Introduction
Osteoarthritis (OA) is the most common form of
arthritis and accounts for pain and disability all over the world
(1). OA is defined as a
degenerative joint disease contributing to the damage of cartilage
and its surrounding tissues (2).
It is generally acknowledged that OA is a severe and debilitating
disease resulting in a poor quality of life, and imposes a great
burden on healthcare systems and costs to society due to the ageing
population (3,4). Notably, patients with OA are at an
elevated risk of cardiovascular mortality (5). The extracellular matrix (ECM) is
regarded as a complex network comprising secreted extracellular
molecules, and its degradation is deemed a central hallmark of OA
(6,7). A number of studies have highlighted
that inflammatory responses and the apoptosis of chondrocytes are
associated with the pathology of OA (8-10).
Despite much progress being made, the pathogenesis behind OA
remains uncertain, as the molecular mechanism implicated in the
degradation of cartilage matrix and the progression of OA is
complicated (11,12). Therefore, more efforts need to be
made to further investigate the underlying mechanism.
Chemokine-like factor 1 (CKLF1) is an unusual member
of the chemokine family that was cloned in 2001, with two
successive cysteine residues in the sequence (13,14).
Mounting evidence has verified that CKLF1 functions as a crucial
modulator in human diseases (15,16).
For instance, the findings of a study by Liu et al (17) suggest that CKLF1 serves an
oncogenic role in the tumorigenesis and metastasis of
hepatocellular carcinoma. CKLF1 contributes to neointimal
hyperplasia via the activation of the NF-κB/vascular cell adhesion
molecule 1 signaling pathway (18). Additionally, Pan et al
(19) proposed that CKLF1 exhibits
higher expression in necrotic cartilage tissues compared with
normal cartilage tissues. However, the relationship between CKLF1
and OA remains elusive.
CC chemokine receptor 5 (CCR5) is a receptor of
CKLF1(20). Extensive research has
revealed that CCR5 participates in the initiation and progression
of breast, gastric, colorectal and pancreatic cancer, in which CCR5
upregulation may elicit pro-tumor effects while CCR5 downregulation
may elicit the opposite effects (21-25).
Furthermore, the CKLF1/CCR5 axis has been reported to promote
neutrophil migration in cerebral ischemia (20). However, whether CKLF1 exerts its
functions in OA by mediating CCR5 expression still requires
investigation.
The aim of the present study was to evaluate the
significance of CKLF1 and to determine the relationship between
CKLF1 and CCR5 in OA.
Materials and methods
Cell culture and treatment
The culture medium for the murine chondrogenic ATDC5
cell line (RIKEN BioResource Center) was DMEM/F12 (HyClone; Cytiva)
with 5% FBS (Cytiva), 100 U/ml penicillin and 100 µg/ml
streptomycin, and cells were routinely maintained in a humidified
incubator with 5% CO2 at 37˚C. To induce inflammatory
injury, untransfected or transfected ATDC5 cells were exposed to 10
ng/ml IL-1β for 24 h at 37˚C (26).
Plasmid transfection
The specific small interfering RNAs (siRNAs)
targeting CKLF1 (siRNA-CKLF1-1 sense, 5'-UUCACAAAGCAUUUCAGAGUA-3'
and antisense, 5'-CUCUGAAAUGCUUUGUGAAGU-3'; and siRNA-CKLF1-2
sense, 5'-UCAUAGAUGUCACAGUUACCA-3' and antisense,
5'-GUAACUGUGACAUCUAUGAUC-3') were provided by Shanghai GenePharma
Co., Ltd., with non-targeting siRNA [siRNA-negative control (NC)
sense, 5'-AUCGCAACAUAGACAGCUAACAG-3' and antisense,
5'-CUGUUAGCUGUCUAUGUUGCGAU-3'] also from Shanghai GenePharma Co.,
Ltd., used as an NC. CCR5 overexpression vector (Ov-CCR5) and the
empty overexpression vector were constructed by Sangon Biotech Co.,
Ltd., using the pBluescript vector. The aforementioned plasmids (20
nM) were transfected into ATDC5 cells using
Lipofectamine® 3000 (Thermo Fisher Scientific, Inc.) at
37˚C for 48 h. Cells were harvested at 48 h post-transfection for
subsequent experiments.
Cell Counting Kit-8 (CCK-8) assay
In brief, following IL-1β stimulation, 10 µl CCK-8
solution (Bioswamp; Wuhan Bienle Biotechnology Co., Ltd.) was added
to the transfected cells (5,000 cells/well) that had been plated
into a 96-well plate. After incubation for an additional 1 h at
37˚C, the optical density (OD) value was recorded at 450 nm using a
microplate reader (BioTek Instruments, Inc.). Cell viability
(%)=(OD treatment-OD blank)/(OD control-OD blank) x100.
TUNEL
Apoptosis was assessed using an in situ cell
death detection kit (Chemicon International; Thermo Fisher
Scientific, Inc.) in accordance with the manufacturer's
instructions. Briefly, ATDC5 cells were immobilized with 4%
paraformaldehyde for 15 min at room temperature, followed by PBS
washing. Then, cells were immersed in 50 µl TUNEL reaction mixture
for 1 h at 37˚C and incubated with 0.5 µg/ml DAPI (Beyotime
Institute of Biotechnology) for 10 min at room temperature.
Finally, after the addition of Antifade Mounting Medium (Beyotime
Institute of Biotechnology), the images were captured under a
fluorescence microscope (Olympus Corporation) in six random fields
of view. The apoptotic rate was calculated as follows: Apoptosis
rate=(average number of apoptotic cells/average number of total
cells) x100.
ELISA
Following indicated transfection or treatment, the
cells were centrifuged at 1,000 x g for 5 min at 4˚C. The
supernatant was collected and used for ELISA. Levels of
inflammatory cytokines TNF-α and IL-6 were examined using mouse
TNF-α ELISA kit (cat. no. ab208348) and mouse IL-6 ELISA kit (cat.
no. ab222503) (both Abcam) according to the manufacturer's
protocol. The OD value at 450 nm was detected using a microplate
reader (BioTek Instruments, Inc.).
Detection of sulfated
glycosaminoglycan (sGAG)
The production of sGAG was assessed using the
dimethylmethylene blue (DMMB) method (27). Cell suspension (20 µl) at the
density of 4x106 cells/cm2 was mixed with 200
µl DMMB reagent (Sigma-Aldrich; Merck KGaA), and the absorbance at
525 nm was recorded using a FlexStation 3 MultiMode Microplate
Reader (Molecular Devices, LLC). Total sGAG was normalized to total
protein for cell division with the application of the BCA protein
assay kit (Pierce; Thermo Fisher Scientific, Inc.).
Reverse transcription-quantitative PCR
(RT-qPCR)
Extraction of total RNA from cells was carried out
using TRI Reagent® (Molecular Research Center, Inc.),
and cDNA was synthesized using the SuperScript RT kit (Invitrogen;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocol. PCR analysis was conducted with FastStart Universal SYBR
Green Master (Roche Diagnostics GmbH) and a 7500c Real-Time PCR
Detection System (Applied Biosystems; Thermo Fisher Scientific,
Inc.). The following thermocycling conditions were used: Initial
denaturation at 95˚C for 10 min, followed by 37 cycles of
denaturation at 95˚C for 15 sec and annealing at 60˚C for 1 min,
then extension for 10 min at 65˚C. The following primers were used
in the present study: CKLF1 forward, 5'-GGCCTTTGCTTATCTCCTTTCC-3'
and reverse, 5'-AGCCTAGCAATCTGCTGTCC-3'; TNF-α forward,
5'-ACCCTCACACTCACAAACCA-3' and reverse, 5'-ACCCTGAGCCATAATCCCCT-3';
IL-6 forward, 5'-GCCTTCTTGGGACTGATGCT-3' and reverse,
5'-TGTGACTCCAGCTTATCTCTTGG-3'; MMP3 forward,
5'-CATCCCCTGATGTCCTCGTG-3' and reverse, 5'-CTTCTTCACGGTTGCAGGGA-3';
MMP13 forward, 5'-ACCCAGCCCTATCCCTTGAT-3' and reverse,
5'-TCTTCCATGTGGTTCCAGCC-3'; type II collagen forward,
5'-ATGAGGGAGCGGTAGAGACC-3' and reverse, 5'-GCCCTAATTTTCGGGCATCC-3';
a disintegrin and metalloproteinase with thrombospondin motifs type
4 (ADAMTS-4) forward, 5'-CCTACCTGGATCAGGCGTTC-3' and reverse,
5'-CTCCCAGAAGGAGCCTTGAC-3'; a disintegrin and metalloproteinase
with thrombospondin motifs type 5 (ADAMTS-5) forward,
5'-GCAGGGAACATAGGCAGGTT-3' and reverse,
5'-ACCAAACTATTCGGTTAGGCTGA-3'; aggrecan forward,
5'-CATGCTTATGCCTTCCGAGC-3' and reverse, 5'-CTTTCTTCTGCCCGAGGGTT-3';
CCR5 forward, 5'-AGCCGGGAAGGTAGTCTCAT-3' and reverse,
5'-GGGGCGTTCTCCAAAACAAC-3'; and GAPDH forward,
5'-GCCTCCTCCAATTCAACCCT-3' and reverse, 5'-CTCGTGGTTCACACCCATCA-3'.
Gene expression was calculated using the 2-ΔΔCq method
(28) and GAPDH acted as the
endogenous control.
Western blotting
The collection and quantification of total protein
from cells were performed using RIPA lysis buffer (Beyotime
Institute of Biotechnology) and a BCA Protein Assay Kit (Pierce;
Thermo Fisher Scientific, Inc.), respectively. Next, protein
samples (25 µg per lane) were electrophoresed on 10% gels using
SDS-PAGE and transferred to PVDF membranes (Merck KGaA). After the
membranes were incubated with 5% non-fat milk for 2 h at room
temperature, non-specific binding was impeded. Subsequently, the
membranes were probed with primary antibodies at 4˚C overnight.
Anti-CKLF1 (1/1,000; cat. no. ab180512), anti-Bcl-2 (1/1,000; cat.
no. ab182858), anti-Bax (1/1,000; cat. no. ab32503), anti-cleaved
caspase 3 (1/5,000; cat. no. ab214430), anti-caspase 3 (1/2,000;
cat. no. ab184787), anti-MMP3 (1/1,000; cat. no. ab52915),
anti-MMP13 (1/3,000; cat. no. ab39012), anti-type II collagen
(1/1,000; cat. no. ab34712), anti-ADAMTS-4 (1/1,000; cat. no.
ab185722), anti-ADAMTS-5 (1/250; cat. no. ab41037), anti-CCR5
(1/1,000; cat. no. ab65850) and anti-GAPDH (1/2,500; cat. no.
ab9485) antibodies were all purchased from Abcam, while the
anti-aggrecan (1/1,000; cat. no. bs-1223R) antibody was obtained
from BIOSS. On the following day, the membranes were incubated with
HRP-conjugated secondary antibody (1/2,000; cat. no. ab6721; Abcam)
for 2 h at room temperature. ECL Prime Western Blotting Detection
Reagent (Amersham; Cytiva) was employed to visualize the protein
bands, and band intensity was determined using Image-Pro Plus
software (version 6.0; Media Cybernetics, Inc.). GAPDH was used as
the loading control.
Co-immunoprecipitation (Co-IP)
ATDC5 cells were rinsed with pre-cooled PBS at 4˚C
for 2 h and dissolved in RIPA lysis buffer (Beyotime Institute of
Biotechnology) on ice for 30 min. After centrifugation at 13,000 x
g for 10 min at 4˚C, the supernatant of cell lysate (500 µg) was
collected and probed with 2 µg IgG antibody, CKLF1 antibody (cat.
no. abs138894; Absin Bioscience, Inc.) or CCR5 antibody (cat. no.
AM20421PU-N; OriGene Technologies, Inc.,) overnight at 4˚C,
followed by incubation with 0.2 mg protein A agarose beads (Pierce;
Thermo Fisher Scientific, Inc.) at room temperature for an
additional 2 h. IgG was used as a negative control. After the IP
reaction, agarose beads were centrifuged at 1,000 x g for 3 min at
4˚C to the bottom of the tube. The supernatant was then carefully
absorbed, and the agarose beads were washed three times with 1 ml
lysis buffer. A total of 15 µl 2X SDS sample buffer was finally
added for boiling at 100˚C for 5 min, followed by western blotting
as aforementioned.
Statistical analysis
All statistical analyses were performed using
GraphPad Prism 8 software (GraphPad Software; Dotmatics) and data
are presented as the mean ± SD of three independent experiments.
Unpaired Student's t-test was used for comparisons between two
groups, while one-way ANOVA followed by Tukey's test was applied
for comparisons among multiple groups. P<0.05 was considered to
indicate a statistically significant difference.
Results
Knockdown of CKLF1 enhances viability
while alleviating inflammation in IL-1β-insulted chondrocytes
A previous study has reported that CKLF1 exhibits
higher expression in necrotic cartilage tissues compared with
normal cartilage tissues (19). To
investigate the role of CKLF1 in IL-1β-stimulated chondrocyte
injury, CKLF1 expression was analyzed by RT-qPCR and western
blotting after ATDC5 cells were stimulated with IL-1β. The results
suggested that IL-1β treatment elevated the expression levels of
CKLF1 in ATDC5 cells (Fig. 1A and
B). To explore the role of CKLF1
in IL-1β-induced ATDC5 cells, CKLF1 was silenced by transfection
with siRNA-CKLF1-1 or siRNA-CKLF1-2 and the interference efficiency
was examined by RT-qPCR. As shown in Fig. 1C, CKLF1 expression was
significantly downregulated in the siRNA-CKLF1-1 and siRNA-CKLF1-2
groups compared with the siRNA-NC group. Further, the elevated
CKLF1 expression in IL-1β-treated chondrocytes were decreased
following transfection of siRNA-CKLF1-1/2 plasmids, and
siRNA-CKLF1-1 was selected for the subsequent loss-of-function
experiments due to an improved interference efficiency (Fig. 1D and E). In a CCK-8 assay, the viability of the
ATDC5 cells was notably reduced following IL-1β treatment and the
suppressed cell viability was restored when CKLF1 was downregulated
(Fig. 1F). TNF-α and IL-6 are
proinflammatory cytokines (29).
The experimental results of ELISA and RT-qPCR analysis demonstrated
that TNF-α and IL-6 levels were significantly increased in
IL-1β-exposed ATDC5 cells, while the levels were significantly
decreased after transfection with siRNA-CKLF1-1 (Fig. 1G and H). In summary, CKLF1 silencing enhanced
the viability and decreased inflammation in IL-1β-challenged
chondrocytes.
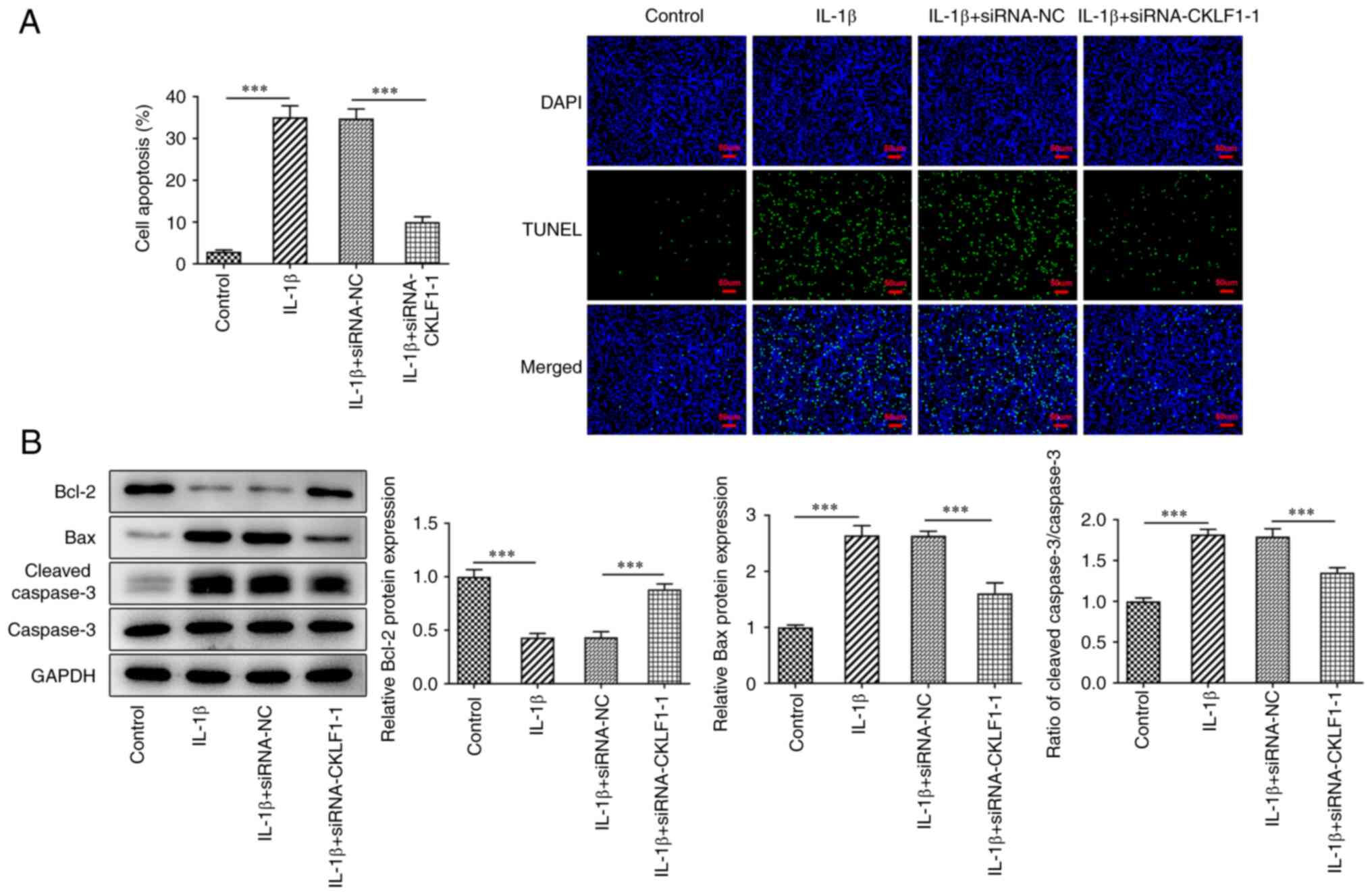
CKLF1 knockdown protects chondrocytes
against IL-1β-triggered apoptosis
Dysregulation of apoptosis occurring in
osteoarthritic cartilage is responsible for the progression of OA
(10). Therefore, apoptosis was
subsequently examined using TUNEL assays and western blot analysis
to explore the effect of CKLF1 silencing on IL-1β-induced apoptosis
in ATDC5 cells. As shown in Fig.
2A, it was observed that IL-1β-challenged ATDC5 cells possessed
a higher apoptotic rate compared with the control group. However,
CKLF1 knockdown impeded the IL-1β-induced apoptosis in ATDC5 cells.
A similar result was also obtained in western blot analysis, which
indicated that the downregulated protein levels of Bcl-2 and the
upregulated levels of Bax and cleaved caspase3/caspase3 in
IL-1β-exposed ATDC5 cells were reversed after CKLF1 was silenced
(Fig. 2B). Overall, CKLF1
interference served a suppressive role in IL-1β-triggered
chondrocyte apoptosis.
CKLF1 depletion alleviates
IL-1β-induced chondrocyte ECM degradation
Type II collagen and aggrecan are the main
components of cartilage ECM (30).
Furthermore, MMP3, MMP13, ADAMTS-4 and ADAMTS-5 are essential for
the degradation of the ECM (31).
To determine whether CKLF1 participates in ECM degradation in OA,
the expression of MMP3, MMP13, type II collagen, ADAMTS-4, ADAMTS-5
and aggrecan at the mRNA and protein levels was assessed by RT-qPCR
and western blot analysis, respectively. The results revealed that
IL-1β treatment significantly increased the expression levels of
MMP3, MMP13, ADAMTS-4 and ADAMTS-5, while it significantly reduced
the expression levels of type II collagen and aggrecan. However,
these effects were all counteracted after CKLF1 was knocked down
(Fig. 3A-D). In addition, using
the DMMB method, sGAG content was revealed to be significantly
reduced in ATDC5 cells following IL-1β treatment and to be
significantly increased again after transfection with siRNA-CKLF1
(Fig. 3E). To conclude, all of
these results suggested that CKLF1 knockdown weakened the
IL-1β-induced ECM degradation in chondrocytes.
CKLF1 binds to its receptor CCR5
CCR5 is a receptor of CKLF1(19). Therefore, subsequent experiments
investigated whether CKLF1 could regulate the IL-1β-induced damage
in chondrocytes by binding to its receptor CCR5. Western blotting
demonstrated that CCR5 was upregulated in IL-1β-stimulated ATDC5
cells. After CKLF1 was silenced, the protein levels of CCR5 were
significantly reduced (Fig. 4A).
Furthermore, a Co-IP assay verified the interaction between CKLF1
and CCR5 (Fig. 4B and C). Overall, CKLF1 bound with its receptor
CCR5.
Silencing CKLF1 reverses the
IL-1β-stimulated viability decrease and inflammatory response in
chondrocytes by downregulating CCR5 expression
To test the hypothesis that CKLF1 was involved in
the development of OA by binding CCR5, CCR5 was first upregulated
by transfection with Ov-CCR5 (Fig.
5A). CCK-8 assays revealed that the increased viability of
IL-1β-treated ATDC5 cells induced by CKLF1 knockdown was reduced
again after CCR5 was overexpressed (Fig. 5B). ELISA and RT-qPCR analysis also
demonstrated that CCR5 overexpression reversed the reduced levels
of TNF-α and IL-6 that were induced by the knockdown of CKLF1
(Fig. 5C and D). In summary, CCR5 elevation restored
the impacts of CKLF1 interference on the viability injury and
inflammation in IL-1β-treated chondrocytes.
CKLF1 depletion mitigates the
IL-1β-evoked apoptosis in chondrocytes by inhibiting CCR5
expression
Whether CKLF1 could regulate IL-1β-induced apoptosis
in chondrocytes by binding to its receptor CCR5 was studied in the
following experiments. TUNEL assays revealed that CKLF1 knockdown
reduced the apoptosis of IL-1β-treated ATDC5 cells, while this
influence was counteracted by CCR5 overexpression (Fig. 6A). As expected, CCR5 overexpression
reversed the increased Bcl-2 protein level and the decreased Bax
and cleaved caspase3/caspase3 levels caused by CKLF1 knockdown
(Fig. 6B). Overall, CKLF1
interference suppressed CCR5 expression to attenuate the
IL-1β-enhanced chondrocyte apoptosis.
CKLF1 knockdown alleviates ECM
degradation in IL-1β-treated chondrocytes via the suppression of
CCR5
To observe the change of ECM degradation in
IL-1β-treated chondrocytes with CKLF1 knockdown and CCR5
overexpression, the expression levels of MMP3, MMP13, type II
collagen, ADAMTS-4, ADAMTS-5 and aggrecan were determined by
RT-qPCR and western blotting. CKLF1 silencing resulted in reduced
MMP3, MMP13, ADAMTS-4 and ADAMTS-5 expression, and increased type
II collagen and aggrecan expression in IL-1β-exposed chondrocytes;
however, this was reversed by overexpression of CCR5 (Fig. 7A-D). Furthermore, the increased
sGAG content following CKLF1 knockdown in IL-1β-exposed
chondrocytes was decreased by CCR5 overexpression (Fig. 7E). Collectively, CKLF1 contributed
to ECM degradation in IL-1β-induced chondrocytes by interacting
with CCR5.
 | Figure 7CKLF1 knockdown alleviates
extracellular matrix degradation in IL-1β-treated chondrocytes via
the suppression of CCR5. (A) RT-qPCR and (B) western blotting were
performed to examine MMP3, MMP13 and type II collagen expression in
IL-1β-exposed ATDC5 cells. (C) RT-qPCR and (D) western blotting
were performed to examine ADAMTS-4, ADAMTS-5 and aggrecan
expression in IL-1β-exposed ATDC5 cells. (E) sGAG content was
examined using the dimethylmethylene blue method.
**P<0.01 and ***P<0.001. CKLF1,
chemokine-like factor 1; CCR5, CC chemokine receptor 5; ADAMTS-4, a
disintegrin and metalloproteinase with thrombospondin motifs type
4; ADAMTS-5, a disintegrin and metalloproteinase with
thrombospondin motifs type 5; Ov-CCR5, CCR5 overexpression vector;
Ov-NC, empty overexpression vector; RT-qPCR, reverse
transcription-quantitative PCR; sGAG, soluble glycosamine sulfate
additive; siRNA, small interfering RNA. |
Discussion
OA has long been acknowledged as a highly prevalent
degenerative joint disease, and it is the most commonly diagnosed
condition of the musculoskeletal system (11,12).
At present, pain management or joint replacement remain the only
available treatments for OA due to the complicated factors involved
in its onset and development, and the incomplete understanding of
the molecular mechanisms (32).
There are increasing reports regarding the pathogenesis of OA in
terms of the inflammatory response (33,34).
The interplay between inflammation and OA has been highlighted by
cartilage injury in OA; thus, exploring effective treatments for
inflammation has also become a research area in OA therapy
(33). Furthermore, dysregulation
of apoptosis occurring in osteoarthritic cartilage is also
responsible for the progression of OA (10). The ECM is an intricate and
specialized three-dimensional macromolecular network (7), and the degradation of the ECM is
viewed as a pivotal hallmark of OA (6). IL-1β is recognized as the major
catabolic factor in the pathogenesis of OA (34,35),
and is frequently utilized to stimulate OA models in vitro
(26). Therefore, 10 ng/ml IL-1β
was employed in the present study to induce an OA model in ATDC5
cells, and then viability, inflammation, apoptosis and degradation
of the ECM in OA were evaluated.
CKLF1 is a cytokine that is widely expressed in the
human body (13). A growing body
of evidence has demonstrated that CKLF1 possesses broad-spectrum
biological functions in human diseases (15,16).
Furthermore, Tao et al (36) demonstrated that CKLF1 has a
distinctly increased expression level in patients with OA. Pan
et al (19) proposed that
CKLF1 exhibits higher expression in necrotic cartilage tissues
compared with normal cartilage tissues. Accumulating research has
revealed that CKLF1 is involved in the inflammation response in
hepatocellular carcinoma (16),
cerebral ischemia injury (37),
psoriasis (38) and renal injury
(39). CKLF1 facilitates the
degradation of the ECM to exacerbate the process of abdominal
aortic aneurysms (40). In the
present study, CKLF1 was revealed to be upregulated in
IL-1β-treated ATDC5 cells. Functional experiments demonstrated that
the attenuated viability of IL-1β-challenged ATDC5 cells was
improved again after CKLF1 was knocked down. Additionally, the
levels and expression of proinflammatory cytokines (TNF-α and IL-6)
were elevated in ATDC5 cells following IL-1β treatment, but
restored by CKLF1 interference, which indicated that the
IL-1β-induced inflammation in chondrocytes was suppressed by the
silencing of CKLF1. Furthermore, the apoptosis of ATDC5 cells
triggered by IL-1β exposure was hampered when CKLF1 was knocked
down, and was accompanied by elevated Bcl-2 protein levels and
decreased Bax and cleaved caspase3/caspase3 levels in the IL-1β +
siRNA-CKLF1-1 group compared with those in the IL-1β + siRNA-NC
group.
Proinflammatory cytokines, including type II
collagen and aggrecan, have been reported to stimulate the
degradation of the ECM (41).
Additionally, MMPs, including MMP3 and MMP13, which have been
determined as main contributors of ECM degradation, are in charge
of type II collagen degradation (42). ADAMTS proteases are zinc-dependent
metalloproteinases that are connected with the degradation of the
ECM, including aggrecan degradation by ADAMTS-4 and ADAMTS-5
(43,44). Furthermore, MMP3, MMP13, ADAMTS-4
and ADAMTS-5 are produced in activated chondrocytes (45). Consistent with these findings, the
present study revealed that the expression levels of MMP3, MMP13,
ADAMTS-4 and ADAMTS-5 were increased, while the expression levels
of type II collagen and aggrecan were decreased in IL-1β-treated
chondrocytes. However, these effects were all mitigated by the
knockdown of CKLF1. It has been reported that sGAG can bind with
ECM-related proteins to direct cellular processes (46). The present study also revealed that
the inhibited sGAG content in IL-1β-exposed chondrocytes was
restored by CKLF1 knockdown.
CCR5, which was first identified as a human
immunodeficiency virus type 1 coreceptor, has been recognized as a
receptor of CKLF1 in transient cerebral ischemia (19,47).
Furthermore, CCR5 is highly expressed in chondrocytes in the
inflammatory environment (48-50).
Based on these findings, the present data confirmed that the
increased expression levels of CCR5 in IL-1β-induced chondrocytes
were decreased after CKLF1 was silenced. Additionally, the present
study confirmed the interaction of CKLF1 with its receptor CCR5. In
addition, after CCR5 was overexpressed, the stimulated viability
and the attenuated inflammation and apoptosis in IL-1β-treated
chondrocytes, that had been caused by CKLF1 knockdown, were all
reversed. Furthermore, the decreased expression levels of MMP3,
MMP13, ADAMTS-4 and ADAMTS-5, and the increased expression levels
of type II collagen and aggrecan in IL-1β-treated chondrocytes due
to CKLF1 interference, were also restored by CCR5
overexpression.
However, there are a number of limitations of the
present study. Only the regulatory effect of CKLF1 and CCR5 on the
damage to chondrocytes exposed to IL-1β in vitro was
discussed. Further in vivo experiments will need to be
performed in future investigations to support the conclusions
obtained in the present study. Additionally, future studies are
required to clarify the expression levels of CKLF1 in OA tissues
and the impacts of CKLF1 overexpression on OA.
In summary, the present study revealed that
silencing of CKLF1 bound to its receptor CCR5 to ameliorate
IL-1β-induced inflammation, apoptosis and degradation of the ECM in
chondrocytes, therefore hindering the progression of OA (Fig. 8). To the best of our knowledge,
this was the first time that the role of CKLF1 in OA was assessed
and the present study was the first to demonstrate an association
between CKLF1 and CCR5 in OA. Overall, the findings of the present
study may have potential implications for OA therapy. However,
future studies are required to further clarify the expression
levels of CKLF1 in OA tissues and the role of CKLF1 in OA in
vivo.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by the Project of
Zhejiang Administration of Traditional Chinese Medicine (grant no.
2020ZA112).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HW and ZW designed the study. HW, ZW and KX
performed the experiments and analyzed the data. HW drafted the
manuscript and interpreted the data. ZW and KX revised the
manuscript for important intellectual content. All authors have
read and approved the final manuscript. HW and ZW confirm the
authenticity of all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
O'Neill TW, McCabe PS and McBeth J: Update
on the epidemiology, risk factors and disease outcomes of
osteoarthritis. Best Pract Res Clin Rheumatol. 32:312–326.
2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Litwic A, Edwards MH, Dennison EM and
Cooper C: Epidemiology and burden of osteoarthritis. Br Med Bull.
105:185–199. 2013.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Pereira D, Ramos E and Branco J:
Osteoarthritis. Acta Med Port. 28:99–106. 2015.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Martel-Pelletier J, Barr AJ, Cicuttini FM,
Conaghan PG, Cooper C, Goldring MB, Goldring SR, Jones G, Teichtahl
AJ and Pelletier JP: Osteoarthritis. Nat Rev Dis Primers.
2(16072)2016.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Palazzo C, Nguyen C, Lefevre-Colau MM,
Rannou F and Poiraudeau S: Risk factors and burden of
osteoarthritis. Ann Phys Rehabil Med. 59:134–138. 2016.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Rahmati M, Nalesso G, Mobasheri A and
Mozafari M: Aging and osteoarthritis: Central role of the
extracellular matrix. Ageing Res Rev. 40:20–30. 2017.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Theocharis AD, Skandalis SS, Gialeli C and
Karamanos NK: Extracellular matrix structure. Adv Drug Deliv Rev.
97:4–27. 2016.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Shen J, Abu-Amer Y, O'Keefe RJ and
McAlinden A: Inflammation and epigenetic regulation in
osteoarthritis. Connect Tissue Res. 58:49–63. 2017.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Chow YY and Chin KY: The role of
inflammation in the pathogenesis of osteoarthritis. Mediators
Inflamm. 2020(8293921)2020.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Hwang HS and Kim HA: Chondrocyte apoptosis
in the pathogenesis of osteoarthritis. Int J Mol Sci.
16:26035–26054. 2015.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Xia B, Di C, Zhang J, Hu S, Jin H and Tong
P: Osteoarthritis pathogenesis: A review of molecular mechanisms.
Calcif Tissue Int. 95:495–505. 2014.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Geyer M and Schönfeld C: Novel insights
into the pathogenesis of osteoarthritis. Current Rheumatol Rev.
14:98–107. 2018.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Liu DD, Song XY, Yang PF, Ai QD, Wang YY,
Feng XY, He X and Chen NH: Progress in pharmacological research of
chemokine like factor 1 (CKLF1). Cytokine. 102:41–50.
2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Han W, Lou Y, Tang J, Zhang Y, Chen Y, Li
Y, Gu W, Huang J, Gui L, Tang Y, et al: Molecular cloning and
characterization of chemokine-like factor 1 (CKLF1), a novel human
cytokine with unique structure and potential chemotactic activity.
Biochem J. 357:127–135. 2001.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Cai X, Deng J, Ming Q, Cai H and Chen Z:
Chemokine-like factor 1: A promising therapeutic target in human
diseases. Exp Biol Med (Maywood). 245:1518–1528. 2020.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Li Y, Yu H and Feng J: Role of
chemokine-like factor 1 as an inflammatory marker in diseases.
Front Immunol. 14(1085154)2023.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Liu Y, Liu L, Zhou Y, Zhou P, Yan Q, Chen
X, Ding S and Zhu F: CKLF1 enhances inflammation-mediated
carcinogenesis and prevents doxorubicin-induced apoptosis via
IL6/STAT3 signaling in HCC. Clin Cancer Res. 25:4141–4154.
2019.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Liu X, Qu C, Zhang Y, Fang J, Teng L,
Zhang R, Zhang X and Shen C: Chemokine-like factor 1 (CKLF1)
aggravates neointimal hyperplasia through activating the
NF-κB/VCAM-1 pathway. FEBS Open Bio. 10:1880–1890. 2020.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Pan Z, Yin H, Wang S, Xiong G and Yin Z:
Bcl-xL expression following articular cartilage injury and its
effects on the biological function of chondrocytes. Eng Life Sci.
20:571–579. 2020.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Chen C, Chu SF, Ai QD, Zhang Z and Chen
NH: CKLF1/CCR5 axis is involved in neutrophils migration of rats
with transient cerebral ischemia. Int Immunopharmacol.
85(106577)2020.PubMed/NCBI View Article : Google Scholar
|
|
21
|
González-Martin A, Mira E and Mañes S:
CCR5 as a potential target in cancer therapy: Inhibition or
stimulation? Anticancer Agents Med Chem. 12:1045–1057.
2012.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Velasco-Velázquez M, Xolalpa W and Pestell
RG: The potential to target CCL5/CCR5 in breast cancer. Expert Opin
Ther Targets. 18:1265–1275. 2014.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Aldinucci D and Casagrande N: Inhibition
of the CCL5/CCR5 axis against the progression of gastric cancer.
Int J Mol Sci. 19(1477)2018.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Pervaiz A, Zepp M, Georges R, Bergmann F,
Mahmood S, Faiza S, Berger MR and Adwan H: Antineoplastic effects
of targeting CCR5 and its therapeutic potential for colorectal
cancer liver metastasis. J Cancer Res Clin Oncol. 147:73–91.
2021.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Singh SK, Mishra MK, Eltoum IA, Bae S,
Lillard JW Jr and Singh R: CCR5/CCL5 axis interaction promotes
migratory and invasiveness of pancreatic cancer cells. Sci Rep.
8(1323)2018.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Xie P, Dan F, Yu G, Ruan W and Yu H:
Laquinimod mitigated IL-1β-induced impairment of the cartilage
extracellular matrix in human ATDC5 chondrocytes. Chem Res Toxicol.
33:933–939. 2020.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Ladner YD, Alini M and Armiento AR: The
dimethylmethylene blue assay (DMMB) for the quantification of
sulfated glycosaminoglycans. Methods Mol Biol. 2598:115–121.
2023.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Landskron G, De la Fuente M, Thuwajit P,
Thuwajit C and Hermoso MA: Chronic inflammation and cytokines in
the tumor microenvironment. J Immunol Res.
2014(149185)2014.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Luo Y, Sinkeviciute D, He Y, Karsdal M,
Henrotin Y, Mobasheri A, Önnerfjord P and Bay-Jensen A: The minor
collagens in articular cartilage. Protein Cell. 8:560–572.
2017.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Malemud CJ: Inhibition of MMPs and
ADAM/ADAMTS. Biochem Pharmacol. 165:33–40. 2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Mathiessen A and Conaghan PG: Synovitis in
osteoarthritis: Current understanding with therapeutic
implications. Arthritis Res Ther. 19(18)2017.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Vilá S: Inflammation in osteoarthritis. P
R Health Sci J. 36:123–129. 2017.PubMed/NCBI
|
|
34
|
Jenei-Lanzl Z, Meurer A and Zaucke F:
Interleukin-1β signaling in osteoarthritis-chondrocytes in focus.
Cell Signal. 53:212–223. 2019.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Blasioli DJ and Kaplan DL: The roles of
catabolic factors in the development of osteoarthritis. Tissue Eng
Part B Rev. 20:355–363. 2014.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Tao K, Tang X, Wang B, Li RJ, Zhang BQ,
Lin JH, Zhang BQ, Lin JH and Li H: Distinct expression of
chemokine-like factor 1 in synovium of osteoarthritis, rheumatoid
arthritis and ankylosing spondylitis. J Huazhong Univ Sci Technolog
Med Sci. 36:70–76. 2016.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Ai Q, Chen C, Chu S, Luo Y, Zhang Z, Zhang
S, Yang P, Gao Y, Zhang X and Chen N: IMM-H004 protects against
cerebral ischemia injury and cardiopulmonary complications via
CKLF1 mediated inflammation pathway in adult and aged rats. Int J
Mol Sci. 20(1661)2019.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Zheng Y, Wang Y, Zhang X, Tan Y, Peng S,
Chen L and He Y: C19, a C-terminal peptide of CKLF1, decreases
inflammation and proliferation of dermal capillaries in psoriasis.
Sci Rep. 7(13890)2017.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Chen C, Ai Q and Wei Y: Hydroxytyrosol
protects against cisplatin-induced nephrotoxicity via attenuating
CKLF1 mediated inflammation, and inhibiting oxidative stress and
apoptosis. Int Immunopharmacol. 96(107805)2021.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Li J, Bao X, Li Y, Wang Y, Zhao Z and Jin
X: Study of the functional mechanisms of osteopontin and
chemokine-like factor 1 in the development and progression of
abdominal aortic aneurysms in rats. Exp Ther Med. 12:4007–4011.
2016.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Risbud MV and Shapiro IM: Role of
cytokines in intervertebral disc degeneration: Pain and disc
content. Nat Rev Rheumatol. 10:44–56. 2014.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Cui N, Hu M and Khalil RA: Biochemical and
biological attributes of matrix metalloproteinases. Prog Mol Biol
Transl Sci. 147:1–73. 2017.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Mead TJ and Apte SS: ADAMTS proteins in
human disorders. Matrix Biol. 71-72:225–239. 2018.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Santamaria S and Yamamoto K: Analysis of
aggrecanase activity using neoepitope antibodies. Methods Mol Biol.
2043:125–136. 2020.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Wang T and He C: Pro-inflammatory
cytokines: The link between obesity and osteoarthritis. Cytokine
Growth Factor Rev. 44:38–50. 2018.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Hintze V, Samsonov SA, Anselmi M, Moeller
S, Becher J, Schnabelrauch M, Scharnweber D and Pisabarro MT:
Sulfated glycosaminoglycans exploit the conformational plasticity
of bone morphogenetic protein-2 (BMP-2) and alter the interaction
profile with its receptor. Biomacromolecules. 15:3083–3092.
2014.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Maeda K, Das D, Nakata H and Mitsuya H:
CCR5 inhibitors: Emergence, success, and challenges. Exp Opin Emerg
Drugs. 17:135–145. 2012.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Hsu YH, Hsieh MS, Liang YC, Li CY, Sheu
MT, Chou DT, Chen TF and Chen CH: Production of the chemokine
eotaxin-1 in osteoarthritis and its role in cartilage degradation.
J Cell Biochem. 93:929–939. 2004.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Yuan GH, Masuko-Hongo K, Sakata M, Tsuruha
J, Onuma H, Nakamura H, Aoki H, Kato T and Nishioka K: The role of
C-C chemokines and their receptors in osteoarthritis. Arthritis
Rheum. 44:1056–1070. 2001.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Alblowi J, Tian C, Siqueira MF, Kayal RA,
McKenzie E, Behl Y, Gerstenfeld L, Einhorn TA and Graves DT:
Chemokine expression is upregulated in chondrocytes in diabetic
fracture healing. Bone. 53:294–300. 2013.PubMed/NCBI View Article : Google Scholar
|