Introduction
Vesicoureteral reflux (VUR) is a common urinary
tract (UT) defect that occurs in approximately 1-2% of young
children (1). High-grade VUR is
seen primarily in male infants and is often associated with
generalised renal damage, i.e. renal hypodysplasia. This rare
condition is regarded as a congenital abnormality associated with
primary VUR (2). Overall, in
children with VUR the most common form of renal damage is a focally
acquired injury caused by ascending urinary tract infections (UTI)
(3). The morbidity seen in
children with VUR is often related to recurrent UTI, which carries
the risk of progressive renal damage.
Familial clustering of VUR is well recognized. The
risk that offspring of affected individuals will have reflux
themselves has been reported to be as high as 66%, while the risk
of a sibling of an affected individual also having the condition is
between 27 and 51% (4-7).
The high frequency of VUR in relatives favours an autosomal
dominant inheritance pattern with reduced penetrance (8-12),
although some studies indicate possible autosomal recessive
inheritance (13) or an X-linked
mode of inheritance (14).
The search for a single gene linked to the
heritability of VUR has so far been unsuccessful, but a large
number of candidate genes have been suggested. These candidates
mainly include genes functioning in the developmental pathways of
the kidney, ureter and ureterovesical junction. During the last
decade, the VUR hypodysplasia anomaly has been included in the
congenital anomalies of the kidney and urinary tract (CAKUT) group,
which comprises a broad spectrum of renal and lower UT structural
and functional abnormalities. The rationale for this approach is
the shared embryological background, starting with an interaction
between the ureteric bud (UB) and metanephric mesenchyme (MM)
(15,16). There has been a rapid increase in
identification of CAKUT-associated genes due to recent advances in
new and more affordable sequencing. Mutations in numerous different
developmental genes have been identified, but there is little
correlation between families and individuals. Despite this
progress, the mutations responsible for the majority of CAKUT
conditions remain unknown (17,18).
The vast majority of genetic studies of VUR and/or
CAKUT have focused on the coding part of the genome. However, in
numerous studies of familial VUR there is a relative paucity of
precise genetic causes, despite indications of a dominant autosomal
inheritance pattern. It may therefore be reasonable to expect that
VUR-related variants are located in the non-coding DNA, such as the
promotor, topologically associating domain (TAD) boundaries, or
regulatory elements. Studies have shown that the regulatory
elements can be located within the gene, or located at a distance
of up to 1 Mb from the gene (19).
Thus, designating variants in non-coding areas as pathological is
far more difficult than identifying variants in protein-coding
genes. Whole-genome scans to identify common regions are the first
step towards locating these regulatory regions. However, genome
scan studies, mainly linkage analyses, show different results with
very few shared genomic regions among hereditary VUR patients.
Besides single nucleotide variants (SNV) and smaller
insertions/deletions (indels), copy number variations (CNV) are
also important contributors to human genetic variation. However,
some CNVs can also be associated with a variety of birth defects
and common diseases (20).
CNV-associated congenital malformations depend on the disruption of
specific genes at breakpoints, and also on which genes are located
within the duplication or deletion. Few studies have investigated
the impact of CNV in relation to VUR/CAKUT (21,22).
For this study we included only families from the
south-western part of Sweden who had the VUR complex (same
phenotype). This was because of the higher levels of heredity in
VUR than in other conditions included in CAKUT. We concentrated on
individuals who had the highest grade of VUR together with renal
hypodysplasia, which is the most severe phenotype. In this study we
explored the possible presence of shared chromosomal areas
(haplotypes) in 14 families with familial VUR. We screened for
unique haplotypes which might be shared by affected individuals in
the families and would therefor indicate a common ancestor.
Screening was performed using high-density single nucleotide
polymorphism (SNP) arrays, followed by genome-wide association
(GWA) and the SNP compatibility matching method recently described
(23,24). As VUR appears to be a genetically
heterogeneous disease we also evaluated shared haplotypes,
including those in subsets of families, for coding and non-coding
genes which could cause the VUR abnormality. In addition, we
searched for CNVs which might be associated with the condition.
Materials and methods
Patients and families
Fourteen families with three or more members with
primary VUR, were recruited at Queen Silvia Children's Hospital (a
tertiary referral center) in Gothenburg, Sweden. The families were
contacted and information on the study was given. Before entering
the study all subjects and/or their parents signed informed consent
forms for genetic screening. Blood samples or buccal smear
(Isohelix SK-2S Buccal Swabs) specimens were collected using
standard procedures. The Regional Ethical Review Board in
Gothenburg approved the study (Dnr 589-05).
A total of 43 patients in 14 families with VUR were
included (49% males). To clarify the relationship and analyse the
pattern of inheritance, pedigrees were constructed for each family
(Fig. 1). Additional members of
the families whose medical histories strongly suggested VUR, but
who had not undergone radiological investigations, were classified
as probable cases. Patients with VUR secondary to a neurogenic
bladder and posterior urethral valves were excluded from the
study.
Clinical data were obtained from medical records. A
VUR grade was obtained from a voiding cystourethrography (VCUG),
levels of kidney damage from dimercaptosuccinic acid (DMSA) or
mercaptoacetyltriglycine (MAG-3) scintigraphy (25), and total kidney function was
measured using glomerular filtration rate (GFR) or was estimated
according to the Schwartz formula (26). When patients had bilateral VUR,
their more severely affected side was measured to establish VUR
grade and kidney damage. Focal kidney damage was defined as one or
more areas with reduced uptake or indentation of the renal outline,
and is caused by postnatally acquired renal scarring (3). Generalised damage, referred to as
congenital renal hypodysplasia, was classified as a small kidney
with reduced tracer uptake or a diffuse parenchymal anomaly. A GFR
of <80% (<2SD) of expected GFR was considered subnormal
(27).
SNP genotyping
Genomic DNA was extracted from blood lymphocytes (in
25 cases) and buccal cells (in 15 cases) using a Qiagen DNeasy
Blood and Tissue Kit (Qiagen) and a Maxwell 16 Buccal Swab LEV DNA
Purification Kit (Promega, Corp.) respectively. For three
individuals from three different families DNA samples were not
available. The samples were genotyped on Affymetrix 250K SNP NspI
arrays, (Affymetrix Inc.), which detects ~262,000 SNPs. The array
experiments were performed either locally (n=34, Department of
Laboratory Medicine at the University of Gothenburg, Gothenburg,
Sweden) or at the BEA Core Facility, (n=6, Karolinska Institute,
Huddinge, Sweden), according to the manufacturer's protocol
(Affymetrix). Briefly, 250 ng of genomic DNA was digested using the
NspI restriction enzyme and ligated to adaptors. After ligation,
the template was subjected to PCR amplification using a generic
primer that recognised the adaptor sequence. The amplified DNA was
fragmented with DNase I, labelled with biotin and hybridised to a
GeneChip® Human Mapping 250 K array. The hybridised
probes were washed using the Affymetrix Fluidics Station 450 and
marked with streptavidin-phycoerythrin. The arrays were scanned
using a confocal laser scanner, GeneChip Scanner 3000 (Affymetrix).
Primary data analysis was performed using GDAS
(GeneChip® DNA Analysis software) and GTYPE (Affymetrix)
for the extraction of genotype calls.
SNP compatibility matching
SNP genotype data for individuals were analysed
using SNP compatibility matching (23,24).
This method can be applied to dominant traits in families where
several members are affected. In the original SNP compatibility
matching method, it was assumed that all affected individuals had a
common ancestor and shared the causal IBD (identical by descent)
variant. Although it was unlikely that all affected individuals in
our 14 families shared the same ancestral variant, we still wanted
to examine this possibility. In a similar way to homozygosity
mapping we also used this method to identify shared haplotypes,
which may act as causal factors for a disease, in different subsets
of affected subjects or families. The aim of the method is to
identify regions free from incompatibilities. For each SNP locus,
individuals can have either genotype ‘AA’, ‘AB’ or ‘BB’. A locus
where at least one affected individual is ‘AA’ and at least one
other affected individual is ‘BB’ is scored as an incompatibility.
A locus of this type cannot by definition be included in the
disease gene haplotype we are trying to locate. However, a
continuous large region of SNP loci, without any incompatibilities
among affected individuals, may include a unique disease haplotype
and, consequently, also the disease gene. Genotypes for our 14
affected families were compared and incompatibilities, as defined
above, for all the 260,000 SNP loci were scored and plotted against
the genome position for each locus. Corresponding genotype data
generated by the Affymetrix 250 K array in four healthy control
individuals were entered into the analysis. Given that an affected
individual and an unaffected control do not have the same disease
phenotype, their DNA must by definition be different. The regions
of the genome that were identical by state (IBS) in both VUR cases
and controls were therefore excluded.
Study strategies
Two different strategies were used for analysing the
study group and the controls (Fig.
2). In the first set of analyses, we tested whether the
disease haplotype was inherited from a common familial ancestor
(causal variant IBD). For this strategy we included one affected
individual per family, since a common ancestor hypothesis presumes
that all affected members of the family share the same haplotype,
which contains the founder gene mutation. We used four controls
added one at a time. Haplotypes found in both the control and the
families were excluded as a non-specific disease haplotype. If the
haplotype region shared by affected individuals, on the other hand,
became covered with incompatibilities when the control data was
added, this indicated that the haplotype was not present in the
control and thus might be specific to the disease. After testing
the controls one after the other, the shared region(s) that
remained could be regarded as identical by reason of descent. In
these analyses the size of haplotype regions we chose was ≥120
SNPs. After this, in a second set of analyses, we searched for
common haplotype regions in subsets of families. All affected
individuals from each family were included and for each family only
haplotypes shared by all family members were selected. We used four
controls to rule out common haplotypes within the general
population. We excluded only those regions that were identical for
VUR cases and all four controls. The haplotype region unique to the
affected families and not present in all four controls could be
regarded as specific to the disease. Given the limited number of
families available for this study, a less stringent filtering
strategy was used with inclusion of a very restricted number of
controls in order to only exclude the most common haplotypes i.e.
haplotypes shared by all four controls. With this strategy,
increase in number of controls would probably mean exclusion of
fewer common haplotypes. The chosen size of haplotype regions was
≥60 SNPs in the second set of analyses.
CNV detection
The R package ‘aroma.affymetrix’ was used for copy
number detection. The CEL files for each sample provided us with
copy number estimates of the intensity values using the CRMA v2
method (28). For copy number
segmentation we used the Circular Binary Segmentation (CBS) method
(29). Criteria for inclusion of
CNVs were: i) number of SNPs per CNV ≥10; ii) CNV size >50 kb
but <3 Mb; iii) CNV frequency <1% in the general population.
The CNVs were also filtered by the log2 values, excluding those
between +/-0.2. We searched for recurrent identical CNVs within
families. CNVs were defined as identical if they had the same state
of duplication or deletion, showed ≤30% difference in length, and
overlapped by >70%.
Data bases
All genomic positions for SNPs and CNVs are given
relative to GRCh37/hg19 genome assembly. Regions shared by seven
families or more were reported in the results. The UCSC genome
browser (https://genome.ucsc.edu) was used to
visualise the regions which could theoretically contain the disease
gene. We recorded the genes, both coding and non-coding DNA
sequences, in these regions. In addition, we examined their
expression and role during kidney development using GUDMAP (the
GenitoUrinary Development Molecular Anatomy Project data;
https://www.gudmap.org) and via an extensive
literature search (https://www.ncbi.nlm.nih.gov/pubmed). The significance
of each CNV detected was determined by comparison with public CNV
databases: DECIPHER (Database of Chromosomal Imbalance and
Phenotype in Humans using Ensembl Resources, https://decipher.sanger.ac.uk/) and DGV (Database of
Genomic Variants, http://dgv.tcag.ca/dgv/app/home).
Results
Clinical characteristics
The study included 43 patients with VUR, from 14
different families (21 males). Two families were nuclear and 12
were extended families. The relationship between the affected
individuals and the pattern of inheritance is shown in pedigrees in
Fig. 1. Phenotypical details of
the study subjects are outlined in Table I. Of the 43 patients with VUR,
high-grade reflux (grades IV to V) was seen in 49%, generalised
kidney damage in 50% and subnormal total renal function in 15% of
the cases. Five cases displayed additional malformations of the UT,
such as bilateral duplex kidney, bladder diverticula and a
unilateral megaureter. One VUR patient with syndromic presentation
was diagnosed with unbalanced translocation, which was inherited
from a healthy father with balanced translocation, the VUR
inheritance being maternal. An additional three cases with
extrarenal manifestations had syndromic features but were
undiagnosed (Table SI).
 | Table IDemographic data, VUR grades, renal
abnormalities and function for the group of individuals included in
the IBD part of the study and for the whole study group. |
Table I
Demographic data, VUR grades, renal
abnormalities and function for the group of individuals included in
the IBD part of the study and for the whole study group.
|
Characteristics | IBD study
cohorta, n=14
(%) | Total VUR cohort,
n=43 (%) |
|---|
| Sex | | |
|
Female | 6(43) | 22(51) |
|
Male | 8(57) | 21(49) |
| Presenting symptom
VUR | | |
|
Pyelonephritis | 9(64) | 29(68) |
|
Pre- and
postnatal screening | 3(22) | 10(23) |
|
Other
symptoms | 2(14) | 4(9) |
| Age at
presentation, months | 11 (0.25-98) | 7 (0.25-98) |
| Grade of
reflux | | |
|
I-III | 4(29) | 22(51) |
|
IV-V | 10(71) | 21(49) |
| Uni- or bilateral
reflux | | |
|
Unilateral | 4(29) | 17(40) |
|
Bilateral | 10(71) | 26(60) |
| Recurrent UTIs | | |
|
No | 4(29) | 13(32) |
|
Yes | 10(71) | 28(68) |
| Renal damage | | |
|
No | 2(14) | 15(36) |
|
Yes,
focal | 2(14) | 6(14) |
|
Yes,
generalizedb | 10(72) | 21(50) |
| Uni- or bilateral
renal damage | | |
|
Unilateral | 9(75) | 22(81) |
|
Bilateral | 3(25) | 5(19) |
| Total renal
function | | |
|
Normal | 10(71) | 34(85) |
|
Subnormal | 4(29) | 6(15) |
The SNP compatibility matching method
for locating risk regions using GWA data GWA data testing for IBD
haplotype (common ancestor)
Regions with no incompatibilities were identified
through SNP genotype data analyses using the method described
above. One affected individual per family (n=14) was included in
these analyses. Since the individual chosen for the analysis was
the most severely affected member of the family, this subgroup
displayed more high-grade VUR (10/14), more generalised kidney
damage, hypodysplasia (10/14) and more frequent subnormal total
renal function (4/14) (Table
I).
In these analyses we excluded shared haplotype
regions in affected individuals that were also found in any of the
controls, i.e. excluded as non-specific disease haplotypes. In the
analyses without controls, the most frequent haplotype was present
in 13 out of 14 VUR families. When tested against four healthy
controls one at a time, a maximum of seven families shared one
haplotype and thus the number of haplotypes shared by ≥7 families
decreased from 34 to only one (Table
II). Thus additional controls were not included as a common
ancestor haplotype already was excluded after used controls. The
conclusion was that we did not find a unique haplotype that was
shared by most or all the families, as the regions of interest
shared by numerous families were also seen in one or more of the
controls.
| Table IINumber of haplotypes shared by ≥
seven families in the different sets of analyses. |
Table II
Number of haplotypes shared by ≥
seven families in the different sets of analyses.
| A, One affected
individual/family |
|---|
| | Number of
haplotypes shared by ≥ seven of the families |
|---|
| Type of
analysis | 7 fam | 8 fam | 9 fam | 10 fam | 11 fam | 12 fam | 13 fam |
|---|
| No control
included | 18 | 8 | 5 | 1 | 1 | 1 | 1 |
| Inclusion of 4
controls | | | | | | | |
|
Haplotypes
found in all 4 controls excludeda,b | 14 | 6 | 4 | 0 | 1 | 1 | 1 |
|
Haplotypes
in any of 4 controls excluded | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| B, All affected
individuals/family |
| | Number of
haplotypes shared by ≥7 of the families |
| Type of
analysis | 7 fam | 8 fam | 9 fam | 10 fam | 11 fam | 12 fam | 13 fam |
| No control
included | 21 | 9 | 5 | 2 | 0 | 0 | 0 |
| Inclusion of 4
controls | | | | | | | |
|
Haplotypes
found in all 4 controls excludeda | 20 | 6 | 5 | 1 | 0 | 0 | 0 |
|
Haplotypes
in any of 4 controls excludedb | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
GWA data testing for disease variant
excluding the common haplotypes
In these analyses we searched for haplotype regions
shared by subsets of VUR families, with the only exclusion being of
haplotypes common in the general population, i.e. haplotypes shared
by all the four controls used (Table
II). Forty out of 43 affected individuals from 14 families were
included in these analyses; DNA was missing from three individuals.
A total of 32 haplotype regions shared by ≥7 families were
identified (Table III). The size
of these possible disease-associated haplotypes varied from 0.20 to
1.93 Mb, with a total of 22.92 Mb, representing 0.76% of the
genome. Twenty regions of haplotypes were shared by seven families,
six regions by eight families and five regions by nine families,
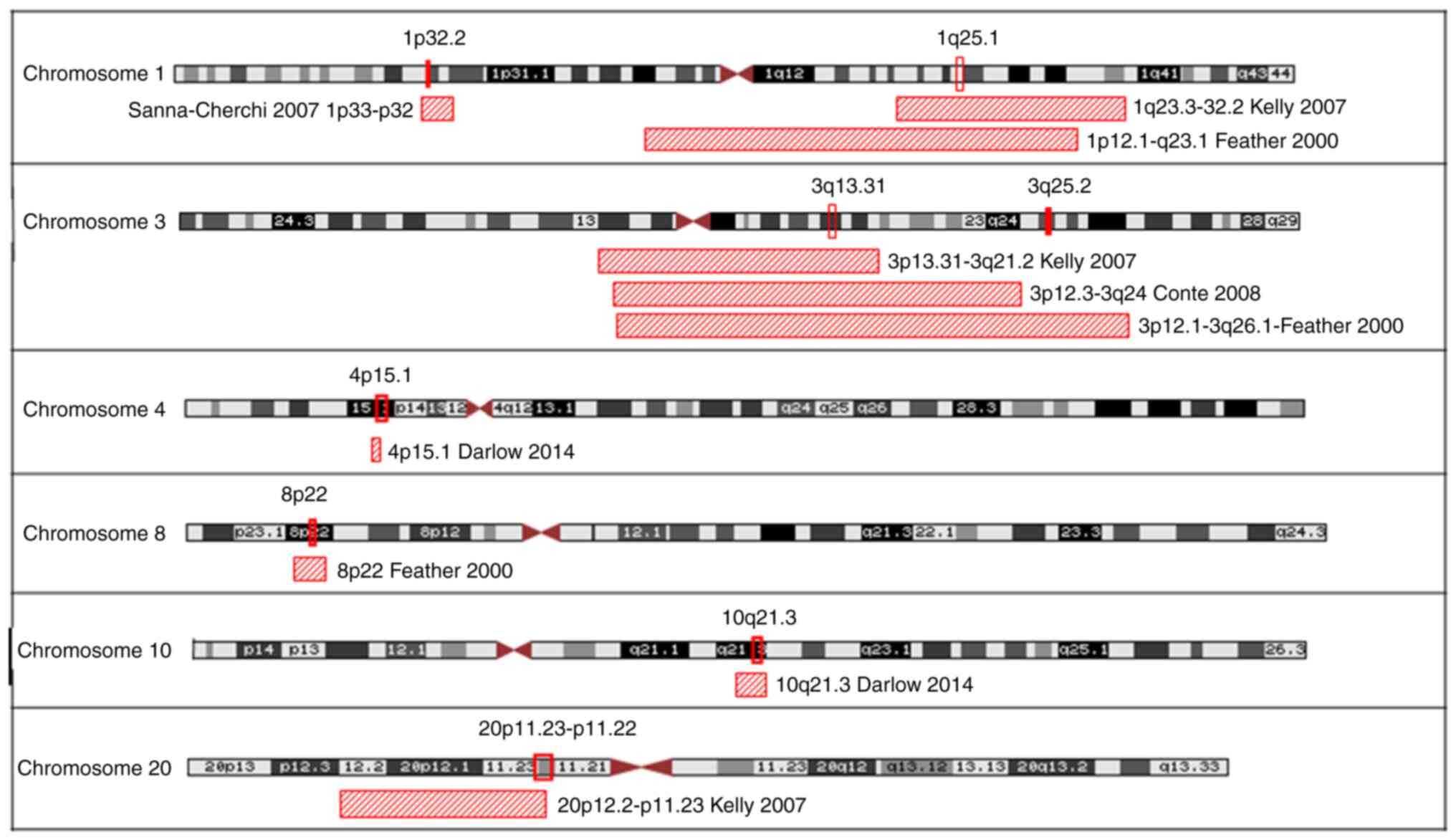
while only one haplotype was shared by 10 families (Table II). Fig. 3 illustrates the genomic locations
of haplotype regions shared by ≥7 families.
| Table IIIHaplotype regions found in ≥ seven
families when including all affected individuals in the 14
families, after exclusion of common haplotypes in the general
population. |
Table III
Haplotype regions found in ≥ seven
families when including all affected individuals in the 14
families, after exclusion of common haplotypes in the general
population.
| Haplotype regions
shared by ≥ seven families | | Genes | Evidence |
|---|
| Genomic
locations | Cytogenetic
band | Size of region,
Mb | Number of families
per haplotype region | Coding | Non-coding | (Refs.) | Region |
|---|
|
Chr1:56,335,237-56,766,727 | 1p32.2 | 0.43 | 8 | 0 | - | (11) | 1p32-33 |
|
Chr1:102,757,243-103,757,081 | 1p21.1 | 1.0 | 7 | - | - | | |
|
Chr1:173,712,120-174,952,226 | 1q25.1 | 1.24 | 7 | - | - | (40) | 1q23.3-q32.2 |
|
Chr3:49,938,758-51,864,849 | 3p21.31-p21.2 | 1.93 | 7 | - | - | | |
|
Chr3:115,953,619-116,316,741 | 3q13.31 | 0.36 | 7 | - | - | (40) (45) | 3p13-q21.2,
3p12.3-q24 |
|
Chr3:153,425,054-153,967,763 | 3q25.2 | 0.54 | 7 | - | - | (9) | 3p12.1-q26.1 |
|
Chr4:33,953,172-34,754,839j | 4p15.1 | 0.80 | 10 | 0 | - | (25) | 4p15.1 |
|
Chr4:43,274,485-43,768,843j | 4p13 | 0.50 | 9 | 0 | - | | |
|
Chr4:81,214,575-82,324,437j | 4q21.21 | 1.11 | 9 | BMP3b, FGF5b | - | | |
|
Chr5:115,384,247-115,845,251 | 5q23.1 | 0.46 | 7 | - | - | | |
|
Chr6:3,982,143-4,279,669 | 6p25.2-p25.1 | 0.36 | 8 | - | - | | |
|
Chr8:9,294,638-9,977,187 | 8p23.1 | 0.68 | 7 | TANKSa,d, SLC9A6a,e | - | | |
|
Chr8:16,029,070-16,489,054 | 8p22 | 0.46 | 7 | - | - | (9) | 8p22 |
|
Chr10:68,938,308-69,975,774 | 10q21.3 | 1.04 | 7 | - | - | (25) | 10q21.3 |
|
Chr10:83,304,656-83,757,199 | 10q23.1 | 0.45 | 7 | - | - | | |
|
Chr10:100,474,570-101,213,280 | 10q24.2 | 0.74 | 8 | - | - | | |
|
Chr11:26,300,178-26,592,685 | 11p14.2 | 0.29 | 7 | - | - | | |
|
Chr11:38,648,159-39,400,252 | 11p12 | 0.75 | 8 | 0 | - | | |
|
Chr11:41,769,608-42,310,568 | 11p12 | 0.54 | 7 | 0 | - | | |
|
Chr12:79,467,497-80,385,649j | 12q21.2-q21.31 | 0.92 | 8 | - | - | | |
|
Chr12:88,355,694-89,207,726 |
12q21.32-q21.33 | 0.85 | 9 | CEP290a,f, KITLGa,g | - | | |
|
Chr13:19,814,247-20,642,012 | 13q12.11 | 0.83 | 8 | - | - | | |
|
Chr13:38,832,645-39,313,967 | 13q13.3 | 0.48 | 7 | FREM2b | - | | |
|
Chr13:83,414,846-83,830,486 | 13q31.1 | 0.42 | 7 | 0 | 0 | | |
|
Chr14:37,462,847-38,176,041 | 14q13.3-q21.1 | 0.71 | 7 | FOXA1b |
RP11-356O9.2c | | |
|
Chr14:66,862,743-67,886,781 | 14q23.3 | 1.02 | 9 | PLEK2a | - | | |
|
Chr15:48,329,542-48,925,115 | 15q21.1 | 0.60 | 7 |
SLC12A1a,h,
FBN1a,i |
RP1-208K4.1c | | |
|
Chr16:78,422,926-78,621,620 | 16q23.1 | 0.20 | 9 | - | - | | |
|
Chr18:26,268,963-26,952,256 | 18q12.1 | 0.68 | 7 | 0 | - | | |
|
Chr19:23,487,250-24,503,985 | 19p12-p11 | 1.02 | 7 | - | - | | |
|
Chr20:21,055,354-22,080,540j |
20p11.23-p11.22 | 1.03 | 7 | PAX1a | - | (40) | 20p12.2-p11.23 |
|
Chr21:30,025,580-30,507,998j | 21q21.3 | 0.48 | 7 | USP16a | - | | |
In these candidate regions where genes causing the
disease might be located, we searched for coding and non-coding
genes of interest in the embryological development of the kidney
and UT. We differentiated between genes with a known function in
the UB and MM, and genes only expressed in the metanephros
according to GUDMAP but without known function (Table III). The haplotype region shared
by the highest number of families (n=10) and located at 4p15.1 did
not contain any protein coding elements. The non-coding RNAs were
not expressed in the post-developmental kidney or UT, although to
our knowledge their expression in the fetal kidney has not yet been
investigated. A haplotype shared by nine families was also located
on chromosome 4, at 4q21.21 (Table
III). In this region, two tentative candidate genes were
located; bone morphogenetic protein 3 (BMP3) in the middle
of the region and fibroblast growth factor 5 (FGF5) at the
lower end of the region, both with known functions in the
embryological development of the kidney and UT. Non-coding RNAs
were also present in this region, but with no known role in the
post-developmental kidney.
Of the other four regions shared by nine families,
two regions (12q21.32-q21.33 and 14q23.3) included genes with
expression in metanephros, but without an identified function
during kidney development (Table
III). In the six haplotype regions shared by eight families no
genes of interest were found. Of the 20 haplotypes shared by seven
families each, one region on cytogenetic band 13q13.3 included the
FRAS1 related extracellular matrix 2 (FREM2) gene with known
functions in the embryonic kidney. Another locus at 14q13.3-21.1,
contained the forkhead box A1 (FOXA1) gene, known to be
involved in early embryological development of numerous organ
systems. Of the remaining haplotypes shared by seven families, four
included genes expressed in the embryological kidney, but without
known function (Table III).
Non-coding RNA genes were present in almost all
haplotype regions, often with detectable expression levels in most
of the tissue samples, as presented by the GTExPortal (https://www.gtexportal.org/home/). A few lncRNA
(14q13.3-q21.1 and 15q21.1, respectively), had expressions that
were exclusive or almost exclusive to the post-natal kidney and UT.
Despite extensive data mining regarding fetal expression of these
lncRNA in the kidney (ENCODE-HaploReg 4.1), no available data could
be found.
A large number of haplotype regions were shared by
six families each (data not shown). A few genes of interest because
of their roles in UB and MM development were located in these
regions. One of these genes was ZFYVE9 at locus 1p32.3.
CNV analysis for locating inherited
chromosomal imbalances
A large number of CNVs were detected in all the
individuals analysed, with the overall CNV distribution (Fig. S1). We searched for recurrent
identical CNVs within the families with log 2 value >0.2 for
gain and log 2 <-0.2 for loss. CNVs shared by several affected
family members were detected in five families, although only one
family (family 32) showed the presence of a shared CNV-a deletion
at 5q31.1-in all affected relatives (Tables IV and SII). This chromosomal region contains
the follistatin like 4 (FSTL4) gene, expressed during renal
tubuli development, according to GUDMAP, but not detected in
earlier embryological phases (in UB or MM). An additional four CNVs
were partially shared, meaning that some but not all affected
family members were carriers of that specific CNV. Of specific
interest is the duplication at 20q13.31, seen in family 80. This
region contains the BMP7 gene, known to have a major
function in kidney and UT development. However, it was only present
in two family members. In addition, eighteen CNVs were shared by ≥2
unrelated individuals among the families. Common CNVs in the
population were excluded.
| Table IVCNV detected in two or more affected
individuals within a family. Segregation with VUR and the genes
located within the region. |
Table IV
CNV detected in two or more affected
individuals within a family. Segregation with VUR and the genes
located within the region.
| A, Family 17 |
|---|
| | Family members | |
|---|
| CNV position | 351 Fa | 369 Fa | 347 Fa | 355 Fe | 368 Fe | Genes |
|---|
|
9:11647401-11913220 | + | - | + | NA | NA | - |
|
12:131734725-131813804 | + | - | + | NA | NA | - |
| B, Family 32 |
| | Family members | |
| CNV position | 236 Fa | 656 Fa | 395 Ma | 235 Fe | 234 M | Genes |
|
5:132581687-132690433 | + | + | + | NA | NA |
FSTL4c |
| C, Family 76 |
| | Family members | |
| CNV position | 650 Fa | 648 Ma | 660 Fa | 644 Fd | 653 M | Genes |
|
14:22855145-23000062 | + | - | + | + | + | TRAC |
| D, Family 77 |
| | Family members | |
| CNV position | 645 Ma | 649 Fa | 690 Fa | 651 M | | Genes |
|
8:18100755-18457679 | + | - | + | NA | |
NAT2c, PSD3b |
|
19:49998299-50281369 | + | - | + | NA | |
PRR12b, IRF3b, BCL2L12b, CPT1Cb, RPS11b, RCN3b, NOSTPb, PRRG2b, SCAF1c, PRMT1c, AP2A1c, TSKSc, FCGRTc, ADM5, RRAS |
| E, Family 80 |
| | Family members | |
| CNV position | 682 Ma | 652 Ma | 658 Ma | 666 Ma | 695 F | Genes |
|
20:54967565-55918939 | - | + | - | - | + |
BMP7b |
|
21:38399356-38712209 | + | + | + | - | - |
DSCR3b, TTC3 |
Discussion
There are numerous association and linkage studies,
mainly genome-wide scans, searching for the chromosomal region(s)
which can explain the heritability of VUR. These studies either
include a large number of small families with ≥2 affected members,
often siblings (30-34),
or a small number of large families with numerous cases (9,11,35-37).
They show few overlapping regions, posing the question of whether
distinctive VUR-associated loci may vary in frequency in different
populations. In our study, the pedigrees of the 14 families with
three or more individuals with primary VUR indicate a dominant
autosomal inheritance pattern with reduced penetrance, in line with
earlier reports (8-11).
Given their origin in a small homogenous region in Sweden, we
wanted to investigate the possibility of shared ancestry among the
families. We used a SNP compatibility matching method, a variant of
linkage analysis, to analyse GWAS data. This data included only
patients with the condition, taking an affected-only approach. This
method was originally designed to detect a disease gene haplotype
derived from a common ancestor, thus establishing a classical
Mendelian dominant inheritance pattern (23,24).
However, in this case the method revealed that there was no
haplotype region shared by all the affected members of the 14
families. This suggested that there was no common ancestral founder
mutation, a finding in line with similar studies of other cohorts
(38,39).
Nevertheless, subsets of the VUR families were shown
to share regions on candidate disease genes. In our study we have
presented haplotype regions shared by seven or more families, and
at least eight of our findings agreed with loci identified in other
studies (on chromosomes 1, 3, 4, 8, 10 and 20, Fig. 4). The haplotype chromosomal region
4p15.1 was the region shared by most families (n=10) and overlaps
with a locus presented in an earlier case/control association study
by Darlow et al (31). The
region contained only non-coding genes, without any records of
expression in the post-developmental kidney. Whether regulatory
elements in this region fulfil functions in the fetal kidney is
unknown and will require further study. Of the other novel
haplotypes shared by several families in this present study, we
found regions 4q21.21 (n=9), 13q13.3 (n=7) and 14q21.1 (n=7) to be
of particular interest given the current knowledge of the role of
genes in fetal development of the kidney and UT. The 4q21.21 region
contains BMP3, encoding a ligand of the growth factor beta
(GFB) superfamily with a role in organogenesis in embryonic kidney
and renal tubuli development (40,41).
This chromosomal region also contains FGF5, which encodes
for a member of the fibroblast growth and differentiation factor
family, which in turn has been shown to have a role in metanephric
development (42). Interaction
between FGF and BMP signaling pathways has been shown to have a
role in the regulation of MM development (43). Interestingly, in a patient with
DiGeorge-like syndrome with unilateral renal agenesis and a
deletion in chromosome 3, BMP3 was suggested as a target
gene through action via the non-coding gene miRNA-4273(44). The role of the BMP receptor family
in VUR is further indicated by Darlow et al, which found an
association with chromosomal region 4q22.3, which contains
BMPR1B (31). The haplotype
region 13q13.3 includes FREM2, which encodes a factor in the
GDNF-RET/BMP signaling pathway, a factor that both affects
expression and has an established function in the UB and MM. In
addition, biallelic mutations in FREM2 cause the recessive
disorder Fraser syndrome type 2, which includes CAKUT anomalies
(45). The 14q21.1 haplotype
includes FOXA1 (HNF3α), a gene involved in early
embryonic development in numerous organ systems. The gene is
expressed in the embryonic kidney (metanephros) and UT, mainly in
epithelia of the ureter. Nevertheless, studies in Foxa1 null
mice did not show any overt malformations in the kidney. However,
the condition led to death due to severe hypoglycaemia and
dehydration, the latter due to nephrogenic diabetes insipidus
(46,47).
In the study of CNV inheritance in the 14 VUR
families, only one family showed segregation of a CNV. This was a
small deletion within chromosome 5q31.1, which included a part of
FSTL4. This gene encodes a calcium ion-binding protein that
is expressed during renal tubuli development, although not detected
in earlier embryological phases (in UB or MM) according to GUDMAP.
The 5q31.1-deletion was not present in the other families and has
not been reported in previous publications (21,22,48).
Whereas the majority of the CNVs detected did not segregate fully
with disease in the families, an overlap between our data and
previously published findings of likely pathogenic de novo
CNVs was seen in three loci; 7p22.1, 12q24(21) and 8q24.13(22). These latter studies included
individuals with CAKUT, mainly renal hypodysplasia, which was also
seen in the majority of our own patients. The CNV at 7p22.1 is
associated with chromosome 7p interstitial duplication syndrome,
which includes developmental delay and intellectual disability.
However, the individual in our study who was found to have this
duplication had neither kidney damage nor an extrarenal phenotype.
The CNV at 12q2 was described as a large pathogenic de novo
duplication associated with congenital kidney malformations
(21).
Using GWA and the SNP compatibility matching method,
we did not identify a unique haplotype IBD for all 14 families in
the south-western part of Sweden with the VUR complex, although
retained haplotypes were identified in subsets of families.
However, a limitation of the present study was the small number of
families with hereditary VUR, and also the limited number of
generations included. The latter limitation was explained mainly by
VUR being a radiological diagnosis not generally available before
the 1970s, and thus the VUR diagnosis is not ideal for this type of
studies of heredity far back. Nevertheless, partially shared
haplotypes on chromosomes 4q and 13q, with possible candidate
genes, were retained as regions of interest after common haplotypes
were eliminated. The genes identified in these regions have known
functions in the embryogenesis of the kidney and UT but the regions
also include non-coding genes. An overwhelming amount of data shows
that the hereditary VUR-hypodysplasia complex is a genetically
heterogeneous disease where less than 10% of VUR patients have an
identified pathogenic causal mutation (18). On the other hand, knowledge of
non-coding regulatory elements and their expression in the UB and
MM at the time points when the VUR anomaly develops, is currently
very limited and thus a limitation of the study. However, this is
an emerging field that aids in the identification of regulatory
elements in the human genome and makes possible the potential
discovery of new mechanisms which govern VUR.
Supplementary Material
Genomic distribution of CNVs detected
in the present study, among all the individuals analysed.
Chromosomal regions for CNVs detected in more one affected family
member are marked. Blue indicates copy number gain; pink indicates
copy number loss. CNVs, copy number variants; chr, chromosome.
Additional malformations of the UT and
other organ systems.
CNV shared by two or more affected
individuals within the families, detailing CNV locus and
characteristics.
Acknowledgements
Mrs. Alice Andersson and Mrs Eva Johansson (The
Queen Silvia Children’s Hospital, Gothenburg, Sweden) assisted in
collecting patient information, samples and data from medical
records.
Funding
Funding: The present study was financed by grants from the
Swedish state under the agreement between the Swedish Government
and county councils, the ALF agreement (grant no.
ALFGBG-830501).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZB, US, TM, MÖ and SF designed the study. AD, RS and
ZB collected the material and performed the SNP genotyping. MÖ
carried out SNP compatibility matching through advanced statistical
analysis. TM, MÖ and SF confirm the authenticity of all the raw
data. All authors have read and approved the final manuscript.
Ethics approval and consent of
participants
The Regional Ethical Review Board in Gothenburg
approved the study (approval no. Dnr 589-05). Written informed
consent was obtained from all patients.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Sargent MA: What is the normal prevalence
of vesicoureteral reflux? Pediatr Radiol. 30:587–593.
2000.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Yeung CK, Godley ML, Dhillon HK, Gordon I,
Duffy PG and Ransley PG: The characteristics of primary
vesico-ureteric reflux in male and female infants with pre-natal
hydronephrosis. Br J Urol. 80:319–327. 1997.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Peters C and Rushton HG: Vesicoureteral
reflux associated renal damage: Congenital reflux nephropathy and
acquired renal scarring. J Urol. 184:265–273. 2010.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Noe HN, Wyatt RJ, Peeden JN Jr and Rivas
ML: The transmission of vesicoureteral reflux from parent to child.
J Urol. 148:1869–1871. 1992.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Jerkins GR and Noe HN: Familial
vesicoureteral reflux: A prospective study. J Urol. 128:774–778.
1982.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Wan J, Greenfield SP, Ng M, Zerin M,
Ritchey ML and Bloom D: Sibling reflux: A dual center retrospective
study. J Urol. 156:677–679. 1996.PubMed/NCBI
|
|
7
|
Parekh DJ, Pope JC IV, Adams MC and Brock
JW III: Outcome of sibling vesicoureteral reflux. J Urol.
167:283–284. 2002.PubMed/NCBI
|
|
8
|
Chapman CJ, Bailey RR, Janus ED, Abbott GD
and Lynn KL: Vesicoureteric reflux: Segregation analysis. Am J Med
Genet. 20:577–584. 1985.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Feather SA, Malcolm S, Woolf AS, Wright V,
Blaydon D, Reid CJ, Flinter FA, Proesmans W, Devriendt K, Carter J,
et al: Primary, nonsyndromic vesicoureteric reflux and its
nephropathy is genetically heterogeneous, with a locus on
chromosome 1. Am J Hum Genet. 66:1420–1425. 2000.PubMed/NCBI View
Article : Google Scholar
|
|
10
|
Eccles MR and Jacobs GH: The genetics of
primary vesico-ureteric reflux. Ann Acad Med Singap. 29:337–345.
2000.PubMed/NCBI
|
|
11
|
Sanna-Cherchi S, Reese A, Hensle T, Caridi
G, Izzi C, Kim YY, Konka A, Murer L, Scolari F, Ravazzolo R, et al:
Familial vesicoureteral reflux: Testing replication of linkage in
seven new multigenerational kindreds. J Am Soc Nephrol.
16:1781–1787. 2005.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Lu W, van Eerde AM, Fan X, Quintero-River
F, Kulkarni S, Ferguson H, Kim HG, Fan Y, Xi Q, Li QG, et al:
Disruption of ROBO2 is associated with urinary tract anomalies and
confers risk of vesicoureteral reflux. Am J Hum Genet. 80:616–632.
2007.PubMed/NCBI View
Article : Google Scholar
|
|
13
|
Weng PL, Sanna-Cherchi S, Hensle T,
Shapiro E, Werzberger A, Caridi G, Izzi C, Konka A, Reese AC, Cheng
R, et al: A recessive gene for primary vesicoureteral reflux maps
to chromosome 12p11-q13. J Am Soc Nephrol. 20:1633–1640.
2009.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Naseri M, Ghiggeri GM, Caridi G and
Abbaszadegan MR: Five cases of severe vesico-ureteric reflux in a
family with an X-linked compatible trait. Pediatr Nephrol.
25:349–352. 2010.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Fillion ML, Watt CL and Gupta IR:
Vesicoureteric reflux and reflux nephropathy: From mouse models to
childhood disease. Pediatr Nephrol. 29:757–766. 2014.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Ichikawa I, Kuwayama F, Pope JC IV,
Stephens FD and Miyazaki Y: Paradigm shift from classic anatomic
theories to contemporary cell biological views of CAKUT. Kidney
Int. 61:889–898. 2002.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Nicolaou N, Pulit SL, Nijman IJ, Monroe
GR, Feitz WF, Schreuder MF, van Eerde AM, de Jong TP, Giltay JC,
van der Zwaag B, et al: Prioritization and burden analysis of rare
variants in 208 candidate genes suggest they do not play a major
role in CAKUT. Kidney Int. 89:476–486. 2016.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Hwang DY, Dworschak GC, Kohl S, Saisawat
P, Vivante A, Hilger AC, Reutter HM, Soliman NA, Bogdanovic R,
Kehinde EO, et al: Mutations in 12 known dominant disease-causing
genes clarify many congenital anomalies of the kidney and urinary
tract. Kidney Int. 85:1429–1433. 2014.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Kleinjan DA and van Heyningen V:
Long-range control of gene expression: Emerging mechanisms and
disruption in disease. Am J Hum Genet. 76:8–32. 2005.PubMed/NCBI View
Article : Google Scholar
|
|
20
|
Southard AE, Edelmann LJ and Gelb BD: Role
of copy number variants in structural birth defects. Pediatrics.
129:755–763. 2012.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Sanna-Cherchi S, Kiryluk K, Burgess KE,
Bodria M, Sampson MG, Hadley D, Nees SN, Verbitsky M, Perry BJ,
Sterken R, et al: Copy-number disorders are a common cause of
congenital kidney malformations. Am J Hum Genet. 91:987–997.
2012.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Caruana G, Wong MN, Walker A, Heloury Y,
Webb N, Johnstone L, James PA, Burgess T and Bertram JF:
Copy-number variation associated with congenital anomalies of the
kidney and urinary tract. Pediatr Nephrol. 30:487–495.
2015.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Ohlsson M, Hedberg C, Bradvik B, Lindberg
C, Tajsharghi H, Danielsson O, Melberg A, Udd B, Martinsson T and
Oldfors A: Hereditary myopathy with early respiratory failure
associated with a mutation in A-band titin. Brain. 135:1682–1694.
2012.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Östensson M: Statistical Methods for
Genome Wide Association Studies. PhD dissertation, University of
Gothenburg. ISBN, 9789173857420, 2012.
|
|
25
|
Piepsz A, Colarinha P, Gordon I, Hahn K,
Olivier P, Roca I, Sixt R and van Velzen J: Paediatric Committee of
the European Association of Nuclear Medicine. Guidelines for
99mTc-DMSA scintigraphy in children. Eur J Nucl Med. 28:BP37–BP41.
2001.PubMed/NCBI
|
|
26
|
Schwartz GJ, Brion LP and Spitzer A: The
use of plasma creatinine concentration for estimating glomerular
filtration rate in infants, children, and adolescents. Pediatr Clin
North Am. 34:571–590. 1987.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Brochner-Mortensen J, Haahr J and
Christoffersen J: A simple method for accurate assessment of the
glomerular filtration rate in children. Scand J Clin Lab Invest.
33:140–143. 1974.PubMed/NCBI
|
|
28
|
Bengtsson H, Wirapati P and Speed TP: A
single-array preprocessing method for estimating full-resolution
raw copy numbers from all Affymetrix genotyping arrays including
GenomeWideSNP 5 & 6. Bioinformatics. 25:2149–2156.
2009.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Olshen AB, Venkatraman ES, Lucito R and
Wigler M: Circular binary segmentation for the analysis of
array-based DNA copy number data. Biostatistics. 5:557–572.
2004.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Kelly H, Molony CM, Darlow JM, Pirker ME,
Yoneda A, Green AJ, Puri P and Barton DE: A genome-wide scan for
genes involved in primary vesicoureteric reflux. J Med Genet.
44:710–717. 2007.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Darlow JM, Dobson MG, Darlay R, Molony CM,
Hunziker M, Green AJ, Cordell HJ, Puri P and Barton DE: A new
genome scan for primary nonsyndromic vesicoureteric reflux
emphasizes high genetic heterogeneity and shows linkage and
association with various genes already implicated in urinary tract
development. Mol Genet Genomic Med. 2:7–29. 2014.PubMed/NCBI View
Article : Google Scholar
|
|
32
|
Marchini GS, Onal B, Guo CY, Rowe CK,
Kunkel L, Bauer SB, Retik AB and Nguyen HT: Genome gender diversity
in affected sib-pairs with familial vesico-ureteric reflux
identified by single nucleotide polymorphism linkage analysis. BJU
Int. 109:1709–1714. 2012.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Cordell HJ, Darlay R, Charoen P, Stewart
A, Gullett AM, Lambert HJ, Malcolm S, Feather SA, Goodship TH,
Woolf AS, et al: Whole-genome linkage and association scan in
primary, nonsyndromic vesicoureteric reflux. J Am Soc Nephrol.
21:113–123. 2010.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Briggs CE, Guo CY, Schoettler C, Rosoklija
I, Silva A, Bauer SB, Retik AB, Kunkel L and Nguyen HT: A genome
scan in affected sib-pairs with familial vesicoureteral reflux
identifies a locus on chromosome 5. Eur J Hum Genet. 18:245–250.
2010.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Sanna-Cherchi S, Caridi G, Weng PL,
Dagnino M, Seri M, Konka A, Somenzi D, Carrea A, Izzi C, Casu D, et
al: Localization of a gene for nonsyndromic renal hypodysplasia to
chromosome 1p32-33. Am J Hum Genet. 80:539–549. 2007.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Casas KA, Mononen TK, Mikail CN, Hassed
SJ, Li S, Mulvihill JJ, Lin HJ and Falk RE: Chromosome 2q terminal
deletion: Report of 6 new patients and review of
phenotype-breakpoint correlations in 66 individuals. Am J Med Genet
A. 130A:331–339. 2004.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Conte ML, Bertoli-Avella AM, de Graaf BM,
Lama G, La Manna A, Grassia C, Rambaldi PF, Oostra BA, Perrotta S
and Punzo F: A genome search for primary vesicoureteral reflux
shows further evidence for genetic heterogeneity. Pediatr Nephrol.
23:587–595. 2008.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Williams G, Fletcher JT, Alexander SI and
Craig JC: Vesicoureteral reflux. J Am Soc Nephrol. 19:847–862.
2008.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Carvas F, Silva A and Nguyen HT: The
genetics of primary, nonsyndromic vesicoureteral reflux. Curr Opin
Urol. 20:336–342. 2010.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Vukicevic S, Helder MN and Luyten FP:
Developing human lung and kidney are major sites for synthesis of
bone morphogenetic protein-3 (osteogenin). J Histochem Cytochem.
42:869–875. 1994.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Takahashi H and Ikeda T: Transcripts for
two members of the transforming growth factor-beta superfamily
BMP-3 and BMP-7 are expressed in developing rat embryos. Dev Dyn.
207:439–449. 1996.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Cancilla B, Ford-Perriss MD and Bertram
JF: Expression and localization of fibroblast growth factors and
fibroblast growth factor receptors in the developing rat kidney.
Kidney Int. 56:2025–2039. 1999.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Dudley AT, Godin RE and Robertson EJ:
Interaction between FGF and BMP signaling pathways regulates
development of metanephric mesenchyme. Genes Dev. 13:1601–1613.
1999.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Cirillo E, Giardino G, Gallo V, Galasso G,
Romano R, D'Assante R, Scalia G, Del Vecchio L, Nitsch L, Genesio R
and Pignata C: DiGeorge-like syndrome in a child with a 3p12.3
deletion involving MIR4273 gene born to a mother with gestational
diabetes mellitus. Am J Med Genet A. 173:1913–1918. 2017.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Kohl S, Hwang DY, Dworschak GC, Hilger AC,
Saisawat P, Vivante A, Stajic N, Bogdanovic R, Reutter HM, Kehinde
EO, et al: Mild recessive mutations in six Fraser syndrome-related
genes cause isolated congenital anomalies of the kidney and urinary
tract. J Am Soc Nephrol. 25:1917–1922. 2014.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Bernardo GM and Keri RA: FOXA1: A
transcription factor with parallel functions in development and
cancer. Biosci Rep. 32:113–130. 2012.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Kaestner KH: The FoxA factors in
organogenesis and differentiation. Curr Opin Genet Dev. 20:527–532.
2010.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Siomou E, Mitsioni AG, Giapros V, Bouba I,
Noutsopoulos D and Georgiou I: Copy-number variation analysis in
familial nonsyndromic congenital anomalies of the kidney and
urinary tract: Evidence for the causative role of a transposable
element-associated genomic rearrangement. Mol Med Rep.
15:3631–3636. 2017.PubMed/NCBI View Article : Google Scholar
|