1. Introduction
Pulmonary fibrosis (PF) is caused by factors
including toxic, autoimmune, drug-induced, traumatic injury and
infectious diseases. As a result, a ‘reparative response’ involving
both fibroblasts and myofibroblasts of lung tissue may be triggered
(1). In certain cases, the failure
to establish normal tissue repair in damaged lung results in marked
alveolar disorganization with imbalanced epithelial cell
proportions, endothelial cell loss or migration to incorrect
locations. Furthermore, sustained alveolar epithelial cell
proliferation (AECs), repeated injury and interstitial fibroblast
proliferation with concomitant deposition of collagenous
extracellular matrix (ECM) result in increased fibrogenesis
(1). Idiopathic PF (IPF) is a
chronic progressive interstitial lung disease of unknown origin.
Histologically, IPF is characterized by massive accumulation of
fibroblasts, myofibroblasts, AECs and macrophages and a significant
deposition of ECM (2). A previous
review showed that AECs, as the main source of pro-fibrogenic
cytokines in IPF, express a variety of cytokines and growth
factors, which can promote the migration, proliferation and
accumulation of extracellular matrix of fibroblasts; these are key
events of cell dysfunction in PF, which involve abnormal wound
healing and participate in the formation of patchy fibroblast
myofibroblast lesions in the pathogenesis of IPF (3). AECs are damaged by pathogenic
microorganisms, dust, drugs, chemicals and oxygen free radicals
which, when coupled with risk factors such as aging and genetics,
may decrease the ability of alveolar epithelial type II (ATII)
cells and lung fibroblasts (LFs) to repair damage to the lung
(4,5). LFs proliferate locally, migrate to
the injury site and differentiate into myofibroblasts, which
produce a large amount of ECM and exhibit contractile function. AS
myofibroblasts typically vanish after successful repair;
dysregulation of the normal repair process can lead to persistent
myofibroblast activation (5).
Therefore, decreasing fibroblast activation can limit the
progression of fibrosis (5).
Myofibroblast apoptosis is key for normal regression of the wound
repair response and impaired myofibroblast apoptosis is associated
with tissue fibrosis (6).
Furthermore, depletion of myofibroblasts by apoptosis is key for
normal wound healing. However, this process does not occur in the
fibroblast foci of IPF (6), as
patients with IPF have abnormal repair processes, including
decreased mesenchymal stem cell (MSC) proliferation,
differentiation and repair capacity. These changes lead to scarring
and subsequent respiratory failure (6). Accordingly, the primary clinical
manifestations of IPF are progressive dyspnea, decreased lung
function and respiratory failure and death. IPF mostly occurs in
middle-aged and elderly people, and aging is a risk factor for IPF
(6). LFs and AECs are senescent in
lung tissue of patients with IPF and lung fibrosis animal models
(7,8). Bone marrow (B-)MSCs are depleted,
indicating that cellular senescence is associated with pathogenesis
of IPF (7).
Aging is an underlying decline in age-related
physiological function, leading to increased age-associated
mortality and reduced reproductive capacity (9). Cellular senescence is irreversible
stagnation of the cell cycle, resulting in loss of intercellular
transport and communication and age-associated intrinsic cellular
functions, such as cell division and replication (9,10).
There are two types of cell senescence: Replicative and premature.
Replicative senescence is caused by telomerase damage (10), not by telomere length, whereas
premature senescence is caused by stress, oncogenes and loss of
tumor suppressor factors (11).
The key characteristic of aging is secretion of a large number of
mediators during the stagnation of the cell cycle. These mediators
are collectively known as the senescence-associated secretory
phenotype (SASP) protein (12).
Two primary aging signaling pathways induce cell senescence
(13). The first is the
cyclin-dependent kinase inhibitor (p16)/retinoblastoma (Rb)
pathway, where p16 can competitively bind to CDK4/6 and inhibit its
kinase activity, thereby decreasing phosphorylation of Rb to
prevent activation of downstream transcription factors, thus
leading to stagnation of the cell cycle. The second pathway is
p53/cyclin-dependent kinase inhibitor 1 (p21). When cells are
stressed, tumor suppressor p53 is activated, p21 is upregulated and
the phosphorylation of Rb is inhibited, leading to cell cycle
arrest (14). The mechanisms of
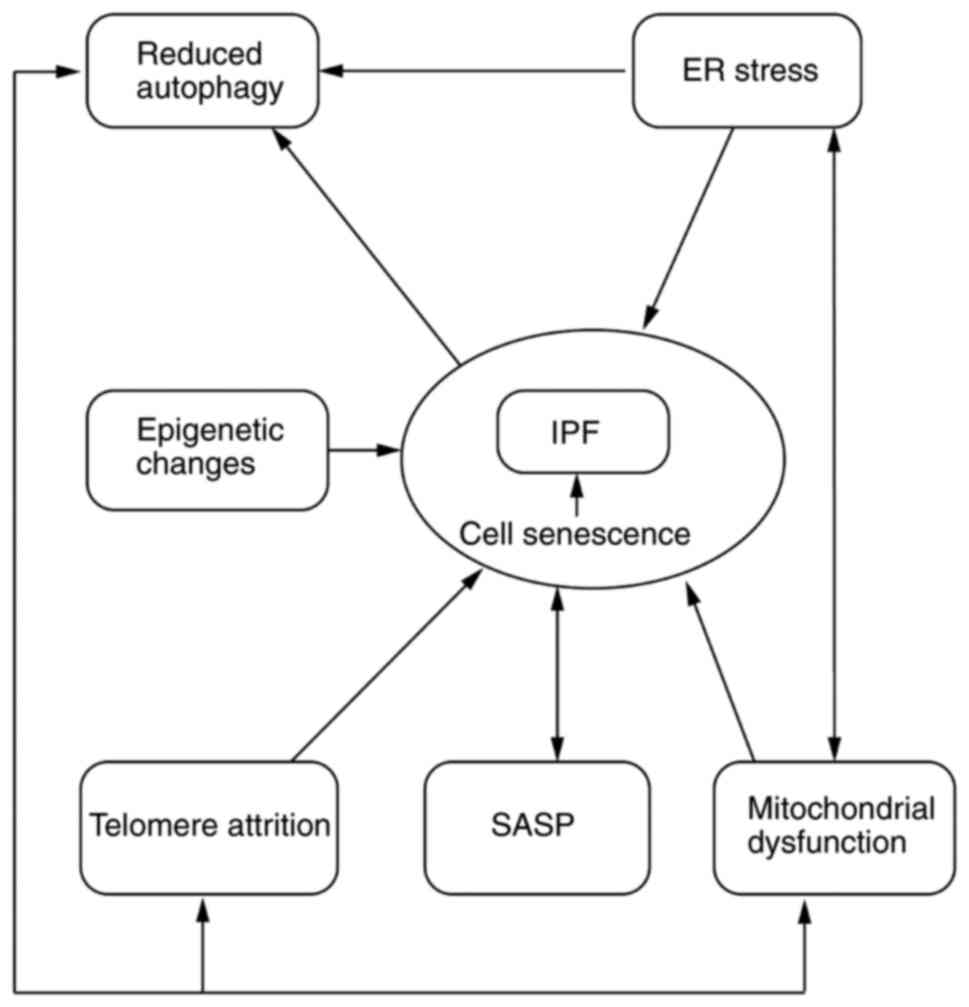
aging associated with development of IPF are illustrated in
Fig. 1.
Studies (3,5,6,15)
have shown that AECs (primarily ATII cells), fibroblasts and
myofibroblasts are involved in the occurrence and development of
PF. Although pirfenidone and nintedanib slow the progression of
IPF, the disease continues to progress and, to the best of our
knowledge, there is no cure other than lung transplantation. To the
best of our knowledge, while IPF is an aging-related disease, the
underlying mechanism linking aging to IPF remains unclear. The
present review summarizes research on the pathogenesis and
treatment of IPF associated with cell aging and provides an
important direction for the future treatment of PF.
2. Senescence of AECs and IPF
ATII cells play an important role in maintaining
pulmonary homeostasis and their main functions include
proliferation, differentiation into ATI cells, secretion of
surfactants and involvement in biological activities such as
pulmonary inflammatory response, immune response, regulation of ECM
and damage repair (15). As AECs
are key cells in the initial phase of IPF, sustained and repeated
AEC injury leads to abnormal changes that promote fibrosis repair.
When the alveolar epithelium is repeatedly damaged, the function
and morphology of ATII cells change and the basal layer shedding
caused by aging and apoptosis may reflect the initial destructive
events in progression of the disease. However, to the best of our
knowledge, the molecular mechanisms involved remain unclear
(2). Studies (16-27)
have shown that aging and apoptosis of ATII cells may be associated
with endoplasmic reticulum (ER) stress and autophagy as well as
telomere damage, mitochondrial dysfunction and epigenetic
changes.
ER stress
In IPF, ER stress is observed in AECs (16). Viral infection and aging trigger a
hyperinflammatory response due to expansion of the ER (28). In addition, susceptibility to ER
stress increases during aging. ER stress initially stimulates an
adaptive unfolded protein response (UPR) to promote cellular
survival but, in the case of persistent chronic stress, UPR
triggers the apoptotic cell death program (16). ER stress is associated with
fibrosis via cell apoptosis, activation/differentiation of
fibroblasts, epithelial-mesenchymal transition (EMT) and activation
or polarization of inflammatory responses (16). In the aging lung, ATII cells are
particularly sensitive to ER stress. ATII cells with knock out of
glucose regulatory protein (GRP) 78, a key protein of ER stress,
show ER stress, injury, senescence and decreased differentiation,
accompanied by abnormal activation of TGF-β1/SMAD signaling
(17). In addition, GRP78 is
reduced in ATII cells in patients with IPF and elderly mice with
IPF. These results suggest that GRP78 reduction is a potential
mechanism underlying the association between ER stress of aging
ATII cells and IPF (17). There is
a dual role of autophagy under ER stress and crosstalk between
autophagy and apoptosis is complicated. Furthermore, ER stress
effectively induces autophagy activation, typically via the Bcl-2
signaling pathway (29). Prolonged
or excessive ER stress induces apoptosis in epithelial cells
through several UPR-dependent downstream mechanisms, including
C/EBP homologous protein induction, activation of the ER-bound
caspase or activation of JNK (30). However, with an increase in age,
sustained ER stress decreases PTEN-induced putative kinase 1
(PINK1) expression and inhibits autophagy (30,31).
In brief, autophagy decreases with aging and accelerated aging may
be attributed to reduced autophagy.
Decreased autophagy
As a cellular protective mechanism, autophagy plays
an important role in cell homeostasis and removal of harmful
substances (18). Autophagy can be
triggered by stress factors, such as reactive oxygen species (ROS),
ER stress and hypoxia. Moreover, insufficient autophagy may
accelerate senescence in epithelial cells and potentiate both EMT
and myofibroblast differentiation. Additionally, inhibition of
autophagy in epithelial cells leads to enhanced EMT and fibrosis
(19). Expression of
multifunctional protein p62 and ubiquitinated protein in the lung
of patients with IPF is increased, indicating insufficient
autophagy in lung tissue (18,20).
Beclin-1, a key autophagic protein, is downregulated in fibroblasts
isolated from patients with IPF (32). Conditional knockout of the tuberous
sclerosis complex 1 gene in mouse epithelial cells renders
bleomycin (BLM)-induced mice more prone to PF; this is reversed by
activation of autophagy by rapamycin (33). Another study (34) has revealed that aging mice with
loss of autophagic proteins [light chain 3β(LC3B)-/- and
autophagy-related 4B cysteine peptidase (ATG4B)-/-] are more
susceptible to BLM-induced lung fibrosis and linked cathepsin A, a
binding partner to LC3B. In addition, ER stress increased apoptosis
of epithelial cells in aging mice with loss of autophagic proteins
(LC3B-/- and ATG4B-/-) (34).
Studies (18-20,32-34)
confirm that autophagy is decreased in AECs of patients with IPF.
Autophagy deficiency leads to epithelial cell dysfunction and
promotes PF, while autophagy activation enhances the repair ability
of epithelial cells and inhibits PF. However, the molecular
mechanism underlying regulation of autophagy and its effect on
epithelial cell function remains unclear and warrants further
investigation.
Telomere attrition
Telomeres are composed of DNA repetitive sequences
and binding proteins that maintain structural integrity of
chromosomes. The binding proteins consist of the protein components
telomeric repeat-binding factor (TRF) 1, TRF2, Ras-related protein
Rap1, TRF1-interacting nuclear factor 2, telomere protective
protein 1 (TPP1) and protection of telomeres 1 gene (35). Telomeric DNA typically contains
clusters of three or four guanines (for example, 5'-TTGGGG-3' in
tetrahymena and 5'-TTAGGG-3' in humans) (35). These telomeric repeats are added by
the enzyme telomerase. Telomerase comprises three parts: Human
telomerase RNA (hTR); telomerase synergistic protein 1 and hT
reverse transcriptase(hTRT). Telomerase performs catalytic reverse
transcription (21). Typically,
normal aging is accompanied by telomere shortening. However,
mutations in the telomerase complex genes hTRT and hTR are found in
8-15% of familial patients with IPF and 1-3% of sporadic cases;
these mutations accelerate telomere shortening and cause
replicative aging of AECs (21-23).
V144M, R865C and R865H mutants of hTRT are key because these
mutants can explain how the hereditary hTERT mutation causes
telomere shortening in IPF patients, so as to further understand
the role of naturally occurring telomerase mutations in the
pathophysiology of some age-related disease states; in vitro
experiments have determined that V144 and R865 in hTRT are key
residues required for the normal function of cell telomerase
(36,37). Moreover, 98 G→A, 37A→G, 108C→U and
325G→U hTR substitution are often noted in familial patients with
IPF. These mutations are predicted to impair base pairing in a
helix in the key pseudoknot domain of hTR (21,38).
The knockout of TRF1 in ATII cells causes severe telomere
dysfunction in the lung of mice and induces PF by inducing DNA
damage and upregulating cell cycle suppressor protein
p21/p53(39). Similarly, specific
knockout of TRF2 in mouse ATII cells is characterized by telomere
dysfunction, resulting in increased p53 and p21 expression. As the
ability of ATII cells to self-renew and differentiate is limited,
these cells are less able to repair BLM-induced lung damage
(40). A recent study (41) suggested that a key role in alveolar
stem cell dysfunction is played by telomere shortening or
uncapping, bridging the gap in telomere abnormality and fibrotic
lung pathology. Failure to regenerate alveoli due to alveolar stem
cell dysfunction may expose lung cells to elevated mechanical
tension, which may activate the TGF-β signaling loop to promote the
fibrotic process (41). In
addition, short telomeres signal and activate p53, which suppresses
phosphatidylglycerol phospholipase C (PGC)-1α and PGC-1β promoters,
leading to mitochondrial dysfunction and cell aging (42). These findings indicate that
telomere shortening serves a key role in the occurrence of IPF.
Mitochondrial dysfunction
Mitochondrial dysfunction drives cell senescence. In
pathogenesis of IPF, mitochondrial dysfunction primarily involves
an imbalance in mitochondrial ROS levels, mitochondrial DNA
changes, mitochondria-mediated reduced autophagy and electron
transport chain imbalance. Mitochondrial dysfunction is associated
with telomere attrition and ER stress (24,25,31).
Epithelial cell damage is associated with increased mitochondrial
ROS in lung tissue of patients with IPF (24). With aging, mitochondrial function
is impaired, leading to increased ROS production, which in turn
causes deterioration of mitochondrial function (24). Changes in mitochondrial DNA
metabolism and lack of phagocytosis often occur in patients with
IPF, leading to increased sensitivity to apoptosis (24). Kim et al (25) showed that Klotho protein plays a
role in protecting AEC mitochondrial DNA integrity. Knockout of
PINK1 in ATII cells of mouse lung tissue leads to mitochondrial
swelling and dysfunction, impaired mitochondrial autophagy and
increased susceptibility to PF (31). Furthermore, there is evidence of a
role played by PINK1/parkin RING-in-between-RING (RBR) E3 ubiquitin
protein ligase-mediated mitochondrial autophagy in IPF.
Furthermore, there is growing evidence supporting the role played
by PINK1-PARK2-mediated mitochondrial autophagy in IPF (43).
The association between mitochondrial dysfunction
and low PINK1 expression indicates that PINK1 may serve an
important role in maintaining the morphology and function of
mitochondria and selectively degrades damaged mitochondria through
autophagy (31). Mitochondrial
dysfunction occurs with and further promotes aging, leading to an
imbalance in the electron transport chain. NAD+ is an
electron acceptor and oxidant, as well as a cofactor in numerous
metabolic and signaling pathways. The ratio of NAD+ to
NADH is key and NAD+ improves mitochondrial function and
longevity in aging mice. One mechanism by which NAD+
affects PF is the regulation of cellular function by regulating the
activity of sirtuins (SIRTs). Deacetylase SIRT is an
NAD+-dependent histone deacetylase (HDAC) and serves an
important role in transcription, cell cycle regulation and
subsequent translation and modification (44-46).
Aging decreases expression of SIRT3, leading to an increase in
acetylation and thereby increasing the levels of mitochondrial ROS
and DNA damage (44). SIRT3
inhibits TGF-β1 signaling and controls myofibroblast transformation
(47). Increased expression levels
of NADPH oxidase 4 (NOX4) have been reported in the lung of
patients with IPF (48). NOX4 is
considered to be a mediator of mitochondrial dysfunction (49). NOX4 enzyme interacts with
mitochondria and affects mitochondrial function and production of
mitochondrial ROS and SIRT, jointly promoting epithelial injury and
lung fibrosis (50). NOX4 also
regulates protein and collagen concentrations of α-smooth muscle
actin (α-SMA) by controlling Smad2/3 and regulating
platelet-derived growth factor (PDGF) to induce fibroblast
migration (48). Telomere
shortening is a feature of premature aging, partly due to increased
mitochondrial ROS (51). There is
an interaction between mitochondria and ER. ER stress can
downregulate PINK1 to regulate mitochondrial function and increase
apoptotic response. Meanwhile, during ER stress, the transfer of
calcium ions from ER to mitochondria leads to mitochondrial
swelling (31).
Epigenetic changes
Epigenetic marks are cell-type specific. A previous
study revealed that microRNA (miRNA or miR)-34a, miR-34b and
miR-34c, members of the aging-associated miR-34 family, are highly
expressed in AECs from patients with IPF (26). The deletion of miR-34a effectively
improves BLM-induced epithelial cell senescence in animal models
(27,52). The mechanism of miR-34 in IPF may
involve p53 activation and downregulation of the transcription
factor early 2 factor to inhibit cell proliferation (26). The activation of p53 induces p21,
leading to cell cycle arrest by inhibiting the expression of cyclin
E and CDK2. Cui et al (27)
showed that miR-34a is increased in AECs but not in LFs. This
suggests that miR-34a expression may be regulated differently in
different types of lung cell. The role of AECs and SASP in IPF is
displayed in Fig. 2.
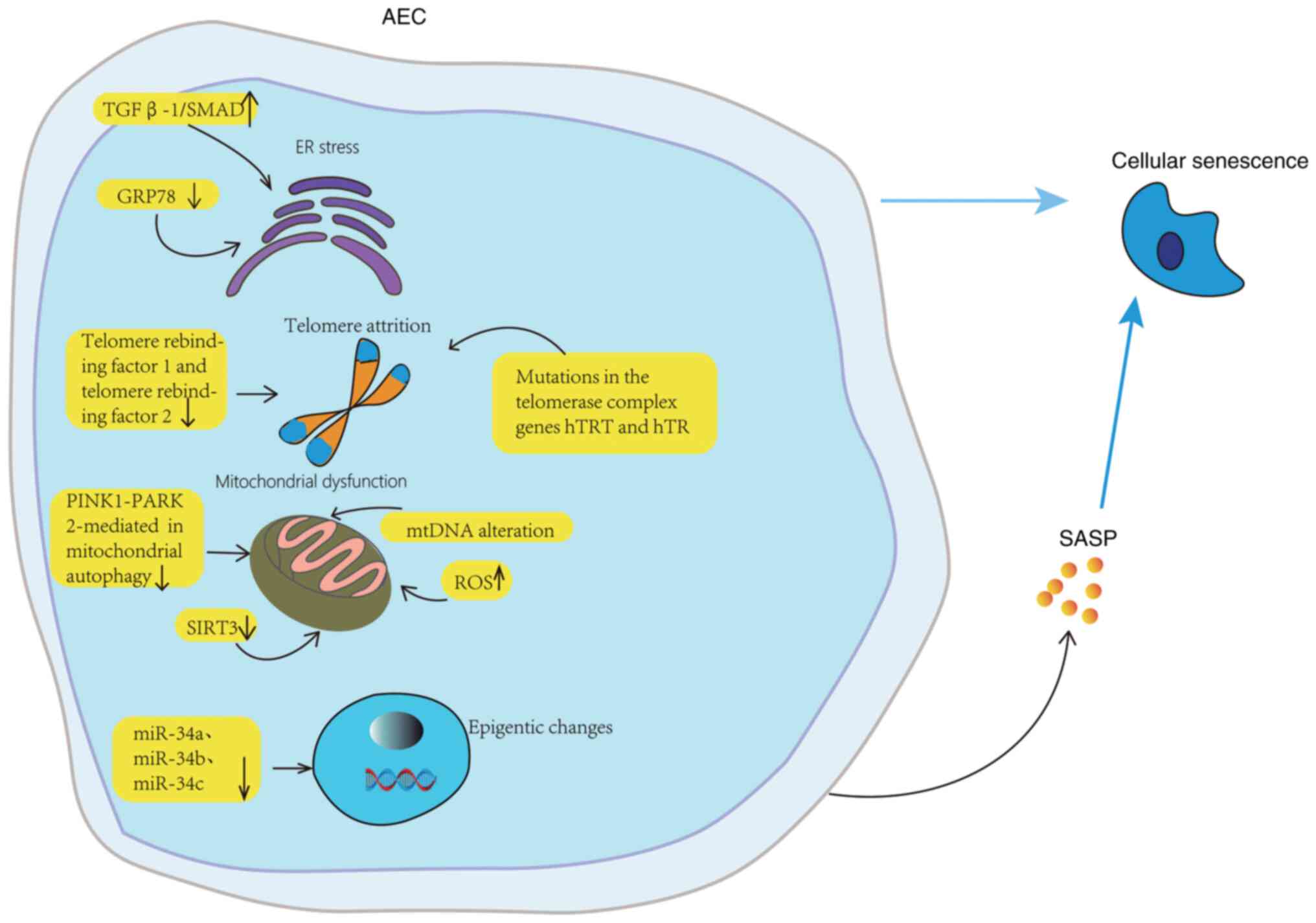 | Figure 2Aging mechanism of AECs. The
senescence and apoptosis of AECs may be related to ER stress,
autophagy, telomere damage, mitochondrial dysfunction, and
epigenetic changes. In aging lungs, AECs are particularly sensitive
to ER stress. GRP78 knockout decreases ER stress, injury, aging and
differentiation, and TGF-β 1/Abnormal activation of SMAD signaling.
Normal aging is accompanied by telomere shortening, while mutations
in hTRT and hTR of telomerase complex gene accelerate telomere
shortening and lead to replicative aging of AECs. In the
pathogenesis of IPF, mitochondrial dysfunction involves the
imbalance of mitochondrial ROS level, the change of mitochondrial
DNA, the downregulation of mitochondrial autophagy mediated by
PINK1-PARK2, and the downregulation of SIRT3 expression. The high
expression of miR-34a, miR-34b and miR-34c can lead to the aging of
AECs. AEC, alveolar epithelial cell; GRP78, glucose regulatory
protein 78; hTRT, human telomerase reverse transcriptase; hTR,
human telomerase RNA; mtDNA, mitochondrial DNA; PINK1, PTEN-induced
putative kinase 1; PARK2, parkin RBR E3 ubiquitin protein ligase;
ROS, reactive oxygen species; SASP, senescence-associated secretion
phenotype; SIRT3, NAD-dependent deacetylase sirtuin-3; miR,
microRNA. |
3. Senescence of fibroblasts and IPF
Fibroblasts serve a central role in the fibrotic
process. Deposition of fibroblasts in the pulmonary interstitium
and generation of ECM serve a key role in the development of IPF
(53). Primary LFs isolated from
the lung tissue of patients with IPF exhibit more senescence
compared with age-matched controls. These senescent fibroblasts
increase levels of senescence-associated β-galactosidase, P16, P21,
P53 and SASP (5). The pathogenesis
of fibroblast senescence in IPF remains unclear but mitochondrial
dysfunction, telomere shortening, epigenetic changes and decreased
autophagy are involved in IPF. To the best of our knowledge,
however, most current research has focused on epigenetics and
autophagy (5,54).
Epigenetic alterations
Epigenetic changes mainly involve DNA methylation,
histone post-translational modifications and miR regulation
(55). Human DNA contains
cytosine-phosphate-guanine(CpG)sites and the methylation of the CpG
island via DNA methyltransferases (DNMTs) prevents RNA polymerase
complex binding to the promoter region and therefore suppresses
gene expression. Additionally, the majority of human CpG sites are
methylated (56). Abnormal DNA
methylation either silences or activates expression of genes that
drive fibrosis and alter the mRNA expression of several genes. For
example, Neveu et al (57)
found that thymocyte differentiation antigen 1 (Thy-1) expression
is epigenetically modified. Treatment with DNMT inhibitor
5-azacytidine attenuates TGF-β1-induced collagen type I α 1 chain
gene and protein expression and α-SMA gene expression in LFs.
Furthermore, inhibiting DNMT1 attenuates TGF-β1-induced DNMT
activity, downstream suppression of Thy-1 expression and decreases
Thy-1 promoter methylation. In addition, DNA methylation induces
differentiation of fibroblasts into myofibroblasts and deposition
of collagen matrix (58).
O6-alkylguanine DNA alkyltransferase (MGMT) is a key DNA repair
enzyme; however, MGMT is hypomethylated with overexpression in IPF
fibroblasts (59). These findings
suggest that inhibition of DNMT might prevent lung fibrosis
(57).
At present, a few histone modifications have been
described. The best-elucidated modifications (60) include acetylation by histone
acetyltransferases (HATs) (61),
deacetylation by HDAC (62),
methylation by histone methyltransferase (HMT), demethylation,
phosphorylation, ubiquitination, sumoylation and poly
ADP-ribosylation (63). Currently,
acetylation is hypothesized to activate cell transcription by
decompression of the chromatin. IPF is characterized by increases
in ‘restrictive’ histone post-translational modifications that
result in decreased anti-fibrotic and pro-apoptotic gene expression
(63). A previous study has shown
that decreased acetylation of histone H3 and H4 in patients with
IPF decreases expression of cyclooxygenase 2 (COX-2) in
fibroblasts, thereby decreasing synthesis of prostaglandin E2 and
effectively inhibiting activation of fibroblasts (64). Moreover, Fas expression is
decreased in IPF and linked to elevated H3K9 trimethylation and
decreased H3 pan-acetylation at the Fas promoter (63). Taken together, the aforementioned
studies indicate an imbalance in both histone acetylation and
methylation in IPF; this imbalance prevents transcriptional
activation of anti-fibrotic and pro-apoptotic genes.
Numerous miRNAs are up- or downregulated in patients
with IPF. For example, increased expression of miR-21 in IPF
fibroblasts inhibits Smad7 activation by TGFβ1, thereby aggravating
PF (65). Additionally, miR-21 is
hypothesized to induce EMT and promote fibrotic processes via
inhibition of Smad7(65).
Furthermore, miR-144-3p is a miRNA that is upregulated >70-fold
in IPF fibroblasts; it can increase α-SMA levels and its mimic has
been shown to downregulate relaxin/insulin-like family peptide
receptor 1in IPF LFs (66).
Decreased autophagy
Transformation of PF fibroblasts into pulmonary
myofibroblasts is a key part of PF. Inhibition of autophagy leads
to increased differentiation of myofibroblasts and enhanced
expression of smooth actin. A lack of autophagy may cause
phenotypical transformation of fibroblasts into myofibroblasts
(67). Beclin-1, a key autophagic
protein, in a complex with class III phosphatidylinositol 3-kinase
and ATG14 serves as a major positive regulator of autophagy
(68). Genetic deletion of
autophagy protein LC3 or Beclin-1 enhances expression of
fibronectin, myofibroblast differentiation marker and SMA in LFs
(69). TGF-β1 is a multifunctional
peptide growth factor with a range of potential effects on cell
proliferation and differentiation and ECM protein production.
TGF-β1 also mediates the downregulation of Caveolin-1 (Cav-1) in
fibroblasts via the MAPK signaling pathway and Cav-1 loss provides
fibroblasts with anti-apoptotic properties (70). Another pathway by which autophagy
regulates the pathological process of IPF is the mammalian target
of rapamycin (mTOR) pathway. This pathway consists of upstream
molecules (PI3K and AKT), tuberous sclerosis complex 1/2 and
downstream molecules (eukaryotic cell translation initiation factor
4E binding protein 1 and ribosomal protein S6 kinase 1) (71). The mTOR pathway inhibits autophagy,
which is characterized by increased apoptotic effector protein
beclin-1 and LC3 levels. Activation of this pathway promotes
differentiation of myofibroblasts and leads to the formation of PF
(72). Similarly, Nho and Hergert
(73) showed that deletion of PTEN
in activated human chromosome 10 PTEN/AKT/mTOR signaling pathway
induces autophagy in myofibroblasts for collagen synthesis. In LFs
of IPF, the PTEN/AKT axis decreases the expression of FOXO3a. The
decreased FOXO3a expression suppresses LC3B transcription and leads
to loss of autophagy on collagen in IPF fibroblasts. Vimentin
intermediate fills and Janus kinase 2/signal transduction activator
3 signaling pathways lead to increased production of the
anti-apoptotic Bcl-2 family protein to inhibit autophagy, thus
causing fibroblast-to-myofibroblast transformation and progression
of PF (74).
A study reported the presence of telomere shortening
in LFs of patients with IPF (5).
This is consistent with telomere length maintained in the
epithelial cells of patients with IPF (67). However, when expression of
telomerase transcriptase in human LF is induced, the telomere
length does not change in BLM-induced lung tissue (75,76).
This suggests that the association between telomere shortening and
cellular senescence occurs only in ATII cells and not in LFs,
showing cell type specificity. In terms of mitochondrial
dysfunction, TGF-β is a pro-fibrosis factor and TGF-β stimulation
of LFs decreases PINK1 levels and promotes mitochondria-mediated
phagocytosis as well as a low levels of myofibroblast
differentiation (44). In
addition, mitochondrial dysfunction of aging fibroblasts results in
mTOR complex 1 (mTORC1) activation, which changes mitochondrial
homeostasis and produces a large amount of ROS. ROS promotes DNA
damage and enhances aging of LFs (77). The role of LFs and SASP in IPF is
illustrated in Fig. 3.
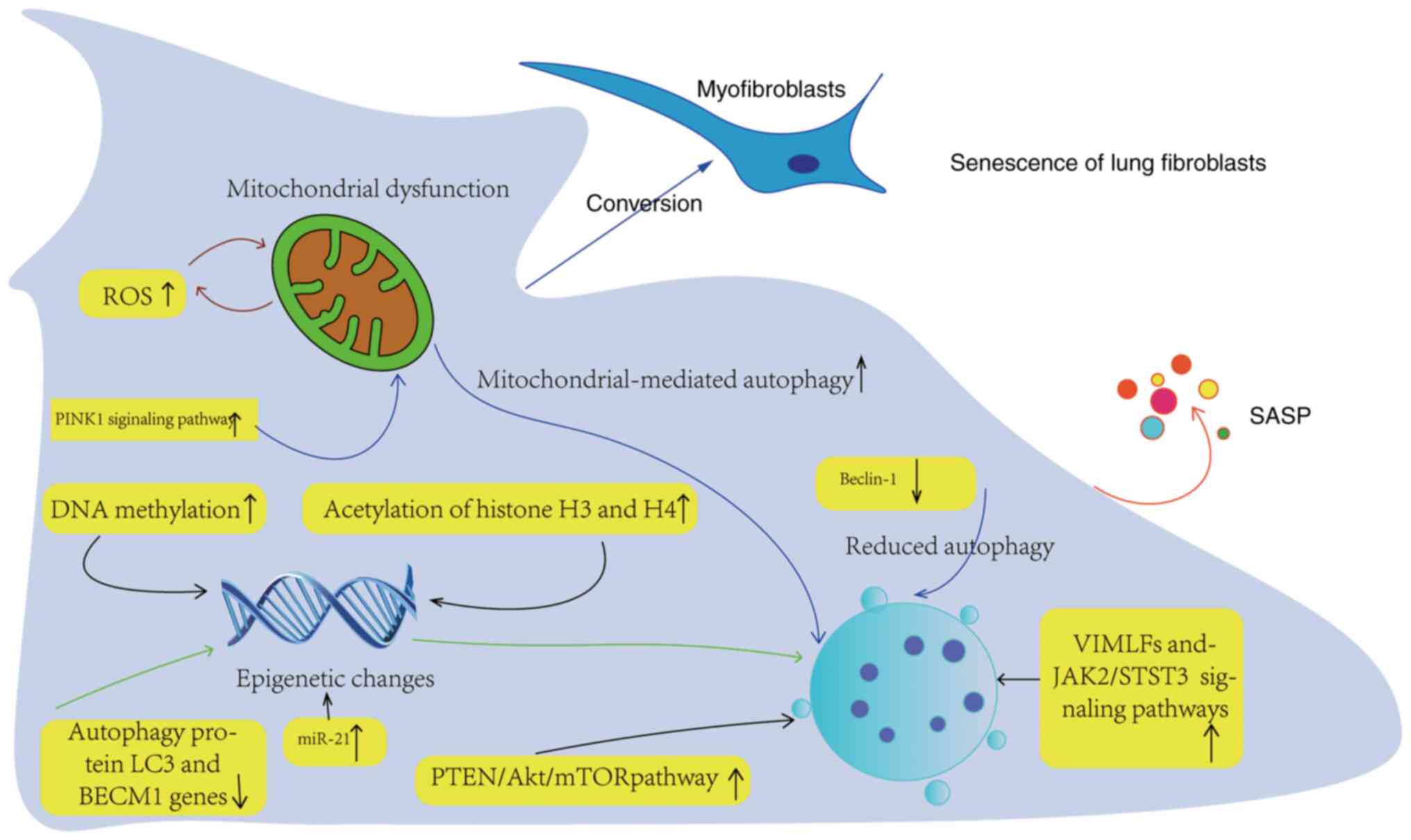 | Figure 3Aging mechanism of lung fibroblasts.
Epigenetic changes mainly involve DNA methylation, histone
post-translational modification and miR regulation. Abnormal DNA
methylation can inhibit or activate the gene expression leading to
fibrosis and change the mRNA expression of genes. The up regulation
of histone H3 and H4 acetylation can promote pulmonary fibroblast
fibrosis. The transformation of PF fibroblasts into pulmonary
muscle fibroblasts is a key part of PF. Inhibition of autophagy may
lead to the transformation of fibroblast phenotype into
myofibroblast, and Beclin-1 is a key autophagic protein and a major
positive regulator of autophagy. Gene deletion of autophagic
protein LC3 or Beclin-1 enhanced the expression of fibronectin,
myofibroblast differentiation marker and SMA in LFs. The
upregulation of PTEN in PTEN/AKT/mTOR signal pathway can
downregulate autophagy of myofibroblasts. Upregulation of ROS and
PINK1 signaling pathway leads to mitochondrial dysfunction related
to autophagy. PINK1, PTEN-induced putative kinase 1; ROS, reactive
oxygen species; SASP, senescence-associated secretion phenotype;
VIMFL, vimentin intermediate fill; JAK2, Janus kinase 2; STST3,
signal transduction activator 3; miR, microRNA. |
4. Stem cell senescence and IPF
Stem cell dysfunction occurs in several
aging-associated diseases, including IPF. MSCs from patients with
IPF are in a transitional state of senescence and induce senescence
paracrine, which can induce senescence in normal fibroblasts. A
study demonstrated that adult stem cells prevent PF progression,
repair lung injury and remodel lung tissue in animal models of PF
(78,79). As MSCs are pluripotent, they can
differentiate into a variety of cell types, including ATII cells,
in response to specific stimuli (80).
Resident stem cells in the adult respiratory system
have been identified, including Clara cell secreted protein (CCSP)
and surfactant protein C-positive bronchioalveolar stem cells,
p63-, Krt5- and/or Krt14-positive basal cells, secretoglobin family
1A member 1-positive Clara cells, tyrosine-protein kinase
Kit-positive lung stem cells, E-cadherin/leucine-rich repeat
containing G protein-coupled receptor 6-positive putative stem
cells and ATⅡ cells that initiate a pro-fibrotic cascade when the
lung is damaged by persistent or repeated noxious stimuli, such as
smoke, viruses, pollutants and genetic factors, and induces an
abnormal environment to promote terminal differentiation of lung
stem/progenitor cells from favoring local alveolar tissue
regeneration to effector or fibroblast/myofibroblast development in
IPF, ultimately promoting fibrosis progression (81).
ATⅡ cells function as alveolar progenitors and
long-term stem cells in the adult lung (82). Adult stem cells undergo dynamic
changes after tissue injury. ATⅡ cells are adult alveolar stem
cells that can differentiate into ATI cells during alveolar
homeostasis and post-injury repair and are involved in the process
of lung repair (83). A previous
study found that alveolar stem cell differentiation involves a
transitional state (84) and a
pre-ATI cell transitional state was identified in lung tissue
regeneration. This unique state is associated with enrichment of
cellular senescence and defective alveolar regeneration pathways,
as well as the prolonged senescence and stress mediated pathway
leading to pathological processes such as fibrosis. Furthermore,
there is a transition between ATI and ATⅡ cells. These transition
states are associated with abnormal epithelial cells, which show
DNA damage response and express aging related genes on the way to
ATI cells and are related to the defects of human pulmonary
fibrosis (84). Metabolic lesions
are associated with progressive PF (84).
Aging affects all cells, including MSCs. B-MSCs
share many features with lung resident progenitor cells and have
anti-inflammatory and immunomodulatory properties (85). A study demonstrated the ability of
MSCs to suppress inflammation, decrease fibrosis and prolong
survival in preclinical PF models (79). Tracking of radiolabeled cells
revealed that when administered intravenously, MSCs primarily
localize to the lung, followed by the liver and other organs
(86). IPF is characterized by
interstitial inflammation and epithelial cell damage, followed by
fibroblast proliferation and collagen deposition. Preferentially
located at sites of inflammation, MSCs inhibit ongoing damage and
contribute to tissue repair (87).
It has been suggested that several pathways are altered in aging
MSCs and lungs, which may increase the risk of IPF (88). Scientists have observed
IPF-associated extrapulmonary effects in B-MSCs. Compared with
B-MSCs from age-matched controls, B-MSCs from patients with IPF
exhibit increased cell size, morphological changes, DNA damage and
telomere shortening with replicative senescence (89). In addition, B-MSCs from patients
with IPF exhibit mitochondrial dysfunction and impaired recovery
under in vitro and in vivo stimulation (89). Senescent B-MSCs have decreased
paracrine capacity by senescent fibroblasts, suggesting a potential
link between senescent B-MSCs and later onset of the IPF (89). In addition, systemic transfer of
MSCs effectively decreases BLM-induced lung injury and fibrosis by
downregulating nitric oxide metabolites, pro-inflammatory factors
and angiogenic cytokines (90).
Elucidating the association between stem cell aging
and PF may provide novel potential treatment options for PF.
5. SASP
There is an association between senescent AECs and
fibroblasts. Senescent AECs promote activation of LFs by increasing
expression of SASP. SASP includes pro-inflammatory cytokines (such
as IL-6 and IL-8), growth factors [such as TGF-β and
granulocyte-macrophage (GM) colony-stimulating factor (CSF)],
chemokines [such as C-X-C motif chemokine ligand (CXCL) 1, CXCL3
and CXCL10] and matrix remodeling enzymes (such as
metalloproteinases) (91). SASP
has potent autocrine and paracrine effects and regulates the tissue
microenvironment through biological processes, including cell
proliferation, migration, inflammation, fibrosis, ECM degradation,
neovascularization, tissue repair and regeneration, senescence
clearance and EMT (92). SASP
creates an inflammatory microenvironment for clearance of senescent
cells and promotes senescence of surrounding cells in a paracrine
manner. The SASP factor varies in different cell types and
senescence-induced stimuli (92).
Fibroblasts and myofibroblasts exhibit stress and senile phenotypes
while secreting a range of cytokines, including pro-inflammatory
cytokines (such as TNF-α, TGF-β, IL-1β, IL-6, IL-8, IL-10 and
IL-18), chemokines (such as CXCL1 and monocyte chemoattractant
protein-1), growth regulators [such as fibroblast growth factors,
connective tissue growth factor (CTGF), GM-CSF and macrophage-CSF],
matrix metalloproteinases (such as MMP-2, MMP-3, MMP-9, MMP-10 and
MMP-12) and leukotrienes (LTs; such as LTA4, LTB4, LTC4, LTD4)
(93,94). These cytokines serve a key role in
regulation of PF and inflammation (95). IL-18 promotes senescence of LFs and
expression of SASP in lung tissues by downregulating Klotho
expression. Furthermore, neutralizing IL-18 by IL-18 binding
protein partially inhibits aging of LFs (94). TGFβ-1 is a key component of SASP.
By upregulating the cell cycle inhibitors p21, p27 and p15, TGFβ
induces the senescence of adjacent cells in a paracrine manner
through the SMAD signaling pathway (96).
Aging ATII cells express SASPs such as PDGF, TNF,
endothelin-1, CTGF, osteopontin, CXCL12 and plasminogen activator
inhibitor 1 (PAI-1), inducing massive proliferation and activation
of IPF LFs and myofibroblasts (97). One salient component is PAI-1 which
is highly expressed in ATII cells. Overexpression of PAI-1 promotes
accumulation of extracellular stroma and acts as a strong inducer
of cell senescence, especially in ATII cells. TGF-β1 increases
PAI-1 expression through a variety of signaling pathways (98), such as TGF-β1, induce ATII-cell
senescence, and that its prosenescent effects are mediated by
PAI-1(99), TGF-β1 induces PAI-1
production in alveolar macrophages through activin receptor-like
kinase 5 activation and Smad3 phosphorylation (100).
Extracellular vesicles (EVs) have been recognized as
a type of SASP. Derived from plasma membranes, EVs have diameters
ranging from 30 nm to 5 µm and are composed of a phospholipid
bilayer. EVs effectively promote cell signaling and differentiation
by transferring bioactive substances, including miRNA, mRNA and
lipids. IPF LF-derived EVs transfer miR-23b-3p and miR-494-3p to
lung epithelial cells (101,102). These miRNAs inhibit production of
mitochondrial ROS, causing mitochondrial dysfunction, DNA damage
response and accelerated cell senescence (101). The expression of miRNAs in
LF-derived EV was correlated with lung function of the donor. The
development of fibrosis is largely regulated by SASP. Targeted
inhibition of specific miRNA species or blocking secretion of
specific EVs from LF may provide new therapeutic options (101).
SASP is involved in lung fibrosis by recruiting a
large number of inflammatory cells into tissue and organs. In
addition, SASP promotes the synthesis of ECM proteins by
stimulating proliferation and transformation of fibroblasts into
myofibroblasts (101).
The senescence of AECs is controlled by the
PTEN/NF-κB pathway; this is a feature of PF. NF-κB is a key target
of the PTEN/PI3K/Akt pathway, a signal transduction pathway that
triggers cytokine release in senescent cells, thereby contributing
to the adjacent cellular microenvironment (103). Other studies have showed that
NF-κB and p38 MAPK pathways are involved in regulation of SASP
(104,105).
In conclusion, SASP directly or indirectly promotes
cell senescence or lung fibrosis via various signaling pathways,
such as blocking the Klotho, SMAD and PTEN/NF-κB pathways. Further
study of this mechanism is key for the treatment of IPF.
6. Interaction between senescent ATII, LF
and other cell types in IPF
Senescent cells have two effects: They can be
physiologically beneficial for tissue repair but they can also be
pathologically harmful in age-associated diseases such as lung
fibrosis (91). One of the key
pathogenic mechanisms of PF is the inability of ATII cells to
repair damaged epithelial cells due to cell death, ineffective
proliferation, migration and differentiation, leading to
interstitial scarring. Apoptosis of ATII cells directly encourages
the progression of PF. Abnormal aging and apoptosis in ATII cells
during acute lung damage are initiators of fibrosis. AECs during
acute lung damage activate fibroblasts to differentiate into
myofibroblasts, which form fibroblast foci and secrete large
amounts of ECM (91). Aging AECs
promote activation of LFs by increasing expression of SASP. One
hypothesis on the origin of myofibroblasts is EMT (91). Cells lose the properties of
epithelial cells and acquire properties of mesenchymal cells
(106). The Wnt pathway is
potentially involved in EMT. In ATII cells with overexpressed
Wnt-1-induced signaling proteins, secretions of certain
pro-fibrosis markers, such as PAI-1, are increased. These markers
induce EMT in adjacent epithelial cells. In turn, abnormally aging
ATII cells activate fibroblasts by secreting chemokines.
Myofibroblasts produce mediators that induce epithelial cell
apoptosis, thereby destabilizing epithelial cells and increasing
alveolar injury. Moreover, apoptosis of AECs leads to formation of
gaps between alveoli (107).
Myofibroblasts are the activated form of fibroblasts that migrate
into the interstitial space and activate further fibroblast
proliferation, leading to increased apoptosis in ATII cells
(108), further promoting lung
fibrosis.
Kadota et al (109) demonstrated accelerated epithelial
cell mitochondrial damage and senescence during IPF. LFs from
patients with IPF were found to induce cellular senescence via
EV-mediated transfer of pathogenic cargo to lung epithelial cells.
The transfer of miR-23b-3p and miR-494-3p via IPF LF-derived EVs
also induced mitochondrial damage and senescence in lung epithelial
cells. In addition, levels of miR-23b-3p and miR-494-3p from IPF
LF-derived EVs correlated positively with disease severity of IPF.
Furthermore, LFs from patients with IPF were found to induce
cellular senescence via EV-mediated transfer of pathogenic
substances to AECs. IPF LF-derived EVs increased mitochondrial ROS
and associated mitochondrial damage in lung epithelial cells,
leading to activation of the DNA damage response and subsequent
epithelial senescence. In addition, miR-23b-3p and miR-494-3p,
released from fibroblasts as EV cargo, were transferred to lung
epithelial cells, where they induced mitochondrial damage and
cellular senescence in HBECs. Scientists have identified a novel
EMT interaction mediated by LF-derived EVs (110). This interaction induces
mitochondrial damage and senescence in epithelial cells during the
pathogenesis of IPF (110). Aging
in ATⅡ cells also contributes to PF. E3 ubiquitin ligase FBW7 binds
TPP1, promotes TPP1 multisite ubiquitination, accelerates
degradation, triggers telomere unpacking and DNA damage responses,
as well as leads to stress-associated telomere dysfunction, which
ultimately inhibits ATⅡ stem cell proliferation and mediates
stress-induced ATⅡ stem cell senescence and PF (111). The proportion of ATⅡ in total
AECs is significantly decreased in patients with IPF (112).
7. Senotherapeutics
Currently, the only FDA-approved medications for
treatment of IPF are pirfenidone and nintedanib (113). While both drugs slow progression
of fibrosis in certain patients with IPF, they do not halt or
reverse progressive fibrosis. Experiments are ongoing and numerous
clinical trials have been conducted (114-116).
A better understanding of cellular senescence lung fibrosis may
provide novel therapeutic strategies for the prevention and
treatment of aging-related PF.
Experiments have focused on the characteristics and
signaling pathways of aging, but the current results are limited.
Most research studies are in their early stages. There is also an
increasing focus on developing anti-aging treatments known as
senotherapy. In principle, aging treatment strategies are broadly
divided into two categories: Anti-aging strategies that selectively
remove senescent cells and homogeneous strategies that block the
aging phenotype by inhibiting SASP without damaging cells (117-119).
These potential therapeutics are summarized in Table I.
 | Table IExperimental potential senolytic
drugs that target senescent alveolar epithelial cells and lung
fibroblasts. |
Table I
Experimental potential senolytic
drugs that target senescent alveolar epithelial cells and lung
fibroblasts.
| First author/s,
year | Target | Potential
therapeutic drugs | Mechanism | (Refs.) |
|---|
| Le Saux et
al, 2013; | Telomere | Raloxifene | Induces telomerase
activity and maintains telomere length | (129) |
| Calado et
al, 2009; | | | | (130) |
| Arish et al,
2019; | | Androgens | | (131) |
| Townsley et
al, 2016 | | GRN510 | The small molecule
telomerase activator GRN510 attenuates fibrosis in mice with
bleomycin- induced PF | (132) |
| | | Danazol | Danazol, a
synthetic androgen, increases telomere length and stabilizes
diffusing capacity for carbon monoxide and forced vital
capacity | |
| Sanders et
al, 2014; | Epigenetics | Vorinostat | Inhibits
ubiquitin-histone deacetylase and induces apoptosis of
fibroblasts | (139) |
| Korfei et
al, 2018; | | | | (140) |
| Mora et al,
2017; | | | | (141) |
| Coward et
al, 2014; | | 5'-azacytidine | Decreases DNA
methylation and fibrosis | (142) |
| Korfei et
al, 2015 | | | | (138) |
| | | BIX-01294 and
3-deazaneplanocin | Inhibitors of
euchromatic histone-lysine N-methyltransferase 2 and enhancer of
zeste homolog 2 histone methyltransferases; increase cyclooxygenase
-2 expression in IPF fibroblasts, which may enhance production of
anti-fibrotic prostanoid PGE2 | |
| | | LBH589 and
SAHA | LBH589
downregulates mRNA expression of ACTA2 and ECM genes COL1A1, COL3A1
and FN in primary IPF fibroblasts and interferes with
fibroblast-to- myofibroblast differentiation. SAHA induces
apoptosis of IPF myofibroblasts, an effect that is mediated, at
least in part, by upregulation of pro-apoptotic gene Bcl-2
antagonist/killer 1 and downregulation of anti- apoptotic gene
Bcl-xL | |
| | | Panobinostat | The pan-histone
deacetylase- inhibitor panobinostat decreases profibrotic
phenotypes, as well as inducing cell cycle arrest and apoptosis in
IPF fibroblasts | |
| Sosulski et
al, 2017; | Mitochondria | Hexafluoro | Increases the
expression of NAD-dependent deacetylase sirtuin-3 | (47) |
| Sato et al,
2016 | | | | (145) |
| | | Metformin | Prevents PF with
NADPH oxidase 4 inhibitors | |
| Liu et al,
2016 | ER |
4-phenylbutyrate | A chemical ER
chaperone that ameliorates ER stress, interstitial damage, collagen
deposition and apoptosis in unilateral ureteral obstruction rat
kidney | (125) |
| Romero et
al, 2016; | Autophagy | Rebamycin | Inhibits mTOR
complex 1 and activates autophagy | (72) |
| Lavieu et
al, 2006; | | | | (121) |
| Lawson et
al, 2008 | | Sphingosine
1-phosphate Berberine | Inhibits activation
of mTOR complex and increases autophagy Antagonizes TGF-β1-mediated
PIK3/Akt/mTOR signaling and increases autophagy | (122) |
| Lehmann et
al, 2017; | SASP | Dasatinib and
quercitin | Inhibit tyrosine
kinases, induce clearance of senescent cells, decrease fibrosis and
SASP production. | (149) |
| Feng et al,
2019 | | | | (151) |
| | | Citrus basic
extract | Downregulates
expression of SASP factors in etoposide- induced fibroblasts by
activating cyclooxygenase 2 | |
| Glassberg et
al, 2017; | Stem cell
transplantation | MSC intravenous
injection | Following in
vivo transplantation, MSCs home in on injured lung tissue,
release paracrine factors and extracellular vesicles, regulate
function of immune cells, decrease the local inflammatory response,
inhibit fibrous proliferation and promote endogenous lung injury
resistance. MSCs decrease bleomycin-induced lung tissue
inflammatory response, cell infiltration and cytokine expression,
extracellular matrix production and collagen deposition and improve
Ashcroft score | (159) |
| Zhao et al,
2021 | | | | (156) |
Senolytics
Therapeutic interventions for autophagy.
Rapamycin attenuates TGF-β-induced differentiation of
myofibroblasts and simultaneously activates autophagy by inhibiting
mTORC1(77). Berberine improves
BLM-induced IPF by antagonizing the TGF-β1-mediated SMAD and the
FAK-dependent PIK3/Akt-mTOR signaling pathways (120). Sphingosine 1-phosphate inhibits
activation of mTORC, increases autophagy and decreases progression
of PF (121).
Therapeutic interventions for ER stress.
Chronic ER stress-mediated apoptosis in ATII cells and macrophages
is a key pathogenic mechanism of IPF (122,123). Perera et al (124) showed that BLM increases ER stress
and apoptosis in ATII cells and lung macrophages. Moreover, this
effect is significantly ameliorated by in vivo blockade of
the calcium-activated potassium channel KCa3.1 with senicapoc, a
KCa3.1 ion channel blocker. Blocking KCA3.1 ion channels decreased
ER stress and apoptosis in ATII cells and macrophages, which may
help to reduce IPF pathology and improve lung function.
Decreasing ER stress and apoptosis in ATII cells
reverse fibrosis. Liu et al (125) showed that 4-phenylbutyrate, a
chemical ER chaperone, ameliorates ER stress, interstitial damage,
collagen deposition and apoptosis in unilateral ureteral
obstruction rat kidney. Research (126) has demonstrated reduced ER
mitochondrial tethering in in vitro experimental ER stress
and in vitro IPF ATII experiments in the presence of
decreased expression of phospholipase acidic cluster classification
protein 2 (PACS2). The levels of PACS2 are affected by its
interaction with transient receptor potential cation channel
subfamily V member 1 (TRPV1) and can be experimentally modified
using capsaicin (CPS) and recovered following CPS treatment. Thus,
therapeutic targeting of the PACS2/TRPV1 axis represents a novel
approach to epithelial protection in IPF (126). Some ER stress inhibitors target
lung cells and most experiments are in vitro (127,128). Therefore, to the best of our
knowledge, the side effects of inhibiting ER stress in treatment of
fibrosis are not mentioned in the literature. This requires further
study on the side effects of ER stress inhibitors on normal
cells.
Therapeutic interventions for telomerase
function. Drugs that target telomeres have therapeutic
potential for lung fibrosis. Le Saux et al (129) demonstrated that activation of
telomerase with the small molecule telomerase activator GRN510
resulted in attenuation of lung fibrosis in BLM-induced fibrosis
mice, showing decreased collagen deposition and loss of lung
function and protecting lung epithelial cells from senescence. The
estrogen receptor modulator raloxifene and androgens have been used
to induce telomerase activity and increase telomere length
(130). In a prospective study,
danazol, a synthetic androgen, was shown to increase telomere
length (131). Danazol was also
found to increase telomere length and stabilize forced vital
capacity and diffusing capacity of carbon monoxide in a small-scale
clinical trial (132). However,
hepatotoxicity and worsening PF associated with long-term use of
danazol has been reported following danazol initiation and
withdrawal (133). Therefore,
more studies need to be conducted for effective interventions
concerning telomerase function in lung fibrosis.
Therapeutic interventions for epigenetic
changes. In lung remodeling and repair, druggable targets
include enzymes that catalyze DNA methylation and demethylation, as
well as enzymes that catalyze post-translational modifications of
histones and non-coding RNAs (miRNAs and long non-coding RNAs)
(134). Investigating the role of
epigenetic factors in development of IPF may provide novel
therapeutic options.
DNMT inhibitor 5-aza-2'-deoxycytidine (decitabine)
enhances miR 17-92 expression and decreases expression of
profibrotic genes including collagen 1A1 and CTGF (134). Its effects are independent of DNA
methylation that enhances apoptosis. Therefore, more selective
inhibitors of DNMTs need to be studied to determine if altered DNA
methylation is involved in the pathogenesis of IPF.
Histone post-translation modifications, such as
acetylation and methylation, are key regulators of transcription
and comprise crucial components of the ‘histone code’ (135). HATs and HDAC regulate histone
acetylation. Histone lysine or arginine methyltransferase and
histone demethylase regulate histone methylation. HDAC alters
differentiation of lung myofibroblasts (136). Moreover, treatment of fibroblasts
with HDAC inhibitors increases histone H3 and histone H4
acetylation close to the promoter of the Fas gene, influences Fas
expression and recovers sensitivity to Fas-mediated apoptosis
(63,137). In fibroblasts from patients with
IPF, application of HDAC inhibitors LBH589 and suberoylanilide
hydroxamic acid (SAHA) increases expression of COX-2 to levels
similar to those observed in controls (64,138). The upregulation of the
pro-apoptotic gene Bcl-2 antagonist/killer 1 and downregulation of
the anti-apoptotic gene Bcl-xL partially mediate SAHA-induced
apoptosis of IPF myofibroblasts, indicating that HDAC inhibitors
may provide a novel therapeutic strategy in IPF by regulating
myofibroblast susceptibility to apoptosis (139). A few studies have demonstrated
that HDAC inhibitors inhibit activation and proliferation of
cultured fibroblasts and attenuate fibrosis in multiple organs in
in vivo animal models (137,139,140). The pan-HDAC-inhibitor
panobinostat decreases profibrotic phenotypes and induces cell
cycle arrest and apoptosis in IPF fibroblasts, thus indicating more
efficiency than pirfenidone in inactivating IPF fibroblasts
(140). The aforementioned study
showed that HDAC inhibitors promote apoptosis of LF, while other
treatments of IPF inhibit apoptosis of epithelial cells.
Vorinostat, a pan-HDAC inhibitor, decreases lung fibrosis by
promoting apoptosis of myofibroblasts, further improving lung
function in mouse model of lung fibrosis (141). Some preclinical evidence showed
that HDAC inhibitors have beneficial effects on IPF, for example,
two inhibitors of the euchromatic histone-lysine
N-methyltransferase 2 and enhancer of zeste homolog 2 HMTs
(BIX-01294 and 3-deazaneplanocin) increase COX-2 expression in IPF
fibroblasts, which may enhance production of PGE2, an anti-fibrotic
prostanoid (142).
As highlighted by a recent review, studies
discovered that non-coding RNAs may be a potential treatment for
IPF (60,65). Many miRNAs(miR-21, miR-133a,
miR-106b-5p) associated with lung fibrosis regulate TGF-β1
expression or function, inflammation, actin expression or cell
signaling (65,143,144). Moreover, blocking upregulated
lung ‘fibro-miRs’ or restoring downregulated ‘anti-fibro-miRs’
attenuates the lung fibrotic response (60).
Epigenetic marks are cell-type specific, yet, to
the best of our knowledge, most research in this area has involved
whole-lung tissue because isolation of sufficient material for
specific cell types is often not feasible in human subjects.
Another challenge with epigenomics is to integrate the epigenetic
mechanisms that affect transcription and translation.
Therapeutic interventions for mitochondrial
dysfunction. Metformin prevents PF via NOX4 inhibition
(145). Metformin-mediated AMPK
activation inhibits TGF-β-induced NOX4 expression. Rangarajan et
al (146) found that
metformin, as well as other AMPK activators, reverse lung fibrosis
by promoting inactivation and apoptosis of myofibroblasts due to a
lack of AMPK activation. The antifibrotic effects of normal SIRT3
expression levels in AECs and fibroblasts have potential
therapeutic applications by decreasing formation of destructive
cellular ROS and may be used in treatment of IPF. Furthermore,
hexafluoro (a novel fluorinated synthetic honokiol analogue)
maintains SIRT3 levels in LFs treated with TGF-β1 cytokines. This
compound partly decreases TGF-ß-induced mitochondrial oxidative
stress and activation of fibroblasts via SIRT3 stimulation. In
addition, hexafluoro decreases levels of profibrotic factors such
as α-SMA and fibronectin, which promote EMT in lung fibrosis
(147).
Senomorphics
Therapeutic interventions for SASP. Removal
of senescent cells decreases expression of SASP factors in
regulating fibrotic pathogenesis of IPF. Targeting SASP components
may be a feasible strategy to block detrimental functions of
senescent cells of IPF. SASP, which is composed of cytokines,
modulates the tissue microenvironment through various biological
processes, including cell proliferation and migration,
inflammation, fibrosis, ECM degradation, neovascularization and
paracrine and autocrine pathways, as well as EMT (103). To investigate if SASP regulators
decrease lung fibrosis, Justice et al (148) conducted a two-center, open-label
study of dasatinib (D) + quercetin (Q), a senolytic, in patients
with IPF to evaluate the feasibility of senolytic intervention. D
is a tyrosine kinase inhibitor, while Q is a natural product that
targets Bcl-2, insulin/insulin-like growth factor 1 and
hypoxia-inducible factor 1α SCAP network components. It selectively
eliminates senescent fibroblasts in BLM-induced fibrosis mouse
models (95,148). A previous study reported that D +
Q inhibits tyrosine kinase, induces clearance of senescent cells,
decreases fibrosis and inhibits production of SASP (149). Furthermore, D + Q eliminates the
anti-apoptosis effect of fibrotic fibroblasts induced by Fas ligand
or TNF-associated apoptosis-inducing ligand, further reversing PF
induced by BLM and decreasing mortality rate, making D + Q a
potential therapeutic target for IPF (150). Citrus basic extract downregulates
expression of SASP factors in etoposide-induced fibroblasts by
activating COX-2(151). Shentu
et al (152) used human
MSC-derived EVs (mEVs) to inhibit TGF-β1 and stimulate both normal
and IPF myofibroblast differentiation. Their findings demonstrated
anti-myofibroblastic effects of Thy-1-mediated mEV uptake and
anti-myofibroblast in IPF fibroblasts and revealed that
mEV-enriched miRs may mediate these effects.
mEVs represent a promising cell-free therapeutic
approach for an array of disorders. Human bronchial epithelial cell
(HBEC)-derived EVs contain a variety of miRNAs, including miR-16,
miR-26a, miR-26b, miR-141, miR-148a and miR-200a (153). By attenuating Wnt signaling,
these miRNAs inhibit TGF-β1-mediated induction of both
myofibroblast differentiation and lung epithelial cellular
senescence. However, this effect in HBEC EVs is more pronounced
than the effects observed in mEVs (153). Kadota et al (153) demonstrated that intratracheal
administration of HBEC EVs attenuates BLM-induced lung fibrosis. To
the best of our knowledge, however, there is no appropriate in
vitro ATII culture system to prepare sufficient quantities of
ATII EVs for injection therapy.
Stem cell transplantation
Cell therapy for lung diseases is developing
rapidly. After tissue damage, MSCs are activated and recruited to
the injured site. MSCs secrete bioactive molecules, regulate local
immune response and establish a microenvironment to promote
regeneration (154). Phase 1b
non-randomized non-placebo clinical trials involving stem cells and
a clinical study (155,156) have been performed using lung
stem/progenitor ATII cells (155,157-159).
Data from these trials support the safety of transplantation of
MSCs or ATII cells in patients with IPF of different severity
levels. Furthermore, Zhao et al (156) systematically reviewed 36
preclinical studies that used MSCs to treat BLM-induced acute lung
injury and PF in rodent models. They found that MSCs significantly
improve BLM-induced PF and suggested that MSCs could be a potential
treatment for both IPF and virus-induced PF. In addition,
intravenous injection of MSCs is feasible and safe for the
treatment of patients with moderate to severe IPF (160). It is challenging to evaluate
parameters such as tissue origin, cell type and delivery method in
large-scale human studies because these parameters determine the
effect of cell therapy. Poggio et al (161) compared intervention strategies of
MSCs administered once and twice weekly and reported that repeated
administration has more potent immunosuppressive effects on T cells
(primarily CD8+ T cells). In a murine model of
BLM-induced PF, repeated administration of MSCs (three times/3
days) had comparable antifibrotic effects to the continuous
administration of pirfenidone (161).
8. Conclusion
IPF is a progressive and fatal diffuse interstitial
lung disease that has a poor prognosis and its primary mechanism
remains unclear. Currently, there is no effective treatment for IPF
other than lung transplantation.
IPF is an aging-associated lung disease in which LF
and AEC senescence play a complex role in pathogenesis. Numerous
studies (16-20,24-28,31-34,41)
have revealed that ATII cell senescence and apoptosis are
associated with ER stress and autophagy, telomere damage,
mitochondrial dysfunction and epigenetic changes, leading to
development of PF. The activation of LF and deposition of ECM
proteins are key steps in the development of IPF. Epigenetic
changes and reduced activation of autophagy promote myofibroblast
differentiation, ultimately leading to PF. Aging AECs promote LF
activation by increasing expression of SASP, thereby increasing
occurrence and development of PF. In short, cell senescence is an
important mechanism of IPF pathogenesis (5,9,54).
AECs (primarily ATII cells) and LF have therapeutic
pathways and the same pathogenic pathways in the process of
mediating PF. These pathways may act as potential new therapeutic
targets for PF in future. More efforts on clinical testing need to
be performed to validate promising therapeutic compounds.
Over the past decades, scientists have explored the
mechanisms that lead to aging, damage and fibrosis of lung cells
and have yielded useful insights (3,5,6,9,12,15,19,53,95).
However, the exact mechanism of cellular senescence remains unclear
and requires further research. Aging affects not only different
types of cell in the lungs but also cells in other organs. The
treatment of lung cell senescence is being actively explored, but
whether the molecular mechanism involved in lung cell senescence
therapy adversely affects normal and aging cells of extrapulmonary
organs remains unclear. It is difficult to treat a specific
pathogenic target accurately, because the expectation of this
treatment is to promote the apoptosis of lung senescent cells and
the proliferation of normal lung cells. It is worth exploring
whether targeted therapy drugs can be developed that accurately
target disease-causing senescent cells in the lung. Stem cell
transplantation has been a hot research topic in recent years.
Existing research has advanced in mouse models of BLM-induced PF
and it may provide new ideas for the treatment of IPF. Novel
therapies targeting cellular senescence may provide new treatment
strategies for IPF and improve survival and quality of life for
patients with IPF.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by the Natural Science
Foundation of Jilin Science and Technology Department (grant no.
20160101089JC) and the Health and Family Planning Research Project
of Jilin Province (grant no. 20152020).
Availability of data and materials
Not applicable.
Authors' contributions
SH and QL both wrote the manuscript. SH, QL and XL
revised the manuscript. SH and QL contributed equally to this work
and should be considered co-first authors. Data authentication is
not applicable. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Rennard SI, Bitterman PB and Crystal RG:
Response of the lower respiratory tract to injury. Mechanisms of
repair of the parenchymal cells of the alveolar wall. Chest.
84:735–739. 1983.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Tian Y, Li H, Qiu T, Dai J, Zhang Y, Chen
J and Cai H: Loss of PTEN induces lung fibrosis via alveolar
epithelial cell senescence depending on NF-κB activation. Aging
Cell. 18(e12858)2019.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Selman M, King TE and Pardo A: Idiopathic
pulmonary fibrosis: Prevailing and evolving hypotheses about its
pathogenesis and implications for therapy. Ann Intern Med.
134:136–151. 2001.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Iwai K, Mori T, Yamada N, Yamaguchi M and
Hosoda Y: Idiopathic pulmonary fibrosis. Epidemiologic approaches
to occupational exposure. Am J Respir Crit Care Med. 150:670–675.
1994.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Lin Y and Xu Z: Fibroblast senescence in
idiopathic pulmonary fibrosis. Front Cell Dev Biol.
8(593283)2020.PubMed/NCBI View Article : Google Scholar
|
|
6
|
King TJ, Pardo A and Selman M: Idiopathic
pulmonary fibrosis. Lancet. 378:1949–1961. 2011.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Rana T, Jiang C, Liu G, Miyata T, Antony
V, Thannickal VJ and Liu RM: PAI-1 regulation of TGF-β1-induced
alveolar type II cell senescence, SASP secretion, and SASP-mediated
activation of alveolar macrophages. Am J Respir Cell Mol Biol.
62:319–330. 2020.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Tashiro J, Rubio GA, Limper AH, Williams
K, Elliot SJ, Ninou I, Aidinis V, Tzouvelekis A and Glassberg MK:
Exploring animal models that resemble idiopathic pulmonary
fibrosis. Front Med (Lausanne). 4(118)2017.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Liu RM and Liu G: Cell senescence and
fibrotic lung diseases. Exp Gerontol. 132(110836)2020.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Mohamad KN, Safuan S, Shamsuddin S and
Foroozandeh P: Aging of the cells: Insight into cellular senescence
and detection Methods. Eur J Cell Biol. 99(151108)2020.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Coppé JP, Desprez PY, Krtolica A and
Campisi J: The senescence-associated secretory phenotype: The dark
side of tumor suppression. Annu Rev Pathol. 5:99–118.
2010.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Kumari R and Jat P: Mechanisms of cellular
senescence: Cell cycle arrest and senescence associated secretory
phenotype. Front Cell Dev Biol. 9(645593)2021.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Sims JT, Ganguly SS, Bennett H, Friend JW,
Tepe J and Plattner R: Imatinib reverses doxorubicin resistance by
affecting activation of STAT3-dependent NF-κB and HSP27/p38/AKT
pathways and by inhibiting ABCB1. PLoS One.
8(e55509)2013.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Lagger G, Doetzlhofer A, Schuettengruber
B, Haidweger E, Simboeck E, Tischler J, Chiocca S, Suske G,
Rotheneder H, Wintersberger E and Seiser C: The tumor suppressor
p53 and histone deacetylase 1 are antagonistic regulators of the
cyclin-dependent kinase inhibitor p21/WAF1/CIP1 gene. Mol Cell
Biol. 23:2669–2679. 2003.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Parimon T, Yao C, Stripp BR, Noble PW and
Chen P: Alveolar epithelial type II cells as drivers of lung
fibrosis in idiopathic pulmonary fibrosis. Int J Mol Sci.
21(2269)2020.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Tanjore H, Blackwell TS and Lawson WE:
Emerging evidence for endoplasmic reticulum stress in the
pathogenesis of idiopathic pulmonary fibrosis. Am J Physiol Lung
Cell Mol Physiol. 302:L721–L729. 2012.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Borok Z, Horie M, Flodby P, Wang H, Liu Y,
Ganesh S, Firth AL, Minoo P, Li C, Beers MF, et al: Grp78 loss in
epithelial progenitors reveals an age-linked role for endoplasmic
reticulum stress in pulmonary fibrosis. Am J Respir Crit Care Med.
201:198–211. 2020.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Araya J, Kojima J, Takasaka N, Ito S,
Fujii S, Hara H, Yanagisawa H, Kobayashi K, Tsurushige C, Kawaishi
M, et al: Insufficient autophagy in idiopathic pulmonary fibrosis.
Am J Physiol Lung Cell Mol Physiol. 304:L56–L69. 2013.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Hill C, Li J, Liu D, Conforti F, Brereton
CJ, Yao L, Zhou Y, Alzetani A, Chee SJ, Marshall BG, et al:
Autophagy inhibition-mediated epithelial-mesenchymal transition
augments local myofibroblast differentiation in pulmonary fibrosis.
Cell Death Dis. 10(591)2019.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Patel AS, Lin L, Geyer A, Haspel JA, An
CH, Cao J, Rosas IO and Morse D: Autophagy in idiopathic pulmonary
fibrosis. PLoS One. 7(e41394)2012.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Armanios MY, Chen JJ, Cogan JD, Alder JK,
Ingersoll RG, Markin C, Lawson WE, Xie M, Vulto I, Phillips JA, et
al: Telomerase mutations in families with idiopathic pulmonary
fibrosis. N Engl J Med. 356:1317–1326. 2007.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Alder JK, Chen JJ, Lancaster L, Danoff S,
Su SC, Cogan JD, Vulto I, Xie M, Qi X, Tuder RM, et al: Short
telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc
Natl Acad Sci USA. 105:13051–13056. 2008.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Armanios M: Telomeres and age-related
disease: How telomere biology informs clinical paradigms. J Clin
Invest. 123:996–1002. 2013.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Kurundkar A and Thannickal VJ: Redox
mechanisms in age-related lung fibrosis. Redox Biol. 9:67–76.
2016.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Kim SJ, Cheresh P, Eren M, Jablonski RP,
Yeldandi A, Ridge KM, Budinger GRS, Kim DH, Wolf M, Vaughan DE and
Kamp DW: Klotho, an antiaging molecule, attenuates oxidant-induced
alveolar epithelial cell mtDNA damage and apoptosis. Am J Physiol
Lung Cell Mol Physiol. 313:L16–L26. 2017.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Disayabutr S, Kim EK, Cha SI, Green G,
Naikawadi RP, Jones KD, Golden JA, Schroeder A, Matthay MA, Kukreja
J, et al: miR-34 miRNAs regulate cellular senescence in type II
alveolar epithelial cells of patients with idiopathic pulmonary
fibrosis. PLoS One. 11(e158367)2016.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Cui H, Ge J, Xie N, Banerjee S, Zhou Y,
Liu RM, Thannickal VJ and Liu G: miR-34a promotes fibrosis in aged
lungs by inducing alveolarepithelial dysfunctions. Am J Physiol
Lung Cell Mol Physiol. 312:L415–L424. 2017.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Wang L, Cheng W and Zhang Z: Respiratory
syncytial virus infection accelerates lung fibrosis through the
unfolded protein response in a bleomycin-induced pulmonary fibrosis
animal model. Mol Med Rep. 16:310–316. 2017.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Pihán P, Carreras-Sureda A and Hetz C:
BCL-2 family: Integrating stress responses at the ER to control
cell demise. Cell Death Differ. 24:1478–1487. 2017.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Burman A, Tanjore H and Blackwell TS:
Endoplasmic reticulum stress in pulmonary fibrosis. Matrix Biol.
68-69:355–365. 2018.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Bueno M, Lai YC, Romero Y, Brands J, St
Croix CM, Kamga C, Corey C, Herazo-Maya JD, Sembrat J, Lee JS, et
al: PINK1 deficiency impairs mitochondrial homeostasis and promotes
lung fibrosis. J Clin Invest. 125:521–538. 2015.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Ricci A, Cherubini E, Scozzi D,
Pietrangeli V, Tabbì L, Raffa S, Leone L, Visco V, Torrisi MR,
Bruno P, et al: Decreased expression of autophagic beclin 1 protein
in idiopathic pulmonary fibrosis fibroblasts. J Cell Physiol.
228:1516–1524. 2013.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Gui YS, Wang L, Tian X, Li X, Ma A, Zhou
W, Zeng N, Zhang J, Cai B, Zhang H, et al: mTOR Overactivation and
compromised autophagy in the pathogenesis of pulmonary fibrosis.
PLoS One. 10(e138625)2015.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Kesireddy VS, Chillappagari S, Ahuja S,
Knudsen L, Henneke I, Graumann J, Meiners S, Ochs M, Ruppert C,
Korfei M, et al: Susceptibility of microtubule-associated protein 1
light chain 3β (MAP1LC3B/LC3B) knockout mice to lung injury and
fibrosis. FASEB J. 33:12392–12408. 2019.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Ma H, Wu X, Li Y and Xia Y: Research
progress in the molecular mechanisms, therapeutic targets, and drug
development of idiopathic pulmonary fibrosis. Front Pharmacol.
13(963054)2022.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Tsang AR, Wyatt HD, Ting NS and Beattie
TL: hTERT mutations associated with idiopathic pulmonary fibrosis
affect telomerase activity, telomere length, and cell growth by
distinct mechanisms. Aging Cell. 11:482–490. 2012.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Tsakiri KD, Cronkhite JT, Kuan PJ, Xing C,
Raghu G, Weissler JC, Rosenblatt RL, Shay JW and Garcia CK:
Adult-onset pulmonary fibrosis caused by mutations in telomerase.
Proc Natl Acad Sci USA. 104:7552–7557. 2007.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Bilgili H, Białas AJ, Górski P and
Piotrowski WJ: Telomere Abnormalities in the Pathobiology of
Idiopathic Pulmonary Fibrosis. J Clin Med. 8(1232)2019.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Povedano JM, Martinez P, Flores JM, Mulero
F and Blasco MA: Mice with pulmonary fibrosis driven by telomere
dysfunction. Cell Rep. 12:286–299. 2015.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Alder JK, Barkauskas CE, Limjunyawong N,
Stanley SE, Kembou F, Tuder RM, Hogan BL, Mitzner W and Armanios M:
Telomere dysfunction causes alveolar stem cell failure. Proc Natl
Acad Sci USA. 112:5099–5104. 2015.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Zhang K, Xu L and Cong YS: Telomere
dysfunction in idiopathic pulmonary fibrosis. Front Med (Lausanne).
8(739810)2021.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Sahin E, Colla S, Liesa M, Moslehi J,
Müller FL, Guo M, Cooper M, Kotton D, Fabian AJ, Walkey C, et al:
Telomere dysfunction induces metabolic and mitochondrial
compromise. Nature. 470:359–365. 2011.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Tsubouchi K, Araya J and Kuwano K:
PINK1-PARK2-mediated mitophagy in COPD and IPF pathogeneses.
Inflamm Regen. 38(18)2018.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Mora AL, Bueno M and Rojas M: Mitochondria
in the spotlight of aging and idiopathic pulmonary fibrosis. J Clin
Invest. 127:405–414. 2017.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Braidy N, Guillemin GJ, Mansour H,
Chan-Ling T, Poljak A and Grant R: Age related changes in NAD+
metabolism oxidative stress and Sirt1 activity in wistar rats. PLoS
One. 6(e19194)2011.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Kwon Y, Kim J, Lee CY and Kim H:
Expression of SIRT1 and SIRT3 varies according to age in mice. Anat
Cell Biol. 48:54–61. 2015.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Sosulski ML, Gongora R, Feghali-Bostwick
C, Lasky JA and Sanchez CG: Sirtuin 3 deregulation promotes
pulmonary fibrosis. J Gerontol A Biol Sci Med Sci. 72:595–602.
2017.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Amara N, Goven D, Prost F, Muloway R,
Crestani B and Boczkowski J: NOX4/NADPH oxidase expression is
increased in pulmonary fibroblasts from patients with idiopathic
pulmonary fibrosis and mediates TGFbeta1-induced fibroblast
differentiation into myofibroblasts. Thorax. 65:733–738.
2010.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Veith C, Boots AW, Idris M, van Schooten
FJ and van der Vliet A: Redox imbalance in idiopathic pulmonary
fibrosis: A role for oxidant Cross-talk between NADPH oxidase
enzymes and mitochondria. Antioxid Redox Signal. 31:1092–1115.
2019.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Zhang Y, Li T, Pan M, Wang W, Huang W,
Yuan Y, Xie Z, Chen Y, Peng J, Li X and Meng Y: SIRT1 prevents
cigarette smoking-induced lung fibroblasts activation by regulating
mitochondrial oxidative stress and lipid metabolism. J Transl Med.
20(222)2022.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Birch J, Barnes PJ and Passos JF:
Mitochondria, telomeres and cell senescence: Implications for lung
ageing and disease. Pharmacol Ther. 183:34–49. 2018.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Shetty SK, Tiwari N, Marudamuthu AS,
Puthusseri B, Bhandary YP, Fu J, Levin J, Idell S and Shetty S: p53
and miR-34a feedback promotes lung epithelial injury and pulmonary
fibrosis. Am J Pathol. 187:1016–1034. 2017.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Chanda D, Otoupalova E, Smith SR,
Volckaert T, De Langhe SP and Thannickal VJ: Developmental pathways
in the pathogenesis of lung fibrosis. Mol Aspects Med. 65:56–69.
2019.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Moss BJ, Ryter SW and Rosas IO: Pathogenic
mechanisms underlying idiopathic pulmonary fibrosis. Annu Rev
Pathol. 17:515–546. 2022.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Poddar S, Kesharwani D and Datta M:
Interplay between the miRNome and the epigenetic machinery:
Implications in health and disease. J Cell Physiol. 232:2938–2945.
2017.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Jones PA: Functions of DNA methylation:
Islands, start sites, gene bodies and beyond. Nat Rev Genet.
13:484–492. 2012.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Neveu WA, Mills ST, Staitieh BS and
Sueblinvong V: TGF-β1 epigenetically modifies Thy-1 expression in
primary lung fibroblasts. Am J Physiol Cell Physiol. 309:C616–C626.
2015.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Rabinovich EI, Kapetanaki MG, Steinfeld I,
Gibson KF, Pandit KV, Yu G, Yakhini Z and Kaminski N: Global
methylation patterns in idiopathic pulmonary fibrosis. PLoS One.
7(e33770)2012.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Huang SK, Scruggs AM, McEachin RC, White
ES and Peters-Golden M: Lung fibroblasts from patients with
idiopathic pulmonary fibrosis exhibit genome-wide differences in
DNA methylation compared to fibroblasts from nonfibrotic lung. PLoS
One. 9(e107055)2014.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Bartczak K, Białas AJ, Kotecki MJ, Górski
P and Piotrowski WJ: More than a genetic code: Epigenetics of lung
fibrosis. Mol Diagn Ther. 24:665–681. 2020.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Roth SY, Denu JM and Allis CD: Histone
acetyltransferases. Annu Rev Biochem. 70:81–120. 2001.PubMed/NCBI View Article : Google Scholar
|
|
62
|
de Ruijter AJ, van Gennip AH, Caron HN,
Kemp S and van Kuilenburg AB: Histone deacetylases (HDACs):
characterization of the classical HDAC family. Biochem J.
370:737–749. 2003.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Huang SK, Scruggs AM, Donaghy J, Horowitz
JC, Zaslona Z, Przybranowski S, White ES and Peters-Golden M:
Histone modifications are responsible for decreased Fas expression
and apoptosis resistance in fibrotic lung fibroblasts. Cell Death
Dis. 4(e621)2013.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Coward WR, Watts K, Feghali-Bostwick CA,
Knox A and Pang L: Defective histone acetylation is responsible for
the diminished expression of cyclooxygenase 2 in idiopathic
pulmonary fibrosis. Mol Cell Biol. 29:4325–4339. 2009.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Liu G, Friggeri A, Yang Y, Milosevic J,
Ding Q, Thannickal VJ, Kaminski N and Abraham E: miR-21 mediates
fibrogenic activation of pulmonary fibroblasts and lung fibrosis. J
Exp Med. 207:1589–1597. 2010.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Bahudhanapati H, Tan J, Dutta JA, Strock
SB, Sembrat J, Àlvarez D, Rojas M, Jäger B, Prasse A, Zhang Y and
Kass DJ: MicroRNA-144-3p targets relaxin/insulin-like family
peptide receptor 1 (RXFP1) expression in lung fibroblasts from
patients with idiopathic pulmonary fibrosis. J Biol Chem.
294:5008–5022. 2019.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Kuwano K, Araya J, Hara H, Minagawa S,
Takasaka N, Ito S, Kobayashi K and Nakayama K: Cellular senescence
and autophagy in the pathogenesis of chronic obstructive pulmonary
disease (COPD) and idiopathic pulmonary fibrosis (IPF). Respir
Investig. 54:397–406. 2016.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Itakura E, Kishi C, Inoue K and Mizushima
N: Beclin 1 forms two distinct phosphatidylinositol 3-kinase
complexes with mammalian Atg14 and UVRAG. Mol Biol Cell.
19:5360–5372. 2008.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Ryter SW, Bhatia D and Choi ME: Autophagy:
A lysosome-dependent process with implications in cellular redox
homeostasis and human disease. Antioxid Redox Signal. 30:138–159.
2019.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Shivshankar P, Brampton C, Miyasato S,
Kasper M, Thannickal VJ and Le Saux CJ: Caveolin-1 deficiency
protects from pulmonary fibrosis by modulating epithelial cell
senescence in mice. Am J Respir Cell Mol Biol. 47:28–36.
2012.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Lawrence J and Nho R: The role of the
mammalian target of Rapamycin (mTOR) in pulmonary fibrosis. Int J
Mol Sci. 19(778)2018.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Romero Y, Bueno M, Ramirez R, Álvarez D,
Sembrat JC, Goncharova EA, Rojas M, Selman M, Mora AL and Pardo A:
mTORC1 activation decreases autophagy in aging and idiopathic
pulmonary fibrosis and contributes to apoptosis resistance in IPF
fibroblasts. Aging Cell. 15:1103–1112. 2016.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Nho RS and Hergert P: IPF fibroblasts are
desensitized to type I collagen matrix-induced cell death by
suppressing low autophagy via aberrant Akt/mTOR kinases. PLoS One.
9(e94616)2014.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Zhao H, Wang Y, Qiu T, Liu W and Yao P:
Autophagy, an important therapeutic target for pulmonary fibrosis
diseases. Clin Chim Acta. 502:139–147. 2020.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Liu T, Ullenbruch M, Young CY, Yu H, Ding
L, Xaubet A, Pereda J, Feghali-Bostwick CA, Bitterman PB, Henke CA,
et al: Telomerase and telomere length in pulmonary fibrosis. Am J
Respir Cell Mol Biol. 49:260–268. 2013.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Nozaki Y, Liu T, Hatano K, Gharaee-Kermani
M and Phan SH: Induction of telomerase activity in fibroblasts from
bleomycin-injured lungs. Am J Respir Cell Mol Biol. 23:460–465.
2000.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Schuliga M, Pechkovsky DV, Read J, Waters
DW, Blokland KEC, Reid AT, Hogaboam CM, Khalil N, Burgess JK, Prêle
CM, et al: Mitochondrial dysfunction contributes to the senescent
phenotype of IPF lung fibroblasts. J Cell Mol Med. 22:5847–5861.
2018.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Banerjee ER, Laflamme MA, Papayannopoulou
T, Kahn M, Murry CE and Henderson WJ: Human embryonic stem cells
differentiated to lung lineage-specific cells ameliorate pulmonary
fibrosis in a xenograft transplant mouse model. PLoS One.
7(e33165)2012.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Li DY, Li RF, Sun DX, Pu DD and Zhang YH:
Mesenchymal stem cell therapy in pulmonary fibrosis: A
meta-analysis of preclinical studies. Stem Cell Res Ther.
12(461)2021.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Cai SX, Liu AR, Chen S, He HL, Chen QH, Xu
JY, Pan C, Yang Y, Guo FM, Huang YZ, et al: Activation of
Wnt/β-catenin signalling promotes mesenchymal stem cells to repair
injured alveolar epithelium induced by lipopolysaccharide in mice.
Stem Cell Res Ther. 6(65)2015.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Liu M, Ren D, Wu D, Zheng J and Tu W: Stem
cell and idiopathic pulmonary fibrosis: Mechanisms and treatment.
Curr Stem Cell Res Ther. 10:466–476. 2015.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Barkauskas CE, Cronce MJ, Rackley CR,
Bowie EJ, Keene DR, Stripp BR, Randell SH, Noble PW and Hogan BL:
Type 2 alveolar cells are stem cells in adult lung. J Clin Invest.
123:3025–3036. 2013.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Wu H, Yu Y, Huang H, Hu Y, Fu S, Wang Z,
Shi M, Zhao X, Yuan J, Li J, et al: Progressive pulmonary fibrosis
is caused by elevated mechanical tension on alveolar stem cells.
Cell. 180:107–121.e17. 2020.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Kobayashi Y, Tata A, Konkimalla A, Katsura
H, Lee RF, Ou J, Banovich NE, Kropski JA and Tata PR: Persistence
of a regeneration-associated, transitional alveolar epithelial cell
state in pulmonary fibrosis. Nat Cell Biol. 22:934–946.
2020.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Jarvinen L, Badri L, Wettlaufer S, Ohtsuka
T, Standiford TJ, Toews GB, Pinsky DJ, Peters-Golden M and Lama VN:
Lung resident mesenchymal stem cells isolated from human lung
allografts inhibit T cell proliferation via a soluble mediator. J
Immunol. 181:4389–4396. 2008.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Gao J, Dennis JE, Muzic RF, Lundberg M and
Caplan AI: The dynamic in vivo distribution of bone marrow-derived
mesenchymal stem cells after infusion. Cells Tissues Organs.
169:12–20. 2001.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Toonkel RL, Hare JM, Matthay MA and
Glassberg MK: Mesenchymal stem cells and idiopathic pulmonary
fibrosis. Potential for clinical testing. Am J Respir Crit Care
Med. 188:133–140. 2013.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Álvarez D, Levine M and Rojas M:
Regenerative medicine in the treatment of idiopathic pulmonary
fibrosis: Current position. Stem Cells Cloning. 8:61–65.
2015.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Cárdenes N, Álvarez D, Sellarés J, Peng Y,
Corey C, Wecht S, Nouraie SM, Shanker S, Sembrat J, Bueno M, et al:
Senescence of bone marrow-derived mesenchymal stem cells from
patients with idiopathic pulmonary fibrosis. Stem Cell Res Ther.
9(257)2018.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Lee SH, Jang AS, Kim YE, Cha JY, Kim TH,
Jung S, Park SK, Lee YK, Won JH, Kim YH and Park CS: Modulation of
cytokine and nitric oxide by mesenchymal stem cell transfer in lung
injury/fibrosis. Respir Res. 11(16)2010.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Margaritopoulos GA, Giannarakis I,
Siafakas NM and Antoniou KM: An update on idiopathic pulmonary
fibrosis. Panminerva Med. 55:109–120. 2013.PubMed/NCBI
|
|
92
|
van Deursen JM: The role of senescent
cells in ageing. Nature. 509:439–446. 2014.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Blokland K, Waters DW, Schuliga M, Read J,
Pouwels SD, Grainge CL, Jaffar J, Westall G, Mutsaers SE, Prêle CM,
et al: Senescence of IPF lung fibroblasts disrupt alveolar
epithelial cell proliferation and promote migration in wound
healing. Pharmaceutics. 12(389)2020.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Zhang LM, Zhang J, Zhang Y, Fei C, Wang L,
Yi ZW and Zhang ZQ: Interleukin-18 promotes fibroblast senescence
in pulmonary fibrosis through down-regulating Klotho expression.
Biomed Pharmacother. 113(108756)2019.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Schafer MJ, White TA, Iijima K, Haak AJ,
Ligresti G, Atkinson EJ, Oberg AL, Birch J, Salmonowicz H, Zhu Y,
et al: Cellular senescence mediates fibrotic pulmonary disease. Nat
Commun. 8(14532)2017.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Muñoz-Espín D and Serrano M: Cellular
senescence: From physiology to pathology. Nat Rev Mol Cell Biol.
15:482–496. 2014.PubMed/NCBI View Article : Google Scholar
|
|
97
|
Bascands JL and Schanstra JP: Obstructive
nephropathy: Insights from genetically engineered animals. Kidney
Int. 68:925–937. 2005.PubMed/NCBI View Article : Google Scholar
|
|
98
|
Adnot S, Breau M and Houssaini A: PAI-1: A
new target for controlling lung-cell senescence and fibrosis? Am J
Respir Cell Mol Biol. 62:271–272. 2020.PubMed/NCBI View Article : Google Scholar
|
|
99
|
Jiang C, Liu G, Luckhardt T, Antony V,
Zhou Y, Carter AB, Thannickal VJ and Liu RM: Serpine 1 induces
alveolar type II cell senescence through activating p53-p21-Rb
pathway in fibrotic lung disease. Aging Cell. 16:1114–1124.
2017.PubMed/NCBI View Article : Google Scholar
|
|
100
|
Ueno M, Maeno T, Nomura M, Aoyagi-Ikeda K,
Matsui H, Hara K, Tanaka T, Iso T, Suga T and Kurabayashi M:
Hypoxia-inducible factor-1α mediates TGF-β-induced PAI-1 production
in alveolar macrophages in pulmonary fibrosis. Am J Physiol Lung
Cell Mol Physiol. 300:L740–L752. 2011.PubMed/NCBI View Article : Google Scholar
|
|
101
|
Goliwas KF and Deshane JS: Extracellular
vesicles: Bidirectional accelerators of cellular senescence in
fibrosis? Am J Respir Cell Mol Biol. 63:547–548. 2020.PubMed/NCBI View Article : Google Scholar
|
|
102
|
Yáñez-Mó M, Siljander PR, Andreu Z, Zavec
AB, Borràs FE, Buzas EI, Buzas K, Casal E, Cappello F, Carvalho J,
et al: Biological properties of extracellular vesicles and their
physiological functions. J Extracell Vesicles.
4(27066)2015.PubMed/NCBI View Article : Google Scholar
|
|
103
|
Kuwano K, Kunitake R, Kawasaki M, Nomoto
Y, Hagimoto N, Nakanishi Y and Hara N: P21Waf1/Cip1/Sdi1 and p53
expression in association with DNA strand breaks in idiopathic
pulmonary fibrosis. Am J Respir Crit Care Med. 154:477–483.
1996.PubMed/NCBI View Article : Google Scholar
|
|
104
|
Chilosi M, Carloni A, Rossi A and Poletti
V: Premature lung aging and cellular senescence in the pathogenesis
of idiopathic pulmonary fibrosis and COPD/emphysema. Transl Res.
162:156–173. 2013.PubMed/NCBI View Article : Google Scholar
|
|
105
|
Alimbetov D, Davis T, Brook AJ, Cox LS,
Faragher RG, Nurgozhin T, Zhumadilov Z and Kipling D: Suppression
of the senescence-associated secretory phenotype (SASP) in human
fibroblasts using small molecule inhibitors of p38 MAP kinase and
MK2. Biogerontology. 17:305–315. 2016.PubMed/NCBI View Article : Google Scholar
|
|
106
|
Willis BC and Borok Z: TGF-beta-induced
EMT: Mechanisms and implications for fibrotic lung disease. Am J
Physiol Lung Cell Mol Physiol. 293:L525–L534. 2007.PubMed/NCBI View Article : Google Scholar
|
|
107
|
He W, Tan R, Dai C, Li Y, Wang D, Hao S,
Kahn M and Liu Y: Plasminogen activator inhibitor-1 is a
transcriptional target of the canonical pathway of Wnt/beta-catenin
signaling. J Biol Chem. 285:24665–24675. 2010.PubMed/NCBI View Article : Google Scholar
|
|
108
|
Liu J, Peng D, You J, Zhou O, Qiu H, Hao
C, Chen H, Fu Z and Zou L: Type 2 alveolar epithelial cells
differentiated from human umbilical cord mesenchymal stem cells
alleviate mouse pulmonary fibrosis through β-catenin-regulated cell
apoptosis. Stem Cells Dev. 30:660–670. 2021.PubMed/NCBI View Article : Google Scholar
|
|
109
|
Kadota T, Yoshioka Y, Fujita Y, Araya J,
Minagawa S, Hara H, Miyamoto A, Suzuki S, Fujimori S, Kohno T, et
al: Extracellular vesicles from fibroblasts induce epithelial-cell
senescence in pulmonary fibrosis. Am J Respir Cell Mol Biol.
63:623–636. 2020.PubMed/NCBI View Article : Google Scholar
|
|
110
|
Yin Y, Chen H, Wang Y, Zhang L and Wang X:
Roles of extracellular vesicles in the aging microenvironment and
age-related diseases. J Extracell Vesicles.
10(e12154)2021.PubMed/NCBI View Article : Google Scholar
|
|
111
|
Wang L, Chen R, Li G, Wang Z, Liu J, Liang
Y and Liu JP: FBW7 mediates senescence and pulmonary fibrosis
through telomere uncapping. Cell Metab. 32:860–877.e9.
2020.PubMed/NCBI View Article : Google Scholar
|
|
112
|
Xu Y, Mizuno T, Sridharan A, Du Y, Guo M,
Tang J, Wikenheiser-Brokamp KA, Perl AT, Funari VA, Gokey JJ, et
al: Single-cell RNA sequencing identifies diverse roles of
epithelial cells in idiopathic pulmonary fibrosis. JCI Insight.
1(e90558)2016.PubMed/NCBI View Article : Google Scholar
|
|
113
|
Shenderov K, Collins SL, Powell JD and
Horton MR: Immune dysregulation as a driver of idiopathic pulmonary
fibrosis. J Clin Invest. 131(e143226)2021.PubMed/NCBI View Article : Google Scholar
|
|
114
|
Ying H, Fang M, Hang QQ, Chen Y, Qian X
and Chen M: Pirfenidone modulates macrophage polarization and
ameliorates radiation-induced lung fibrosis by inhibiting the
TGF-β1/Smad3 pathway. J Cell Mol Med. 25:8662–8675. 2021.PubMed/NCBI View Article : Google Scholar
|
|
115
|
Spagnolo P, Kropski JA, Jones MG, Lee JS,
Rossi G, Karampitsakos T, Maher TM, Tzouvelekis A and Ryerson CJ:
Idiopathic pulmonary fibrosis: Disease mechanisms and drug
development. Pharmacol Ther. 222(107798)2021.PubMed/NCBI View Article : Google Scholar
|
|
116
|
Wang J, Hu K, Cai X, Yang B, He Q, Wang J
and Weng Q: Targeting PI3K/AKT signaling for treatment of
idiopathic pulmonary fibrosis. Acta Pharm Sin B. 12:18–32.
2022.PubMed/NCBI View Article : Google Scholar
|
|
117
|
Birch J and Gil J: Senescence and the
SASP: Many therapeutic avenues. Genes Dev. 34:1565–1576.
2020.PubMed/NCBI View Article : Google Scholar
|
|
118
|
Veret D and Brondello JM: Senotherapy:
Advances and new clinical perspectives. Med Sci (Paris).
36:1135–1142. 2020.PubMed/NCBI View Article : Google Scholar : (In French).
|
|
119
|
Liao Z, Yeo HL, Wong SW and Zhao Y:
Cellular Senescence: Mechanisms and Therapeutic Potential.
Biomedicines. 9(1769)2021.PubMed/NCBI View Article : Google Scholar
|
|
120
|
Chitra P, Saiprasad G, Manikandan R and
Sudhandiran G: Berberine inhibits Smad and non-Smad signaling
cascades and enhances autophagy against pulmonary fibrosis. J Mol
Med (Berl). 93:1015–1031. 2015.PubMed/NCBI View Article : Google Scholar
|
|
121
|
Lavieu G, Scarlatti F, Sala G, Carpentier
S, Levade T, Ghidoni R, Botti J and Codogno P: Regulation of
autophagy by sphingosine kinase 1 and its role in cell survival
during nutrient starvation. J Biol Chem. 281:8518–8527.
2006.PubMed/NCBI View Article : Google Scholar
|
|
122
|
Lawson WE, Crossno PF, Polosukhin VV,
Roldan J, Cheng DS, Lane KB, Blackwell TR, Xu C, Markin C, Ware LB,
et al: Endoplasmic reticulum stress in alveolar epithelial cells is
prominent in IPF: Association with altered surfactant protein
processing and herpesvirus infection. Am J Physiol Lung Cell Mol
Physiol. 294:L1119–L1126. 2008.PubMed/NCBI View Article : Google Scholar
|
|
123
|
Korfei M, Ruppert C, Mahavadi P, Henneke
I, Markart P, Koch M, Lang G, Fink L, Bohle RM, Seeger W, et al:
Epithelial endoplasmic reticulum stress and apoptosis in sporadic
idiopathic pulmonary fibrosis. Am J Respir Crit Care Med.
178:838–846. 2008.PubMed/NCBI View Article : Google Scholar
|
|
124
|
Perera UE, Organ L, Dewage S, Derseh HB,
Stent A and Snibson KJ: Increased levels of ER stress and apoptosis
in a sheep model for pulmonary fibrosis are alleviated by in vivo
blockade of the KCa3.1 Ion Channel. Can Respir J.
2021(6683195)2021.PubMed/NCBI View Article : Google Scholar
|
|
125
|
Liu SH, Yang CC, Chan DC, Wu CT, Chen LP,
Huang JW, Hung KY and Chiang CK: Chemical chaperon 4-phenylbutyrate
protects against the endoplasmic reticulum stress-mediated renal
fibrosis in vivo and in vitro. Oncotarget. 7:22116–22127.
2016.PubMed/NCBI View Article : Google Scholar
|
|
126
|
Knoell J, Chillappagari S, Knudsen L,
Korfei M, Dartsch R, Jonigk D, Kuehnel MP, Hoetzenecker K, Guenther
A and Mahavadi P: PACS2-TRPV1 axis is required for ER-mitochondrial
tethering during ER stress and lung fibrosis. Cell Mol Life Sci.
79(151)2022.PubMed/NCBI View Article : Google Scholar
|
|
127
|
Pao HP, Liao WI, Tang SE, Wu SY, Huang KL
and Chu SJ: Suppression of endoplasmic reticulum stress by 4-PBA
protects against hyperoxia-induced acute lung injury via
Up-regulating Claudin-4 expression. Front Immunol.
12(674316)2021.PubMed/NCBI View Article : Google Scholar
|
|
128
|
Qin X, Lin X, Liu L, Li Y, Li X, Deng Z,
Chen H, Chen H, Niu Z, Li Z and Hu Y: Macrophage-derived exosomes
mediate silica-induced pulmonary fibrosis by activating fibroblast
in an endoplasmic reticulum stress-dependent manner. J Cell Mol
Med. 25:4466–4477. 2021.PubMed/NCBI View Article : Google Scholar
|
|
129
|
Le Saux CJ, Davy P, Brampton C, Ahuja SS,
Fauce S, Shivshankar P, Nguyen H, Ramaseshan M, Tressler R, Pirot
Z, et al: A novel telomerase activator suppresses lung damage in a
murine model of idiopathic pulmonary fibrosis. PLoS One.
8(e58423)2013.PubMed/NCBI View Article : Google Scholar
|
|
130
|
Calado RT, Yewdell WT, Wilkerson KL, Regal
JA, Kajigaya S, Stratakis CA and Young NS: Sex hormones, acting on
the TERT gene, increase telomerase activity in human primary
hematopoietic cells. Blood. 114:2236–2243. 2009.PubMed/NCBI View Article : Google Scholar
|
|
131
|
Arish N, Petukhov D and Wallach-Dayan SB:
The role of telomerase and telomeres in interstitial lung diseases:
From molecules to clinical implications. Int J Mol Sci.
20(2996)2019.PubMed/NCBI View Article : Google Scholar
|
|
132
|
Townsley DM, Dumitriu B, Liu D, Biancotto
A, Weinstein B, Chen C, Hardy N, Mihalek AD, Lingala S, Kim YJ, et
al: Danazol treatment for telomere diseases. N Engl J Med.
374:1922–1931. 2016.PubMed/NCBI View Article : Google Scholar
|
|
133
|
Chambers DC, Lutzky VP, Apte SH, Godbolt
D, Feenstra J and Mackintosh J: Successful treatment of
telomeropathy-related interstitial lung disease with
immunosuppression and danazol. Respirol Case Rep.
8(e607)2020.PubMed/NCBI View Article : Google Scholar
|
|
134
|
Dakhlallah D, Batte K, Wang Y,
Cantemir-Stone CZ, Yan P, Nuovo G, Mikhail A, Hitchcock CL, Wright
VP, Nana-Sinkam SP, et al: Epigenetic regulation of miR-17~92
contributes to the pathogenesis of pulmonary fibrosis. Am J Respir
Crit Care Med. 187:397–405. 2013.PubMed/NCBI View Article : Google Scholar
|
|
135
|
Escargueil AE, Soares DG, Salvador M,
Larsen AK and Henriques JA: What histone code for DNA repair? Mutat
Res. 658:259–270. 2008.PubMed/NCBI View Article : Google Scholar
|
|
136
|
Guo W, Shan B, Klingsberg RC, Qin X and
Lasky JA: Abrogation of TGF-beta1-induced fibroblast-myofibroblast
differentiation by histone deacetylase inhibition. Am J Physiol
Lung Cell Mol Physiol. 297:L864–L870. 2009.PubMed/NCBI View Article : Google Scholar
|
|
137
|
Pang M and Zhuang S: Histone deacetylase:
A potential therapeutic target for fibrotic disorders. J Pharmacol
Exp Ther. 335:266–272. 2010.PubMed/NCBI View Article : Google Scholar
|
|
138
|
Korfei M, Skwarna S, Henneke I, MacKenzie
B, Klymenko O, Saito S, Ruppert C, von der Beck D, Mahavadi P,
Klepetko W, et al: Aberrant expression and activity of histone
deacetylases in sporadic idiopathic pulmonary fibrosis. Thorax.
70:1022–1032. 2015.PubMed/NCBI View Article : Google Scholar
|
|
139
|
Sanders YY, Hagood JS, Liu H, Zhang W,
Ambalavanan N and Thannickal VJ: Histone deacetylase inhibition
promotes fibroblast apoptosis and ameliorates pulmonary fibrosis in
mice. Eur Respir J. 43:1448–1458. 2014.PubMed/NCBI View Article : Google Scholar
|
|
140
|
Korfei M, Stelmaszek D, MacKenzie B,
Skwarna S, Chillappagari S, Bach AC, Ruppert C, Saito S, Mahavadi
P, Klepetko W, et al: Comparison of the antifibrotic effects of the
pan-histone deacetylase-inhibitor panobinostat versus the IPF-drug
pirfenidone in fibroblasts from patients with idiopathic pulmonary
fibrosis. PLoS One. 13(e207915)2018.PubMed/NCBI View Article : Google Scholar
|
|
141
|
Mora AL, Rojas M, Pardo A and Selman M:
Emerging therapies for idiopathic pulmonary fibrosis, a progressive
age-related disease. Nat Rev Drug Discov. 16:755–772.
2017.PubMed/NCBI View Article : Google Scholar
|
|
142
|
Coward WR, Feghali-Bostwick CA, Jenkins G,
Knox AJ and Pang L: A central role for G9a and EZH2 in the
epigenetic silencing of cyclooxygenase-2 in idiopathic pulmonary
fibrosis. FASEB J. 28:3183–3196. 2014.PubMed/NCBI View Article : Google Scholar
|
|
143
|
Wei P, Xie Y, Abel PW, Huang Y, Ma Q, Li
L, Hao J, Wolff DW, Wei T and Tu Y: Transforming growth factor
(TGF)-β1-induced miR-133a inhibits myofibroblast differentiation
and pulmonary fibrosis. Cell Death Dis. 10(670)2019.PubMed/NCBI View Article : Google Scholar
|
|
144
|
Liu S, Chen X, Zhang S, Wang X, Du X, Chen
J and Zhou G: miR-106b-5p targeting SIX1 inhibits TGF-β1-induced
pulmonary fibrosis and epithelial-mesenchymal transition in asthma
through regulation of E2F1. Int J Mol Med. 47(04855)2021.PubMed/NCBI View Article : Google Scholar
|
|
145
|
Sato N, Takasaka N, Yoshida M, Tsubouchi
K, Minagawa S, Araya J, Saito N, Fujita Y, Kurita Y, Kobayashi K,
et al: Metformin attenuates lung fibrosis development via NOX4
suppression. Respir Res. 17(107)2016.PubMed/NCBI View Article : Google Scholar
|
|
146
|
Rangarajan S, Bone NB, Zmijewska AA, Jiang
S, Park DW, Bernard K, Locy ML, Ravi S, Deshane J, Mannon RB, et
al: Metformin reverses established lung fibrosis in a bleomycin
model. Nat Med. 24:1121–1127. 2018.PubMed/NCBI View Article : Google Scholar
|
|
147
|
Akamata K, Wei J, Bhattacharyya M, Cheresh
P, Bonner MY, Arbiser JL, Raparia K, Gupta MP, Kamp DW and Varga J:
SIRT3 is attenuated in systemic sclerosis skin and lungs, and its
pharmacologic activation mitigates organ fibrosis. Oncotarget.
7:69321–69336. 2016.PubMed/NCBI View Article : Google Scholar
|
|
148
|
Justice JN, Nambiar AM, Tchkonia T,
LeBrasseur NK, Pascual R, Hashmi SK, Prata L, Masternak MM,
Kritchevsky SB, Musi N, et al: Senolytics in idiopathic pulmonary
fibrosis: Results from a first-in-human, open-label, pilot study.
Ebiomedicine. 40:554–563. 2019.PubMed/NCBI View Article : Google Scholar
|
|
149
|
Lehmann M, Korfei M, Mutze K, Klee S,
Skronska-Wasek W, Alsafadi HN, Ota C, Costa R, Schiller HB, Lindner
M, et al: Senolytic drugs target alveolar epithelial cell function
and attenuate experimental lung fibrosis ex vivo. Eur Respir J.
50(1602367)2017.PubMed/NCBI View Article : Google Scholar
|
|
150
|
Hohmann MS, Habiel DM, Coelho AL, Verri WJ
and Hogaboam CM: Quercetin enhances ligand-induced apoptosis in
senescent idiopathic pulmonary fibrosis fibroblasts and reduces
lung fibrosis in vivo. Am J Respir Cell Mol Biol. 60:28–40.
2019.PubMed/NCBI View Article : Google Scholar
|
|
151
|
Feng F, Wang Z, Li R, Wu Q, Gu C, Xu Y,
Peng W, Han D, Zhou X, Wu J and He H: Citrus alkaline extracts
prevent fibroblast senescence to ameliorate pulmonary fibrosis via
activation of COX-2. Biomed Pharmacother.
112(108669)2019.PubMed/NCBI View Article : Google Scholar
|
|
152
|
Shentu TP, Huang TS, Cernelc-Kohan M, Chan
J, Wong SS, Espinoza CR, Tan C, Gramaglia I, van der Heyde H, Chien
S and Hagood JS: Thy-1 dependent uptake of mesenchymal stem
cell-derived extracellular vesicles blocks myofibroblastic
differentiation. Sci Rep. 7(18052)2017.PubMed/NCBI View Article : Google Scholar
|
|
153
|
Kadota T, Fujita Y, Araya J, Watanabe N,
Fujimoto S, Kawamoto H, Minagawa S, Hara H, Ohtsuka T, Yamamoto Y,
et al: Human bronchial epithelial cell-derived extracellular
vesicle therapy for pulmonary fibrosis via inhibition of TGF-β-WNT
crosstalk. J Extracell Vesicles. 10(e12124)2021.PubMed/NCBI View Article : Google Scholar
|
|
154
|
Yang S, Liu P, Jiang Y, Wang Z, Dai H and
Wang C: Therapeutic applications of mesenchymal stem cells in
idiopathic pulmonary fibrosis. Front Cell Dev Biol.
9(639657)2021.PubMed/NCBI View Article : Google Scholar
|
|
155
|
Chambers DC, Enever D, Ilic N, Sparks L,
Whitelaw K, Ayres J, Yerkovich ST, Khalil D, Atkinson KM and
Hopkins PM: A phase 1b study of placenta-derived mesenchymal
stromal cells in patients with idiopathic pulmonary fibrosis.
Respirology. 19:1013–1018. 2014.PubMed/NCBI View Article : Google Scholar
|
|
156
|
Zhao Y, Yan Z, Liu Y, Zhang Y, Shi J, Li J
and Ji F: Effectivity of mesenchymal stem cells for
bleomycin-induced pulmonary fibrosis: A systematic review and
implication for clinical application. Stem Cell Res Ther.
12(470)2021.PubMed/NCBI View Article : Google Scholar
|
|
157
|
Tzouvelekis A, Paspaliaris V, Koliakos G,
Ntolios P, Bouros E, Oikonomou A, Zissimopoulos A, Boussios N,
Dardzinski B, Gritzalis D, et al: A prospective, non-randomized, no
placebo-controlled, phase Ib clinical trial to study the safety of
the adipose derived stromal cells-stromal vascular fraction in
idiopathic pulmonary fibrosis. J Transl Med. 11(171)2013.PubMed/NCBI View Article : Google Scholar
|
|
158
|
Serrano-Mollar A, Gay-Jordi G,
Guillamat-Prats R, Closa D, Hernandez-Gonzalez F, Marin P, Burgos
F, Martorell J, Sánchez M, Arguis P, et al: Safety and tolerability
of alveolar type ii cell transplantation in idiopathic pulmonary
fibrosis. Chest. 150:533–543. 2016.PubMed/NCBI View Article : Google Scholar
|
|
159
|
Glassberg MK, Minkiewicz J, Toonkel RL,
Simonet ES, Rubio GA, DiFede D, Shafazand S, Khan A, Pujol MV,
LaRussa VF, et al: Allogeneic human mesenchymal stem cells in
patients with idiopathic pulmonary fibrosis via intravenous
delivery (AETHER): A phase I safety clinical trial. Chest.
151:971–981. 2017.PubMed/NCBI View Article : Google Scholar
|
|
160
|
Serrano-Mollar A, Nacher M, Gay-Jordi G,
Closa D, Xaubet A and Bulbena O: Intratracheal transplantation of
alveolar type II cells reverses bleomycin-induced lung fibrosis. Am
J Respir Crit Care Med. 176:1261–1268. 2007.PubMed/NCBI View Article : Google Scholar
|
|
161
|
Poggio HA, Antunes MA, Rocha NN, Kitoko
JZ, Morales MM, Olsen PC, Lopes-Pacheco M, Cruz FF and Rocco PRM:
Impact of one versus two doses of mesenchymal stromal cells on lung
and cardiovascular repair in experimental emphysema. Stem Cell Res
Ther. 9(296)2018.PubMed/NCBI View Article : Google Scholar
|