Introduction
Liver cancer is one of the deadliest types of cancer
globally (1) and is ranked fourth
and second regarding incidence and mortality in China (2). Hepatitis B and C viral infection,
alcohol consumption and non-alcoholic fatty liver disease are the
risk factors of liver cancer (3).
Although surgical resection remains the most effective treatment
for liver cancer, only patients with unilobar tumours with
preserved liver function and without hepatic vascular invasion
metastases qualify for surgery. However, surgical resection alone
cannot eradicate the tumour completely (4). Currently, chemotherapy remains an
important adjuvant therapy for liver cancer. Nevertheless, its
clinical use is often seriously restricted owing to poor
selectivity and adverse reactions (5). In addition, despite the rapid
progress in molecular-targeted therapies and immunotherapy
triggered by the emergence of small-molecule targeted drugs,
obstacles such as a narrow treatment window, low response rate and
multidrug resistance seriously hinder the clinical application of
these agents (6,7). Globally, although the morbidity and
mortality of liver cancer are increasing, the improvement and
efficacy of its major treatments remain seriously limited. Thus,
there is a pressing need to formulate novel curative protocols for
liver cancer that are safer and more effective.
Over the past two decades, nano-targeted delivery
systems have shown promise for tumour treatment (8,9).
Nano-targeted drug-delivery systems (DDSs) can not only passively
target the tumour by enhancing the penetration and retention (EPR)
effect (10,11) but also actively target tumour blood
vessels and cells via the targeted modification of carrier
materials to promote drug accumulation in tumours, improve drug
safety and reduce adverse events incidence (12-14).
Moreover, using a nano-targeted delivery system, chemotherapeutic
drugs can be made more soluble and stable and their affinity toward
blood proteins can be reduced (15,16).
More importantly, polymer-based nanoparticles (NPs) exhibit high
biocompatibility, biodegradability and structural generality,
thereby serving as additional alternatives to custom-made
drug-delivery vehicles according to specific requirements with
excellent treatment efficacy and safety performance (17-19).
In addition, NP systems can be administered orally, locally, via
injection, systemically and through the lungs, according to the
specific demands (20-22).
Numerous nano-systems have afforded improved therapeutic results in
animal models, with some of them entering clinical trials and some
even being translated to clinical practice (23-26).
However, complete translation of success in vivo experiments
into clinical use remains challenging. Therefore, previously,
‘variable-size’ and ‘stimulus-response’ strategies, such as the
core-shell strategy, surface carrying tactic and Trojan horse
tactic have been proposed (27-29).
The use of new functional carrier materials or targeted chemical
modifications has become a breakthrough in designing new
drug-delivery systems.
Targeted drug delivery can occur in two forms: A
passive targeting pattern, which is based on an enhanced EPR
action; and an active targeting pattern, which depends on the
introduction of targeting ligands into the NPs, for drug
distribution to a specific site (30). The combination of the two patterns
into one NP can significantly improve the selective accumulation
effectiveness of the targeted drug-delivery system and enhance its
medication delivery. Regarding active targeting, NP surfaces can be
coated with certain specific parts aimed at specific receptors that
are overexpressed in the tumour microenvironment or on the exterior
of cancerous tissues (31).
Glycyrrhetinic acid (GA) is mostly obtained from the
underground part of Glycyrrhiza glabra L. Negishi et
al (32) first reported that
rat hepatocyte membranes contained a GA-binding site, which was
later demonstrated to be the protein kinase Cα, which is highly
expressed in liver tumour cells at a level that is 1.5- to 5-fold
higher than that in normal hepatocytes (33,34).
Owing to the specific recognition ability of GA receptors and their
protective effects on normal liver tissues, GA-modified drugs and
NPs exhibit considerable liver-targeting ability and targeted
induction of apoptosis in liver cancer cells and have been used to
manage liver-related diseases in numerous countries and regions
(35-39).
Artesunate (ART) is an artemisinin derivative that
has been a focus of antitumour drug research and has shown
favorable inhibitory effects on liver cancer (40,41).
However, the low solubility, easy degradation in water, short
half-life, poor selectivity for tumours and low bioavailability of
ART significantly limits its antitumour effects and subsequent
clinical applications (42,43).
Thus, the incorporation of ART into novel nano-preparations is
proposed. GA-coated and ART-loaded polyethylene glycol (PEG)-poly
(lactic-co-glycolic acid) (PLGA) (ART/GA-PEG-PLGA) NPs were
previously prepared (42). In the
present study, the cellular uptake of these NPs mediated by the GA
receptor as well as their intracellular mechanisms, pro-apoptotic
effects, potential mechanisms, pharmacokinetics, biological
distribution and antitumour effects, were examined. Thus, the
collective results of the present study may provide insights into
the development of a drug-delivery system for effective liver
cancer treatment.
Materials and methods
Materials
Eagle's Minimal Essential Medium (EMEM) and F-12K
were purchased from Gibco; Thermo Fisher Scientific, Inc.;
parenzyme was also procured from Gibco; Thermo Fisher Scientific,
Inc.; 3,3'-dioctadecyloxacarbocyanine perchlorate (DiO) was
purchased from Beyotime Institute of Biotechnology. Chlorpromazine,
wortmannin, genistein and methyl-β-cyclodextrin were obtained from
Selleck Chemicals. Reactive oxygen species (ROS, cat. no. S0033M),
JC-1 (cat. no. C2005) and cell apoptosis and cell cycle detection
kits (cat. no. C1052) were purchased from Beyotime Institute of
Biotechnology. The Annexin V-FITC/PI double staining apoptosis
detection kit (cat. no. KGA-108) was obtained from Nanjing KeyGen
Biotech Co., Ltd.; marker10-180 (BE2666-250) was procured from
EASYBIO. Ponceaux (cat. no. A100860) and phenylmethanesulfonyl
fluoride (cat. no. 100754) were provided by Sangon Biotech Co.,
Ltd. ProBlott membrane regenerative fluid ZN1923 was purchased from
Beijing Biolab Technology Co., Ltd. Anti-caspase-3 (cat. no. 9662),
anti-caspase-7 (cat. no. 9492), anti-poly-(ADP-ribose)-polymerase
(PARP) (cat. no. 9542), anti-phosphorylated-p38 mitogen-activated
protein kinase (MAPK) (cat. no. 9211) and anti-p38 MAPK (cat. no.
9212) antibodies were obtained from Cell Signaling Technology,
Inc.
Cell culture
The HepG2 cell line (cat. no. HB-8065; cultured from
liver cancer, cells with high GA receptor expression) and A549 cell
line (cat. no. CCL-185; cultured from lung cancer, cells with low
GA receptors expression) were purchased from the American Tissue
Culture Collection (ATCC). Hep3B-luc was purchased from Wuxi Apptec
Co., Ltd. The HepG2 cell line was authenticated by Procell Life
Science & Technology Co., Ltd. using short tandem repeat (STR)
analysis. DNA was extracted from the HepG2 cells using Chelex100
(cat. no. 1432832; Bio-Rad Laboratories, Inc.). The 20 STRs,
including the Amelogenin locus, were amplified using the 21 CELLID
system and separated using ABI 3130X1 Genetic Analyzer (Thermo
Fisher Scientific, Inc.). The signals were subsequently analysed
using GeneMapper IDX software (v1.6; Applied Biosystems) and
compared via the ATCC, DSMZ (https://www.dsmz.de/), JCRB (https://cellbank.nibiohn.go.jp/) and Cellosaurus
(https://www.cellosaurus.org/) databases.
The authentication results revealed that the DNA of the cell line
matched perfectly with the type of cell lines in a cell line
retrieval.
Animal studies
Female BALB/c nude mice (total number, 24; age, 6-8
weeks; weight, 18-22 g) were obtained from Zhejiang Vitonolihua
Experimental Animal Technology Co., Ltd. Male Sprague Dawley (SD)
rats (total number, 8; age, 6-8 weeks; weight, 244-254 g) were
obtained from Jihui Laboratory Animal Co. Ltd. All mice and rats
were kept in specific pathogen-free (SPF) environment with a
temperature and humidity of 20-26˚C and 40-70%, respectively, under
a 12-h light/dark cycle. All mice were provided water and food
ad libitum. The rats were fasted overnight and fed 4 h after
dose administration, while they had free access to water. All
animal tests were performed according to the National Research
Council's Guide for the Care and Use of Laboratory Animals.
Uptake of NPs into HepG2 cells and
their intracellular localization
NPs were prepared and characterized as previously
reported (42). The uptake of NPs
into HepG2 cells and their intracellular mechanisms were
investigated.
Cellular uptake determination
To estimate the hepatoma-targeting ability of
GA-coated NPs, the affinity of NPs toward HepG2 and A549 cells was
compared. The fluorescent probe Nile red (NR) was loaded onto the
GA-modified NP to trace its uptake. Control groups were generated
using normal cells, free-NR and NR-encapsulated unmodified NPs
(NR/PEG-PLGA). The cells were seeded into each well at a density of
2x105 cells. Following an additional 24-h incubation
period, the cells were co-incubated with each group of drugs at the
equal NR of 50 µg/ml. Subsequently, they were washed with cold
phosphate-buffered solution (PBS) thrice, and then incubated for an
additional 0.5, 1 or 4 h. The fluorescence intensity in cells was
analysed via flow cytometry (FCM; Beckman DxFlex Flow Cytometer;
with Cytexpert 2.5 software; Beckman Coulter, Inc.).
Intracellular localization
HepG2 cells were seeded at a density of
2x104 cells/well of microscope slides and incubated at
37˚C in an atmosphere containing 5% CO2 for 24 h. Next,
they were incubated with each drug group (free-NR, NR/PEG-PLGA or
NR/GA-PEG-PLGA NPs) with an equivalent NR of 50 µg/ml for 4 h. The
cells were washed thrice using cold PBS and the cell membranes were
stained with DiO at 10 µM. Subsequently, the cells were washed
thrice and fixed on ice with 4% paraformaldehyde for 15 min
followed by three washes and staining with DAPI (10 µg/ml) for 5
min at room temperature. A confocal laser scanning microscope
(CLSM; LSCM 780; Carl Zeiss AG) was used to acquire fluorescence
images.
Uptake pathway identification
Pharmacological inhibitors were used to date the
uptake pathway. The pharmacological inhibitors were used at the
following concentrations: free-GA, 10 µM; 2-deoxyglucose (cat. no.
D8375; Sigma-Aldrich; Merck KGaA), 20 mM; chlorpromazine, 10 µM;
wortmannin, 10 µM; genistein, 50 µM; and methyl-β-cyclodextrin, 5
mM. HepG2 cells from each group were pre-incubated with their
indicated inhibitors for 1 h followed by the administration of
NR/GA-PEG-PLGA NPs at a NR concentration of 50 µg/ml and incubation
for an additional 4 h. Subsequently, the cells were washed with
cold PBS thrice and analysed using FCM.
Pro-apoptotic effects and relevant
mechanisms. Determination of intracellular ROS
The oxidative conversion of
2',7'-dichlorodihydrofluorescein diacetate (DCFH-DA) to
dichlorofluorescein (DCF; 488 nm excitation wavelength) is the
foundation for measuring ROS emergence in cells. The HepG2 cells
(3x105/ml) were treated with blank NPs, free-ART,
ART/PEG-PLGA or ART/GA-PEG-PLGA at an ART concentration of 5 mg/ml.
After 72 h of culture at 37˚C with 5% CO2, the cells
were digested with trypsin without EDTA and centrifuged for 5 min
at 400 x g and 4˚C. The cells were subsequently cultured for 20 min
in the dark in medium containing 5 µM DCFH-DA at 37˚C.
Intracellular ROS interacts with DCFH-DA to produce green
fluorescent DCF, whose strength was determined via FCM over 0.5
h.
Mitochondrial membrane potential measurement.
For the quantitation assay, the HepG2 cells were seeded at a
density of 1x105 cells/well. After 12 h of culture at
37˚C with 5% CO2, the cells were washed with PBS and
continuously cultured for 72 h with free-ART, ART/PEG-PLGA or
ART/GA-PEG-PLGA at an ART concentration of 5 mg/ml. Then, the cells
were washed, collected and mixed in a JC-1 dye working solution
according to the manufacturer's protocol, followed by FCM
analysis.
Cell cycle distribution. The effects of
ART/GA-PEG-PLGA NPs on HepG2 cell block were evaluated using FCM.
The cells were treated with free-ART, ART/PEG-PLGA or
ART/GA-PEG-PLGA at an ART concentration of 5 mg/ml for 72 h, then
collected and fixed in 70% ethanol overnight at 4˚C. Next, the
HepG2 cells were harvested and treated with RNase at 50 µg/ml at
37˚C for 0.5 h followed by incubation with propidium iodide (PI) at
65 µg/ml in an ice bath for 0.5 h in the dark and analysis using
FCM.
Annexin V-FITC/PI double staining. Necrosis
and apoptosis in the ART/GA-PEG-PLGA NP-treated HepG2 cells were
determined using Annexin V-FITC. The cells were seeded at a density
of 6x105 cells/well and treated with free-ART,
ART/PEG-PLGA or ART/GA-PEG-PLGA at an equal ART concentration of 5
mg/ml for 72 h. The cells were subsequently collected, re-suspended
in 500 µl binding buffer, dyed with 5 µl Annexin V-FITC and 5 µl PI
solution at room temperature in the dark for 15 min and finally
assessed using FCM.
Western blot analyses
The mechanisms underlying the apoptosis induced by
the different ART preparations were investigated via western
blotting. The HepG2 cells were treated with free-ART, ART/PEG-PLGA
or ART/GA-PEG-PLGA at an ART concentration of 5 mg/ml for 72 h. The
total proteins were extracted via a radio-immuno-precipitation
assay lysis buffer (Beyotime Institute of Biotechnology) and
concentration was measured using a BCA protein assay kit (Abcam).
The extract was separated by 10% SDS-PAGE; sample size, 60 µg) and
then transferred onto a polyvinylidene fluoride (PVDF) membrane
(Merck KGaA). Following blocking with 5% BSA for 60 min at room
temperature, the membrane was incubated with a specific primary
antibody (dilution ratio, 1:1,000) overnight at 4˚C. The PVDF
membrane was subsequently washed thrice with TBST (0.1% Tween-20,
10 min each time) and incubated with the corresponding secondary
antibody [goat anti-rabbit IgG H&L (HRP) and rabbit anti-mouse
IgG H&L (HRP); cat. no. ab6721 and ab6728, respectively;
1:10,000 dilution; Abcam] at room temperature for 1 h. Protein
bands were revealed using an ECL luminescent reagent (cat. no.
32209; Thermo Fisher Scientific, Inc.). The density of the bands
was quantified using Image-Pro Plus 6.0 (Tanon, Science and
Technology Co., Ltd.).
Pharmacokinetics studies
Male SD rats were randomized into two groups (i.e.,
free-ART and ART/GA-PEG-PLGA NPs). Free-ART was dissolved in a 0.5%
carboxymethylcellulose sodium (CMC-Na) solution (v/v). The two
formulations were administered intragastrically at an ART dose of
50 mg/kg. At the indicated time points (5 and 15 min; and 0.5, 1,
2, 4, 8, 12, 24 and 48 h), blood samples were collected and
separated to obtain the plasma. ART was detected using liquid
chromatography-mass-mass-22 (LC/MS/MS-22) spectrometry analysis
[Triple Quad 6500; Shanghai AB SCIEX Analytical Instrument Trading
Co; ionisation mode used: Negative. Parent (m/z)/daughter (m/z):
Artesunate-Q1/Q3 masses, 407.20/261.00 Da; dihydroartemisinin-Q1/Q3
masses, 307.30/261.10 Da; Glipizide-Q1/Q3 masses, 446.20/321.10 Da.
Flow rate, 0.40 ml/min; column temperature, 60˚C; pressure range
(pump A/B), 0.0-100.0 MPa]. The Data Analysis System v3.0
(BioGuider Co.) was used to analyze the pharmacokinetics
parameters. Carbon dioxide euthanasia was performed for rats: Rats
were placed into the euthanasia box and carbon dioxide was flushed
in and adjusted to 50% vol/min. The state of the rats was carefully
observed and input was continued for 1 min after loss of
consciousness. Death of rats was confirmed through cervical
dislocation.
Tumour cell inoculation
A suspension of 0.02 ml Hep3B-luc tumour cells
(including 3x106 cells; ratio of PBS to
Matrigel®, 1:1) was seeded in situ on the left
hepatic lobe of each mouse. On day 7 following the cell
inoculation, the tumour bio-fluorescence signal was measured using
IVIS Lumina III, and nine mice were selected based on the intensity
of the fluorescence signal and body weight.
Hepatoma-targeted and bio-distribution
detection
NR was used as the fluorescent probe to detect the
hepatoma-targeting ability and bio-distribution of GA-PEG-PLGA NPs.
The tumour model mice were randomized into three groups and
administered the different NR preparations intragastrically at an
NR dose of 20 mg/kg. The fluorescence signal distribution of NR was
detected in vivo at 0.5, 2, 4 and 8 h following
administration. Subsequently, the mice were euthanized, the main
organs (heart, liver, spleen, lung and kidney) were harvested and
the intensity of the NR signal was detected in each organ.
Antitumour activity
The antitumour activity of ART/GA-PEG-PLGA NPs was
investigated in female BALB/c nude mice (age, 6-8 weeks) bearing
Hep3B-luc in situ transplanted tumours. When the
fluorescence signal intensity of the implanted tumour cells reached
~107, the mice were randomized into four groups (i.e.,
control, free-NR, NR/PEG-PLGA NPs and NR/GA-PEG-PLGA NPs) and
treated intragastrically with different ART preparations at an ART
concentration of 50 mg/kg once daily for 24 days, with the
exception of the control group. The tumour fluorescence signal and
body weight of the mice were measured twice per week. The mice were
humanely euthanized when they lost >20% body weight because of
weakness or near-death conditions. The inhibition rate of tumor
(IRT) was calculated using the following formula: i) Based on the
fluorescence signal:
IRT=[(ROIcontrol-ROIdrug)/ROIcontrol]
x100% (where ROIdrug: Mean tumour fluorescence value of
the treated mice; ROIcontrol: Mean tumour fluorescence
value of the control mice); ii) Based on the tumour weight:
IRT=[(Wcontrol-Wdrug)/Wcontrol]
x100% (where Wdrug: mean tumour weight of the treated
mice; Wcontrol: Mean tumour weight of the control
mice).
The survival rates of the mice were calculated over
a 24-day period. Their survival time was calculated based on the
grouping day (pg-d0) and the corresponding survival curves were
drawn.
The euthanasia method of mice was the same as that
of rats aforementioned. Lastly, the tumours and major organs
(heart, liver, spleen, lungs and kidneys) were collected at 4 h
after the last drug administration and immersed in formalin and
paraffin followed by haematoxylin and eosin staining for 5 min and
7 sec, respectively, at room temperature. The tissues were observed
using light microscope (ECLIPSE E100; Nikon Corp.). The tumours
were also weighted.
Statistical analysis
All data are expressed as the mean ± standard
deviation and were analysed via one-way analysis of variance
followed by a Tukey's post-hoc test using SPSS (version 19; IBM
Corp.). For all the results, P<0.05 and P<0.01 were
considered to indicate a statistically significant and extremely
significant difference, respectively.
Results
Uptake of NPs and intracellular
mechanism
To identify the specific hepatoma target function of
GA-coated NPs, the uptake of NR-labelled NPs into HepG2 and A549
cells, which express GA receptors at high and low levels,
respectively, was assessed. As shown in Fig. 1A and B, the untreated cells or those incubated
with free-NR exhibited little fluorescence. The HepG2 and A549
cells incubated with NR/GA-PEG-PLGA NPs or NR/PEG-PLGA NPs
expressed significantly higher fluorescence compared with those
treated with free-NR. Increased cellular uptake of NR/GA-PEG-PLGA
and NR/PEG-PLGA NPs was attributed to energy-dependent active
transportation rather than the passive diffusion of free-NR. In
addition, the encapsulation of NR in NPs could easily avoid its
excretion from cells. At all time-points, the fluorescence
intensity was significantly higher in the GA-coated NP groups than
in the non-GA-coated groups. Of note, similar results were also
observed in A549 cells, indicating that the affinity of GA
modification for liver cancer cells was not significantly different
between these two types of cells. This suggests that the
experimental design may need to be improved. Next, the cellular
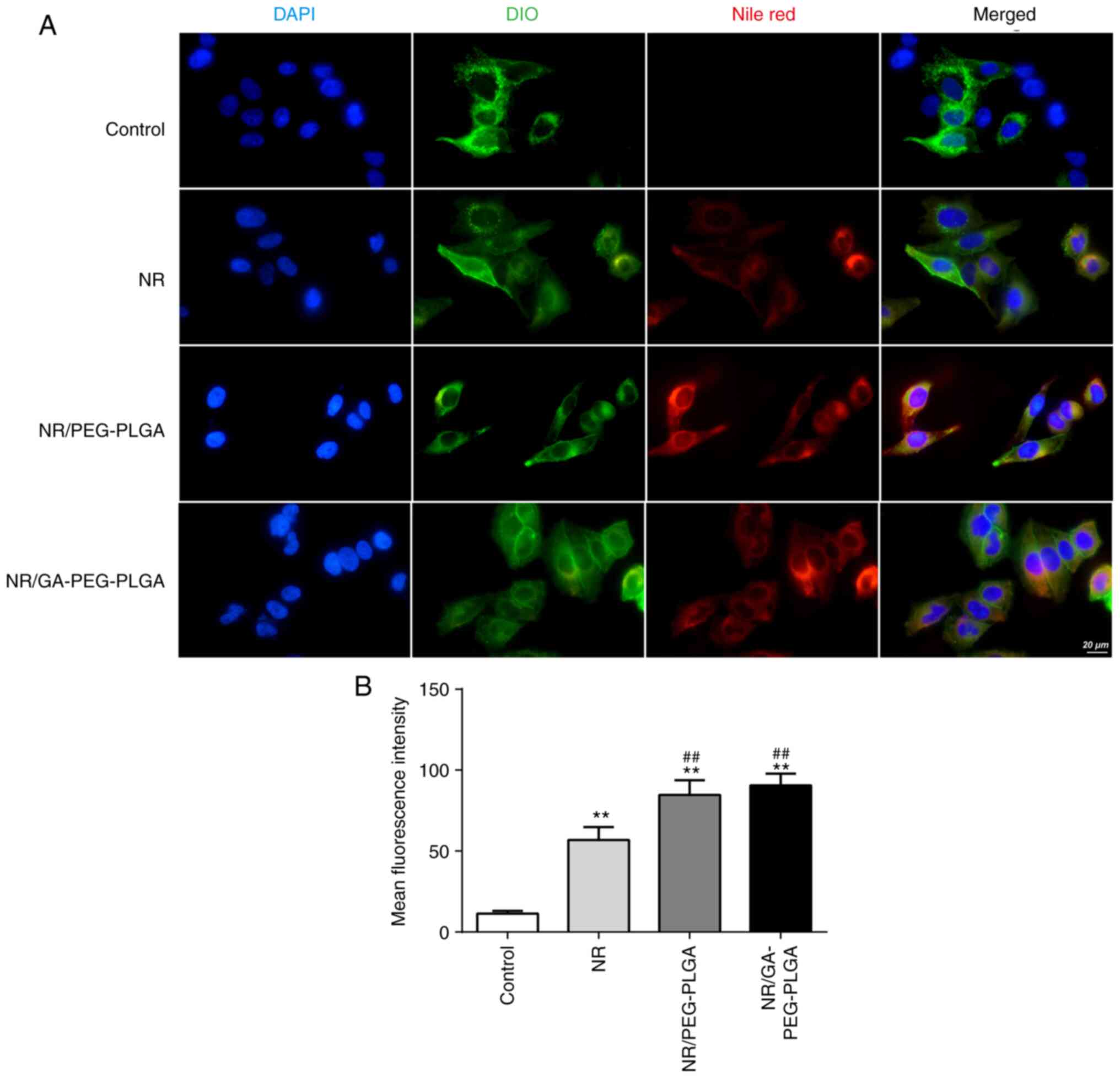
internalization of GA-modified NPs into HepG2 cells was examined
using a CLSM. Both NP groups exhibited higher levels of NR cellular
internalization compared with the free-NR group (Fig. 2). The uptake of GA-coated NPs was
higher than that of nano GA-coated NPs in the cytoplasm and nucleus
of HepG2 cells, suggesting that GA decoration was effective in
delivering drugs to cells with high GA receptor expression. In
accordance with the FCM data, these results demonstrated that GA
receptor-mediated endocytosis efficiently and selectively transmits
GA-coated NPs to HepG2 cells, thus improving targeted drug delivery
to tumour tissues.
Various endocytic inhibitors were employed to probe
the internalization channels of GA-coated NPs. The significantly
lower uptake observed following 2-deoxyglucose exposure indicated
that it was relevant to the energy-dependent process of endocytosis
(Fig. 1C). Pre-incubation with
wortmannin, genistein and methyl-β-cyclodextrin significantly
inhibited the intracellular uptake of NPs, suggesting that the
macropinocytosis and caveolae-mediated pathways were the
internalization channels. In comparison, the chlorpromazine-treated
group exhibited no significant decline in NP uptake, indicating
that the specific clathrin-mediated pathway was not the uptake
pathway in this case. Hence, micropinocytosis and caveolae-mediated
endocytosis were the internalization channels of the GA-modified
NPs. In addition, the uptake of GA-coated NPs was significantly
reduced by free-GA pre-treatment through a competitive combination
of the GA receptors. Surplus free-GA can saturate these binding
sites, ensuring that the GA-coated NPs can no longer utilize them.
The results revealed that the uptake of GA-coated NPs by liver
cancer cells depend largely on these GA-binding sites.
Pro-apoptotic effects of
ART/GA-PEG-PLGA NPs and the underlying mechanisms
The cells were incubated with blank NPs or other
formulations at an ART concentration of 5 mg/ml. Compared with
normal cells, blank NP-treated cells exhibited no change in
intracellular ROS levels, indicating that the blank NPs demonstrate
favorable biocompatibility (Fig.
3A). ART-loaded NPs triggered a higher ROS level than free-ART.
Moreover, compared with none-GA-modified NPs, the GA-modified NPs
exhibited higher ROS levels. This increase in ROS levels resulted
in cell growth arrest and apoptosis. These data demonstrated that
the GA-coated NPs were superior to normal NPs in causing oxidative
damage to liver cancer cells.
The mitochondrial membrane potential (MMP) is an
important parameter for mitochondrial function and its decrease is
often considered to promote apoptosis. An increase in the green
fluorescence ratio indicates the occurrence of mitochondrial
depolarization. It was observed that blank NPs did not affect the
MMP, whereas the ART-loaded NPs decreased it in the HepG2 cells
(Fig. 3B). The levels of the JC-1
green ratio were significantly increased in the HepG2 cells
following GA-coated NPs treatment compared with that in the cells
treated with non-GA-coated NPs.
The effects of ART/GA-PEG-PLGA NPs on cell cycle
progression in the HepG2 cells were assessed via FCM. The ART
formulations induced cell cycle arrest in the HepG2 cells and not
in the untreated or blank NP-treated cells (Fig. 3C). The cell distribution decreased
in the G1 phase and increased in the S and G2 phases following the
treatment. In all the preparations, the ART/GA-PEG-PLGA NPs
exhibited the strongest effect on cell cycle inhibition in the S
phase. Therefore, these results suggested that ART/GA-PEG-PLGA NPs
can effectively inhibit HepG2 cell proliferation.
In the present study, the effects of ART-loaded NPs
on apoptosis in HepG2 cells were assessed. Following culture,
cellular apoptosis was determined via Annexin V-FITC and PI double
staining. ART-loaded NPs exhibited significantly stronger
apoptosis-inducing effects on HepG2 cells than free-ART; moreover,
compared with the other groups, ART/GA-PEG-PLGA NP-treated HepG2
cells exhibited the highest apoptotic rate (Fig. 3D). These data indicated that
ART/GA-PEG-PLGA NPs demonstrated promising apoptotic effects in the
HepG2 cells.
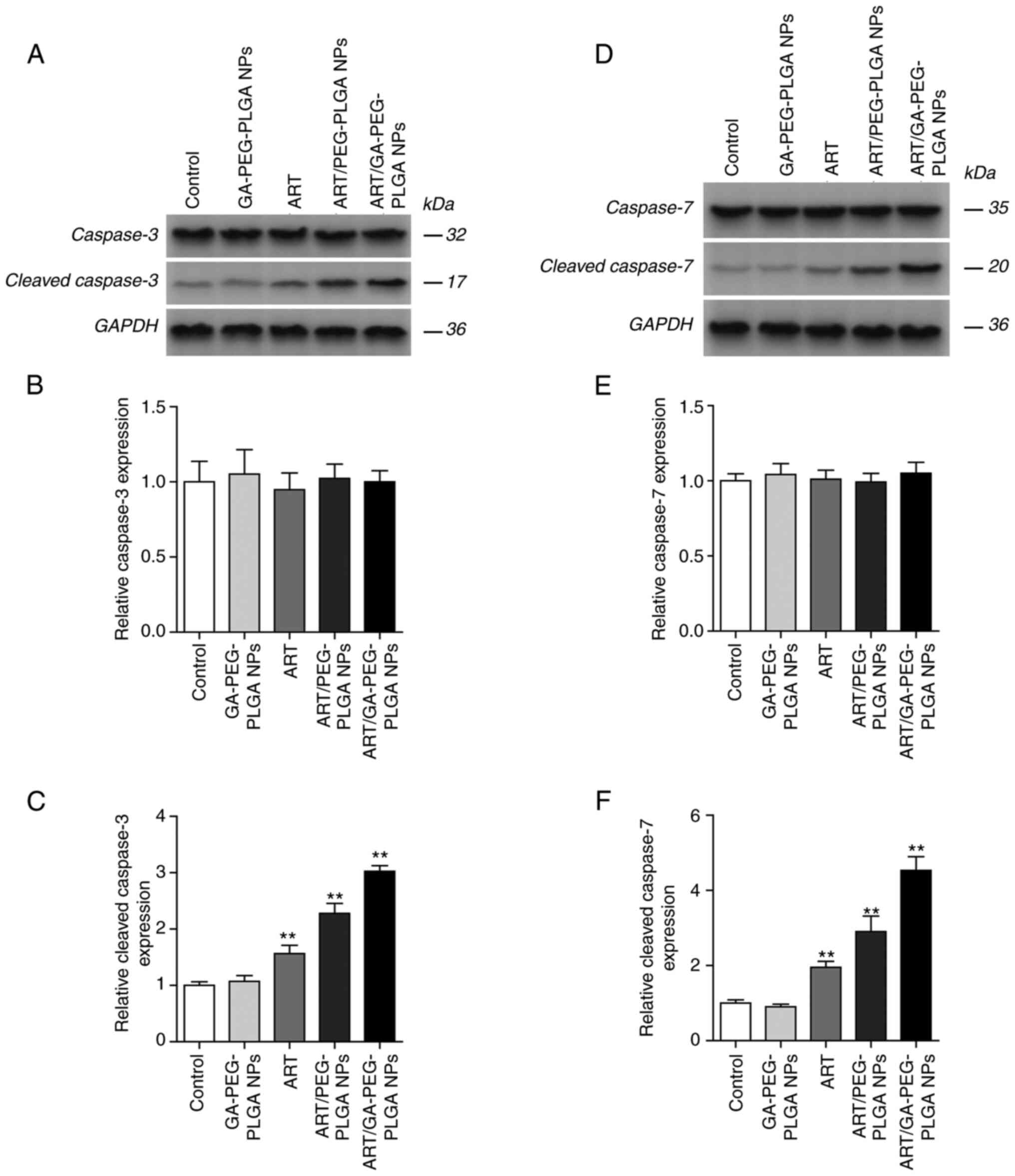
Western blot analyses revealed that ART-loaded NPs
induced internal apoptosis pathways in HepG2 cells (Figs. 4 and 5). Caspase-3/7 is directly involved in
the cleavage of important intracellular substrates to disintegrate
cellular structures during apoptosis (44). P38 reportedly functions as an
antitumour factor and regulates the cell cycle at several
transition points (45). PARP is
related to cell death and is cleaved specifically and rapidly
during apoptosis (46,47). Compared with the free-ART group and
ART/PEG-PLGA NP group, cleaved caspase-3/7 expression increased
following treatment with ART/GA-PEG-PLGA NPs (Fig. 4C and F), whereas negligible variation was
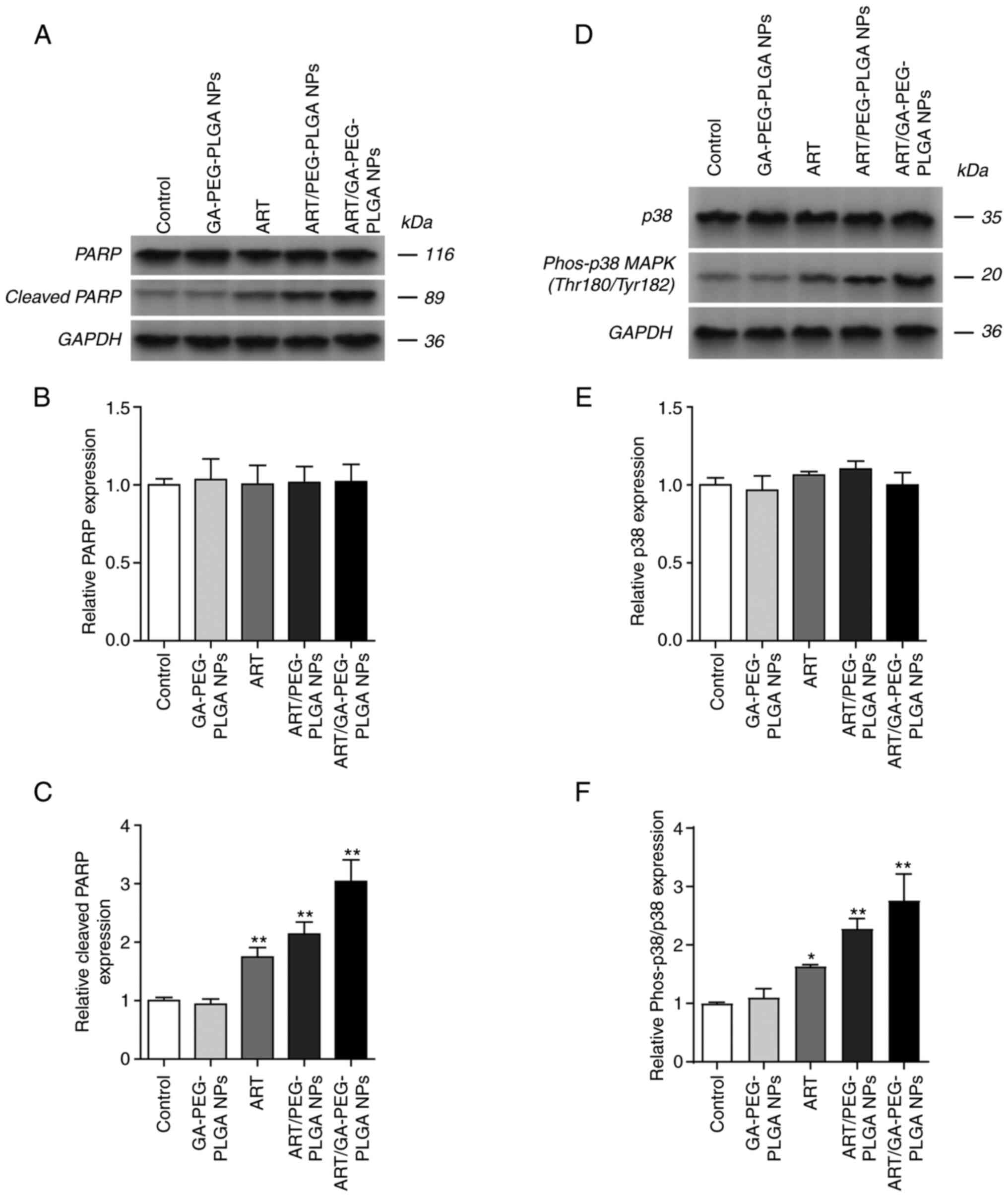
observed for caspase-3/7 expression (Fig. 4B and E). Furthermore, ART/GA-PEG-PLGA NPs
notably upregulated cleaved PARP and Phos-p38/p38 MAPK
(Thr180/tyr182) in HepG2 cells compared with free-ART and
ART/PEG-PLGA NPs (Fig. 5C and
F), whereas negligible variation
was observed for PARP and p38 MAPK (Thr180/tyr182) expression
(Fig. 5B and E). Similar outcomes were obtained in the
cell apoptosis assay. Because p38 MAPK is an important modulator of
cell death, increased cell apoptosis triggered by ART/GA-PEG-PLGA
NPs is considered to be mediated by the p38 MAPK/caspase-3/7
signaling pathway.
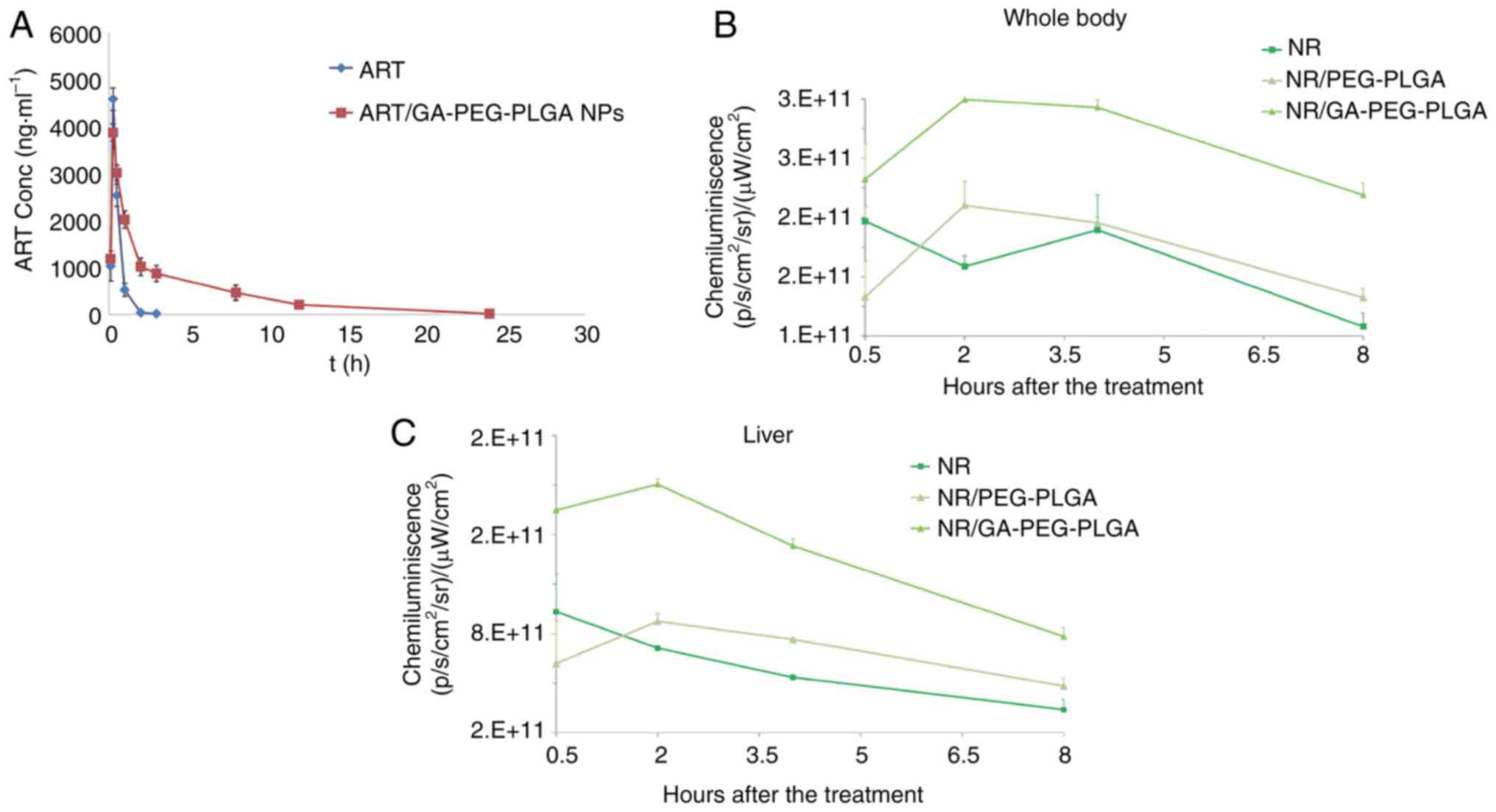
Pharmacokinetics studies. Following drug
administration, a sharp increase and then a sharp decrease in the
plasma concentrations of ART was observed in the free-ART group.
ART was almost undetectable at 3 h following drug intragastrical
administration (Fig. 6A).
Conversely, the ART cycles were longer in the ART/GA-PEG-PLGA NP
group than in the free-ART group, and its plasma concentrations
could still be detected at 12 h after drug administration. There
was a large difference in the time courses of plasma ART
concentrations between the free-ART and GA-coated NP groups,
indicating that nanorization could prolong the peripheral
circulation of ART. The main pharmacokinetics parameters of
free-ART and ART-loaded NPs are summarized in Table I. The total area under the curve
(AUC0-∞) of the ART-loaded NP group was significantly
greater than that of the free-ART group, indicating that
bioavailability was enhanced after the encapsulation of ART in NPs.
The extended mean retention time (MRT) and elimination half-life
(t1/2z) of ART-loaded NPs contributed to the higher
AUC0-∞ values. Interestingly, the pharmacokinetic
parameters of the ART/GA-PEG-PLGA NP group were different from
those of the free-ART group. Although the Cmax of the NP
group was lower than that of the free-ART group, its
AUC0-∞ value and MRT were higher than those of the
free-ART group. It was hypothesized that these results could be
attributed to the liver-targeting feature of ART/GA-PEG-PLGA NPs,
which promoted the liver accumulation of ART and decreased its
level in the blood circulation.
 | Table IPharmacokinetic parameters in rats
treated with a free-ART suspension or ART-loaded NPs at an
equivalent dose of 5 mg ART per kg (n=3). |
Table I
Pharmacokinetic parameters in rats
treated with a free-ART suspension or ART-loaded NPs at an
equivalent dose of 5 mg ART per kg (n=3).
| | Parameters |
|---|
| Groups |
Cmax |
Tmax | AUC0–t,
h/ng/ml | AUC0-∞,
h/ng/ml | MRT, h | T1/2z, h |
|---|
| ART | 4,578 | 0.25 | 2,539.49 | 2,539.54 | 0.50 | 0.25 |
| ART/GA-PEG-PLGA
NPs | 3,897 | 0.25 |
12,527.96a |
13,477.30a | 5.54a | 6.67a |
Hepatoma-targeting and bio-distribution of
GA-coated NPs. The fluorescence signal distribution of NR was
analysed in the whole body and liver at different time-points
(Figs. 6B and C and 7).
Compared with the NR-loaded NP group, the free-NR group exhibited
lower fluorescence levels at 2 h following the drug
administrations, implying that free-NR was eliminated faster in
vivo. Although the NR levels of the free-NR-treated group were
higher than those of the NR/PEG-PLGA group at 0.5 h, higher levels
of NR were observed in the NR-loaded NP groups at 2, 4 and 8 h in
the whole body and liver. The liver fluorescence of NR in the
GA-modified NP group was markedly higher than that detected in the
non-GA-modified group, suggesting that GA modification upregulated
the liver-targeting effect of the NPs. Thus, these data
demonstrated that GA-PEG-PLGA NPs can promote the liver-targeting
distribution of the drug and prolong its retention time.
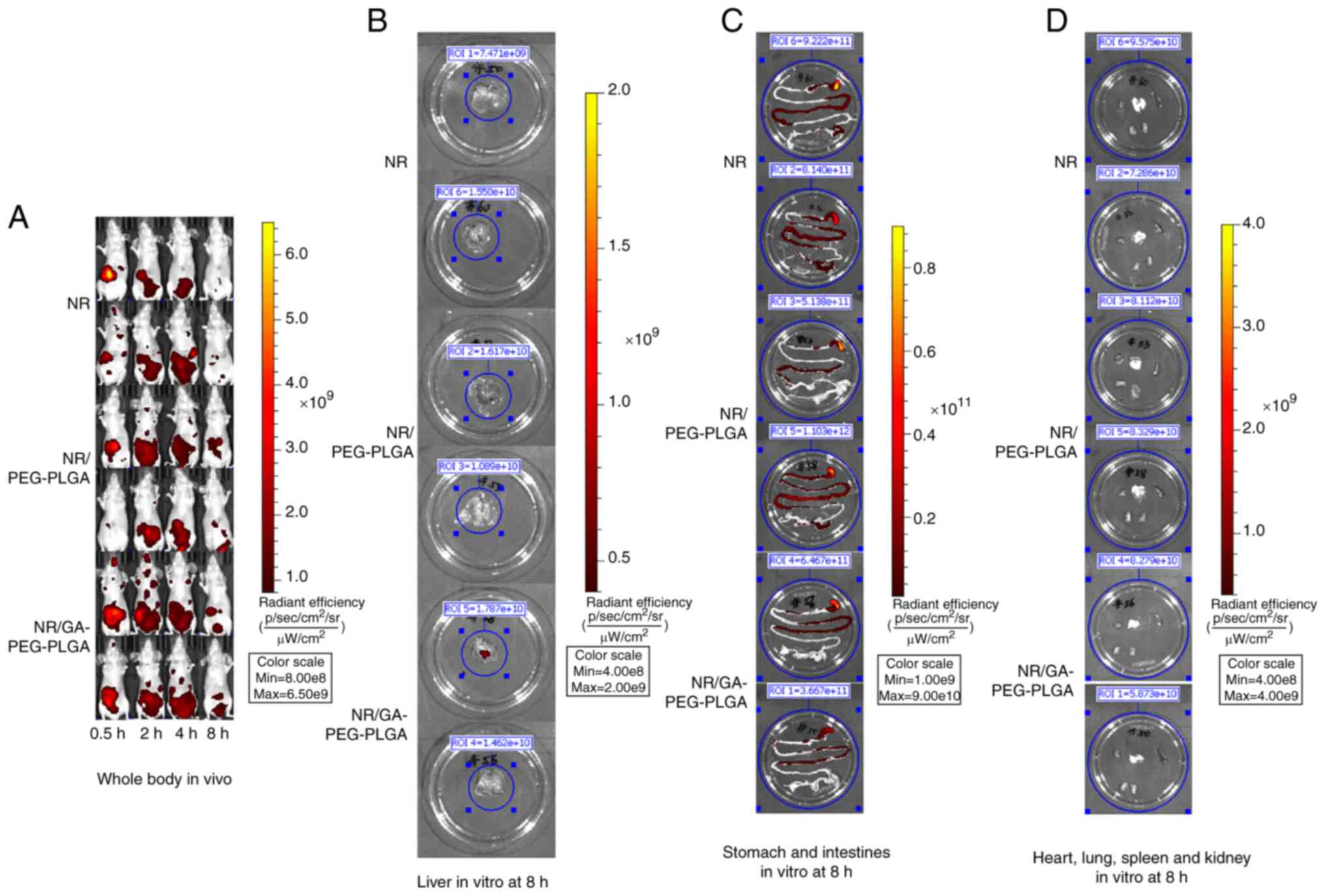 | Figure 7Fluorescence signal distribution in
the whole body at 0.5, 2, 4 and 8 h and in the main organs at 8 h
in vitro (n=2). NR was used as the fluorescent probe to
detect the hepatoma-targeting ability and bio-distribution of
GA-PEG-PLGA NPs. The tumour model mice were randomized into three
groups and administered intragastrically the different NR
preparations at an NR dose of 20 mg/kg. The mice were euthanized
after 8 h, the main organs (heart, liver, spleen, lungs and
kidneys) were dissected and the NR signal intensity in each organ
was detected. Compared with the NR-loaded NP group, the free-NR
group exhibited lower fluorescence levels at 2 h after drug
administration. Higher levels of NR were observed in the NR-loaded
NP groups at 2, 4 and 8 h compared with the free-NR treated group
in the whole body and liver. The liver fluorescence of NR in the
GA-modified NP group was remarkably higher than that observed in
the non-GA-modified group (P<0.05). Fluorescence of the (A)
whole body; (B) liver; (C) stomach and intestine; and (D) heart,
spleen, lungs and kidneys. NR, Nile red; GA, glycyrrhetinic acid;
NPs, liver-targeted nanoparticles; PEG-PLGA, polyethylene
glycol-poly (lactic-co-glycolic acid). |
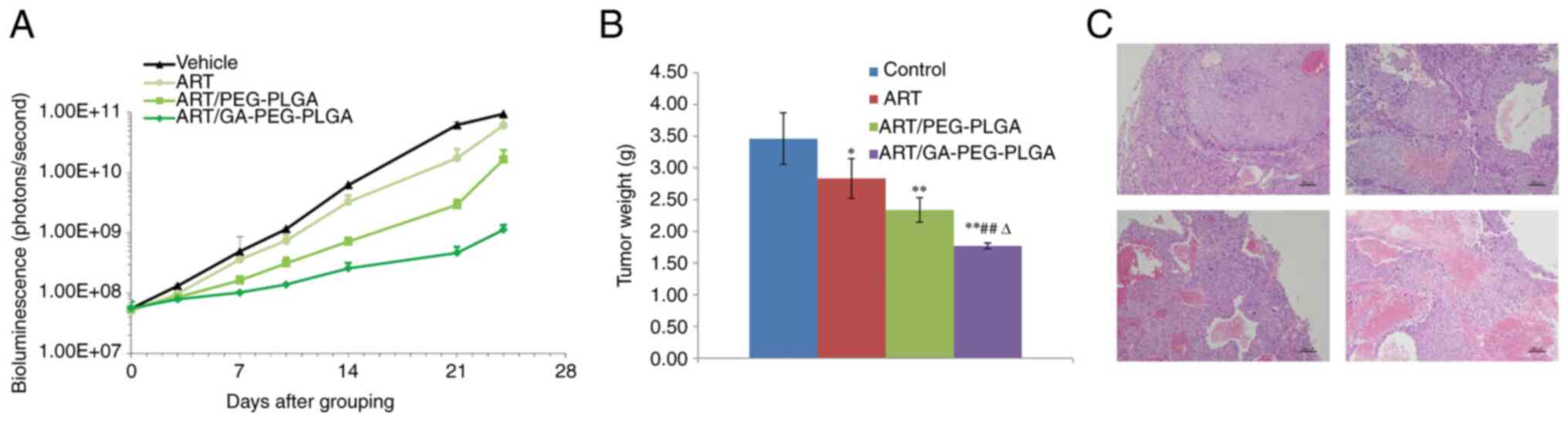
Antitumour activity
A xenograft tumour model was generated in BALB/c
nude mice with Hep3B-luc cell embedment, to explore the antitumour
abilities of ART/GA-PEG-PLGA NPs. Tumours removed from each group
are presented in Fig. 8. The
tumour growth, weight and survival curves of each group were
recorded and calculated (Fig. 9A
and B). Similar to patients with
liver cancer, it was observed that tumour-bearing mice succumbed
because of inadequate nutrition during long-term treatment, whereas
no deaths were observed in the different ART formulation-treated
groups during the test period. Following the completion of the
experiment, the in vitro liver bioluminescence and tumour
weight data revealed that the trend of the IRT according to the
different ART formulations was ART/GA-PEG-PLGANPs > ART/PEG-PLGA
NPs > free-ART (Table II). An
additional analysis revealed that the IRT results of the NP and
free-ART groups coincided in the two calculation approaches. In
addition, a histological analysis of tumour sections revealed that
compared with the other groups the tumour necrosis area was larger
and apoptosis was increased in tumours treated with ART/GA-PEG-PLGA
NPs (Fig. 9C). These outcomes
revealed that ART/GA-PEG-PLGA NPs had a more promising antitumour
activity than free-ART or ART-loaded non-functional NPs.
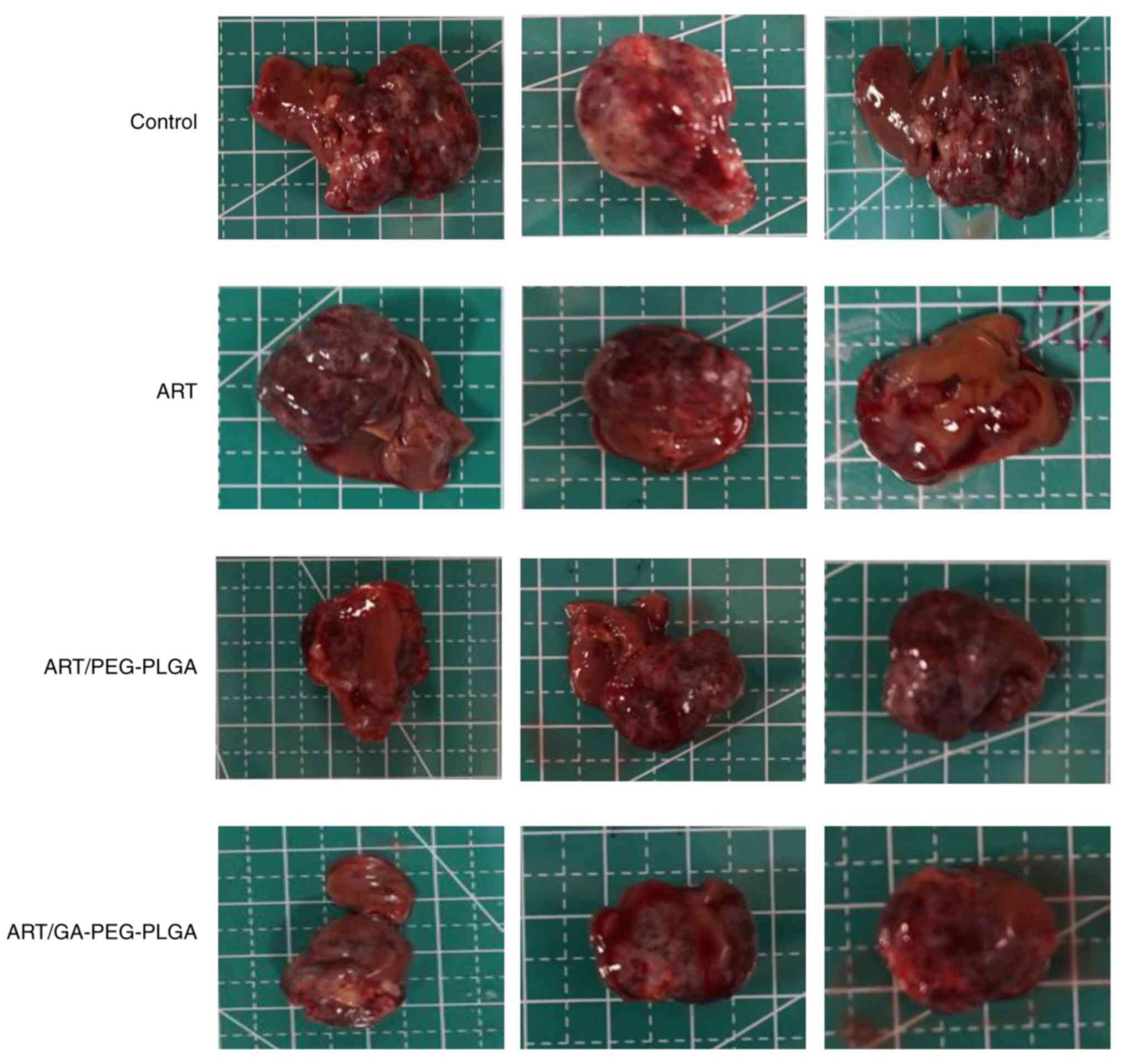 | Figure 8Tumours removed from each group. Mice
bearing Hep3B-luc in situ transplanted tumours were randomized into
four groups (i.e., control, free-NR, NR/PEG-PLGA NPs and
NR/GA-PEG-PLGA NPs) and treated intragastrically with different ART
preparations at an ART concentration of 50 mg/kg once daily for 24
days, with the exception of the control group. The tumours were
collected at 4 h after the last drug administration. The distance
between the solid lines, the solid lines and the dashed lines on
the anatomical board is 1 cm and 0.5 cm, respectively. NR, Nile
red; GA, glycyrrhetinic acid; NPs, liver-targeted nanoparticles;
PEG-PLGA, polyethylene glycol-poly (lactic-co-glycolic acid); ART,
artesunate. |
| Table IITherapeutic effects in the Hep3B-luc
xenograft model. |
Table II
Therapeutic effects in the Hep3B-luc
xenograft model.
| Group | Bioluminescence
(ROI, x109 photons/sec) (PG-21) |
IRTBioluminescence, % | Tumour weight,
g | IRTTumour
weight,% |
|---|
| Control | 5.72±1.38 | - | 3.78±0.15 | - |
| ART | 4.49±0.55 | 21.50 |
30.3±0.36a | 19.84 |
| ART/PEG-PLGA
NPs |
2.94±0.37a,b | 48.60 |
1.92±0.23a,c | 49.21 |
| ART/GA-PEG-PLGA
NPs |
1.03±0.29a,c,d | 81.99 |
0.74±0.11a,c,d | 80.42 |
Discussion
Significant progress has been made in the
nano-targeted delivery systems used for liver cancer treatment over
the last two decades. However, NPs designed to target liver cancer
continue facing certain challenges, such as the low effective
concentration of drugs in tumour tissues (48), with a mediocre 4% passive
targeted-drug delivery and 8% active targeted-drug delivery
(49). These findings were mainly
attributed to an elaborate reticuloendothelial system and the
trapping and interference of NPs in the delivery pathway. Regarding
active targeted delivery, although various ligands (such as
peptides, antibodies and galactose) are currently in use (50), the possible immunoreaction of
exogenous biomolecular targeting ligands and the instability of the
galactose-targeting effect continue to hinder the efficiency of
their delivery. Therefore, there remain high demands for novel
liver cancer specific delivery targeting ligands. The emergence of
GA renewed hope in this field since it not only yielded a higher
expression of receptors on liver cancer cells but also exerted
numerous pharmacological activities, such as anti-ulcer,
anti-allergy, immune-modulating, antiviral, antitumour,
liver-preservation and antioxidant effects, suggesting that the
GA-coated NPs proposed in the present study could target hepatoma
tissue, improve treatment outcomes and reduce adverse events.
Because of the generally serious toxicity of
chemotherapeutics, the active ingredients of traditional Chinese
medicine have become a hot topic in antitumour research as they
reportedly possess various curative effects against tumour
occurrence, development, metastasis and immune modulation via
multiple ways and multi-targets (51). ART, an artemisinin derivative
isolated from the traditional herb Artemisia annua, can
induce the apoptosis and differentiation of various human tumour
cells and is regarded as a potential anticancer agent (39,52,53).
However, its clinical application is seriously affected due to the
poor pharmaceutic properties aforementioned. In the present study,
ART was encapsulated in GA-coated NPs to overcome the existing
limitations and enhance its treatment effect. The present data
illustrated that the ART/GA-PEG-PLGA NPs could significantly
increase the liver distribution of ART and prolong MRT to enhance
its therapeutic effects compared with free-ART.
GA-modified DDSs have garnered considerable
attention regarding the treatment of liver cancer. The main reasons
for this include their capability of promoting GA receptor-mediated
endocytosis and enhancing liver targeting. These observations are
consistent with the present research results. Most studies
regarding GA-modified DDSs used HepG2 cells to study in
vitro cellular uptake (35,54-56),
mainly for the following reasons: High GA receptor expression, high
differentiation degree, relatively complete biotransformation
characteristics of metabolic enzymes, retention of the stability of
metabolic enzymes in studies related to drug effects (absence of
changes caused by increased number of passages), and homology
between the contained biotransformation metabolic enzymes and
normal human liver parenchyma cells. In these studies that aimed at
improving the cellular uptake of GA-modified DDSs, A549 cells are
commonly used as a control because of their low GA receptor
expression (50,55). Therefore, the present study also
selected HepG2 and A549 cells to study the specific liver
cancer-targeting function of the system and improve the cellular
internalization of GA-modified NPs. The HepG2 cell line was
authenticated before the study began.
Induction of apoptosis is one of the most important
targets of cancer research; therefore, the pro-apoptotic effects of
ART/GA-PEG-PLGA NPs and their possible underlying mechanisms were
investigated. The pre-set results revealed that ART/GA-PEG-PLGA NPs
remarkably increased cell apoptosis by enhancing cellular uptake
mediated by the GA ligand. As expected, the in vivo
antitumour effects were consistent with the in vitro results
(40), indicating the potent
anti-liver cancer efficiency of ART/GA-PEG-PLGA NPs.
Of note, the present study also has certain
shortcomings. Due to limited research funding, insufficient
research samples and short research time, there are shortcomings in
the reliability and universality of the study's conclusions. In
future research, we will improve new research methods, such as
using gene knockout technology to prepare GA receptor knockout
HepG2 cells, to even out these shortcomings and improve the quality
and reliability of the research.
In the present study, innovative hepatoma-targeting
ART-loaded and GA-coated PEG-PLGA NPs were successfully developed
to deliver ART to hepatoma cells. GA modification enabled the
selective delivery to and the accumulation of ART in liver cells.
ART/GA-PEG-PLGA NPs exhibited improved tumour-targeting abilities
and in vivo treatment efficiency, which were attributed to
the tumour-targeting ability exhibited by GA. Overall,
ART/GA-PEG-PLGA NPs may have promising prospects for treating
hepatoma.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by the National Natural
Science Foundation of China (grant no. 82104699), the Research
Project on the Application of Public Welfare Technology supported
by the Science and Technology Department of Zhejiang (grant no.
LGF22H290003), the Medical Health Science and Technology Major
Project of Hangzhou Health Commission (grant no. Z20230014) and the
Hepatology (Traditional Chinese and Western Medicine) of Hangzhou
Medical Peak Subject.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
XWP, JSH, SRL, RXZ and JFB participated in research
design. XWP, JJX, YDS, RYH and TTS conducted experiments. YDS, RYH
and TTS contributed reagents or analytic tools. XWP, JJX, YDS, RYH,
RXZ and JFB performed data analysis. XWP, JSH, SRL, RXZ and JFB
discussed and edited the manuscript. XWP, JJX and YDS wrote or
contributed to the writing of the manuscript. All authors have read
and approved the final version of the manuscript. JJX and YDS
confirm the authenticity of all the raw data.
Ethics approval and consent to
participate
All procedures performed in studies involving animal
participants were in accordance with the national ethical
standards. The present study was approved (approval no.
ON-01-QD003-2020v1.0) by the Institutional Animal Care and Use
Committee of WuXi AppTec (Nantong) Co., Ltd.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249.
2021.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Zhou M, Wang H, Zeng X, Yin P, Zhu J, Chen
W, Li X, Wang L, Wang L, Liu Y, et al: Mortality, morbidity, and
risk factors in China and its provinces, 1990-2017: A systematic
analysis for the global burden of disease study 2017. Lancet.
394:1145–1158. 2019.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Kulik L and El-Serag HB: Epidemiology and
management of hepatocellular carcinoma. Gastroenterology.
156:477–491.e1. 2019.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Sugawara Y and Hibi T: Surgical treatment
of hepatocellular carcinoma. Biosci Trends. 15:138–141.
2021.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Piñero F, Silva M and Iavarone M:
Sequencing of systemic treatment for hepatocellular carcinoma:
Second line competitors. World J Gastroenterol. 26:1888–1900.
2020.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Colquhoun SD and Wan YY: Hepatocellular
carcinoma diagnosis and treatment: An overview. Liver Res.
4:159–160. 2020.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Yang JD and Heimbach JK: New advances in
the diagnosis and management of hepatocellular carcinoma. BMJ.
371(m3544)2020.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Malla RR, Kumari S, Kgk D, Momin S and
Nagaraju GP: Nanotheranostics: Their role in hepatocellular
carcinoma. Crit Rev Oncol Hematol. 151(102968)2020.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Wiwatchaitawee K, Quarterman JC, Geary SM
and Salem AK: Enhancement of therapies for glioblastoma (GBM) using
nanoparticle-based delivery systems. AAPS PharmSciTech.
22(71)2021.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Kalyane D, Raval N, Maheshwari R, Tambe V,
Kalia K and Tekade RK: Employment of enhanced permeability and
retention effect (EPR): Nanoparticle-based precision tools for
targeting of therapeutic and diagnostic agent in cancer. Mater Sci
Eng C Mater Biol Appl. 98:1252–1276. 2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Yang C, Tan Y, Qi H, Zhou J, Long L, Zhan
Q, Wang Y, Yuan X and Kang C: Boosting of the enhanced permeability
and retention effect with nanocapsules improves the therapeutic
effects of cetuximab. Cancer Biol Med. 17:433–443. 2020.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Yang M, Li J, Gu P and Fan X: The
application of nanoparticles in cancer immunotherapy: Targeting
tumor microenvironment. Bioact Mater. 6:1973–1987. 2020.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Baker A and Khan MS, Iqbal MZ and Khan MS:
Tumor-targeted drug delivery by nanocomposites. Curr Drug Metab.
21:599–613. 2020.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Hu X, Zhang J, Deng L, Hu H, Hu J and
Zheng G: Galactose-modified pH-sensitive niosomes for controlled
release and hepatocellular carcinoma target delivery of tanshinone
IIA. AAPS PharmSciTech. 22(96)2021.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Severino P, da Silva CF, Andrade LN, de
Lima Oliveira D, Campos J and Souto EB: Alginate nanoparticles for
drug delivery and targeting. Curr Pharm Des. 25:1312–1334.
2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Wathoni N, Rusdin A, Motoyama K, Joni IM,
Lesmana R and Muchtaridi M: Nanoparticle drug delivery systems for
α-mangostin. Nanotechnol Sci Appl. 13:23–36. 2020.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Essa D, Kondiah PPD, Choonara YE and
Pillay V: The design of poly (lactide-co-glycolide) nanocarriers
for medical applications. Front Bioeng Biotechnol.
8(48)2020.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Begines B, Ortiz T, Pérez-Aranda M,
Martínez G, Merinero M, Argüelles-Arias F and Alcudia A: Polymeric
nanoparticles for drug delivery: Recent developments and future
prospects. Nanomaterials (Basel). 10(1403)2020.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Du M, Ouyang Y, Meng F, Zhang X, Ma Q,
Zhuang Y, Liu H, Pang M, Cai T and Cai Y: Polymer-lipid hybrid
nanoparticles: A novel drug delivery system for enhancing the
activity of Psoralen against breast cancer. Int J Pharm.
561:274–282. 2019.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Anderson CF, Grimmett ME, Domalewski CJ
and Cui H: Inhalable nanotherapeutics to improve treatment efficacy
for common lung diseases. Wiley Interdiscip Rev Nanomed
Nanobiotechnol. 12(e1586)2020.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Carter P, Narasimhan B and Wang Q:
Biocompatible nanoparticles and vesicular systems in transdermal
drug delivery for various skin diseases. Int J Pharm. 555:49–62.
2019.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Wei T, Cheng Q, Min YL, Olson EN and
Siegwart DJ: Systemic nanoparticle delivery of CRISPR-Cas9
ribonucleoproteins for effective tissue specific genome editing.
Nat Commun. 11(3232)2020.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Anselmo AC and Mitragotri S: Nanoparticles
in the clinic: An update. Bioeng Transl Med.
4(e10143)2019.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Krauss AC, Gao X, Li L, Manning ML, Patel
P, Fu W, Janoria KG, Gieser G, Bateman DA, Przepiorka D, et al: FDA
approval summary: (Daunorubicin and Cytarabine) Liposome for
injection for the treatment of adults with high-risk acute myeloid
leukemia. Clin Cancer Res. 25:2685–2690. 2019.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Naqvi S, Panghal A and Flora SJS:
Nanotechnology: A promising approach for delivery of
neuroprotective drugs. Front Neurosci. 14(494)2020.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Gu Z, Wang Q, Shi Y, Huang Y, Zhang J,
Zhang X and Lin G: Nanotechnology-mediated immunochemotherapy
combined with docetaxel and PD-L1 antibody increase therapeutic
effects and decrease systemic toxicity. J Control Release.
286:369–380. 2018.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Chen B, Dai W, He B, Zhang H, Wang X, Wang
Y and Zhang Q: Current multistage drug delivery systems based on
the tumor microenvironment. Theranostics. 7:538–558.
2017.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Zhang YR, Lin R, Li HJ, He WL, Du JZ and
Wang J: Strategies to improve tumor penetration of nanomedicines
through nanoparticle design. Wiley Interdiscip Rev Nanomed
Nanobiotechnol. 11(e1519)2019.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Niu Y, Zhu J, Li Y, Shi H, Gong Y, Li R,
Huo Q, Ma T and Liu Y: Size shrinkable drug delivery nanosystems
and priming the tumor microenvironment for deep intratumoral
penetration of nanoparticles. J Control Release. 277:35–47.
2018.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Lim S, Park J, Shim MK, Um W, Yoon HY, Ryu
JH, Lim DK and Kim K: Recent advances and challenges of repurposing
nanoparticle-based drug delivery systems to enhance cancer
immunotherapy. Theranostics. 9:7906–7923. 2019.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Gonda A, Zhao N, Shah JV, Calvelli HR,
Kantamneni H, Francis NL and Ganapathy V: Engineering
tumor-targeting nanoparticles as vehicles for precision
nanomedicine. Med One. 4(e190021)2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Negishi M, Irie A, Nagata N and Ichikawa
A: Specific binding of glycyrrhetinic acid to the rat liver
membrane. Biochim Biophys Acta. 1066:77–82. 1991.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Sun Y, Dai C, Yin M, Lu J, Hu H and Chen
D: Hepatocellular carcinoma-targeted effect of configurations and
groups of glycyrrhetinic acid by evaluation of its
derivative-modified liposomes. Int J Nanomedicine. 13:1621–1632.
2018.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Wu F, Li X, Jiang B, Yan J, Zhang Z, Qin
J, Yu W and Gao Z: Glycyrrhetinic acid functionalized nanoparticles
for drug delivery to liver cancer. J Biomed Nanotechnol.
14:1837–1852. 2018.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Li YL, Zhu XM, Liang H, Orvig C and Chen
ZF: Recent advances in asialoglycoprotein receptor and
glycyrrhetinic acid receptor-mediated and/or pH-responsive
hepatocellular carcinoma-targeted drug delivery. Curr Med Chem.
28:1508–534. 2021.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Stecanella LA, Bitencourt APR, Vaz GR,
Quarta E, Silva Júnior JOC and Rossi A: Glycyrrhizic acid and its
hydrolyzed metabolite 18β-glycyrrhetinic acid as specific ligands
for targeting nanosystems in the treatment of liver cancer.
Pharmaceutics. 13(1792)2021.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Kim J, Lee S and Na K: Glycyrrhetinic
acid-modified silicon phthalocyanine for liver cancer-targeted
photodynamic therapy. Biomacromolecules. 22:811–822.
2021.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Li L, Han S, Yang C, Liu L, Zhao S, Wang
X, Liu B, Pan H and Liu Y: Glycyrrhetinic acid modified MOFs for
the treatment of liver cancer. Nanotechnology.
31(325602)2020.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Huang S, Ren D, Wu X, Li M, Yu X, Nie X
and Wang Y and Wang Y: Glycyrrhetinic acid and TAT peptide modified
dual-functional liposomes for treatment of hepatocellular cancer.
Curr Top Med Chem. 20:2493–2505. 2020.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Li ZJ, Dai HQ, Huang XW, Feng J, Deng JH,
Wang ZX, Yang XM, Liu YJ, Wu Y, Chen PH, et al: Artesunate
synergizes with sorafenib to induce ferroptosis in hepatocellular
carcinoma. Acta Pharmacol Sin. 42:301–310. 2021.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Yin S, Yang H, Zhao X, Wei S, Tao Y, Liu
M, Bo R and Li J: Antimalarial agent artesunate induces G0/G1 cell
cycle arrest and apoptosis via increasing intracellular ROS levels
in normal liver cells. Hum Exp Toxicol. 39:1681–1689.
2020.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Pan X, Liu S, Ju L, Xi J, He R, Zhao Y,
Zhuang R and Huang J: Preparation, evaluation, and in vitro
cytotoxicity studies of artesunate-loaded glycyrrhetinic acid
decorated PEG-PLGA nanoparticles. Drug Dev Ind Pharm. 46:1889–1897.
2020.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Ye RR, Peng W, Chen BC, Jiang N, Chen XQ,
Mao ZW and Li RT: Mitochondria-targeted artesunate conjugated
cyclometalated iridium(iii) complexes as potent anti-HepG2
hepatocellular carcinoma agents. Metallomics. 12:1131–1141.
2020.PubMed/NCBI View Article : Google Scholar
|
|
44
|
McComb S, Chan PK, Guinot A,
Hartmannsdottir H, Jenni S, Dobay MP, Bourquin JP and Bornhauser
BC: Efficient apoptosis requires feedback amplification of upstream
apoptotic signals by effector caspase-3 or -7. Sci Adv.
5(eaau9433)2019.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Martínez-Limón A, Joaquin M, Caballero M,
Posas F and de Nadal E: The p38 pathway: From biology to cancer
therapy. Int J Mol Sci. 21(1913)2020.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Slade D: PARP and PARG inhibitors in
cancer treatment. Genes Dev. 34:360–394. 2020.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Zhu H, Wei M, Xu J, Hua J, Liang C, Meng
Q, Zhang Y, Liu J, Zhang B, Yu X and Shi S: PARP inhibitors in
pancreatic cancer: Molecular mechanisms and clinical applications.
Mol Cancer. 19(49)2020.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Wilhelm S, Tavares AJ, Qin D, Ohta S,
Audet J, Dvorak HF and Chan WCW: Analysis of nanoparticle delivery
to tumours. Nat Rev Mater. 1(16014)2016.
|
|
49
|
von Maltzahn G, Park JH, Lin KY, Singh N,
Schwöppe C, Mesters R, Berdel WE, Ruoslahti E, Sailor MJ and Bhatia
SN: Nanoparticles that communicate in vivo to amplify tumour
targeting. Nat Mater. 10:545–552. 2011.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Chen F, Zhang J, He Y, Fang X, Wang Y and
Chen M: Glycyrrhetinic acid-decorated and reduction-sensitive
micelles to enhance the bioavailability and anti-hepatocellular
carcinoma efficacy of tanshinone IIA. Biomater Sci. 4:167–182.
2016.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Rahman M, Almalki WH, Alrobaian M, Iqbal
J, Alghamdi S, Alharbi KS, Alruwaili NK, Hafeez A, Shaharyar A,
Singh T, et al: Nanocarriers-loaded with natural actives as newer
therapeutic interventions for treatment of hepatocellular
carcinoma. Expert Opin Drug Deliv. 18:489–513. 2021.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Yang X, Zheng Y, Liu L, Huang J, Wang F
and Zhang J: Progress on the study of the anticancer effects of
artesunate. Oncol Lett. 22(750)2021.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Zhao F, Vakhrusheva O, Markowitsch SD,
Slade KS, Tsaur I, Cinatl J Jr, Michaelis M, Efferth T, Haferkamp A
and Juengel E: Artesunate impairs growth in cisplatin-resistant
bladder cancer cells by cell cycle arrest, apoptosis and autophagy
induction. Cells. 9(2643)2020.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Zhao L, Liang L, Guo M, Li M, Yu X and
Wang Y and Wang Y: Hepatocellular carcinoma targeting and
pharmacodynamics of paclitaxel nanoliposomes modified by
glycyrrhetinic acid and ferric tetroxide. Curr Top Med Chem.
21:1268–1284. 2021.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Cao M, Gao Y, Zhan M, Qiu N, Piao Y, Zhou
Z and Shen Y: Glycyrrhizin acid and glycyrrhetinic acid modified
polyethyleneimine for targeted DNA delivery to hepatocellular
carcinoma. Int J Mol Sci. 20(5074)2019.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Sang L, Li J, Zhang F, Jia J, Zhang J,
Ding P, Sun T and Wang D: Glycyrrhetinic acid modified chlorambucil
prodrug for hepatocellular carcinoma treatment based on DNA
replication and tumor microenvironment. Colloids Surf B
Biointerfaces. 220(112864)2022.PubMed/NCBI View Article : Google Scholar
|