf2 pathway and
HO-1 in the effect of Eda on airway responsiveness to Mch, normal,
Nrf2−/− and HO-1−/− mice were treated with
Eda and their airway responsiveness to Mch was measured. As shown
in Table IV, the airway
responsiveness of Nrf2−/− and HO-1−/− mice to
Mch was significantly higher than that of normal mice treated with
OVA and Eda (P<0.05). The airway responsiveness of
Nrf2−/− and HO-1−/− mice treated with Eda was
increased to a similar level as the OVA + saline group. The airway
responsiveness of Nrf2−/− mice and HO-1−/−
mice without OVA treatment was not significantly different from
control mice.
| Table IV.Airway responsiveness to Mch in
Nrf2−/− or HO-1−/− mice. |
Table IV.
Airway responsiveness to Mch in
Nrf2−/− or HO-1−/− mice.
|
| RR, % of
baseline |
|---|
|
|
|
|---|
| Group | NS | Mch (1 mg/ml) | Mch (2 mg/ml) | Mch (4 mg/ml) |
|---|
| Control | 103.6±11.5 |
119.3±12.8 | 124.5±16.7 | 158.9±18.2 |
| OVA + saline | 110.1±15.2 | 135.1±14.2 | 174.3±10.9 |
219.3±14.5 |
| OVA + Eda | 104.5±12.3 | 117.2±10.6 | 128.6±12.4 | 156.9±17.1 |
| OVA + Eda+
Nrf2−/− | 113.6±15.7 |
136.9±13.5a |
176.75±12.4a |
216.8±14.1a |
| OVA + Eda+
HO-1−/− | 115.2±16.5 |
136.4±13.2a |
171.4±10.7a |
214.2±13.3a |
|
Nrf2−/− | 103.7±12.4 | 111.5±11.5 | 120.4±10.7 | 153.4±15.2 |
|
HO-1−/− | 106.4±11.4 | 115.2±13.2 | 121.1±12.1 | 141.1±12.1 |
Discussion
In recent years, the hypotheses of free radical cell
injury and oxidant/antioxidant imbalance in asthma have attracted
widespread attention (12–14). It was indicated that the increased
ROS activated NF-κB activity, enhanced asthmatic inflammatory
response and induced glucocorticoid resistance (36). The basic features of bronchial asthma
include chronic airway inflammation, airway hyper-responsiveness,
and airway wall structural changes induced by long-term
inflammatory conditions (37).
Frossi et al (38) found that
oxidative stress upregulated Th2-mediated inflammatory responses,
increased the severity of asthma, enhanced bronchial
hyperresponsiveness and promoted airway remodeling. Therefore, the
use of antioxidants to correct an imbalance in the
oxidant/antioxidant system in asthma patients may protect the lung
from oxidative damage and mitigate inflammatory reactions and
airway remodeling. The present study established an animal model of
asthma with OVA and examined the effect of Eda against it. It was
shown that Eda reduced airway responsiveness to Mch when compared
with normal saline. The numbers of total cells, eosinophils,
lymphocytes and neutrophils in BALF were also significantly reduced
by Eda compared with saline, as were the levels of cytokines (IL-6,
IL-13, IFN-γ and TNF-α) in BALF. Histopathology showed that Eda
alleviated perivascular edema, peribronchial inflammation and
macrophage infiltration in the alveolar space caused by OVA
compared with saline. These results revealed that Eda effectively
reduced the degree of asthma and inflammatory reaction in lungs.
Other studies have shown the anti-inflammatory effects of Eda in
the lung. In LPS-induced lung injury, Eda markedly reduced
polymorphonuclear leukocyte infiltration and TNF-α and IL-6
concentrations in both serum and BALF (20,39). It
was also shown to suppress LPS-induced NF-κB activation and
cyclo-oxygenase-2 expression (20).
In an animal model of lung injury induced by black gunpowder smog,
Eda significantly reduced myeloperoxidase activity and alleviated
neutrophil infiltration of the lung (40). In the lung tissues of rats with
myocardial ischemia reperfusion injury, Eda significantly decreased
the concentration of serum creatine kinase isoenzyme, the lung
permeability index, β-defensin-2 mRNA expression and TNF-α protein
expression (41). Taken together,
these results suggest that the protective effect of Eda against
lung asthma may be related to a suppression of the inflammatory
response.
Oxidative stress has been shown to be closely
related to the development of asthma and is involved in airway
inflammation, airway hyper-responsiveness, increased airway
vascular permeability, tissue damage and airway remodeling
(39–44). Multiple ROS have strong oxidative
activity and may cause tissue damage, leading to smooth muscle
contraction and airway hyper-responsiveness (45). Oxidative stress can preferentially
induce T cell activation to Th2, resulting in Th1/Th2 unbalance
(46). Frossi et al (38) found that oxidative stress aggravated
the severity of asthma, increased bronchial hyper-responsiveness,
and promoted airway remodeling. In mice where oxidative
stress-related genes were knocked out, an asthma and airway
remodeling model could not be established (47). After antioxidant enzymes were
administered for 28 days, the lung function of asthma patients was
significantly improved (48).
Studies have confirmed that alveolar epithelial cells in asthma
patients are more susceptible to oxidative stress, apoptosis and
increased permeability (49). A
growing body of evidence supports the theory that oxidative stress
mediated by exogenous and endogenous ROS is an important factor in
the initiation of inflammatory mediators and cell damage (50). To measure oxidative stress in the
lung, the present study examined the levels of ROS, T-AOC and MDA
and antioxidative enzymes in lung tissue. The analyses revealed
that Eda significantly decreased the levels of ROS, T-AOC and MDA
compared with controls, and restored the levels of the
antioxidative enzyme, SOD. A study has shown that levels of
peroxide, MDA and conjugated diene were increased in the bronchial
secretions of patients with asthma, while catalase activity was
significantly reduced (51). As a
ROS metabolite, MDA reflects lipid peroxidation, protein
denaturation and impaired integrity of endothelial cells (52–54).
Under asthma attacks, more peroxide, MDA, conjugated diene and
other substances were found, and SOD activity decreased
significantly (13,55). SOD is an important enzyme in the
endogenous antioxidative system in lung tissue (56). SOD levels in lung tissues indirectly
reflect the degree of damage to lung cells by ROS (56). The results of oxidative stress and
antioxidative enzymes in the present study indicated that Eda may
alleviate the severity of asthma by suppressing oxidative injury
and restoring the antioxidative enzyme, SOD.
It has been shown that Eda may exert a protective
effect through the Nrf2/HO-1 pathway (28–30). Li
et al (57) revealed that Eda
protects against hyperosmolarity-induced oxidative stress and
apoptosis in primary human corneal epithelial cells by increasing
the expression of Nrf2 and its target genes, such as HO-1 and
GPx-1. Eda was also found to upregulate the gene expression of
Nrf2/HO-1 and alleviate cisplatin-induced neurobehavioral deficits
(58). To explore the effect of Eda
on the Keap1/Nrf2 pathway and HO-1 expression in lungs, the present
study measured the expression levels of Keap1, Nrf2 and HO-1 by
western blotting. The results showed that Eda significantly
decreased the Keap1/Nrf2 ratio, when compared with controls,
indicating that the Keap1/Nrf2 pathway was activated. Eda also
greatly increased HO-1 expression, suggesting that HO-1 may also
participate in the anti-asthmatic action of Eda. It was
hypothesized that the levels of Nrf2 and HO-1 may be decreased in
response to OVA, but the results showed that they were not
significantly changed by it. The reason for this may be that the
levels of Nrf2 and HO-1, which is an ARE, can be activated in
response to stress and inflammatory insults. Under oxidative or
chemical stress, activated Nrf2 translocates into the nucleus and
interacts with AREs (59). The
expression of cytoprotective target genes, including phase II
detoxifying enzymes, antioxidant proteins and molecular
proteasomes/chaperones, is then transactivated in response to the
stress (60). As a result, in the
present study, there may be a balance between the consumption and
production of Nrf2 and HO-1 during OVA insult, making it appear
that the levels of Nrf2 and HO-1 were unchanged. Treatment with
Eda, however, may further activate the Nrf2/ARE pathway, increasing
the levels of Nrf2 and HO-1. To confirm the role of the Keap1/Nrf2
pathway and HO-1 on the effect of Eda, normal, Nrf2−/−
and HO-1−/− mice were treated with Eda and the effects
on airway responsiveness to Mch were measured. The airway
responsiveness of Nrf2−/− or HO-1−/− mice to
Mch was increased to a similar level as the OVA + saline group and
was significantly higher compared with that of normal mice treated
with Eda. This result suggested that Nrf2 or HO-1 deficiency
abolished the effect of Eda on airway responsiveness to Mch.
Combined with the western blot results, these findings indicated
that the activated Keap1/Nrf2 pathway and HO-1 were essential in
the anti-asthmatic effect of Eda.
In conclusion, Eda effectively reduced the severity
of symptoms in a mouse model of asthma, which may be related to
suppression of the inflammatory response and its antioxidative
abilities. The activated Keap1/Nrf2 pathway and HO-1 may also be
involved in the anti-asthmatic effects of Eda.
Acknowledgements
Not applicable.
Funding
The current study was funded by Shanghai Jing'an
District Health Planning Commission (grant no. JWRC2014G02).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HC designed the study, provided the funding and
revised the manuscript; YP performed the airway responsiveness
experiments, cell count and cytokines measurement in BALF and wrote
the manuscript. WL performed the lung histopathological changes and
oxidative stress experiments; YF performed the western blot
analysis. JX analyzed the data.
Ethical approval and consent to
participate
The present study was approved by the Institutional
Animal Care and Use Committee of the Jing'an District Centre
Hospital of Fudan University (approval no. AS-17-3652).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Verhasselt V, Milcent V, Cazareth J, Kanda
A, Fleury S, Dombrowicz D, Glaichenhaus N and Julia V: Breast
milk-mediated transfer of an antigen induces tolerance and
protection from allergic asthma. Nat Med. 14:170–175. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kuipers H and Lambrecht BN: The interplay
of dendritic cells, Th2 cells and regulatory T cells in asthma.
Curr Opin Immunol. 16:702–708. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mabalirajan U, Dinda AK, Kumar S, Roshan
R, Gupta P, Sharma SK and Ghosh B: Mitochondrial structural changes
and dysfunction are associated with experimental allergic asthma. J
Immunol. 181:3540–3548. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Aguilera-Aguirre L, Bacsi A,
Saavedra-Molina A, Kurosky A, Sur S and Boldogh I: Mitochondrial
dysfunction increases allergic airway inflammation. J Immunol.
183:5379–5387. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Platts-Mills TA: The allergy epidemics:
1870-2010. J Allergy Clin Immunol. 136:3–13. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li Y and Li GP: Oxidative stress in
asthma: A distinct clinical and pathologic feature? J Biol Regul
Homeost Agents. 30:1053–1057. 2016.PubMed/NCBI
|
|
7
|
Mishra V, Banga J and Silveyra P:
Oxidative stress and cellular pathways of asthma and inflammation:
Therapeutic strategies and pharmacological targets. Pharmacol Ther.
181:169–182. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bullone M and Lavoie JP: The contribution
of oxidative stress and inflamm-aging in human and equine asthma.
Int J Mol Sci. 18:E26122017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Swerdlow RH: Treating neurodegeneration by
modifying mitochondria: Potential solutions to a ‘complex’ problem.
Antioxid Redox Signal. 9:1591–1603. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jessie BC, Sun CQ, Irons HR, Marshall FF,
Wallace DC and Petros JA: Accumulation of mitochondrial DNA
deletions in the malignant prostate of patients of different ages.
Exp Gerontol. 37:169–174. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hirai K, Aliev G, Nunomura A, Fujioka H,
Russell RL, Atwood CS, Johnson AB, Kress Y, Vinters HV, Tabaton M,
et al: Mitochondrial abnormalities in Alzheimer's disease. J
Neurosci. 21:3017–3023. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Servais S, Boussouar A, Molnar A, Douki T,
Pequignot JM and Favier R: Age-related sensitivity to lung
oxidative stress during ozone exposure. Free Radic Res. 39:305–316.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jang HY, Kim SM, Yuk JE, Kwon OK, Oh SR,
Lee HK, Jeong H and Ahn KS: Capsicum annuum L. methanolic extract
inhibits ovalbumin-induced airway inflammation and oxidative stress
in a mouse model of asthma. J Med Food. 14:1144–1151. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jung WK, Lee DY, Choi YH, Yea SS, Choi I,
Park SG, Seo SK, Lee SW, Lee CM, Kim SK, et al: Caffeic acid
phenethyl ester attenuates allergic airway inflammation and
hyperresponsiveness in murine model of ovalbumin-induced asthma.
Life Sci. 82:797–805. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Eisner MD, Yelin EH, Katz PP, Earnest G
and Blanc PD: Exposure to indoor combustion and adult asthma
outcomes: Environmental tobacco smoke, gas stoves, and woodsmoke.
Thorax. 57:973–978. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Emelyanov A, Fedoseev G, Abulimity A,
Rudinski K, Fedoulov A, Karabanov A and Barnes PJ: Elevated
concentrations of exhaled hydrogen peroxide in asthmatic patients.
Chest. 120:1136–1139. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ganas K, Loukides S, Papatheodorou G,
Panagou P and Kalogeropoulos N: Total nitrite/nitrate in expired
breath condensate of patients with asthma. Respir Med. 95:649–654.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kharitonov SA, Yates D, Springall DR,
Buttery L, Polak J, Robbins RA and Barnes PJ: Exhaled nitric oxide
is increased in asthma. Chest. 107 (Suppl 3):S156–S157. 1995.
View Article : Google Scholar
|
|
19
|
Noor JI, Ikeda T, Ueda Y and Ikenoue T: A
free radical scavenger, edaravone, inhibits lipid peroxidation and
the production of nitric oxide in hypoxic-ischemic brain damage of
neonatal rats. Am J Obstet Gynecol. 193:1703–1708. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang Z, Luo Z, Bi A, Yang W, An W, Dong
X, Chen R, Yang S, Tang H, Han X and Luo L: Compound edaravone
alleviates lipopolysaccharide (LPS)-induced acute lung injury in
mice. Eur J Pharmacol. 811:1–11. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Han CH, Guan ZB, Zhang PX, Fang HL, Li L,
Zhang HM, Zhou FJ, Mao YF and Liu WW: Oxidative stress induced
necroptosis activation is involved in the pathogenesis of hyperoxic
acute lung injury. Biochem Biophys Res Commun. 495:2178–2183. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang X, Lai R, Su X, Chen G and Liang Z:
Edaravone attenuates lipopolysaccharide-induced acute respiratory
distress syndrome associated early pulmonary fibrosis via
amelioration of oxidative stress and transforming growth
factor-β1/Smad3 signaling. Biochem Biophys Res Commun. 495:706–712.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Uchiyama M, Tojo K, Yazawa T, Ota S, Goto
T and Kurahashi K: Edaravone prevents lung injury induced by
hepatic ischemia-reperfusion. J Surg Res. 194:551–557. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu S, Liu K, Sun Q, Liu W, Xu W, Denoble
P, Tao H and Sun X: Consumption of hydrogen water reduces
paraquat-induced acute lung injury in rats. J Biomed Biotechnol.
2011:3050862011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cheng ZQ, Han JY, Sun P, Weng YY, Chen J,
Wu GY and Ma HX: Edaravone attenuates paraquat-induced lung injury
by inhibiting oxidative stress in human type II alveolar epithelial
cells. World J Emerg Med. 3:55–59. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xu JZ, Shen BZ, Li Y, Zhang T, Xu WH, Liu
XW and Lu HG: Edaravone attenuates ischemia-reperfusion injury by
inhibiting oxidative stress in a canine lung transplantation model.
Chin Med J (Engl). 121:1583–1587. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Keum YS, Owuor ED, Kim BR, Hu R and Kong
AN: Involvement of Nrf2 and JNK1 in the activation of antioxidant
responsive element (ARE) by chemopreventive agent phenethyl
isothiocyanate (PEITC). Pharm Res. 20:1351–1356. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang J, Shi X, Chen Z, Geng J, Wang Y,
Feng H, Zhu G and Chen Q: Edaravone reduces iron-mediated
hydrocephalus and behavioral disorder in rat by activating the
Nrf2/HO-1 pathway. J Stroke Cerebrovasc Dis. 27:3511–3520. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang D, Xiao Y, Lv P, Teng Z, Dong Y, Qi
Q and Liu Z: Edaravone attenuates oxidative stress induced by
chronic cerebral hypoperfusion injury: Role of ERK/Nrf2/HO-1
signaling pathway. Neurol Res. 40:1–10. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu Z, Yang C, Meng X, Li Z, Lv C and Cao
P: Neuroprotection of edaravone on the hippocampus of
kainate-induced epilepsy rats through Nrf2/HO-1 pathway. Neurochem
Int. 112:159–165. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ogata-Suetsugu S, Yanagihara T, Hamada N,
Ikeda-Harada C, Yokoyama T, Suzuki K, Kawaguchi T, Maeyama T,
Kuwano K and Nakanishi Y: Amphiregulin suppresses epithelial cell
apoptosis in lipopolysaccharide-induced lung injury in mice.
Biochem Biophys Res Commun. 484:422–428. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Feng J, Li P, Li X and Zhou H: Anatomic
Distribution and morphology of common tracheal/bronchial/pulmonary
cells. Rapid On-Site Evaluation (ROSE) in Diagnostic Interventional
Pulmonology. Feng J, Li X, Li P, Li Q and Shi Y: Springer;
Singapore: 2019, View Article : Google Scholar
|
|
33
|
Guedes AG, Jude JA, Paulin J, Rivero-Nava
L, Kita H, Lund FE and Kannan MS: Airway responsiveness in
CD38-deficient mice in allergic airway disease: Studies with bone
marrow chimeras. Am J Physiol Lung Cell Mol Physiol. 308:L485–L493.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yin N, Peng Z, Li B, Xia J, Wang Z, Yuan
J, Fang L and Lu X: Isoflurane attenuates
lipopolysaccharide-induced acute lung injury by inhibiting
ROS-mediated NLRP3 inflammasome activation. Am J Transl Res.
8:2033–2046. 2016.PubMed/NCBI
|
|
35
|
Zhao P, Zhou WC, Li DL, Mo XT, Xu L, Li
LC, Cui WH and Gao J: Total glucosides of Danggui Buxue Tang
attenuate BLM-induced pulmonary fibrosis via regulating oxidative
stress by inhibiting NOX4. Oxid Med Cell Longev. 2015:6458142015.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Barnes PJ, Ito K and Adcock IM:
Corticosteroid resistance in chronic obstructive pulmonary disease:
Inactivation of histone deacetylase. Lancet. 363:731–733. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bousquet J, Jeffery PK, Busse WW, Johnson
M and Vignola AM: Asthma. From bronchoconstriction to airways
inflammation and remodeling. Am J Respir Crit Care Med.
161:1720–1745. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Frossi B, De Carli M, Piemonte M and
Pucillo C: Oxidative microenvironment exerts an opposite regulatory
effect on cytokine production by Th1 and Th2 cells. Mol Immunol.
45:58–64. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yang T, Zhang J, Sun L, Zhu X, Li J, Wang
J, Chen H, Bao R, Deng X, Hou J and Liu Y: Combined effects of a
neutrophil elastase inhibitor (sivelestat sodium) and a free
radical scavenger (edaravone) on lipopolysaccharide-induced acute
lung injury in rats. Inflamm Res. 61:563–569. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wang Z, Li R, Liu Y, Liu X, Chen W, Xu S,
Guo Y, Duan J, Chen Y and Wang C: Protective effects of edaravone
combined puerarin on inhalation lung injury induced by black
gunpowder smog. Int Immunopharmacol. 26:125–132. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhang W, Guo Y, Yu S, Wei J and Jin J:
Effects of edaravone on the expression of β-defensin-2 mRNA in lung
tissue of rats with myocardial ischemia reperfusion. Mol Med Rep.
7:1683–1687. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Sugiura H and Ichinose M: Oxidative and
nitrative stress in bronchial asthma. Antioxid Redox Signal.
10:785–797. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hoshino T, Okamoto M, Takei S, Sakazaki Y,
Iwanaga T and Aizawa H: Redox-regulated mechanisms in asthma.
Antioxid Redox Signal. 10:769–783. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Castillo SS, Levy M, Thaikoottathil JV and
Goldkorn T: Reactive nitrogen and oxygen species activate different
sphingomyelinases to induce apoptosis in airway epithelial cells.
Exp Cell Res. 313:2680–2686. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Katsumata U, Miura M, Ichinose M, Kimura
K, Takahashi T, Inoue H and Takishima T: Oxygen radicals produce
airway constriction and hyperresponsiveness in anesthetized cats.
Am Rev Respir Dis. 141:1158–1161. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
King MR, Ismail AS, Davis LS and Karp DR:
Oxidative stress promotes polarization of human T cell
differentiation toward a T helper 2 phenotype. J Immunol.
176:2765–2772. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Imaoka H, Hoshino T, Takei S, Sakazaki Y,
Kinoshita T, Okamoto M, Kawayama T, Yodoi J, Kato S, Iwanaga T and
Aizawa H: Effects of thioredoxin on established airway remodeling
in a chronic antigen exposure asthma model. Biochem Biophys Res
Commun. 360:525–530. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Mitsunobu F, Yamaoka K, Hanamoto K, Kojima
S, Hosaki Y, Ashida K, Sugita K and Tanizaki Y: Elevation of
antioxidant enzymes in the clinical effects of radon and thermal
therapy for bronchial asthma. J Radiat Res. 44:95–99. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Bucchieri F, Puddicombe SM, Lordan JL,
Richter A, Buchanan D, Wilson SJ, Ward J, Zummo G, Howarth PH,
Djukanović R, et al: Asthmatic bronchial epithelium is more
susceptible to oxidant-induced apoptosis. Am J Respir Cell Mol
Biol. 27:179–185. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Crapo JD: Oxidative stress as an initiator
of cytokine release and cell damage. Eur Respir J Suppl. 44:S4–S6.
2003. View Article : Google Scholar
|
|
51
|
Boljevic S, Daniljak IG and Kogan AH:
Changes in free radicals and possibility of their correction in
patients with bronchial asthma. Vojnosanit Pregl. 50:3–18. 1993.(In
Russian, Serbian). PubMed/NCBI
|
|
52
|
Fang WT, Li HJ and Zhou LS: Protective
effects of prostaglandin E1 on human umbilical vein endothelial
cell injury induced by hydrogen peroxide. Acta Pharmacol Sin.
31:485–492. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Yamamoto H, Yamamoto Y, Yamagami K, Kume
M, Kimoto S, Toyokuni S, Uchida K, Fukumoto M and Yamaoka Y:
Heat-shock preconditioning reduces oxidative protein denaturation
and ameliorates liver injury by carbon tetrachloride in rats. Res
Exp Med (Berl). 199:309–318. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Zhao X, Jin L, Shen N, Xu B, Zhang W, Zhu
H and Luo Z: Salidroside inhibits endogenous hydrogen peroxide
induced cytotoxicity of endothelial cells. Biol Pharm Bull.
36:1773–1778. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Zhao SF, Zhang QR, Xia SJ, Lin SR and Gu
F: Influence of Yiqi Dingchuan decoction on antioxidant ability of
alvoelar macrophages in Guinea pigs with asthma. Chin J Basic Med
Tradit Chin Med. 6:31–34. 2000.
|
|
56
|
Mach WJ, Thimmesch AR, Pierce JT and
Pierce JD: Consequences of hyperoxia and the toxicity of oxygen in
the lung. Nurs Res Pract. 2011:2604822011.PubMed/NCBI
|
|
57
|
Li Y, Liu H, Zeng W and Wei J: Edaravone
protects against hyperosmolarity-induced oxidative stress and
apoptosis in primary human corneal epithelial cells. PLoS One.
12:e01744372017. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Jangra A, Kwatra M, Singh T, Pant R,
Kushwah P, Ahmed S, Dwivedi D, Saroha B and Lahkar M: Edaravone
alleviates cisplatin-induced neurobehavioral deficits via
modulation of oxidative stress and inflammatory mediators in the
rat hippocampus. Eur J Pharmacol. 791:51–61. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Li AL, Shen T, Wang T, Zhou MX, Wang B,
Song JT, Zhang PL, Wang XL, Ren DM, Lou HX and Wang XN: Novel
diterpenoid-type activators of the Keap1/Nrf2/ARE signaling pathway
and their regulation of redox homeostasis. Free Radic Biol Med.
141:21–33. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Kobayashi M and Yamamoto M: Molecular
mechanisms activating the Nrf2-Keap1 pathway of antioxidant gene
regulation. Antioxid Redox Signal. 7:385–394. 2005. View Article : Google Scholar : PubMed/NCBI
|