Introduction
Colorectal cancer (CRC) is one of the three most
common cancer types worldwide, causing high morbidity and mortality
(1-3).
An estimated 151,030 new cases and 52,580 deaths occurred in the
United States in 2022, rendering CRC one of the top three causes of
cancer-related mortality (4). In
China, CRC ranks third in incidence and is a common cause of
cancer-related mortality (5), with
555,477 new CRC cases and 286,162 deaths in 2020 (6,7).
Notably, these numbers are increasing. The overall 5-year survival
rate in China is lower than that in the United States, but the
number of patients with metastatic CRC in China is higher than that
in the United States (8-10).
An estimated 20-30% of patients are further diagnosed with
unresectable metastatic CRC and 50-60% of patients develop
metastatic CRC (11), which
substantially jeopardizes therapeutic potential and impairs
successful clinical outcomes. Therefore, the development of
state-of-the-art screening approaches and new therapeutic
strategies for CRC is required.
Oncogenic mutations often cause tumorigenesis and
RAS mutations [Kirsten rat sarcoma virus (KRAS), HRas
proto-oncogene, GTPase (HRAS), and NRAS proto-oncogene, GTPase
NRAS)] are found in 20-30% of malignant human tumors and in ~45% of
CRC cases (12). RAS mutations
cause tumor initiation and drive uncontrollable tumor cell
proliferation (13,14); they are also associated with poor
prognosis (15-17).
RAS proteins are guanosine triphosphatases (GTPases) that function
as binary switches cycling between inactive (guanosine
diphosphate-bound) and active [guanosine-5'-triphosphate
(GTP)-bound] states (18,19). Activated RAS proteins can bind to
numerous downstream effectors, such as RAF and PI3K, which regulate
critical cellular processes, including metabolism, proliferation
and survival (20). RAS proteins
are subject to a number of regulatory factors and this regulation
is often tightly controlled in cells; however, oncogenic mutations
in RAS proteins alter this tightly regulated process, leading to
the constitutive activation of RAS proteins. Consequently, RAS
mutations cause the aberrant activation of the RAS signaling
network, including the two dominant pathways: The
RAF/mitogen-activated protein kinase kinase (MEK)/extracellular
signal-regulated kinase (ERK) pathway and the PI3K/Akt pathway. In
CRC, RAS is a prognostic and predictive biomarker; however,
therapeutic strategies targeting RAS-driven CRC are still
lacking.
Notably, RAS signaling pathways can be modulated by
Aurora kinases A, B and C (AURKA/B/C), which may lead to the
development of a promising therapeutic strategy for RAS-driven
cancer. Aurora kinases regulate cell mitosis, including centrosome
duplication, spindle assembly, chromosome alignment, chromosome
segregation and the fidelity-monitoring spindle checkpoint
(21,22). Accumulating evidence shows an
association between aberrant AURKA expression and cancer
development, including breast, pancreatic, ovarian and gastric
cancer (23), which can be
ascribed to the development of aneuploidy, supernumerary
centrosomes, defective mitotic spindles and resistance to
apoptosis. Notably, it has been shown that aberrant AURKA
expression is associated with poor prognosis and chemotherapy
response in CRC (24,25). Aurora kinases are considered a
promising cancer target and several Aurora kinase inhibitors,
including alisertib (ALS) and barasertib, have been developed and
evaluated at various preclinical and clinical stages (26). Furthermore, the co-operation
between Aurora kinases and RAS proteins has been reported in
numerous types of cancer (27),
and targeting Aurora kinases and RAS signaling alone or combined
results in various responses (28-33);
suggesting that further analysis of this co-operation is needed. In
particular, RAS allele specificity in cancer treatment should be
considered.
In a previous study by our team, the regulatory
effects of ALS, a selective inhibitor of AURKA, on cell
proliferation, migration, apoptosis and autophagy were evaluated in
KRAS wild-type (WT) and BRAF V600E-mutant CRC cell lines using
stable isotope labeling by amino acids in a cell culture-based
approach (34). However,
comparison of the effects of AURKA inhibition was not conducted
with regard to different RAS mutants in CRC, such as KRAS G12D,
G12V, G13D and A146T. In the present study, the regulatory effects
of ALS on PI3K/Akt and mitogen-activated protein kinase (MAPK)
signaling pathways, apoptosis, autophagy and cell proliferation
were assessed against a panel of human CRC cell lines and
engineered Flp-In T-REx stable cell lines.
Materials and methods
Chemicals and reagents
ALS and selumetinib (Sel) were purchased from
Selleck Chemicals and stored at 100 mM in DMSO at -20˚C. DMSO, FBS,
ammonium persulfate for western blotting, protease and phosphatase
inhibitor cocktails, doxycycline (DOX) and Dulbecco's PBS were
purchased from MilliporeSigma. All required cell culture media,
including Eagle's Minimum Essential Medium for Caco-2 and SK-CO-1
cells, McCoy's 5A for HT29 cells, Roswell Park Memorial Institute
(RPMI)1640 for Colo-678 and CCCL-18 cells, and Dulbecco's Modified
Eagle Medium for HCT116 cells were obtained from Corning, Inc. A
CellTiter-Glo™ luminescent cell viability assay kit was
purchased from Promega Corporation. Pierce™
bicinchoninic acid (BCA) protein assay kit and
radioimmunoprecipitation assay (RIPA) buffer were sourced from
Thermo Fisher Scientific, Inc. Western blotting substrate (20X
LumiGLO® Reagent and 20X Peroxide; cat. no. 7003) was
purchased from Cell Signaling Technology, Inc. Skimmed milk and
nitrocellulose membrane were purchased from Bio-Rad Laboratories,
Inc. Primary antibodies for cleaved poly ADP-ribose polymerase
[PARP, (cat. no. 5625S)], phosphorylated (p-)Akt (Ser473) (cat. no.
4060S), Akt (cat. no. 9272S), p-Erk1/2 (Thr202/Tyr204) (cat. no.
4370S), Erk1/2 (cat. no. 4695S), RAS (cat. no. 3339S) and LC3B-I/II
(cat. no. 3868S), and secondary antibodies for rabbit (cat. no.
7074S) and mouse (cat. no. 7076S) were purchased from Cell
Signaling Technology, Inc., and β-actin (cat. no. sc-47778) was
purchased from Santa Cruz Biotechnology, Inc. All primary
antibodies were diluted at 1:1,000 and secondary antibodies were
diluted at 1:4,000.
Cell lines
The well-recognized and commonly used CRC cell lines
Caco-2KRAS WT, Colo-678KRAS G12D,
SK-CO-1KRAS G12V, HCT116KRAS G13D,
CCCL-18KRAS A146T and HT29BRAF V600E were
purchased from American Type Culture Collection (ATCC). Cells were
cultured in ATCC-recommended complete medium supplemented with 10%
FBS and maintained in a humidified incubator at 37˚C with 5%
CO2. The Flp-In T-REx 293 cell line (cat. no. R78007)
with the Flp-In system (cat. no K601001) was purchased from Thermo
Fisher Scientific, Inc., maintained in DMEM with zeocin (100 µg/ml)
and blasticidin (15 µg/ml) and stored at 37˚C with 5%
CO2 as instructed by the manufacturer. This cell line is
designed for efficient generation of stable cell lines that ensures
homogenous expression of the protein of interest (KRAS). Stable
cell lines expressing KRAS WT, G12D and A146T were generated
according to the manufacturer's instructions (Thermo Fisher
Scientific, Inc.). Briefly, co-transfection of the Flp-In cell line
with a Flp-In expression vector and the Flp recombinase vector
results in targeted integration of the expression vector to the
same locus in every cell, ensuring homogeneous levels of gene
expression. In the present study, the KRAS mutation was generated
using the PfuUItra II Hotstart PCR Master Mix (cat. no. 600850;
Agilent Technologies, Inc.) following the manufacturer's
instructions. The in-house constructs containing
mCherry-H2B-P2A-linked KRAS WT, KRAS G12D and A146T were
co-transfected with pOG44 into Flp-In T-REx 293 cells using
Lipofectamine™ 3000 Transfection Reagent (cat. no.
L3000001; Invitrogen; Thermo Fisher Scientific, Inc.), followed by
2 weeks of hygromycin (100 µg/ml) and blasticidin (15 µg/ml)
selection in DMEM supplemented with 10% Tet-free FBS. Subsequently,
the selected clones were further seeded into 96-well plates at a
density of 50 cells/plate to achieve a monoclonal cell population
for 4-6 weeks. Doxycycline (2 ng) was used to induce KRAS protein
expression. Protein expression was verified by western blotting and
imaging. Mycoplasma Plus PCR Primer Set (Agilent Technologies,
Inc.) was used to detect mycoplasma in all cell lines. Cells were
treated with ALS or Sel with 0.05% DMSO.
Cell viability and proliferation
assessment
To evaluate the different inhibitory effects of ALS
on the cell viability of CRC cell lines, the CellTiter-Glo assay
was performed according to the manufacturer's instructions.
Briefly, a panel of CRC cell lines, including Caco-2, Colo-678,
SK-CO-1, HCT116, CCCL-18 and HT29, were plated and treated with ALS
for 48 and 96 h at concentrations of 0.001, 0.01, 0.1, 1 and 10 µM
in clear-bottomed, white 96-well plates. The CellTiter-Glo
substrate (1:1) was added prior to the assay. The luminescence was
measured using a BioTek Synergy NEO plate reader instrument (BioTek
Instruments, Inc.) equipped with upper EM 620/665 LUM and lower EX
330 LUM filters.
To assess the effects of AURKA and MEK inhibitors on
cell proliferation, engineered Flp-In T-REx 293 stable cell lines
expressing KRAS WT, G12D and A146T (1x103 cells/well)
were cultured in the presence of 2 ng DOX upon exposure to ALS at
0.001, 0.01, 0.1, 1 and 10 µM and Sel at 0.001, 0.01, 0.1, 1 and 10
µM. Cell proliferation was monitored using an IncuCyte®
live-cell analysis system, and images were captured every 4 h in
three fields per well in clear-bottomed, black 96-well plates. To
determine the synergistic effect of ALS and Sel, the combination
index (CI) was calculated via the Chou-Talalay method using
CompuSyn (35). CI<1 was
considered to indicate synergism. Data were analyzed and plotted
using Prism 9 (GraphPad Software; Dotmatics).
RAS-GTP pull-down
Caco-2, Colo-678, SK-CO-1, HCT116, CCCL-18 and HT29
CRC cell lines were grown in 10% FBS in 60-mm dishes and treated
with ALS at 0.1, 1 and 5 µM for 48 h. Then, cells were washed in
cold PBS twice and harvested in SDS-free lysis buffer (25 mM
Tris-HCl, pH 7.2; 150 mM NaCl; 5 mM MgCl2; 1% NP-40; 5%
glycerol; and 1% protease inhibitor cocktails) to assess the
RAS-GTP level using the active RAS detection kit (cat. no. 8821;
Cell Signaling Technology, Inc.) according to the manufacturer's
instructions. Briefly, protein samples were incubated with RAF1
RAS-binding domain (RAF1-RBD) for 4 h in a cold room at 4˚C in the
presence of glutathione resin. Subsequently, the samples were
pelleted by centrifugation at 6,000 x g for 15 sec and washed with
lysis buffer twice before western blotting.
Western blotting
The effects of ALS and Sel on protein expression and
the RAS signaling pathway were examined using western blotting.
Caco-2, Colo-678, SK-CO-1, HCT116, CCCL-18 and HT29 CRC cell lines
were seeded into 6-well plates at 2x105 cells/well. The
following day, cells were treated with either ALS alone (0.1, 1 and
5 µM) for 48 h or first treated with Sel (0.1 µM) for 24 h followed
by ALS (0.1, 1 and 5 µM) for another 24 h. Subsequently, cells were
washed once with ice-cold PBS and cell protein samples were
collected and processed in RIPA buffer containing phosphatase and
protease inhibitor cocktails. The protein concentration was
measured via Pierce BCA protein assay and 20 µg protein/lane were
separated by SDS-PAGE on a 10% gel containing ammonium persulfate
(10%) and transferred using Trans-Blot Turbo Mini 0.2 µm
Nitrocellulose Transfer Packs (cat. no. 1704158; Bio-Rad
Laboratories, Inc.). Subsequently, the membrane was blocked with 5%
skim milk in TBS-Tween (1%) for 1 h at room temperature and
incubated with primary antibody (1:1,000) in a cold room (4˚C)
overnight. The next day, the membrane was incubated with secondary
antibodies at room temperature for 1 h before film development in a
dark room. Protein expression levels were normalized to the
densitometric value of the internal control, β-actin, using Image J
1.54b (National Institutes of Health).
Statistical analysis
Data are presented as the mean ± standard deviation.
Multiple comparisons were assessed through a one-way ANOVA followed
by Tukey's multiple comparison post hoc test using GraphPad Prism
9. P<0.05 was considered to indicate a statistically significant
difference. All assays were repeated a minimum of three times
(n≥3).
Results
ALS modulates CRC cell proliferation
and the active form of RAS in a KRAS allele-specific manner
In order to determine the effects of AURKA
inhibition on RAS signal output, the effects of ALS on cell
viability were examined against a panel of CRC cell lines bearing
KRAS WT, G12D, G12V, G13D, A146T and BRAF V600E mutations. The KRAS
G13D-expressing cell line, HCT116, was the most susceptible to ALS
treatment over 48 and 96 h (Fig.
1A and B). Colo-678KRAS
G12D, SK-CO-1KRAS G12V and CCCL-18KRAS
A146T were also susceptible but to a lesser extent. KRAS WT
and BRAF V600E-expressing cell lines exhibited a moderate response
to ALS treatment (Fig. 1A and
B). Subsequently, the RAS-GTP
level was evaluated following the treatment of cells with ALS.
RAF1-RBD was used to pull down the active form of RAS from
Caco-2KRAS WT, Colo-678KRAS G12D,
SK-CO-1KRAS G12V, HCT116KRAS G13D,
CCCL-18KRAS A146T and HT29BRAF V600E cells.
In KRAS WT-expressing Caco-2 cells, ALS increased the level of
RAS-GTP in a concentration-dependent manner (Fig. 2). In KRAS mutant-expressing cells,
ALS increased RAS-GTP level in SK-CO-1KRAS G12V,
HCT116KRAS G13D and CCCL-18KRAS A146T cells,
whereas ALS decreased the RAS-GTP level in Colo-678KRAS
G12D cells. Notably, in HT29 cells expressing BRAF V600E, a
constitutively active BRAF mutant commonly found in CRC that does
not rely on RAS activation to activate the RAS-RAF-MEK-ERK signal,
ALS decreased the level of the active form of RAS. In combination,
these results suggested that the inhibition of AURKA exerts
different inhibitory effects on cell proliferation and regulates
the active form of RAS in a KRAS allele-specific manner.
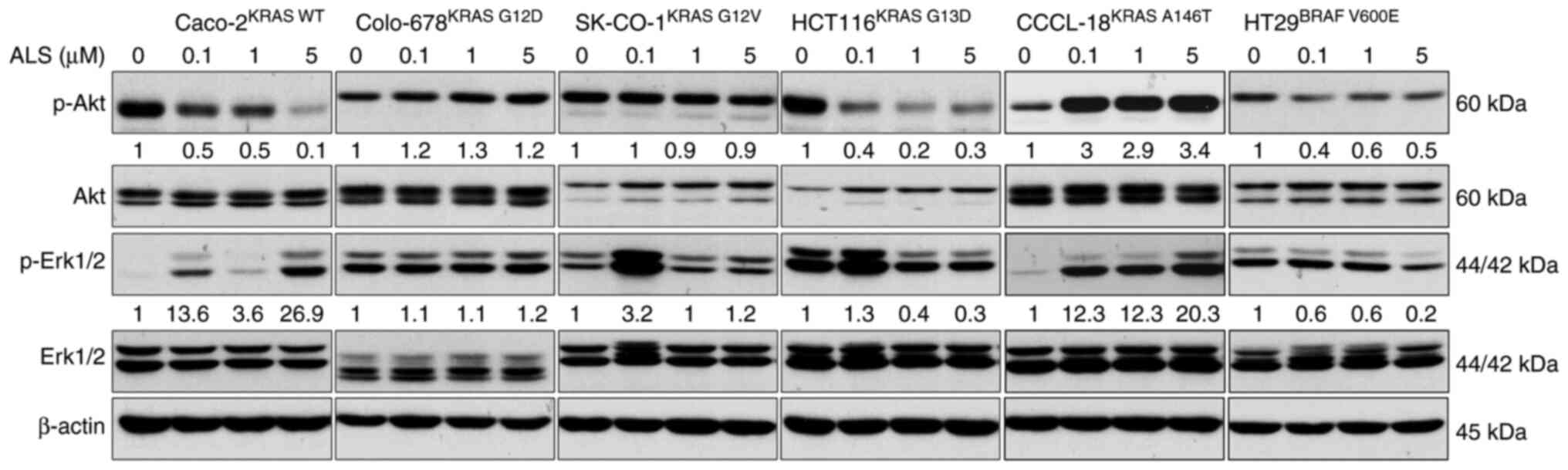
ALS affects RAS signaling in a KRAS
allele-specific manner
Following the examination of RAS-GTP level, the
phosphorylation levels of Akt and Erk were assessed in CRC cell
lines. The PI3K/Akt and MAPK signaling pathways are the two
dominant downstream pathways of RAS. The phosphorylation levels of
Akt and Erk are the two major indicators for RAS activation. CRC
cells were treated with 0.1, 1 and 5 µM ALS, and the levels of
p-Akt and p-Erk were analyzed. ALS inhibited the phosphorylation of
Akt but enhanced the phosphorylation of Erk in KRAS WT-expressing
Caco-2 cells (Figs. 3 and S1). There was no marked change in the
levels of p-Akt and p-Erk in Colo-678KRAS G12D and
SK-CO-1KRAS G12V cells. In KRAS G13D-expressing HCT116
cells, ALS suppressed the phosphorylation of Akt and Erk.
Similarly, ALS suppressed the activation of Akt and Erk in BRAF
V600E-expressing HT29 cells, although the effect on Erk
phosphorylation was not significant. However, ALS enhanced the
phosphorylation of Akt and Erk in CCCL-18KRAS A146T
cells. These results suggested that the inhibition of AURKA by ALS
leads to a RAS allele-specific modulation in the PI3K/Akt and MAPK
signaling pathways.
ALS manipulates apoptosis and
autophagy in a KRAS allele-specific manner
To test the influence of the RAS allele-specific
regulatory effect of ALS on cell death, the expression levels of
cleaved PARP and LC3I and II, as surrogate markers of apoptosis and
autophagy, were examined. The panel of CRC cell lines was treated
with ALS, and the expression levels of cleaved PARP and LC3I and II
were measured. As shown in Fig. 4,
KRAS WT-expressing Caco-2 cells underwent apoptosis and autophagy
upon exposure to ALS, which was evident from the increase in the
levels of cleaved PARP and the ratio of LC3II/I. Notably, apoptosis
and autophagy-related proteins were only measured as an indicator
instead of direct examination of apoptosis and autophagy, but the
results are consistent with our previous study (34). There was no marked alteration in
apoptosis and autophagy in Colo-678KRAS G12D cells
following treatment with ALS. However, there was a marked increase
in the expression levels of cleaved PARP and the ratio of LC3II/I
in SK-CO-1KRAS G12V cells. Furthermore, in
HCT116KRAS G13D, CCCL-18KRAS A146T and
HT29BRAF V600E cells, ALS markedly enhanced the
expression levels of cleaved PARP and, to a lesser extent, also
increased the ratio of LC3II/I in CCCL-18KRAS A146T and
HT29BRAF V600E cells. Collectively, the pharmacological
inhibition of AURKA via ALS resulted in different regulatory
effects on apoptosis and autophagy in a RAS allele-specific
manner.
MEK inhibitor displays different
regulatory effects on RAS signals in a KRAS allele-specific
manner
As the present study observed the RAS
allele-specific regulatory effects of ALS on RAS signaling pathways
in CRC cell lines, it was subsequently examined whether a MEK
inhibitor could have similar effects. To test this hypothesis, the
effects of Sel, a MEK inhibitor, on PI3K/Akt and MAPK signaling
pathways were first evaluated. Figs.
5 and S2 show that Sel
inhibited the MAPK signaling pathway in all tested cell lines,
evident from the suppression of Erk phosphorylation; however, there
were different responses to Sel treatment in the PI3K/Akt signaling
pathway. The expression levels of p-Akt were decreased in
Caco-2KRAS WT, Colo-678KRAS G12D and
SK-CO-1KRAS G12V cell lines, but were increased in the
remaining cell lines. Furthermore, the effects of Sel on apoptosis
and autophagy were tested across the CRC cell lines. Sel did not
markedly affect apoptosis in Caco-2KRAS WT,
Colo-678KRAS G12D and CCCL-18KRAS A146T cell
lines, as indicated by the unaffected PARP cleavage (Fig. 6). However, Sel induced cell
apoptosis in SK-CO-1KRAS G12V, HCT116KRAS
G13D and HT29BRAF V600E cell lines, evident from
the increase in the expression levels of cleaved PARP.
Additionally, Sel promoted cell autophagy by increasing the
conversion of LC3I to LC3II in Caco-2KRAS WT,
HCT116KRAS G13D, CCCL-18KRAS A146T and
HT29BRAF V600E cell lines. In combination, these results
suggested that the MEK inhibitor exerted different regulatory
effects on RAS signaling pathways, resulting in different responses
to apoptosis and autophagy.
Combination of ALS and a MEK inhibitor
regulates RAS signals, apoptosis and autophagy in a KRAS
allele-specific manner
Following evaluation of the effects of ALS and Sel
on RAS signals, the outcome of the dual inhibition of AURKA and MEK
via ALS and Sel on RAS signaling pathways, and on apoptosis and
autophagy, was examined. Caco-2KRAS WT,
Colo-678KRAS G12D, SK-CO-1KRAS G12V,
HCT116KRAS G13D, CCCL-18KRAS A146T and
HT29BRAF V600E cells were first treated with Sel (0.1
µM) for 24 h followed by treatment with 0.1, 1 or 5 µM ALS for
another 24 h. The RAS signaling output was evaluated by determining
the levels of p-Akt and p-Erk, and apoptosis and autophagy were
examined by determining the levels of cleaved PARP and LC3I/II. ALS
counteracted the inhibitory effect of Sel on the PI3K/Akt signaling
pathway in Caco-2KRAS WT, CCCL-18KRAS A146T
and HT29BRAF V600E cells, with an increase in the
expression levels of p-Akt, whereas ALS enhanced the suppressive
effect of Sel on the PI3K/Akt signaling pathway in
Colo-678KRAS G12D and SK-CO-1KRAS G12V cells,
with a further reduction in the expression levels of p-Akt
(Figs. 7 and S3). In addition, ALS strengthened the
inhibitory effect of Sel on the MAPK signaling pathway in
Colo-678KRAS G12D, SK-CO-1KRAS G12V and
HT29BRAF V600E cells, with further suppression of Erk
phosphorylation, although in the effects on Erk phosphorylation in
SK-CO-1KRAS G12V cells were not significant, whereas ALS
was counteractant to Sel in the remaining cell lines. Furthermore,
ALS enhanced the apoptotic effects in the presence of Sel in all
cell lines except for Colo-678 cells (Fig. 8). In particular, ALS enhanced the
cleavage of PARP in KRAS WT-expressing Caco-2 cells, which did not
occur in the presence of Sel alone. ALS enhanced autophagy in the
presence of Sel in Caco-2KRAS WT, Colo-678KRAS
G12D, HCT116KRAS G13D and HT29BRAF
V600E cells, but there was no marked alteration in
SK-CO-1KRAS G12V and CCCL-18KRAS A146T cells.
The combination of ALS and Sel regulated RAS signal output,
apoptosis and autophagy in a RAS allele-specific manner.
Combination of ALS and a MEK inhibitor
exerts a synergistic cell proliferation inhibitory effect
Since the commonly occurring KRAS mutants (G12D and
A146T) exhibit distinct biological features in CRC, such as
intermediate hyperproliferative phenotype of KRAS A146T in colons
and high hyperproliferative phenotype of KRAS G12D in colons
(36), Flp-In T-REx cells were
engineered to express KRAS WT, G12D and A146T along with mCherry to
assess the effects of ALS and Sel, alone or in combination, on cell
proliferation. In a dose escalation assay, ALS and Sel monotherapy
exerted a stronger inhibitory effect on cell proliferation in the
context of KRAS G12D and A146T compared with KRAS WT (Fig. 9A and B). Notably, combination treatment with
ALS and Sel displayed a synergistic effect on cell proliferation
inhibition in KRAS WT, G12D and A146T-expressing cells at
concentrations from 0.1-10 µM (Fig.
9C), indicated by CI<1. Collectively, these results
suggested that the dual inhibition of AURKA and MEK could generate
an enhanced effect.
Discussion
The notion that each RAS protein is unique has been
acknowledged, and increasing evidence has shown that the RAS
allele-specific approach for cancer monotherapy or combination
therapy may result in encouraging outcomes in preclinical and
clinical settings (37-39).
In particular, a KRASG12C-targeted therapy in lung
cancer treatment has suggested that the RAS allele can be directly
and specifically targeted (40).
However, this KRASG12C-targeted therapy is only
applicable to a cysteine mutation and causes drug resistance to
sotorasib and adagrasib (41-43),
which limits the application of this therapy in other RAS
mutant-driven cancers. The rationale of the present study stems
from the concept of a ‘RAS allele-specific therapeutic approach for
cancer therapy’. Given the increasing evidence showing that RAS
proteins are biochemically and structurally unique (37), it has been suggested that each RAS
protein represents a unique biological function. Therefore, the
present study aimed to assess the effects of ALS against a panel of
CRC cell lines bearing different KRAS mutations. Whilst the present
pilot study has shown a KRAS allele-specific response, further
in vivo studies are needed.
KRAS mutations are present in ~45% of CRC cases,
with G12D, G12V, G13D and A146T being the most common. KRAS-driven
CRC is associated with substantial morbidity and mortality
(44), and therefore requires
urgent, novel therapeutic strategies with reduced side effects.
Increasing evidence has shown that Aurora kinases are promising
cancer therapeutic targets (45-52),
and the selective inhibition of these targets in a RAS
allele-specific manner may have profound therapeutic advantages for
cancer treatment. It has previously been shown that AURKA knockdown
decreases MAPK signal output, whereas AURKA overexpression promotes
MAPK signaling in nasopharyngeal carcinoma (33). In the present study, the RAS
allele-specific regulatory effects of ALS on RAS signaling pathways
were observed against a panel of CRC cell lines bearing different
KRAS mutations. Furthermore, Davis et al (31) showed a varied response in the
combined inhibition of MEK and AURKA in
KRAS/phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic
subunit α double-mutant CRC. The present study also found that ALS
alone or in combination with a MEK inhibitor exerted different
regulatory effects on cell proliferation, and PI3K/Akt and MAPK
signaling pathways, and differentially induced apoptosis and
autophagy, suggesting that the KRAS mutational profile is important
for ALS treatment. Notably, the inhibitory effect on the PI3K/Akt
signaling pathway differed in the Caco-2 cell line when the cells
were treated with ALS alone or in combination with Sel. This
differential inhibitory effect on Caco-2 cells could suggest that
the PI3K/Akt signaling pathway is the main route for RAS signaling,
compared to the MAPK signaling pathway, and that AURKA interplays
with the PI3K/Akt signaling pathway more than the MAPK signaling
pathway in a WT KRAS setting. Together, the data suggested that WT
KRAS-mediated PI3K/Akt signaling pathway is more susceptible to ALS
inhibition. However, simultaneous inhibition with ALS and Sel
showed the opposite effect, suggesting that there may be a feedback
activation loop between MAPK and the PI3K/Akt signaling pathway.
Indeed, the feedback activation loop has been reported in
regulation of the RAS signaling pathway, which has been proposed as
a potential therapeutic targeting strategy in CRC (36). However, this observation in the
context of WT KRAS needs further study.
The RAS subfamily consists of KRAS, NRAS and HRAS,
all of which are mutated in human cancer (53). These isoforms demonstrate a high
degree of sequence identity, except at the C-terminus.
Nevertheless, they are mutated in cancer in a non-random
distribution, suggesting context-dependent differences in
biological function. NRAS mutations are most common in malignant
melanoma, hematopoietic malignancies and thyroid cancer; while HRAS
mutations are most common in head and neck, and bladder cancers
(53). KRAS mutations are the most
common mutations in cancer, occurring in up to 22% of all human
cancer cases, predominately in lung cancer, CRC and pancreatic
cancer (53). KRAS mutations
typically occur in exons 2 and 3, at codons 12, 13 and 61 (37,53).
KRAS G12D, G13D, G12V and A146T mutations often occur in CRC, and
these mutants have unique biological features, such as
tissue-specific effects on homeostasis (36), which further supports the notion of
RAS allele specificity. For example, KRAS A146T-driven CRC may be
more susceptible to a negative feedback regulation by ribosomal S6
kinases (36). Various responses
were also observed in different KRAS-mutated CRC cell lines.
Epidemiological and prospective clinical studies
have shown that cancer expressing different mutant forms of KRAS
exhibits distinct clinical behaviors (37,54).
These differences are believed to arise from the rewiring of signal
transduction networks in a RAS mutation-dependent manner, which is
encouraging studies on the RAS context dependency in signal output.
For example, a comparison of KRAS G12C, G12V and G12D mutations in
patient-derived non-small cell lung cancer cell lines demonstrated
the activation of MAPK and PI3K/Akt in G12D lines, whereas G12C and
G12V exhibited little PI3K/Akt signaling and prominent RAL
activation (55). Another study
reported similar RAS allele-specific rewiring (56). While these studies are critical for
establishing that isoform or allele-specific effects occur, the
mechanisms and translational implications have not been explored.
Given this lack of understanding, RAS allele-specific biology is
currently a major research focus, and RAS context dependency in
monotherapy or combination therapy of other key node inhibitors
with RAS signaling pathway inhibitors has been a dominant topic in
the RAS research community.
The present study is inspired by the aforementioned
notion. In particular, previous studies on the preferential
signaling output of KRAS G12D vs. KRAS Q61H in lung cancer
(57) and distinct biological
features of KRAS G12D vs. KRAS A146T in CRC (36) clearly demonstrate the role of RAS
allele specificity in cancer treatment. For example, the present
study demonstrated that the KRAS G13D-expressing CRC cell line was
the most susceptible to treatment with ALS, which could be ascribed
to the dual suppression of the PI3K/Akt and MAPK signaling
pathways. In the KRAS A146T-expressing CRC cell line, ALS enhanced
both the PI3K/Akt and MAPK signaling pathways, which could disturb
the cell viability, resulting in over-activated signal outputs
(58,59). However, ALS suppressed both
PI3K/Akt and MAPK signal outputs in the HT29 cell line, which only
showed moderate sensitivity in cell proliferation. This moderate
sensitivity could be ascribed to the BRAF V600E mutant. In
addition, the crosstalk between other key nodes and RAS signaling
pathways may be involved in the response to ALS treatment.
Furthermore, there are limitations in the present
study. First, the lack of cell line authentication by STR profiling
could affect the accuracy of the results. Second, the HT29 cell
line may not be the most appropriate as a CRC cell model, because
it has been indicated that the HT29 cell line originated from an
adenocarcinoma of the rectosigmoid part of the intestine. Third,
the lack of an AURKA knockdown experiment is also a limitation,
although it has been reported in other studies (26-30).
Furthermore, in vivo studies are needed to support the
effects observed in vitro in future.
In conclusion, the present findings revealed a
potential RAS allele-specific therapeutic role for AURKA inhibition
and RAS signaling modulation in CRC treatment. Given that KRAS is
the most common oncogene in human cancer, including CRC, novel
therapeutic strategies are urgently needed. Such a strategy could
not only target a single kinase but also be therapeutically
effective by harnessing oncogenic KRAS with an allele-specific
approach.
Supplementary Material
Histogram showing the relative
expression levels of p-Akt/Akt and p-Erk1/2/Erk1/2 in the western
blot images shown in Fig. 3.
*P<0.05, **P<0.01,
***P<0.001, ****P<0.0001. ALS,
alisertib; p, phosphorylated.
Histogram showing the relative
expression levels of p-Akt/Akt and p-Erk1/2/Erk1/2 in the western
blot images shown in Fig. 5.
*P<0.05, **P<0.01,
***P<0.001, ****P<0.0001. p,
phosphorylated; Sel, selumetinib.
Histogram showing the relative
expression levels of p-Akt/Akt and p-Erk1/2/Erk1/2 in the western
blot images shown in Fig. 7.
*P<0.05, **P<0.01,
***P<0.001, ****P<0.0001. ALS,
alisertib; p, phosphorylated; Sel, selumetinib.
Acknowledgements
Not applicable.
Funding
Funding: The present study was funded by the Medical Science and
Technology Research Project of Foshan City (grant. nos.
2017AB003533, 2220001004234, 2020001005635), the Medical Science
and Technology Research Foundation of Guangdong Province (grant.
no. A2018150), the Scientific Research Start Plan of Shunde
Hospital, Southern Medical University (grant. no. SRSP2018002), the
Medical Science and Technology Research Project of Foshan City
(grant. nos. 2220001004234 and 2020001005635) and the Foshan
Medical Key Talent Project.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
BR, YG and SC developed the study concept and
design. BR, YG, SC, ZG, KZ, YY, QL, JF and ZL acquired the data.
Analysis and interpretation of data was performed by BR, YG, SC,
ZG, KZ, YY, QL, JF and ZL. BR, YG and SC drafted the manuscript,
and statistical analysis was performed by BR, YG and SC. Technical
and material support was provided by QL, JF and ZL. YJ and ZH
designed and supervised the study, and finalized the manuscript.
BR, YJ and ZH confirm the authenticity of all the raw data. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Colorectal cancer burden in EU-27. Source:
ECIS-European Cancer Information System. https://ecis.jrc.ec.europa.eu, accessed
15/01/2021.
|
|
3
|
Siegel RL, Miller KD, Goding Sauer A,
Fedewa SA, Butterly LF, Anderson JC, Cercek A, Smith RA and Jemal
A: Colorectal cancer statistics, 2020. CA Cancer J Clin.
70:145–164. 2020.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Cancer Stat Facts: Colorectal Cancer,
2022.
|
|
5
|
Global Cancer Observatory, World Health
Organization. https://gco.iarc.fr/.
|
|
6
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249.
2021.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Chen H, Lu B and Dai M: Colorectal cancer
screening in China: Status, challenges, and prospects-China, 2022.
China CDC Wkly. 4:322–328. 2022.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Chu E: An update on the current and
emerging targeted agents in metastatic colorectal cancer. Clin
Colorectal Cancer. 11:1–13. 2012.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Li N, Lu B, Luo C, Cai J, Lu M, Zhang Y,
Chen H and Dai M: Incidence, mortality, survival, risk factor and
screening of colorectal cancer: A comparison among China, Europe,
and northern America. Cancer Lett. 522:255–268. 2021.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Lombardi L, Morelli F, Cinieri S, Santini
D, Silvestris N, Fazio N, Orlando L, Tonini G, Colucci G and
Maiello E: Adjuvant colon cancer chemotherapy: Where we are and
where we'll go. Cancer Treat Rev. 36 (Suppl 3):S34–S41.
2010.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Zhu G, Pei L, Xia H, Tang Q and Bi F: Role
of oncogenic KRAS in the prognosis, diagnosis and treatment of
colorectal cancer. Mol Cancer. 20(143)2021.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Jackson EL, Willis N, Mercer K, Bronson
RT, Crowley D, Montoya R, Jacks T and Tuveson DA: Analysis of lung
tumor initiation and progression using conditional expression of
oncogenic K-ras. Genes Dev. 15:3243–3248. 2001.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Pylayeva-Gupta Y, Grabocka E and Bar-Sagi
D: RAS oncogenes: Weaving a tumorigenic web. Nat Rev Cancer.
11:761–774. 2011.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Graziano SL, Gamble GP, Newman NB, Abbott
LZ, Rooney M, Mookherjee S, Lamb ML, Kohman LJ and Poiesz BJ:
Prognostic significance of K-ras codon 12 mutations in patients
with resected stage I and II non-small-cell lung cancer. J Clin
Oncol. 17:668–675. 1999.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Tsao MS, Aviel-Ronen S, Ding K, Lau D, Liu
N, Sakurada A, Whitehead M, Zhu CQ, Livingston R, Johnson DH, et
al: Prognostic and predictive importance of p53 and RAS for
adjuvant chemotherapy in non small-cell lung cancer. J Clin Oncol.
25:5240–5247. 2007.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Andreyev HJ, Norman AR, Cunningham D,
Oates JR and Clarke PA: Kirsten ras mutations in patients with
colorectal cancer: The multicenter ‘RASCAL’ study. J Natl Cancer
Inst. 90:675–684. 1998.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Simanshu DK, Nissley DV and McCormick F:
RAS proteins and their regulators in human disease. Cell.
170:17–33. 2017.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Schubbert S, Shannon K and Bollag G:
Hyperactive Ras in developmental disorders and cancer. Nat Rev
Cancer. 7:295–308. 2007.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Stephen AG, Esposito D, Bagni RK and
McCormick F: Dragging ras back in the ring. Cancer Cell.
25:272–281. 2014.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Nigg EA: Mitotic kinases as regulators of
cell division and its checkpoints. Nat Rev Mol Cell Biol. 2:21–32.
2001.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Bolanos-Garcia VM: Aurora kinases. Int J
Biochem Cell Biol. 37:1572–1577. 2005.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Gautschi O, Heighway J, Mack PC, Purnell
PR, Lara PN Jr and Gandara DR: Aurora kinases as anticancer drug
targets. Clin Cancer Res. 14:1639–1648. 2008.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Dotan E, Meropol NJ, Zhu F, Zambito F,
Bove B, Cai KQ, Godwin AK, Golemis EA, Astsaturov I and Cohen SJ:
Relationship of increased aurora kinase A gene copy number,
prognosis and response to chemotherapy in patients with metastatic
colorectal cancer. Br J Cancer. 106:748–755. 2012.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Baba Y, Nosho K, Shima K, Irahara N, Kure
S, Toyoda S, Kirkner GJ, Goel A, Fuchs CS and Ogino S: Aurora-A
expression is independently associated with chromosomal instability
in colorectal cancer. Neoplasia. 11:418–425. 2009.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Du R, Huang C, Liu K, Li X and Dong Z:
Targeting AURKA in cancer: Molecular mechanisms and opportunities
for cancer therapy. Mol Cancer. 20(15)2021.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Umstead M, Xiong J, Qi Q, Du Y and Fu H:
Aurora kinase A interacts with H-Ras and potentiates Ras-MAPK
signaling. Oncotarget. 8:28359–28372. 2017.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Dauch D, Rudalska R, Cossa G, Nault JC,
Kang TW, Wuestefeld T, Hohmeyer A, Imbeaud S, Yevsa T, Hoenicke L,
et al: A MYC-aurora kinase A protein complex represents an
actionable drug target in p53-altered liver cancer. Nat Med.
22:744–753. 2016.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Xie Y, Zhu S, Zhong M, Yang M, Sun X, Liu
J, Kroemer G, Lotze M, Zeh HJ III, Kang R and Tang D: Inhibition of
Aurora kinase A induces necroptosis in pancreatic carcinoma.
Gastroenterology. 153:1429–1443.e5. 2017.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Wang-Bishop L, Chen Z, Gomaa A, Lockhart
AC, Salaria S, Wang J, Lewis KB, Ecsedy J, Washington K, Beauchamp
RD and El-Rifai W: Inhibition of AURKA reduces proliferation and
survival of gastrointestinal cancer cells with activated KRAS by
preventing activation of RPS6KB1. Gastroenterology. 156:662–675.e7.
2019.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Davis SL, Robertson KM, Pitts TM, Tentler
JJ, Bradshaw-Pierce EL, Klauck PJ, Bagby SM, Hyatt SL, Selby HM,
Spreafico A, et al: Combined inhibition of MEK and Aurora A kinase
in KRAS/PIK3CA double-mutant colorectal cancer models. Front
Pharmacol. 6(120)2015.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Dos Santos EO, Carneiro-Lobo TC, Aoki MN,
Levantini E and Bassères DS: Aurora kinase targeting in lung cancer
reduces KRAS-induced transformation. Mol Cancer.
15(12)2016.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Wan XB, Long ZJ, Yan M, Xu J, Xia LP, Liu
L, Zhao Y, Huang XF, Wang XR, Zhu XF, et al: Inhibition of Aurora-A
suppresses epithelial-mesenchymal transition and invasion by
downregulating MAPK in nasopharyngeal carcinoma cells.
Carcinogenesis. 29:1930–1937. 2008.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Ren BJ, Zhou ZW, Zhu DJ, Ju YL, Wu JH,
Ouyang MZ, Chen XW and Zhou SF: Alisertib induces cell cycle
arrest, apoptosis, autophagy and suppresses EMT in HT29 and Caco-2
cells. Int J Mol Sci. 17(41)2015.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Chou TC: Drug combination studies and
their synergy quantification using the Chou-Talalay method. Cancer
Res. 70:440–446. 2010.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Poulin EJ, Bera AK, Lu J, Lin YJ, Strasser
SD, Paulo JA, Huang TQ, Morales C, Yan W, Cook J, et al:
Tissue-Specific Oncogenic Activity of KRASA146T. Cancer
Discov. 9:738–755. 2019.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Haigis KM: KRAS alleles: The devil is in
the detail. Trends Cancer. 3:686–697. 2017.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Lito P, Solomon M, Li LS, Hansen R and
Rosen N: Allele-specific inhibitors inactivate mutant KRAS G12C by
a trapping mechanism. Science. 351:604–608. 2016.PubMed/NCBI View Article : Google Scholar
|
|
39
|
McFall T, Diedrich JK, Mengistu M,
Littlechild SL, Paskvan KV, Sisk-Hackworth L, Moresco JJ, Shaw AS
and Stites EC: A systems mechanism for KRAS mutant allele-specific
responses to targeted therapy. Sci Signal.
12(eaaw8288)2019.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Hong DS, Fakih MG, Strickler JH, Desai J,
Durm GA, Shapiro GI, Falchook GS, Price TJ, Sacher A, Denlinger CS,
et al: KRASG12C inhibition with sotorasib in advanced
solid tumors. N Engl J Med. 383:1207–1217. 2020.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Zhao Y, Murciano-Goroff YR, Xue JY, Ang A,
Lucas J, Mai TT, Da Cruz Paula AF, Saiki AY, Mohn D, Achanta P, et
al: Diverse alterations associated with resistance to KRAS(G12C)
inhibition. Nature. 599:679–683. 2021.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Tanaka N, Lin JJ, Li C, Ryan MB, Zhang J,
Kiedrowski LA, Michel AG, Syed MU, Fella KA, Sakhi M, et al:
Clinical acquired resistance to KRASG12C inhibition
through a novel KRAS switch-II pocket mutation and polyclonal
alterations converging on RAS-MAPK reactivation. Cancer Discov.
11:1913–1922. 2021.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Awad MM, Liu S, Rybkin II, Arbour KC,
Dilly J, Zhu VW, Johnson ML, Heist RS, Patil T, Riely GJ, et al:
Acquired resistance to KRASG12C Inhibition in cancer. N
Engl J Med. 384:2382–2393. 2021.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Ferlay J, Colombet M, Soerjomataram I,
Dyba T, Randi G, Bettio M, Gavin A, Visser O and Bray F: Cancer
incidence and mortality patterns in Europe: Estimates for 40
countries and 25 major cancers in 2018. Eur J Cancer. 103:356–387.
2018.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Hong X, O'Donnell JP, Salazar CR, Van
Brocklyn JR, Barnett KD, Pearl DK, deCarvalho AC, Ecsedy JA, Brown
SL, Mikkelsen T and Lehman NL: The selective Aurora-A kinase
inhibitor MLN8237 (alisertib) potently inhibits proliferation of
glioblastoma neurosphere tumor stem-like cells and potentiates the
effects of temozolomide and ionizing radiation. Cancer Chemother
Pharmacol. 73:983–990. 2014.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Zhou N, Singh K, Mir MC, Parker Y, Lindner
D, Dreicer R, Ecsedy JA, Zhang Z, The BT, Almasan A and Hansel DE:
The investigational Aurora kinase A inhibitor MLN8237 induces
defects in cell viability and cell-cycle progression in malignant
bladder cancer cells in vitro and in vivo. Clin Cancer Res.
19:1717–1728. 2013.PubMed/NCBI View Article : Google Scholar
|
|
47
|
D'Assoro AB, Liu T, Quatraro C, Amato A,
Opyrchal M, Leontovich A, Ikeda Y, Ohmine S, Lingle W, Suman V, et
al: The mitotic kinase Aurora-a promotes distant metastases by
inducing epithelial-to-mesenchymal transition in ERα(+) breast
cancer cells. Oncogene. 33:599–610. 2014.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Boss DS, Beijnen JH and Schellens JH:
Clinical experience with aurora kinase inhibitors: A review.
Oncologist. 14:780–793. 2009.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Yuan CX, Zhou ZW, Yang YX, He ZX, Zhang X,
Wang D, Yang T, Wang NJ, Zhao RJ and Zhou SF: Inhibition of mitotic
Aurora kinase A by alisertib induces apoptosis and autophagy of
human gastric cancer AGS and NCI-N78 cells. Drug Des Devel Ther.
9:487–508. 2015.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Ding YH, Zhou ZW, Ha CF, Zhang XY, Pan ST,
He ZX, Edelman JL, Wang D, Yang YX, Zhang X, et al: Alisertib, an
Aurora kinase A inhibitor, induces apoptosis and autophagy but
inhibits epithelial to mesenchymal transition in human epithelial
ovarian cancer cells. Drug Des Devel Ther. 9:425–464.
2015.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Wang F, Li H, Yan XG, Zhou ZW, Yi ZG, He
ZX, Pan ST, Yang YX, Wang ZZ, Zhang X, et al: Alisertib induces
cell cycle arrest and autophagy and suppresses
epithelial-to-mesenchymal transition involving PI3K/Akt/mTOR and
sirtuin 1-mediated signaling pathways in human pancreatic cancer
cells. Drug Des Devel Ther. 9:575–601. 2015.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Niu NK, Wang ZL, Pan ST, Ding HQ, Au GH,
He ZX, Zhou ZW, Xiao G, Yang YX, Zhang X, et al: Pro-apoptotic and
pro-autophagic effects of the Aurora kinase A inhibitor alisertib
(MLN8237) on human osteosarcoma U-2 OS and MG-63 cells through the
activation of mitochondria-mediated pathway and inhibition of p38
MAPK/PI3K/Akt/mTOR signaling pathway. Drug Des Devel Ther.
9:1555–1584. 2015.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Prior IA, Lewis PD and Mattos C: A
comprehensive survey of Ras mutations in cancer. Cancer Res.
72:2457–2467. 2012.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Imamura Y, Lochhead P, Yamauchi M, Kuchiba
A, Qian ZR, Liao X, Nishihara R, Jung S, Wu K, Nosho K, et al:
Analyses of clinicopathological, molecular, and prognostic
associations of KRAS codon 61 and codon 146 mutations in colorectal
cancer: Cohort study and literature review. Mol Cancer.
13(135)2014.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Ihle NT, Byers LA, Kim ES, Saintigny P,
Lee JJ, Blumenschein GR, Tsao A, Liu S, Larsen JE, Wang J, et al:
Effect of KRAS oncogene substitutions on protein behavior:
Implications for signaling and clinical outcome. J Natl Cancer
Inst. 104:228–239. 2012.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Hammond DE, Mageean CJ, Rusilowicz EV,
Wickenden JA, Clague MJ and Prior IA: Differential reprogramming of
isogenic colorectal cancer cells by distinct activating KRAS
mutations. J Proteome Res. 14:1535–1546. 2015.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Zhou ZW, Ambrogio C, Bera AK, Li Q, Li XX,
Li L, Son J, Gondi S, Li J, Campbell E, et al: KRASQ61H
preferentially signals through MAPK in a RAF dimer-dependent manner
in non-small cell lung cancer. Cancer Res. 80:3719–3731.
2020.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Ecker V, Stumpf M, Brandmeier L, Neumayer
T, Pfeuffer L, Engleitner T, Ringshausen I, Nelson N, Jücker M,
Wanninger S, et al: Targeted PI3K/AKT-hyperactivation induces cell
death in chronic lymphocytic leukemia. Nat Commun.
12(3526)2021.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Burgess MR, Hwang E, Mroue R, Bielski CM,
Wandler AM, Huang BJ, Firestone AJ, Young A, Lacap JA, Crocker L,
et al: KRAS allelic imbalance enhances fitness and modulates MAP
kinase dependence in cancer. Cell. 168:817–829.e15. 2017.PubMed/NCBI View Article : Google Scholar
|