Introduction
Atherosclerosis, a chronic vascular disorder of the
arterial wall, has become a predominant cause of a variety of
cardiovascular (CV) disorders, including ischemic stroke,
myocardial infarction, heart attack and aortic aneurysm (1). Despite the advances in novel
therapeutic strategies against atherosclerosis, this disorder
remains the leading cause of death and disability worldwide
(2). Endothelial-cell (EC)
apoptosis serves a critical role in the pathogenesis of
atherosclerosis via multiple mechanisms: i) Disrupting normal
function of the endothelium to dysregulate lipid homeostasis,
immunity and inflammation (3); ii)
breaking the integrity and barrier functions of the endothelium to
facilitate lipid deposition, leading to atherogenesis (4); and iii) destabilizing plaque to
predispose patients to arterial thrombosis (5). Therefore, elucidating the molecular
mechanism underlying EC apoptosis is required to further understand
the disorder.
MicroRNAs (miRNAs/miRs) are small, non-coding RNA
molecules which are 17-22 nucleotides in length that negatively
regulate the expression of their target genes by promoting mRNA
degradation or by repressing their translation (6). miRNAs participate in various cellular
functions, including proliferation, differentiation, senescence and
apoptosis (7). Accumulating
evidence has demonstrated that abnormally expressed miRNAs are
implicated in the initiation and progression of atherosclerosis
(8,9). Various miRNAs, including miR-34a,
miR-122, miR-210 and miR-876, have been reported to be involved in
the pathogenesis of atherosclerosis by controlling EC apoptosis
(10-13).
Human aortic endothelial cells (HAECs) are
implicated in the pathogenesis of atherosclerosis (4). The dysfunction of HAECs can be
affected by oxidized low-density lipoprotein (ox-LDL), which are a
critical risk factor in atherosclerosis (12). Therefore, in the present study,
ox-LDL-induced HAECs were used as atherosclerosis cell models to
explore the molecular mechanism underlying atherosclerosis.
Although the role of miR-454-3p in tumorigenesis is well documented
(14-17),
its involvement in the regulation of HAEC apoptosis remains to be
elucidated. The present study investigated the role of miR-454-3p
in cell apoptosis in atherosclerosis.
Materials and methods
Antibodies and reagents
TRPC3 antibodies (cat. no. ab241343; 1:1,000) were
purchased from Abcam. Cleaved caspase-3 antibodies (cat. no. 9661;
1:1,000) and tubulin antibodies (cat. no. 2128; 1:2,000) were
purchased from Cell Signaling Technology, Inc. Horseradish
peroxidase (HRP) conjugated secondary antibody (anti-rabbit IgG,
HRP-linked Antibody; cat. no. 7074; 1:5,000) was purchased from
Cell Signaling Technology, Inc. miR-454-3p mimics and
antagomiR-454-3p were obtained from Guangzhou RiboBio' HAECs were
treated with 0, 25, 50 or 100 µg/ml of ox-LDL to elucidate the
effects of ox-LDL, which were provided by Beijing Xiesheng
BioTechnology Co., Ltd.
Cell culture
HAECs were obtained from ScienCell Research
Laboratories, Inc. (cat. no. 6100) and cultured in Endothelial Cell
Growth Medium (Sigma-Aldrich; Merck KGaA) supplemented with
Endothelial Cell Growth Supplement (BD Biosciences), 10% FBS
(Gibco; Thermo Fisher Scientific, Inc.) and 100 U/ml
penicillin-streptomycin (Invitrogen; Thermo Fisher Scientific,
Inc.). HAECs were cultured in a humidified atmosphere with 5%
CO2 at 37˚C. To investigate the potential role of
miR-45-3p in atherosclerotic progression, HAECs were exposed to
ox-LDL at various concentrations (0, 25, 50 or 100 µg/l) for 24 h
or to 50 µg/ml ox-LDL at various durations (0, 12, 24 or 48 h).
Reverse transcription quantitative PCR
(RT-qPCR)
Total RNA was isolated from HAECs using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.), according to the manufacturer's protocol. miR-454-3p was
quantified by synthesizing complementary (c)DNA using a TaqMan
Advanced miRNA cDNA Synthesis kit (Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. RNA was reverse
transcribed into cDNA using M-MLV Reverse Transcriptase (Promega
Corporation) according to the manufacturer's protocol to detect
TRPC3 expression. RT-qPCR was performed using a GeneAmp PCR System
9700 (Applied Biosystems; Thermo Fisher Scientific, Inc.) and a
PrimeScript miRNA RT-PCR kit or Takara SYBR RT-PCR kit (both,
Takara Biotechnology Co., Ltd.), according to the manufacturer's
protocol. For the measurement of miR-454-3p, the thermocycling
conditions were as follows: Initial denaturation at 95˚C for 5 min;
followed by 40 cycles of denaturation at 95˚C for 15 sec, annealing
at 56˚C for 25 sec and elongation at 72˚C for 40 sec. For the
measurement of TRPC3, Initial denaturation at 95˚C for 2 min,
followed by 35 cycles of denaturation at 95˚C for 30 sec, annealing
at 56˚C for 30 sec and elongation at 72˚C for 1 min, and a final
elongation at 72˚C for 2 min. Relative gene expression was
calculated via the 2-ΔΔCq method (18) and normalized to endogenous controls
U6 or GAPDH. The following primer pairs were used for the qPCR:
miR-454-3p forward, 5'-GCGCGTAGTGCAATATTGCTTA-3' and reverse,
5'-AGTGCAGGGTCCGAGGTATT-3'; U6 forward,
5'-CGCTTCGGCAGCACATATACTAA-3' and reverse,
5'-TATGGAACGCTTCACGAATTTGC-3'; TRPC3 forward,
5'-AGCAGCTCTTGACGATCTGG-3' and reverse,
5'-GCACAACGAGACACTTGATAGC-3' and GAPDH forward,
5'-AATCCCATCACCATCTTC-3' and reverse, 5'-AGGCTGTTGTCATACTTC-3'.
Transient transfection of miR-454-3p
mimics or inhibitors
miR-454-3p mimics (50 nM) or antagomiR-454-3p (100
nM) were transiently transfected into HAECs using Lipofectamine
RNAiMAX Transfection reagent (Invitrogen; Thermo Fisher Scientific,
Inc.), according to the manufacturer's protocol. The sequences are
listed as follows: miR-454-3p mimic, 5'-UAGUGCAAUAUUGCUUAUAGGGU-3';
mimic control miR-NC, 5'-UCACAACCUCCUAGAAAGAGUAGA-3'.
antagomiR-454-3p, 5'-ACCCUAUAAGCAAUAUUGC ACUA-3'; negative control
antagomiR-NC, 5'-UUGUACUAC ACAAAAGUACUG-3'. Cells were used for
further experiments 24 h post-transfection. For the luciferase
reporter assay, cells were collected for the detection 48 h
post-transfection.
Transfection with siRNA and
plasmids
SiRNA (50 nM) or plasmids was transfected were
transiently transfected into HAECs using Lipofectamine RNAiMAX
Transfection reagent (Invitrogen; Thermo Fisher Scientific, Inc.),
according to the manufacturer's protocol. SiRNA targeting TRPC3
(si-TRPC3) and siRNA negative control (si-NC) were synthesized by
GenePharma Co. Ltd. (Shanghai, China). The sequences are listed as
follows: si-TRPC3: 5'-CCCAGTTTACATGGACTGAAA-3'); si-NC
(5'-CAACAAGATGAAGAGCACCAA-3'). The full length cDNA sequences of
TRPC3 were cloned into pcDNA3.1 vector (Invitrogen) to construct
3'-UTR-deleted TRPC3 plasmid.
Cell viability assay
Cell viability of HAECs was examined using a Cell
Counting Kit-8 (CCK-8; Dojindo Laboratories) assay according to the
manufacturer's protocol. Briefly, cells were seeded into 96-well
plate at a density of 2,500 cells/well. Prior to analysis, CCK-8
reagent was added to each well and HAECs were cultured at 37˚C for
4 h. Absorbance at 450 nm was detected using a microplate reader
(Molecular Devices, LLC). Experiments were conducted in
triplicate.
Apoptosis analysis
Cell apoptosis was detected using an Annexin V-FITC
Apoptosis Detection kit (BD Biosciences) according to the
manufacturer's protocol. Harvested cells were washed twice with PBS
and then stained with 1 µg/ml Annexin V-FITC for 15 min in the dark
at room temperature. Following washing with PBS, cells were stained
with 1 µg/ml propidium iodide (PI) for 10 min in the dark at room
temperature. Cells were then subjected to flow cytometric (FCM)
analysis using a FACSCanto FCM flow cytometer (BD Biosciences). A
total of 1x104 cells were collected using the
forward-scatter/side-scatter scatterplot method to exclude mutually
adherent cells and cell debris (19). Data analysis was conducted using
FlowJo software (version 7.6.5; FlowJo LLC).
Lactate dehydrogenase (LDH) release
assay
Cell culture medium was collected and LDH activity
was measured using a commercial LDH Activity Assay kit (Nanjing
Jiancheng Bioengineering Institute), according to the
manufacturer's protocol. Briefly, 25 µl cell supernatant was mixed
with 25 µl substrate in the kit and the solution was incubated at
37˚C for 15 min. A total of 25 µl 2,4-dinitrophenylhydrazine in the
kit was then added into the samples. Following incubation at 37˚C
for 15 min in the dark, 250 µl 0.4 mol/l NaOH solution was added
and the mixture was further incubated at room temperature for 5
min. Absorbance was recorded at a wavelength of 450 nm using a
microplate reader.
Western blotting
Collected cells were lysed with RIPA buffer (Pierce;
Thermo Fisher Scientific, Inc.) supplemented with protease
inhibitors (Roche Diagnostics). The protein concentration was
measured using the Pierce BCA protein assay kit (Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocols. The
detailed protocol is described previously (20). Following centrifugation at 12,000 x
g, 4˚C for 10 min, the supernatant was harvested and quantified
using a BCA Protein Assay kit (Thermo Fisher Scientific, Inc.).
Equal amounts of protein (50 µg/lane) were then separated on 10%
SDS-PAGE and transferred to nitrocellulose membranes. Following
blocking with 5% non-fat milk at room temperature for 1 h, the
membranes were probed with primary antibodies overnight at 4˚C,
followed by incubation with horseradish peroxidase-conjugated
secondary antibodies at room temperature for 1 h. Bands were
visualized using an Enhanced Chemiluminescence Western Blotting
Analysis kit (Invitrogen; Thermo Fisher Scientific, Inc.). Tubulin
was used as a loading control.
Caspase-3 activity assay
Caspase-3 activity was determined using a Caspase-3
Activity Assay kit (Beyotime Institute of Biotechnology), according
to the manufacturer's protocol. Cells were harvested and lysed for
15 min at 4˚C using the cell lysis buffer supplied by the kit.
Suspensions were then centrifuged at 12,000 x g for 10 min at 4˚C.
Supernatants were collected and 10 µl caspase-3 substrate
Ac-LEHD-pNA (from kit) and 10 µl supernatant was added to 80 µl
reaction buffer (from kit). Following incubation at 37˚C for 2 h,
absorbance was measured at a wavelength of 405 nm using a
microplate reader.
Bioinformatics analysis and Luciferase
reporter assay
The bioinformatics tool TargetScan (http://www.targetscan.org/vert_72/) was utilized
to predict potential target genes of miR-454-3p. HAECs were seeded
into 24-well plates at a density of 7.5x104 cells/per
well. After 24 h, cells were co-transfected with pGL3 luciferase
reporter vectors (Promega Corporation) containing wild-type (WT) or
mutated 3'-untranslated region (3'-UTR) sequences of TRPC3
alongside miR-454-3p mimics or antagomiR-454-3p using Lipofectamine
2000 (Invitrogen; Thermo Fisher Scientific, Inc.). pRL-TK vectors
(Promega Corporation) were co-transfected as an internal control of
transfection efficiency. At 48 h post-transfection, cells were
harvested and subjected to luciferase activity analysis using a
Dual-Luciferase Reporter Assay system (Promega Corporation),
according to the manufacturer's protocol. Relative reporter gene
activity was measured by normalizing to Renilla luciferase
activity.
Statistical analysis
All data are presented as the mean ± standard
deviation (SD). Statistical analyses were conducted using a
two-tailed Student's t-test or one-way ANOVA followed by Dunnett's
test with SPSS software (version 17.0; SPSS, Inc.). P<0.05 was
considered to indicate a statistically significant difference.
Results
Ox-LDL-treatment downregulates
miR-454-3p in HAECs
Ox-LDL is a key atherosclerotic risk factor that
induces EC apoptosis and contributes to the initiation and
progression of atherosclerosis (12). HAECs were exposed to ox-LDL at
various concentrations (0, 25, 50 or 100 µg/l) for 24 h or to 50
µg/ml ox-LDL at various durations (0, 12, 24 or 48 h) to
investigate the potential role of miR-45-3p in atherosclerotic
progression. Ox-LDL treatment reduced cell viability and increased
cell apoptosis in a concentration- (Fig. 1A-B) and time-dependent (Fig. 1D-E) manner, as assessed by CCK-8 and
apoptosis analyses. Furthermore, RT-qPCR results demonstrated that
ox-LDL treatment reduced miR-454-3p expression in a dose- (Fig. 1C) and time-dependent manner
(Fig. 1F).
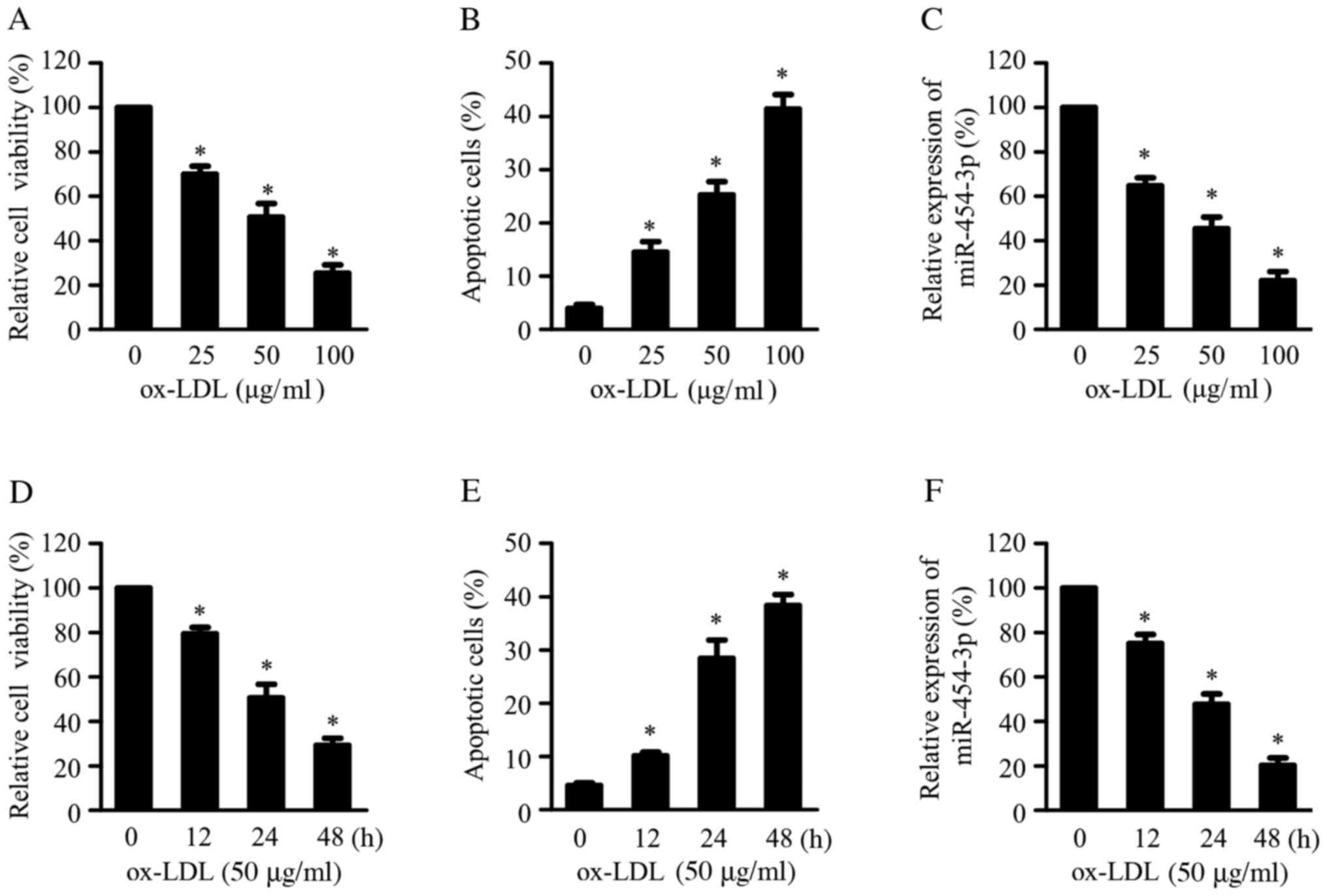 | Figure 1miR-454-3p is downregulated in
ox-LDL-treated HAECs. HAECs treated with 0, 25, 50 or 100 μg/ml
ox-LDL for 24 h, followed by (A) CCK-8 and (B) cell apoptosis
analyses. (C) RT-qPCR analysis of miR-454-3p expression in HAECs
treated with 0, 25, 50 or 100 μg/ml ox-LDL for 24 h. HAECs treated
with 50 μg/ml ox-LDL for 0, 12, 24 or 48 h, followed by (D) CCK-8
and (E) cell apoptosis analyses. (F) Following treatment with 50
μg/ml ox-LDL of HAECs for 0, 12, 24 or 48 h, miR-454-3p expression
was measured by RT-qPCR. The expression of miR-454-3p was
normalized to U6 small nuclear RNA. *P<0.05, compared
with the group treated with 50 µg/ml ox-LDL for 0 h. miR, microRNA;
ox-LDL, oxidized low-density lipoprotein; HAEC, human aortic
endothelial cells; CCK-8, Cell Counting Kit-8; RT-qPCR, reverse
transcription-quantitative PCR. Experiments were conducted in
triplicate and data are presented as mean ± standard deviation
(SD). |
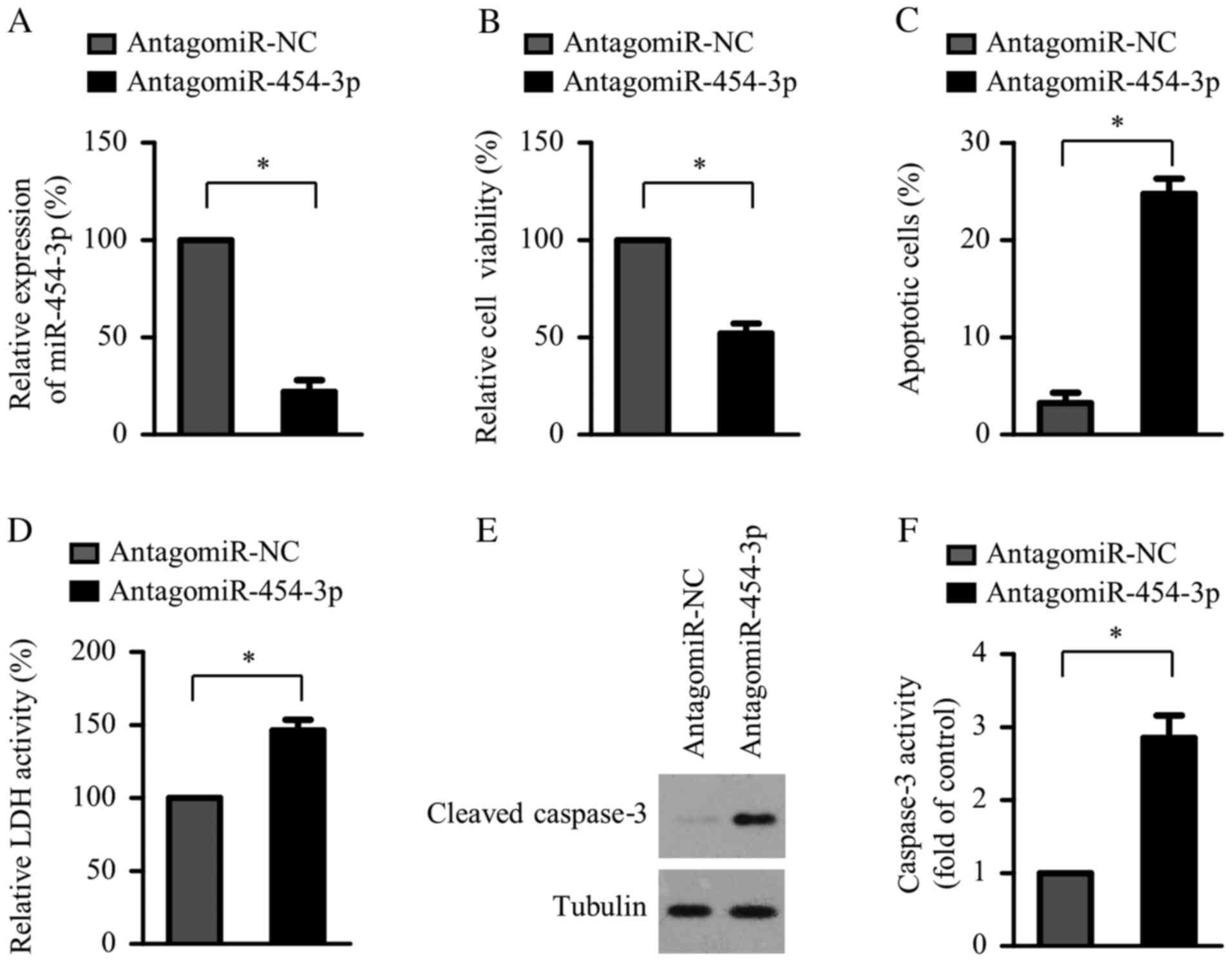
miR-454-3p inhibition induces HAEC
apoptosis
The effect of miR-454-3p suppression on cell
viability and apoptosis of HAECs was determined. miR-454-3p
expression significantly decreased in the miR-454-3p inhibitor
transfection group compared with the negative control group
(Fig. 2A). CCK-8 assay results
demonstrated that miR-454-3p suppression significantly reduced the
viability of HAECs compared with the negative control group
(Fig. 2B). Consistent with cell
viability inhibition, compared with negative control groups, HAEC
apoptosis was significantly increased by miR-454-3p suppression
(Fig. 2C) and the LDH assay
confirmed that this suppression significantly induced HAEC cell
death (Fig. 2D). Furthermore,
compared with negative control groups, miR-454-3p suppression
significantly increased cleaved caspase-3 protein expression and
caspase-3 activities in HAECs (Fig.
2E and F), confirming increased
cell apoptosis.
miR-454-3p attenuates ox-LDL-induced
apoptosis in HAECs
Based on the previous results of ox-LDL treatment on
miR-454-3p and the suppressive role of miR-454-3p on HAEC
apoptosis, it was determined whether miR-454-3p overexpression
could rescue ox-LDL-induced apoptosis of HAECs. HAECs were
transfected with miR-454-3p or miR-negative control (miR-NC),
followed by treatment with 50 µg/ml ox-LDL for 24 h. RT-qPCR
results demonstrated successful miR-454-3p overexpression in HAECs
transfected with miR-454-3p compared with miR-NC-transfected cells
(Fig. 3A). CCK-8 assay revealed
that miR-454-3p overexpression reversed the inhibitory effect of
ox-LDL on the viability of HAECs (Fig.
3B). Additionally, Annexin V-FITC/PI double-staining analysis
and LDH release assay revealed that ox-LDL-induced apoptosis of
HAECs was significantly attenuated by miR-454-3p overexpression
compared with miR-NC-transfected cells (Fig. 3C-D). Consistently, ox-LDL-induced
increase in cleaved caspase-3 protein expression and activity were
significantly reduced by miR-454-3p overexpression (Fig. 3E and F).
TRPC3 is a direct target of miR-454-3p
in HAECs
To elucidate the molecular mechanism by which
miR-454-3p regulated HAEC apoptosis, the bioinformatics tool
TargetScan (David Bartel Lab; Whitehead Institute for Biomedical
Research; Massachusetts Institute of Technology) was utilized to
predict potential target genes of miR-454-3p. Among the potential
target genes (Table SI), TRPC3 has
been reported to be a key gene contributing to the pathogenesis and
development of atherosclerosis (21,22).
The present study then explored whether TRPC3 might be used as a
potential target gene of miR-454-3p. The possible binding site of
miR-454-3p in TRPC3 3'-UTR is shown in Fig. 4A. To determine whether miR-454-3p
directly targeted TRPC3, a Dual-Luciferase Reporter Assay system
containing WT or mutated miR-454-3p binding sites in the 3'-UTR was
used. miR-454-3p overexpression significantly inhibited WT 3'-UTR
luciferase activity compared with miR-NC-transfected cells
(Fig. 4B); however, there was no
significant effect on mutant 3'-UTR between miR-454-3p
overexpression and miR-NC group. Consistently, miR-454-3p
suppression significantly increased WT 3'-UTR luciferase activity
compared with the negative control group but showed no significant
effect on the mutant 3'-UTR of TRPC3 (Fig. 4C). RT-qPCR and Western blotting
analyses revealed that miR-454-3p transfection decreased TRPC3 mRNA
and protein expression (Fig. 4D and
E). Furthermore, miR-454-3p
inhibition increased TRPC3 mRNA and protein expression (Fig. 4F and G). In summary, these results revealed that
TRPC3 was a direct target of miR-454-3p.
 | Figure 4TRPC3 is a direct target of miR-454-3p
in HAECs. (A) Target sites of miR-454-3p in TRPC3 3'-UTR mRNA. WT
or mut TRPC3 3'-UTR luciferase reporter vectors alongside miR-NC or
miR-454-3p were co-transfected into HAECs. After 48 h, luciferase
activities were determined in HAECs transfected with (B) miR-NC or
miR-454-3p or with (C) antagomiR-NC or antagomiR-454-3p. Reverse
transcription-quantitative PCR analysis of TRPC3 mRNA levels in
HAECs transfected with (D) miR-NC or miR-454-3p or with (F)
antagomiR-NC or antagomiR-454-3p. Expression of TRPC3 mRNA was
normalized to GAPDH. Western blotting results of TRPC3 protein
levels in HAECs transfected with (E) miR-NC or miR-454-3p or with
(G) antagomiR-NC or antagomiR-454-3p. Tubulin was used as a loading
control. Data are presented as the mean ± standard deviation (SD)
and experiments were performed in triplicate.
*P<0.05. TRPC3, transient receptor potential
canonical 3; miR, microRNA; HAEC, HAEC, human aortic endothelial
cells; UTR, untranslated region; WT, wild-type; mut, mutant; NC,
normal control; NS, not significant. |
miR-454-3p inhibits EC apoptosis via
inhibition of TRPC3 expression
To address the role of TRPC3 in mediating the
anti-apoptotic effect of miR-454-3p in HAECs, TRPC3 knockdowns were
used to determine whether they reversed HAEC apoptosis mediated by
miR-454-3p suppression. As shown in Fig. 5A, the expression of TRPC3
significantly decreased in the siTRPC3 transfection group compared
with the si-NC group. Compared with their respective negative
control groups, antagomiR-454-3p transfection suppressed the
viability of HAECs, whereas TRPC3 silencing reversed this
repression of cell viability (Fig.
5B). Accordingly, the increased apoptosis caused by miR-454-3p
inhibition was reversed by TRPC3 silencing (Fig. 5C-D), as assessed by Annexin
V-FITC/PI double-staining analysis and an LDH release assay. To
further validate the role of TRPC3 in mediating the anti-apoptotic
action of miR-454-3p in HAECs, a rescue experiment was performed by
introducing a 3'-UTR-deleted TRPC3 plasmid into HAECs transfected
with miR-454-3p. Western blotting indicated that miR-454-3p
decreased TRPC3 protein levels, which were partially restored by
co-transfection with a TRPC3 plasmid (Fig. 5E). The CCK-8 assay demonstrated that
cell viability was significantly decreased in ox-LDL-treated HAECs
compared with control cells, while miR-454-3p transfection restored
cell viability (Fig. 5F). However,
TRPC3 overexpression significantly decreased the beneficial effect
of miR-454-3p. Additionally, ox-LDL treatment significantly
increased HAEC apoptosis compared with controls (Fig. 5G-H). This effect was significantly
attenuated following miR-454-3p overexpression. However, TRPC3
overexpression inhibited the ability of miR-454-3p to suppress
ox-LDL-induced apoptosis. Thus, these results indicated that
miR-454-3p inhibited EC apoptosis by targeting TRPC3.
 | Figure 5miR-454-3p inhibits endothelial
apoptosis via inhibition of TRPC3 expression. Western blotting of
TRPC3 expression in HAECs transfected with (A) antagomiR-454-3p or
antagomiR-454-3p + siTRPC3. Tubulin was used as a loading control.
HAECs were transfected with antagomiR-454-3p or antagomiR-454-3p +
siTRPC3, followed by (B) CCK-8, (C) cell apoptosis and (D) LDH
release analyses. HAECs transfected with miR-454-3p or miR-454-3p +
TRPC3 were treated with 50 μg/ml ox-LDL for 24 h, followed by (E)
western blotting of TRPC3 expression, (F) CCK-8, (G) cell apoptosis
and (H) LDH release analyses. Data are presented as the mean ±
standard deviation (SD) and experiments were performed in
triplicate. *P<0.05. miR, microRNA; TRPC3, transient
receptor potential canonical 3; HAEC, human aortic endothelial
cells; si, small interfering RNA; CCK-8, Cell Counting Kit-8; LDH,
lactate dehydrogenase; NC, negative control; ox-LDL, oxidized
low-density lipoprotein. |
Discussion
EC apoptosis manifests in the early stages of
atherosclerosis and contributes to the pathogenesis of the disorder
(4). Accumulating evidence has
demonstrated that aberrantly-expressed miRNAs serve critical roles
in EC apoptosis by targeting crucial factors or key pathways
regulating cell apoptosis (8). For
instance, the lethal-7g gene was demonstrated to suppress EC
apoptosis by directly targeting caspase-3(23). miR-429 suppressed
atherosclerosis-associated EC apoptosis by inhibiting B-cell
lymphoma 2(24). Li et al
(12) reported that miR-210
upregulation induced EC apoptosis by suppressing pyruvate
dehydrogenase lipoamide kinase isozyme 1 during atherosclerosis
development. It was reported that downregulation of miR-142-3p
suppressed EC apoptosis and atherosclerotic progression by
increasing the expression of rapamycin-insensitive companion of
mTOR and activating protein kinase B signaling (25). Therefore, identifying novel miRNAs
that participate in EC apoptosis will expand the understanding of
the molecular mechanism underlying the pathogenesis of
atherosclerosis.
miR-454-3p, which has been reported to be abnormally
expressed in various types of cancers, may have an oncogenic role.
miR-454-3p promoted breast cancer metastasis by suppressing the
regulation of nuclear pre-mRNA domain containing 1A and by
activating Wingless/Integrated signaling (26). Furthermore, miR-454-3p may act as a
tumor suppressor; a previous study demonstrated miR-454-3p
downregulation in glioblastoma and that it exerted
tumor-suppressive functions by targeting nuclear factor of
activated T cells, cytoplasmic 1(17). By inhibiting signal transducer and
activator of transcription 3 and autophagy-related protein 12,
miR-454-3p negatively regulated the growth of chondrosarcoma
(16). Although its role in
carcinogenesis has been well documented, its roles and underlying
mechanisms in EC apoptosis remain to be elucidated.
In the present study, ox-LDL-induced HAECs were used
as an in vitro cell model of atherosclerosis and the
potential role of miR-454-3p in ox-LDL-induced EC apoptosis was
investigated. The results demonstrated that ox-LDL suppressed
miR-454-3p expression in a dose- and time-dependent manner. This
suppression significantly attenuated EC viability, mimicking the
apoptosis-inducing effects of ox-LDL treatment. miR-454-3p
overexpression almost completely reversed ox-LDL-induced EC
apoptosis and relieved ox-LDL-elicited suppression of cell
viability, revealing the anti-arteriosclerotic effects of
miR-454-3p. Previous studies demonstrated that plasma miR-454-3p is
upregulated in gliomas and may act as a sensitive biomarker for the
diagnosis of gliomas (27,28). In future studies, determining the
difference between miR-454-3p in plasma in patients with
atherosclerosis and in plasma from healthy subjects will be
beneficiary in order to investigate whether miR-454-3p acts as a
novel potential diagnostic biomarker for atherosclerosis.
Through in silico algorithm analyses and
experimental verification, the present study demonstrated that
TRPC3 was a direct target of miR-454-3p. TRPC3, a member of the
TRPC family of calcium-permeable, non-selective cation channels,
participates in diverse functions in CV and hematopoietic systems
(24). Microarrays have
demonstrated significant increases of TRPC3 mRNA levels in plaques
obtained from patients with atherosclerosis (29). Consistent with this observation,
TRPC3 was reported to exhibit higher expression in the aortic roots
of atherosclerotic mice compared with aortic cross-sections from
non-atherosclerotic animals (30),
indicating an association between TRPC3 overexpression and the
presence of atherosclerosis (30).
Additionally, TRPC3 deficiency impaired atherosclerotic-lesion
formation in a mouse model of atherosclerosis (31). Apoe knockout mice, a unique mouse
model of atherosclerosis, with endothelial-specific overexpression
of human TRPC3 exhibited increased size and cellularity of advanced
atherosclerotic lesions (32),
supporting a pro-atherogenic role of endothelial TRPC3.
Additionally, a previous study reported that TRPC3 is required for
EC apoptosis induced by endoplasmic-reticulum stress (33). Therefore, the present study
hypothesized that miR-454-3p may exert its anti-apoptotic effect by
suppressing TRPC3. The results demonstrated that TRPC3 silencing
completely blocked the increased apoptosis caused by miR-454-3p
suppression, while TRPC3 overexpression reversed the inhibitory
effect of miR-454-3p in ox-LDL-induced HAEC cell apoptosis. These
results indicated that miR-454-3p inhibited EC apoptosis by
directly targeting TRPC3 expression.
In conclusion, the results of the present study
revealed that miR-454-3p is a mediator for EC apoptosis. In HAECs,
miR-454-3p overexpression reversed ox-LDL-induced apoptosis by
repressing TRPC3. Additionally, the results demonstrated that the
miR-454-3p/TRPC3 pathway was required for the survival of HAECs
under normal culture conditions. These findings further elucidated
the molecular mechanisms underlying ox-LDL-induced apoptosis in
HAECs.
Supplementary Material
The potential targets of miR-454-3p
predicted by TargetScan.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by Guanddong Provincial
Medical Science and Technology Research Fund (grant no. 2018270),
Guanddong Provincial Traditional Chinese Medical Bureau Scientific
Research fund (grant no. 20181013) and The Youth Scientific
Research Project of Guangdong Second Provincial General Hospital,
(grant no. YQ2017-018).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
LL and YL designed the present study. LL, QY, HL and
RM performed the experiments and analyzed data. LL and YL wrote the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Tabas I, García-Cardeña G and Owens GK:
Recent insights into the cellular biology of atherosclerosis. J
Cell Biol. 209:13–22. 2015.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Hosin AA, Prasad A, Viiri LE, Davies AH
and Shalhoub J: MicroRNAs in atherosclerosis. J Vasc Res.
51:338–349. 2014.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Menghini R, Casagrande V, Marino A,
Marchetti V, Cardellini M, Stoehr R, Rizza S, Martelli E, Greco S,
Mauriello A, et al: miR-216a: A link between endothelial
dysfunction and autophagy. Cell Death Dis. 5(e1029)2014.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Gimbrone MA Jr and García-Cardeña G:
Endothelial Cell Dysfunction and the Pathobiology of
Atherosclerosis. Circ Res. 118:620–636. 2016.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Mallat Z and Tedgui A: Apoptosis in the
vasculature: Mechanisms and functional importance. Br J Pharmacol.
130:947–962. 2000.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Bushati N and Cohen SM: microRNA
functions. Annu Rev Cell Dev Biol. 23:175–205. 2007.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Hammond SM: An overview of microRNAs. Adv
Drug Deliv Rev. 87:3–14. 2015.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Sun X, Belkin N and Feinberg MW:
Endothelial microRNAs and atherosclerosis. Curr Atheroscler Rep.
15(372)2013.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Menghini R, Stöhr R and Federici M:
MicroRNAs in vascular aging and atherosclerosis. Ageing Res Rev.
17:68–78. 2014.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Li Y, Zhang K and Mao W: Inhibition of miR
34a prevents endothelial cell apoptosis by directly targeting HDAC1
in the setting of atherosclerosis. Mol Med Rep. 17:4645–4650.
2018.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Li Y, Yang N, Dong B, Yang J, Kou L and
Qin Q: MicroRNA-122 promotes endothelial cell apoptosis by
targeting XIAP: Therapeutic implication for atherosclerosis. Life
Sci. 232(116590)2019.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Li Y, Yang C, Zhang L and Yang P:
MicroRNA-210 induces endothelial cell apoptosis by directly
targeting PDK1 in the setting of atherosclerosis. Cell Mol Biol
Lett. 22(3)2017.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Xu K, Liu P and Zhao Y: Upregulation of
microRNA-876 Induces Endothelial Cell Apoptosis by Suppressing
Bcl-Xl in Development of Atherosclerosis. Cell Physiol Biochem.
42:1540–1549. 2017.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Wu X, Ding N, Hu W, He J, Xu S, Pei H, Hua
J, Zhou G and Wang J: Down-regulation of BTG1 by miR-454-3p
enhances cellular radiosensitivity in renal carcinoma cells. Radiat
Oncol. 9(179)2014.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Song Y, Guo Q, Gao S and Hua K: miR-454-3p
promotes proliferation and induces apoptosis in human cervical
cancer cells by targeting TRIM3. Biochem Biophys Res Commun.
516:872–879. 2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Bao X, Ren T, Huang Y, Sun K, Wang S, Liu
K, Zheng B and Guo W: Knockdown of long non-coding RNA HOTAIR
increases miR-454-3p by targeting Stat3 and Atg12 to inhibit
chondrosarcoma growth. Cell Death Dis. 8(e2605)2017.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Zuo J, Yu H, Xie P, Liu W, Wang K and Ni
H: miR-454-3p exerts tumor-suppressive functions by down-regulation
of NFATc2 in glioblastoma. Gene. 710:233–239. 2019.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Mukasa A, Lahn M, Fleming S, Freiberg B,
Pflum E, Vollmer M, Kupfer A, O'Brien R and Born W: Extensive and
preferential Fas/Fas ligand-dependent death of gammadelta T cells
following infection with Listeria monocytogenes. Scand J Immunol.
56:233–247. 2002.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Roberts KP, Ensrud KM and Hamilton DW: A
comparative analysis of expression and processing of the rat
epididymal fluid and sperm-bound forms of proteins D and E. Biol
Reprod. 67:525–533. 2002.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Tano JY, Smedlund K and Vazquez G:
Endothelial TRPC3/6/7 proteins at the edge of cardiovascular
disease. Cardiovasc Hematol Agents Med Chem. 8:76–86.
2010.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Feng M, Xu D and Wang L: miR-26a inhibits
atherosclerosis progression by targeting TRPC3. Cell Biosci.
8(4)2018.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Zhang Y, Chen N, Zhang J and Tong Y:
Hsa-let-7g miRNA targets caspase-3 and inhibits the apoptosis
induced by ox-LDL in endothelial cells. Int J Mol Sci.
14:22708–22720. 2013.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Zhang T, Tian F, Wang J, Jing J, Zhou SS
and Chen YD: Atherosclerosis-associated endothelial cell apoptosis
by miR-429-mediated down regulation of Bcl-2. Cell Physiol Biochem.
37:1421–1430. 2015.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Qin B, Shu Y, Long L, Li H, Men X, Feng L,
Yang H and Lu Z: MicroRNA-142-3p Induces Atherosclerosis-Associated
Endothelial Cell Apoptosis by Directly Targeting Rictor. Cell
Physiol Biochem. 47:1589–1603. 2018.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Ren L, Chen H, Song J, Chen X, Lin C,
Zhang X, Hou N, Pan J, Zhou Z, Wang L, et al: miR-454-3p-mediated
Wnt/β-catenin signaling antagonists suppression promotes breast
cancer metastasis. Theranostics. 9:449–465. 2019.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Shao N, Wang L, Xue L, Wang R and Lan Q:
Plasma miR-454-3p as a potential prognostic indicator in human
glioma. Neurol Sci. 36:309–313. 2015.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Shao N, Xue L, Wang R, Luo K, Zhi F and
Lan Q: miR-454-3p Is an Exosomal Biomarker and Functions as a Tumor
Suppressor in Glioma. Mol Cancer Ther. 18:459–469. 2019.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Cagnin S, Biscuola M, Patuzzo C, Trabetti
E, Pasquali A, Laveder P, Faggian G, Iafrancesco M, Mazzucco A,
Pignatti PF, et al: Reconstruction and functional analysis of
altered molecular pathways in human atherosclerotic arteries. BMC
Genomics. 10(13)2009.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Smedlund K, Tano JY and Vazquez G: The
constitutive function of native TRPC3 channels modulates vascular
cell adhesion molecule-1 expression in coronary endothelial cells
through nuclear factor kappaB signaling. Circ Res. 106:1479–1488.
2010.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Tano JY, Solanki S, Lee RH, Smedlund K,
Birnbaumer L and Vazquez G: Bone marrow deficiency of TRPC3 channel
reduces early lesion burden and necrotic core of advanced plaques
in a mouse model of atherosclerosis. Cardiovasc Res. 101:138–144.
2014.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Smedlund KB, Birnbaumer L and Vazquez G:
Increased size and cellularity of advanced atherosclerotic lesions
in mice with endothelial overexpression of the human TRPC3 channel.
Proc Natl Acad Sci USA. 112:E2201–E2206. 2015.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Ampem PT, Smedlund K and Vazquez G:
Pharmacological evidence for a role of the transient receptor
potential canonical 3 (TRPC3) channel in endoplasmic reticulum
stress-induced apoptosis of human coronary artery endothelial
cells. Vascul Pharmacol. 76:42–52. 2016.PubMed/NCBI View Article : Google Scholar
|