Introduction
Biliary atresia (BA) is a rare destructive
inflammatory disease that occurs in infancy and affects the
intrahepatic and extrahepatic bile duct system to varying degrees,
resulting in intrahepatic cholestasis, intrahepatic and
extrahepatic bile duct obstruction, progressive liver fibrosis and
malignant progression to liver cirrhosis (1,2). It
has a high incidence in Asia, occurring in ~1:5,000 live births
(3), while in Western countries
the incidence is relatively low, ~1:15,000-19,000 live births
(4). According to the clinical
manifestations, BA can be divided into perinatal and fetal types.
The perinatal type accounts for ~90% and the majority of patients
have no concomitant malformations. The fetal type accounts for ~10%
with jaundice occurring in the early postnatal period, and the
majority of patients are also accompanied by congenital
malformations, such as BA and splenic malformation syndrome
(5). BA can also be divided into
three types according to the level of proximal biliary obstruction.
In type I BA (accounting for 5%), atresia occurs at the common bile
duct, and there is often a cyst structure in the proximal end of
the atresia. In type II BA (accounting for 2%), obstruction occurs
at the common hepatic duct. In type III BA (accounting for
>90%), the extrahepatic bile duct is completely atretic, and the
hepatic hilum is a fibrotic solid structure (6). At present, the etiology of BA is not
clear, and it is considered to be the final result of multiple
conditions, such as sclerosing occlusive inflammatory biliary
disease. Possible causes include congenital genetic factors,
infection factors accompanied by inflammation and immune response,
maternal factors and vascular factors (7-9).
Therefore, studying the molecular mechanism of BA is a key
scientific issue in the clinic that needs to be solved.
Circular RNA (circRNA) is a type of non-coding RNA
molecule that does not have a 5'-terminal cap or a 3'-terminal poly
(A) tail and forms a ring structure with covalent bonds. As circRNA
molecules have a closed ring structure, they are not affected by
RNA exonucleases in cells, they are not easy to degrade and their
expression is more stable (10,11).
Previous studies (12-14)
have shown that circRNA molecules contain binding sites for
microRNA (miRNA/miR) or RNA binding proteins, which act as miRNA
sponges and trans-acting factors in cells, suggesting that circRNA
may influence and regulate human diseases by regulating
disease-associated miRNAs (15).
Furthermore, a number of previous studies have shown that circRNAs
are associated with numerous diseases, such as systemic lupus
erythematosus (16), coronary
artery disease (17), several
types of cancer (such as breast and stomach cancer) (18) and nervous system disease (19). However, there have been only a
small number of studies on the circRNA regulatory network in BA,
and the mechanism of most circRNAs in BA is still in its infancy.
With the development of next-generation sequencing and
bioinformatics analysis, circRNA research is progressing. Numerous
circRNAs are demonstrated to be involved in the progression of a
number of diseases, and because of their conservation, stability,
specificity, richness and easy detection (20) they not only point out a new
direction for clinical treatment, but also provide new markers for
the early diagnosis of BA. A number of circRNAs also provide novel
ideas for clarifying the mechanism of the circRNA-miRNA axis in the
process of liver fibrosis (21,22).
In the present study, DECs between BA and CC tissues
were identified based on high-throughput RNA sequencing.
Subsequently, eight candidate circRNAs were selected and their
expression levels in the liver tissues of patients with BA and
control patients with choledochal cyst (CC) were detected using
reverse transcription-quantitative polymerase chain reaction
(RT-qPCR). The miRNAs that can bind to the eight circRNAs and their
downstream target genes were predicted using bioinformatics
technology, and Gene Ontology (GO) and Kyoto Encyclopedia of Genes
and Genomes (KEGG) pathway enrichment analysis were carried out.
The results of the present study provided an important theoretical
basis for the molecular mechanism of the circRNA network in
regulating the occurrence and development of BA.
Materials and methods
Sample preparation
Between April 2018 and May 2020, 38 patients with BA
and 54 patients with choledochal cysts (CCs) were enrolled in the
present study. All patients were diagnosed via laparoscopic bile
duct exploration by the same surgical team at Shenzhen Children's
Hospital (Shenzhen, China), and liver biopsy tissues were obtained
at the time of surgery. The mean age of the patients in the BA
group was 72.58±27.31 days, and the group included 15 male and 23
female patients (Table SII). The
mean age of the patients in the CC group was 40.32±38.62 months,
and the group included 14 male and 40 female patients (Table SIII). The liver tissues were
immersed in RNA sample preservation solution (cat. no. R916331;
Macklin, Inc.) and cryopreserved at -80˚C. The patients did not
receive any treatment before surgery. The present study was
approved by the Ethics Committee of Shenzhen Children's Hospital
(approval no. SUMC2017-026). The parents of all the subjects
provided written informed consent.
RNA isolation
TRIzol® reagent (Invitrogen; Thermo
fisher Scientific, Inc.) was used to extract total RNA from BA
liver tissues as well as CC tissues. The concentration and purity
of RNA was detected using a Nanodrop-1000 (Thermo Fisher
Scientific, Inc.) and Qubit RNA HS Assay Kit (cat. no. Q32852;
Thermo Fisher Scientific, Inc.), then the yield and quality was
evaluated using an Agilent 2100 Bioanalyzer (Agilent Technologies,
Inc.) to test the integrity of the RNA. All RNA integrity numbers
were >7 to ensure RNA quality.
Library construction and
sequencing
Before the construction of the library, ribosomal
RNA (rRNA) was removed using a Ribo-Zero Plus rRNA Depletion Kit
(cat. no. 20037135; Illumina, Inc.). NEBNext® Multiplex
Small RNA Library Prep Set (cat. no. E7330S; Illumina, Inc.) was
used to generate a sequencing library. To map the sequence to each
sample, a barcode had to be added. Subsequently, the quality of the
library was examined using a RNA high sensitivity chip on an
Agilent 2100 Bioanalyzer (Agilent Technologies, Inc.). VAHTS
Library Quantification Kit for Illumina (cat. no. NQ101; Vazyme,
Inc.) was used to accurately quantify the effective concentration
of the library with an ABI StepOnePlus Real-Time PCR system (cat.
no. 4376600; Applied Biosystems; Thermo Fisher Scientific, Inc.).
The library was diluted to 20 pM as the final concentration. Then a
sample cluster was performed on the cBot cluster generation system
(cat. no. SY-312-2001; Illumina, Inc.) with TruSeq PE Cluster kit
v3-cBot-HS (cat. no. 20015963; Illumina, Inc.), followed by
paired-end sequencing of 125-bp reads with the Illumina HiSeq 2500
platform (Illumina, Inc.). All steps followed the manufacturer's
protocols.
Sequencing data analysis and circRNA
analysis
The accuracy of the sequencing results of the
extracted RNA from liver BA and CC tissues was verified by
filtering the sequencing data. Using Trimmomatic (23), the reads containing adapters were
removed, then the low-quality sequences at the 5' and 3' ends were
trimmed, and the reads containing >5% N bases were removed. This
produced high-quality clean reads for all downstream analyses. The
reference genome as well as gene annotation files were downloaded
from the Ensembl genome browser (Ensembl GRCh37 release 110-July
2023; https://grch37.ensembl.org/index.html). The sequencing
reads were mapped to the human genome using the HISAT2 software,
and circRNAs were identified using ‘circRNA_finder’ analysis.
Additionally, the expression of known miRNAs was compared with the
precursor and mature miRNA sequences in miRbase (version 22)
(24) using default parameters
(25). The differentially
expressed mRNAs, miRNAs and circRNAs were identified using the
edgeR software package (26), with
P<0.05 and log2 fold-change (FC)>1 as selection parameters.
The pheatmap R package (https://cran.r-project.org/web/packages/pheatmap/pheatmap.pdf)
was used to cluster the samples. CircRNA sequencing was performed
by Vazyme Biotech Co., Ltd. qPCR was performed to verify the
expression of circRNA in tissue samples and all primer sequences
are presented in Table SI.
Functional and pathway enrichment
analysis
R software (version 4.0.2; https://cran.r-project.org/doc/contrib/Liu-R-refcard.pdf)
was used to estimate DECs between BA and CC samples. GO (https://geneontology.org/) term enrichment analysis
was performed, including molecular function, cellular component and
biological process, along with KEGG (http://www.genome.ad.jp/kegg/) analysis. DECs were
identified using the clusterProfiler (http://www.bioconductor.org/packages/release/bioc/html/clusterProfiler.html),
org.Hs.eg.db (http://www.bioconductor.org/packages/release/data/annotation/html/org.Hs.eg.db.html),
enrichplot (http://www.bioconductor.org/packages/release/bioc/html/enrichplot.html)
and ggplot2 packages (https://cran.rstudio.com/bin/windows/contrib/4.2/ggplot2_3.4.2.zip)
in Bioconductor, which is an R package used to perform GO
functional and KEGG pathway enrichment analysis.
RT-qPCR
The expression of the circRNAs was validated using
BA and CC tissues. Liver tissues were frozen in liquid nitrogen and
then crushed into a homogenate. Total RNA was extracted using
TRIzol® reagent and transcribed into cDNA using an
rtSTAR™ First-Strand cDNA synthesis kit (cat. no.
AS-FS-003-02; Arraystar, Inc.). Specific primers (presented in
Table SI) were designed with
Primer Premier 5.0 (Premier Biosoft), and synthesized by Vazyme
Biotech Co., Ltd. Following the manufacturer's protocols, Arraystar
SYBR® Green Real-time qPCR Master Mix (Arraystar Inc.)
was used for qPCR. The cycling conditions were 5 min at 95˚C for
the initial denaturation period, then 15 sec at 95˚C for
denaturation and 1 min at 60˚C for annealing and extension,
repeated for 40 cycles. Expression levels were normalized to
endogenous control (taqman endogenous controls FG, Human GAPDH;
cat. no. 4352934E; Applied Biosystem; Thermo Fisher Scientific,
Inc.), and the FC relative to circRNA expression levels in the CC
group was calculated using the 2-ΔΔCq method according
to a previous study (27). All
steps followed the manufacturers' protocols.
Target gene prediction and functional
enrichment analysis
StarBase (v2.0) (28), TargetScan (https://www.targetscan.org/vert_80/) and miRanda
software (https://cbio.mskcc.org/miRNA2003/miranda.html) were
used to predict the downstream miRNAs of the circRNAs. The target
mRNAs of candidate miRNAs were further analyzed using the miRDB
(https://mirdb.org/custom.html),
miRTarBase (https://mirtarbase.cuhk.edu.cn) and TargetScan
databases. Subsequently, functional enrichment analysis of these
mRNAs was carried out as a Venn diagram using R software. All
databases were used according to default parameters.
Network visualization
In the present study, the online tool Search Tool
for the Retrieval of Interacting Genes/Proteins (https://string-db.org/) was used to analyze the
protein-protein interaction (PPI) of the predicted target genes,
and Cytoscape software (version 3.4.0) (29,30)
was used to construct the PPI network. Through node observation,
the key nodes of the PPI network were examined. The Wilcoxon rank
sum test was used to test for significant differences in
topological properties between the BA and CC groups.
Receiver operating characteristic
(ROC) curve analysis
To evaluate the impact of differential gene
expression on the disease status of BA, ROC curve analysis was
used. This was conducted by plotting the ROC curve using gene
expression data juxtaposed with the sample state (with or without
BA), thereby enabling an assessment of gene expression accuracy
(31). The pROC package in R
software was used to generate these ROC curves (31). The entropy weight method was used
to determine the entropy weight of each gene, following which the
ROC curves for five genes with significant differences between the
BA and CC groups were plotted. The accuracy of each biomarker was
determined by the area under the curve (AUC) derived from the ROC
curve analysis.
Statistical analysis
R software was used to integrate and analyze the
data. Continuous variables are expressed as the mean ± standard
deviation (at least 3 experimental repeats). An independent samples
t-test was used to compare the continuous variables between BC and
CC groups, as the samples were independent from each other. The
figures were prepared using GraphPad Prism 8.0 (GraphPad Software;
Dotmatics). P<0.05 was considered to indicate a statistically
significant difference.
Results
Identification and annotation of
circRNAs
In the present study, the sequencing reads were
first mapped to the human genome, and the circRNAs were then
systematically identified and annotated using ‘circRNA_finder’
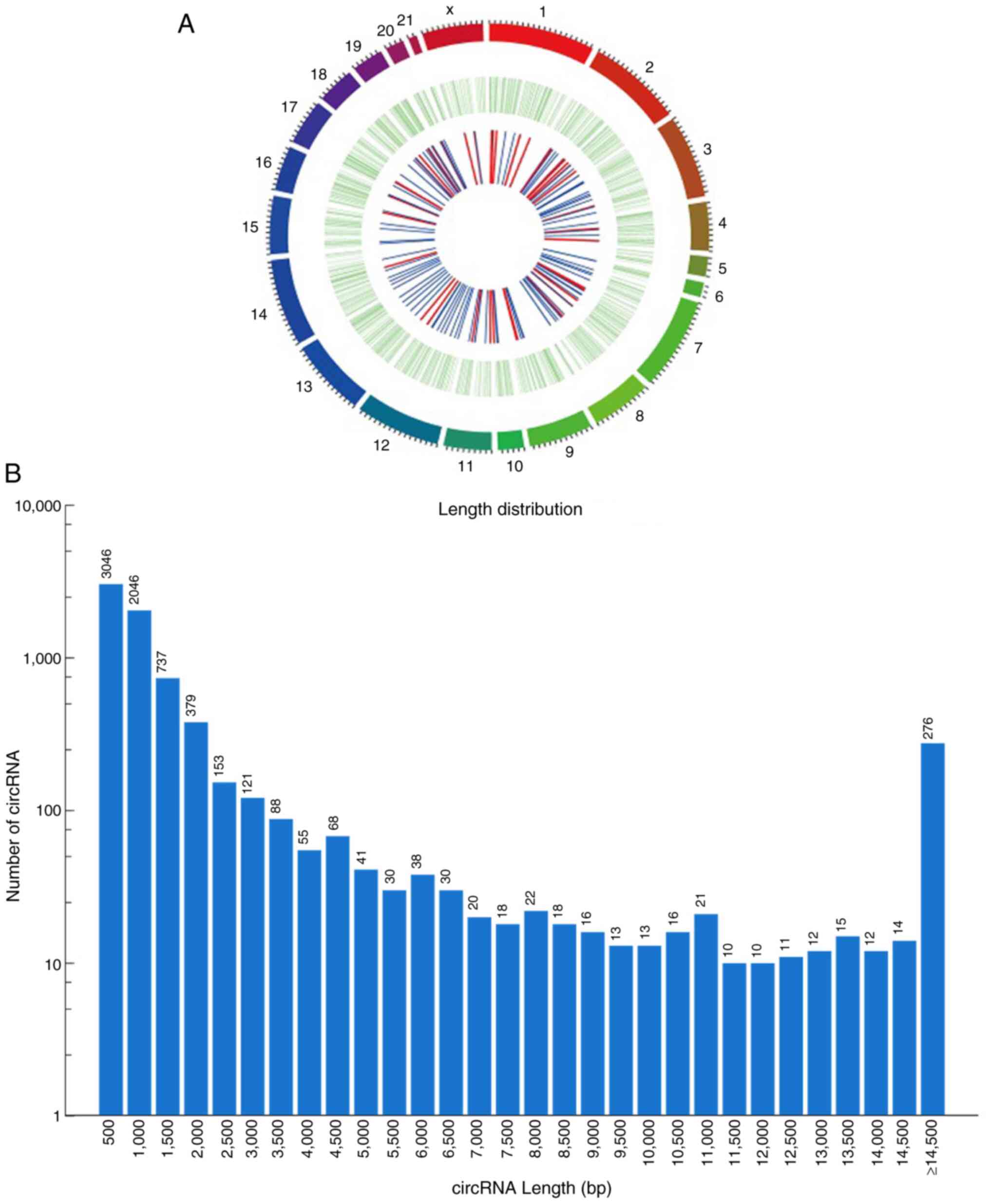
analysis. In total, 7,349 circRNAs were identified and three types
of circRNAs were revealed, including exon, intergenic and intron
circRNAs. Among them, most circRNAs (83.4%) were of the exon type,
6.4% were intergenic and 7.9% were intron type (a small percentage
were not annotated). circRNA transcripts were distributed in the
majority of chromosomes (Chr) (Fig.
1A). circRNAs from Chr1, Chr2, Chr3 and Chr13 accounted for
9.49, 7.48, 6.18 and 5.58%, respectively. These chromosomes
corresponded to more than half of the RNAs of interest. In
addition, the length of the circRNAs from these four choromosomes
ranged from 152-9,637 bp, and the distribution frequency was 69.3%
for circRNAs ranging from 152-1,000 bp and 16.4% for circRNAs
>2,000 bp (Fig. 1B).
Identification of DECs in BA
To identify the DECs associated with BA and CC,
high-throughput analysis was performed on liver tissues from BA and
CC cases (3 cases each). The R software package was used to analyze
the differential expression, and a list of upregulated and
downregulated DECs was obtained. According to the criteria of
log2FC >2 and P<0.05, there were 78 DECs, including 16
upregulated circRNAs and 62 downregulated circRNAs (Fig. 2A). The top five upregulated genes
and the top three downregulated genes (using the volcano map in
Fig. 2B) were selected for
subsequent RT-qPCR verification. Table
I presents the names of all upregulated and downregulated
circRNAs.
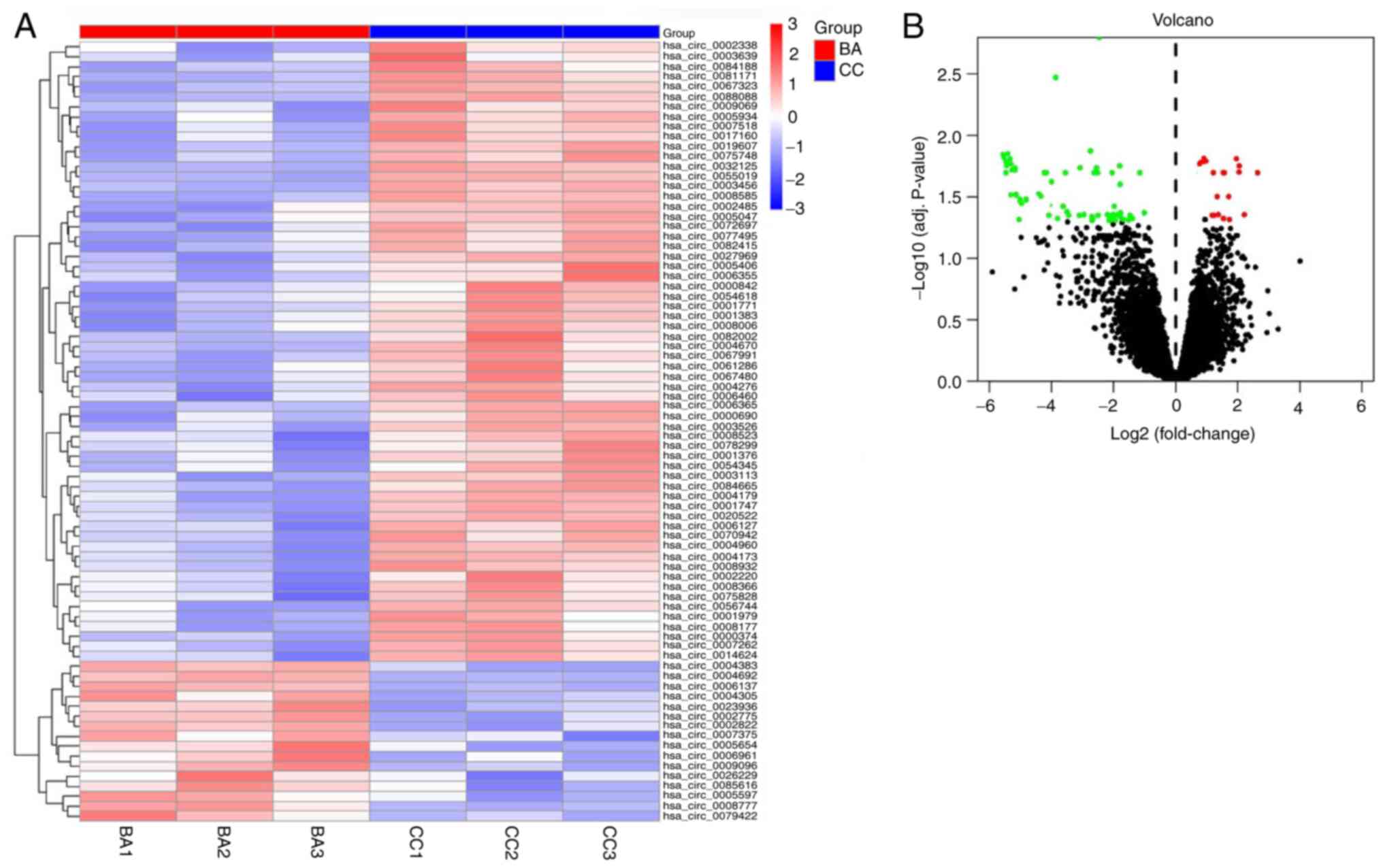 | Figure 2Heatmap and volcano plot of DECs. (A)
Heatmap indicating 16 upregulated circRNAs and 62 downregulated
circRNAs; different rows represent the different genes. Red,
upregulated genes; blue, downregulated genes. (B) Volcano plot
representing circRNA expression in BA. Black points, normally
expressed circRNAs; red points, upregulated DECs; and green points,
downregulated DECs. BA, biliary atresia; CC, choledochal cyst;
circRNA, circular RNA; DECs, differentially expressed circRNAs;
adj., adjusted. |
 | Table IDECs between biliary atresia
choledochal cyst tissues (16 upregulated and 62 downregulated
circRNAs). |
Table I
DECs between biliary atresia
choledochal cyst tissues (16 upregulated and 62 downregulated
circRNAs).
| Type of DECs | circRNAs |
|---|
| Upregulated | hsa_circ_0006137,
hsa_circ_0079422, hsa_circ_0007375, hsa_circ_0005597,
hsa_circ_0006961, hsa_circ_0004305, hsa_circ_0002775,
hsa_circ_0009096, hsa_circ_0085616, hsa_circ_0026229,
hsa_circ_0005654, hsa_circ_0004692, hsa_circ_0023936,
hsa_circ_0004383, hsa_circ_0002822, hsa_circ_0008777 |
| Downregulated | hsa_circ_0081171,
hsa_circ_0084665, hsa_circ_0075828, hsa_circ_0000374,
hsa_circ_0006460, hsa_circ_0005934, hsa_circ_0001747,
hsa_circ_0054345, hsa_circ_0067991, hsa_circ_0005047,
hsa_circ_0008177, hsa_circ_0003526, hsa_circ_0056744,
hsa_circ_0008523, hsa_circ_0002338, hsa_circ_0088088,
hsa_circ_0008932, hsa_circ_0001383, hsa_circ_0002485,
hsa_circ_0055019, hsa_circ_0061286, hsa_circ_0003113,
hsa_circ_0000690, hsa_circ_0004173, hsa_circ_0070942,
hsa_circ_0006355, hsa_circ_0054618, hsa_circ_0082002,
hsa_circ_0008585, hsa_circ_0008366, hsa_circ_0017160,
hsa_circ_0067323, hsa_circ_0005406, hsa_circ_0007518,
hsa_circ_0003639, hsa_circ_0032125, hsa_circ_0082415,
hsa_circ_0027969, hsa_circ_0008006, hsa_circ_0078299,
hsa_circ_0004670, hsa_circ_0004960, hsa_circ_0009069,
hsa_circ_0075748, hsa_circ_0020522, hsa_circ_0007262,
hsa_circ_0006365, hsa_circ_0019607, hsa_circ_0002220,
hsa_circ_0006127, hsa_circ_0001376, hsa_circ_0067480,
hsa_circ_0072697, hsa_circ_0084188, hsa_circ_0003456,
hsa_circ_0000842, hsa_circ_0001979, hsa_circ_0001771,
hsa_circ_0004276, hsa_circ_0014624, hsa_circ_0004179,
hsa_circ_0077495 |
Functional and pathway enrichment
analysis of DECs
GO functional and KEGG pathway enrichment analysis
were performed on the host genes of the 78 DECs using R software.
The results of GO analysis indicated that the main biological
processes of these host genes were ‘positive regulation of
catabolic process’, ‘negative regulation of catabolic processes’,
‘regulation of microtubule motor activity’ and ‘cellular response
to alcohol’. The primary cellular component category consisted of
the categories ‘intracellular part’, ‘organelle part’, ‘plasma
membrane region’ and ‘cytoplasm’. Finally, the main molecular
functions included ‘enzyme binding’, ‘GTPase activating protein
binding’, ‘glucocorticoid receptor binding’ and various enzyme
activities, such as ‘transferase activity’ and ‘phosphotransferase
activity, alcohol group as acceptor’ (Fig. 3A). The KEGG pathway analysis of
these genes was primarily enriched in ‘pyruvate metabolism’, ‘ABC
transporters’, ‘intestinal immune network for IgA production’,
‘viral myocarditis’, ‘leishmaniasis’, ‘Staphylococcus aureus
infection’, ‘hematopoietic cell lineage’, ‘toxoplasmosis’, ‘cell
adhesion molecules’, ‘systemic lupus erythematosus’ and ‘phagosome’
(Fig. 3B).
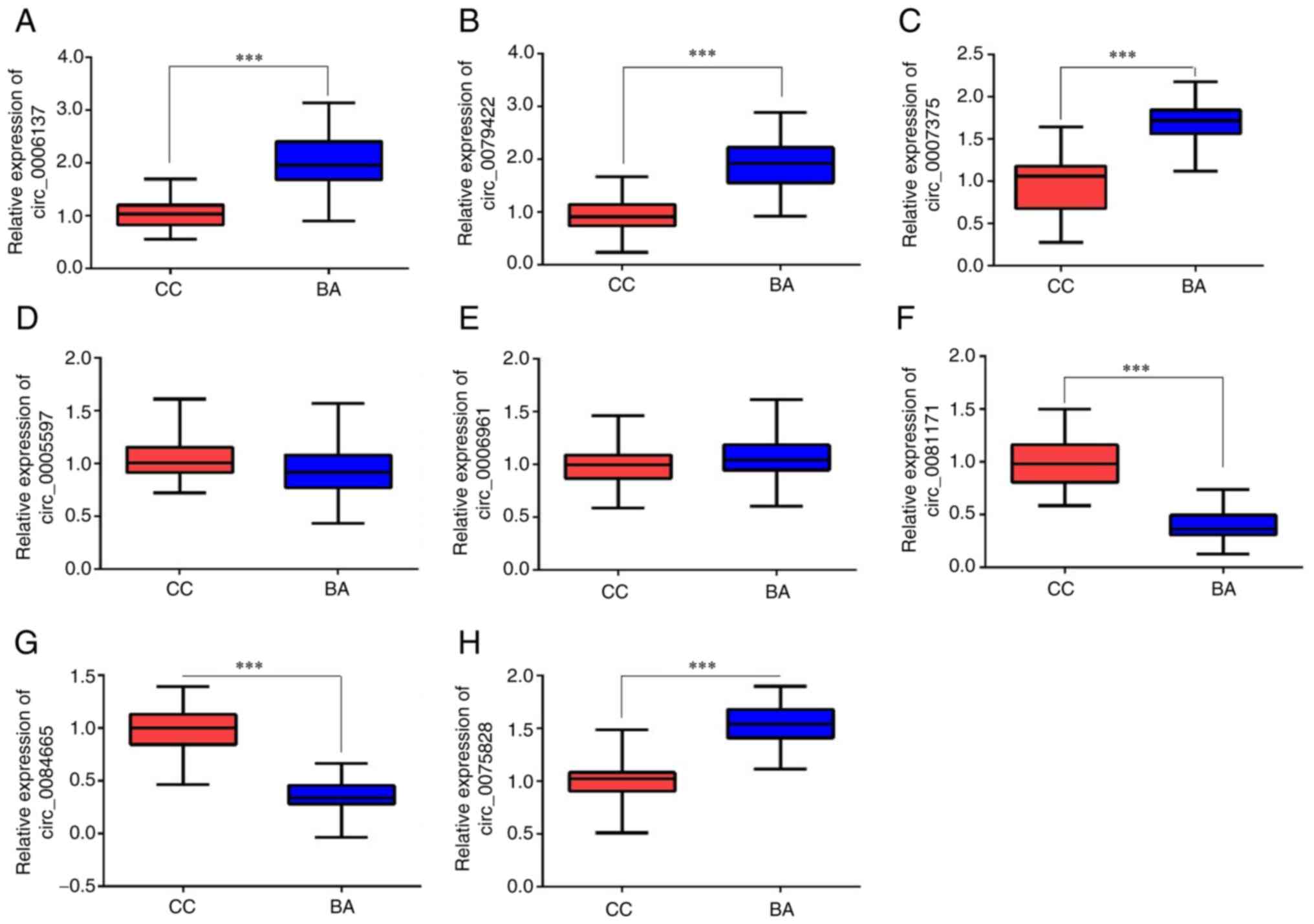
Validation of DECs using RT-qPCR
Liver tissue samples from 38 patients in the BA
group and 54 patients in the CC group were analyzed using RT-qPCR.
A total of five significantly upregulated circRNAs
(hsa_circ_0006137, hsa_circ_0079422, hsa_circ_0007375,
hsa_circ_0005597 and hsa_circ_0006961) and three significantly
downregulated circRNAs (hsa_circ_0081171, hsa_circ_0084665 and
hsa_circ_0075828) from the R software analysis were selected for
RT-qPCR to verify the expression of these DECs. The RT-qPCR results
demonstrated that the expression levels of hsa_circ_0006137,
hsa_circ_0079422 and hsa_circ_0007375 were significantly increased
(Fig. 4A-C), while the expression
levels of hsa_circ_0081171 and hsa_circ_0084665 were significantly
reduced (Fig. 4F and G) in patients with BA compared with the
CC group. However, there was no significant difference in the
expression levels of hsa_circ_0005597 and hsa_circ_0006961 between
the two groups (Fig. 4D and
E). Hsa_circ_0075828 also
exhibited a significant increase in patients with BA compared with
the CC group (Fig. 4H), contrary
to the previous screening results. Therefore, five validated DECs
were used for bioinformatics analysis.
Construction of the circRNA regulatory
network
An increasing number of studies have demonstrated
that circRNAs can increase the expression levels of downstream
genes by binding to miRNAs as molecular sponges (32,33).
Therefore, 244 potential target miRNAs of hsa_circ_0006137,
hsa_circ_0079422, hsa_circ_0007375, hsa_circ_0081171 and
hsa_circ_0084665 were predicted through starBase (v2.0). According
to competitive endogenous RNA (ceRNA) theory, there is a negative
correlation between a circRNA and its target miRNAs (34). Therefore, through a literature
search, seven miRNAs were selected as the target miRNAs of the
circRNAs (hsa_circ_0006137/miR-26a-5p, hsa_circ_0006137/miR-145-5p,
hsa_circ_0079422/miR-593-3p, hsa_circ_0007375/miR-1206,
hsa_circ_0007375/miR-1208, hsa_circ_0081171/miR-18a-5p and
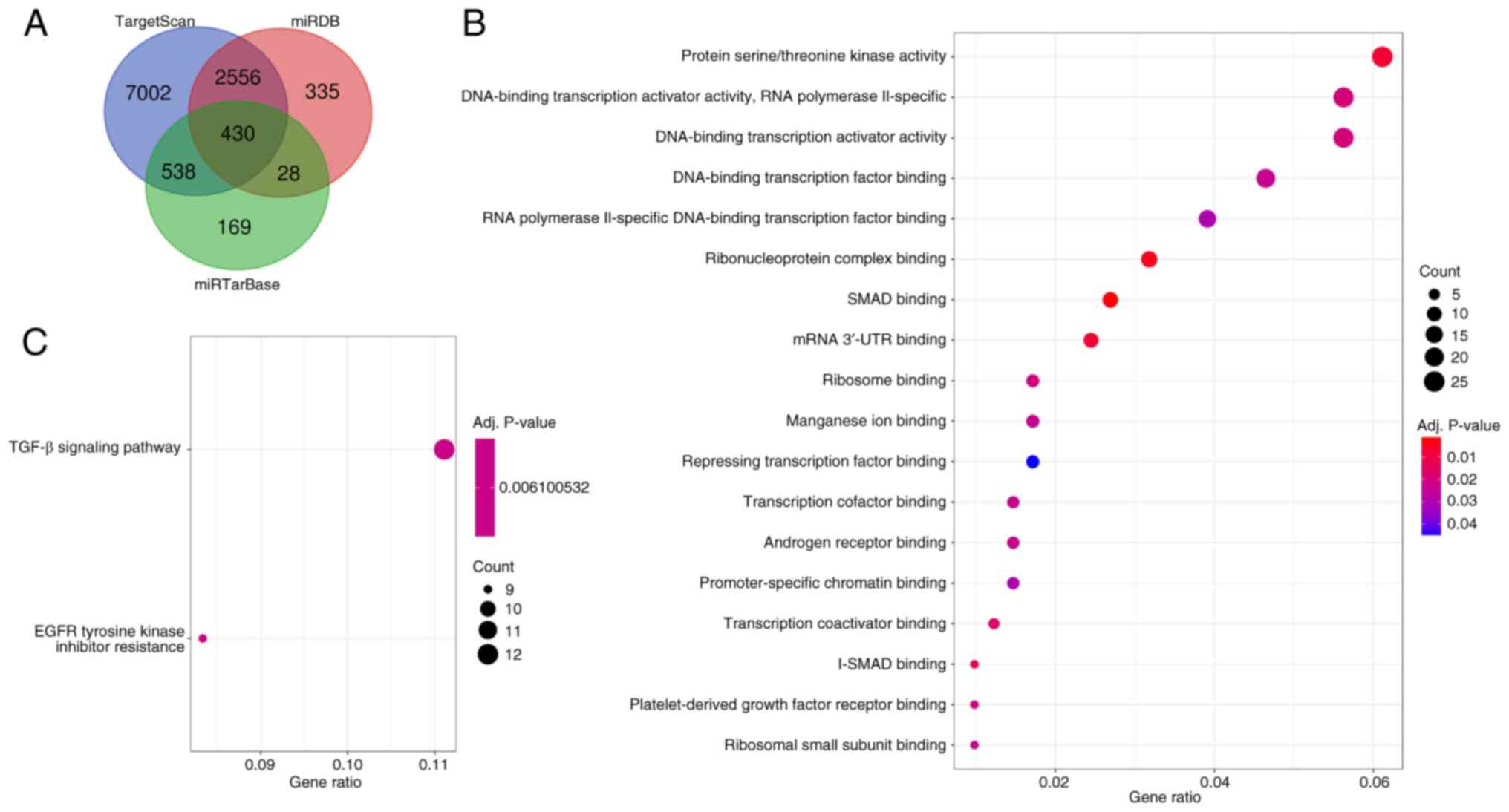
hsa_circ_0084665/miR-22-5p) for further analysis. Subsequently, 430
target mRNAs were predicted to correspond to these seven miRNAs
through the miRDB, miRTarBase and TargetScan databases (Fig. 5A).
Functional analysis of mRNAs
To examine the potential functional role of the five
circRNAs, GO and KEGG pathway enrichment analysis on the target
mRNAs was carried out. As presented in Fig. 5B, these genes were significantly
enriched in the forward transcriptional regulation of ‘protein
serine/threonine kinase activity’, ‘RNA polymerase II promoter’,
‘DNA-binding transcription activator activity, RNA polymerase
II-specific’, ‘DNA-binding transcription factor binding’ and ‘SMAD
binding’. The associated pathways obtained using KEGG analysis were
fewer, but ‘TGF-β signaling pathway’ and ‘EGFR tyrosine kinase
inhibitor resistance’ were included in the enriched pathways
(Fig. 5C). In summary, these
functional analysis results suggested that the circRNA network may
regulate the development of BA through the TGF-β and EGFR signaling
pathways, which supports previous study results.
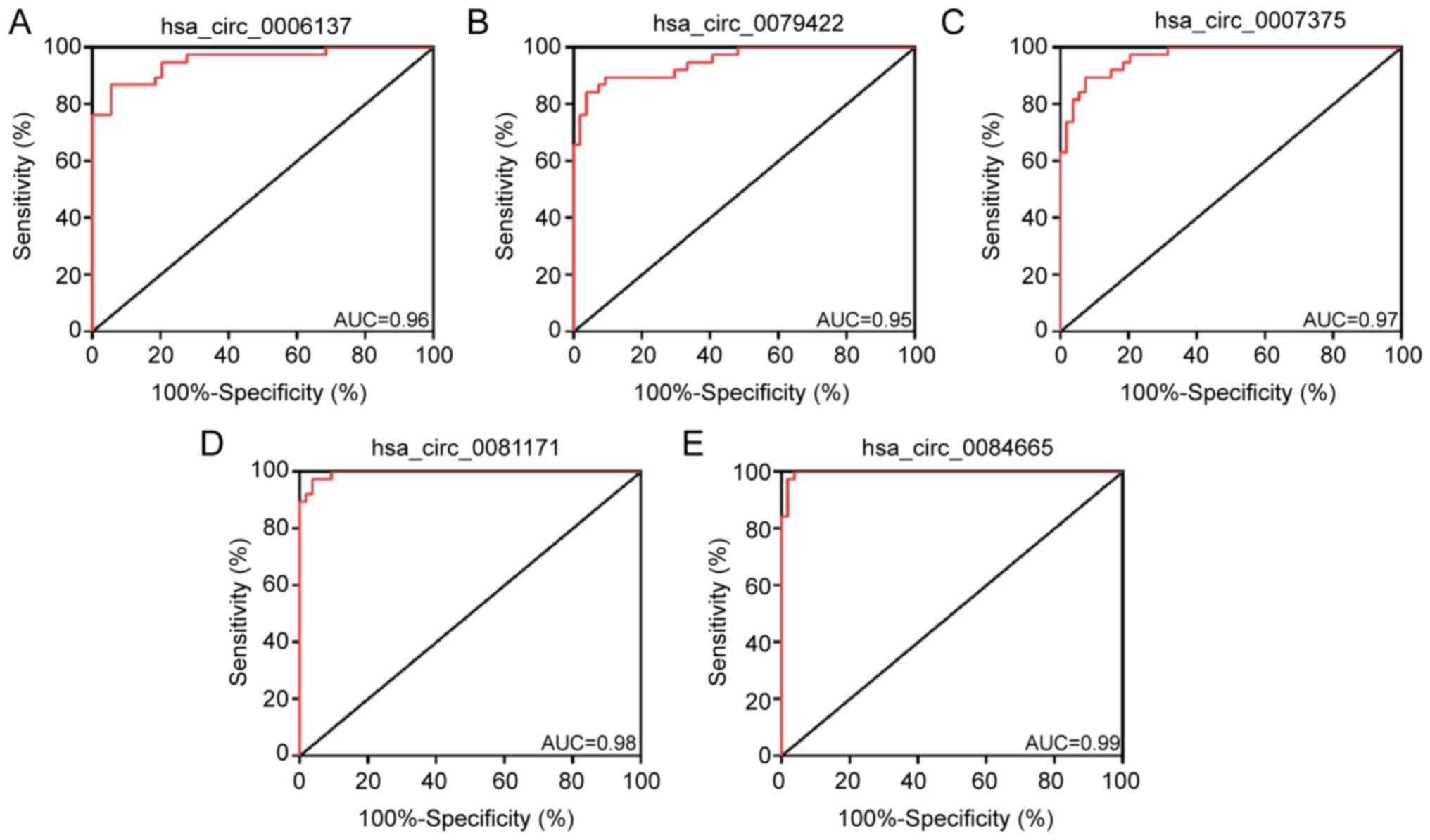
Evaluation of DECs using ROC
analysis
To further investigate the diagnostic potential of
the aforementioned circRNAs, ROC analysis was used to evaluate the
detection sensitivity and specificity. As presented in Fig. 6A-E, the AUC of hsa_circ_0006137,
hsa_circ_0079422, hsa_circ_0007375, hsa_circ_0081171 and
hsa_circ_0084665 in the differential diagnosis of BA compared with
CC was >0.8, indicating that these circRNAs have a relatively
high sensitivity and specificity for BA. These findings suggested
that these circRNAs may serve as potential indicators for
distinguishing BA from CC and could offer important insights for
clinical research.
Discussion
BA is a destructive inflammatory disease,
Lakshminarayanan and Davenport (35) demonstrated that viral infection,
toxicological effects and gene mutations may be associated with it.
In previous years, research has been devoted to investigating new
therapeutic targets and biomarkers of BA. For example, Girard and
Panasyuk (36) revealed an
abnormal expression of a number of genes (such as GPC1 and TCF4) in
BA. High-throughput sequencing technology has broadened the
understanding of gene regulatory networks. Genome-wide sequencing
demonstrated that ~93% of the genome is transcribed into RNA, but
only 2% encodes proteins (37).
Although the total number of nucleotides in the human genome is 30
times that of the nematode genome, the number of protein coding
sequences is similar, which highlights the importance of non-coding
RNA (ncRNA) sequences in regulating eukaryotic gene expression
(38). With the widespread
acceptance of the concept of ceRNA suggested by Salmena et
al (39), miRNA has become the
core of the ncRNA regulatory network. Calvopina et al
(40) revealed that numerous types
of miRNAs are specifically expressed in the tissues of children
with BA, which proves that the gene regulatory network centered on
miRNA may serve an important role in the pathogenesis of BA.
Previously, circRNA has been revealed to serve as an important
ceRNA that can regulate gene expression at the posttranscriptional
level by binding to target miRNAs (41). Due to further research, an
increasing number of circRNAs have been revealed to be new
diagnostic markers for diseases, including cancer (42,43).
However, there have only been a small number of reports on circRNAs
associated with BA.
To the best of our knowledge, the present study is
the first to analyze the circRNA regulatory network of BA,
revealing 16 upregulated circRNAs and 62 downregulated circRNAs.
The function of the DECs was investigated using GO and KEGG
enrichment analysis. In addition, three upregulated circRNAs and
two downregulated circRNAs were verified using RT-qPCR. GO
enrichment analysis of the DECs indicated that ‘regulation of
catabolic process’, ‘regulation of cellular catabolic process’ and
‘positive regulation of biological process’ were mostly enriched in
the biological process category, indicating that the disturbance of
energy metabolism may promote the occurrence of BA. In terms of
cell component and molecular function, the membrane region and
transferase activity indict that intercellular junction and the
enrichment of extracellular matrix were involved, which may be
associated with the damage of bile duct epithelial cells and the
inflammatory infiltration of the bile duct in BA. KEGG analysis
demonstrated that ‘myocarditis’ was significantly enriched. A
previous study demonstrated that bacteremia caused by golden
Staphylococci can be complicated with endocarditis,
metastatic infection or septicemia syndrome (44). Furthermore, patients with liver
disease can experience lesions of the biliary tract or gallbladder
(45,46).
In the present study, three upregulated circRNAs
were identified to bind to five miRNAs. According to previous
studies, miR-26a-5p can increase the transcriptional level of THAP
domain-containing protein 2 and induce apoptosis in endometrial
cancer cells (47). In a mouse
model of myocardial infarction, the expression of miR-26a-5p was
downregulated in myocardial cells following ischemia-reperfusion
injury, and myocardial ischemia-reperfusion injury was regulated by
the expression level of PTEN gene through the PI3K/AKT signaling
pathway (48). In our previous
study, it was revealed that the expression level of miR-145 was
significantly decreased in BA (49), while in the present study, it was
revealed that the upregulated hsa_circ_0006137 had a binding site
for miR-145, which may be the reason for the downregulation of the
latter in BA. In osteosarcoma, miR-593-3p can inhibit tumorigenesis
by promoting the upregulation of zinc finger E-box binding homeobox
2(50). SNHG14(51) and MAP3K2(52) genes have been shown to serve as
targets of miR-1206 and miR-1208 respectively, and miR-1206 and
miR-1208 can act as targets for tumor suppression.
To fully understand the effects of
circRNA-associated regulatory networks on BA, the miRNA-circRNA and
miRNA-mRNA interaction was predicted. GO and KEGG enrichment
analyses of the genes in this network were carried out and revealed
that the enrichment terms were associated with the pathogenesis of
BA. GO enrichment analysis suggested that these mRNAs were involved
in the ‘DNA-binding transcription activator activity, RNA
polymerase II-specific’. The results of the KEGG pathway enrichment
analysis demonstrated that these downstream target genes were
significantly enriched in the ‘TGF-β signaling pathway’, while
enrichment of ‘EGFR tyrosine kinase inhibitor resistance’ was also
observed. The EGFR family is one of the most studied receptor
protein tyrosine kinases, because it serves a universal role in
signal transduction and tumorigenesis (53). Activation of the TGF-β signaling
pathway can increase the expression levels of extracellular matrix
proteins (such as SMAD and PI3K) (54), cause an imbalance between
extracellular matrix production and degradation, and promote the
occurrence of BA. For example, Chung-Davidson et al
(55) revealed that BA
cholangiopathy can be delayed by blocking the TGF-β signaling
pathway.
Therefore, the present study suggests that the
identified DECs may be associated with BA by regulating gene
expression. Further investigations should be performed by
experimental methods such as dual-luciferase activity experiment
and PCR tests. Further verification of the interaction of circRNAs
with miRNAs and in-depth study of the function of circRNAs and
their effect on cell regulation should be performed. While the
present study revealed important insights into the circRNA
regulatory network of BA, it should acknowledge certain
limitations. One such limitation was the use of the same samples
for both identification and validation of circRNAs. Using the same
samples for both stages of the study can introduce bias, as the
validation stage was not independent of the identification stage.
However, the findings of the present study offered a foundation for
future research, and further studies with independent validation
cohorts to validate and expand upon the present results should be
performed.
In conclusion, the present study obtained a circRNA
map of BA liver tissue based on RNA high-throughput sequencing and
identified 78 DECs. Subsequently, the expression of three
upregulated circRNAs and two downregulated circRNAs in BA liver
tissues were further verified. Moreover, circRNA regulatory
networks in BA were constructed for the first time and their
potential biological functions were analyzed. The study of the
circRNA-miRNA pathway may provide further insights for examining
the pathogenesis of BA. Thus, the potential molecular mechanism of
circRNAs in BA require further elucidation. However, it is
important to note that these results were not directly associated
with prognosis, as no clinical data were considered in the present
analysis. Future studies should incorporate relevant clinical data
to evaluate the prognostic potential of these circRNAs.
Supplementary Material
Primer sequences used for
circRNA.
Clinical information of patients with
biliary atresia.
Clinical information of choledochal
cyst patients.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by the Shenzhen Medical
and Health Project (grant no. SZSM201812055), the National Natural
Science Foundation of China (grant no. 81770512) and the Medical
Science and Technology Research Foundation of Guangdong Province
(grant no. A2019541).
Availability of data and materials
The sequencing datasets generated and/or analyzed
during the current study are available in Gene Expression Omnibus
(https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE240795).
All other datasets used and/or analyzed during the current study
are available from the corresponding author on reasonable
request.
Authors' contributions
DL and YD designed the study. DL, YD and JG
conducted the experiments. ZW, LZ and BW analyzed the data and
wrote the manuscript. All authors read and approved the final
version of the manuscript. DL, YD, JG, ZW, LZ and BW confirm the
authenticity of all the raw data.
Ethics approval and consent to
participate
All procedures performed in studies involving human
participants were approved by the Ethics Committee of Shenzhen
Children's Hospital (Shenzhen, China; approval no. SUMC2017-026).
The parents/guardians of all subjects signed a written informed
consent form.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kawano Y, Yoshimaru K, Uchida Y, Kajihara
K, Toriigahara Y, Shirai T, Takahashi Y and Matsuura T: Biliary
atresia in a preterm and extremely low birth weight infant: A case
report and literature review. Surg Case Rep. 6(321)2020.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Bezerra JA, Wells RG, Mack CL, Karpen SJ,
Hoofnagle JH, Doo E and Sokol RJ: Biliary atresia: Clinical and
research challenges for the twenty-first century. Hepatology.
68:1163–1173. 2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Hsiao CH, Chang MH, Chen HL, Lee HC, Wu
TC, Lin CC, Yang YJ, Chen AC, Tiao MM, Lau BH, et al: Universal
screening for biliary atresia using an infant stool color card in
Taiwan. Hepatology. 47:1233–1240. 2008.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Hartley JL, Davenport M and Kelly DA:
Biliary atresia. Lancet. 374:1704–1713. 2009.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Davenport M, Tizzard SA, Underhill J,
Mieli-Vergani G, Portmann B and Hadzić N: The biliary atresia
splenic malformation syndrome: A 28-year single-center
retrospective study. J Pediatr. 149:393–400. 2006.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Hartley JL, O'Callaghan C, Rossetti S,
Consugar M, Ward CJ, Kelly DA and Harris PC: Investigation of
primary cilia in the pathogenesis of biliary atresia. J Pediatr
Gastroenterol Nutr. 52:485–488. 2011.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Fabris L, Cadamuro M, Guido M, Spirli C,
Fiorotto R, Colledan M, Torre G, Alberti D, Sonzogni A, Okolicsanyi
L and Strazzabosco M: Analysis of liver repair mechanisms in
Alagille syndrome and biliary atresia reveals a role for notch
signaling. Am J Pathol. 171:641–653. 2007.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Muraji T: Biliary atresia: New lessons
learned from the past. J Pediatr Gastroenterol Nutr. 53:586–587.
2011.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Edom PT, Meurer L, da Silveira TR, Matte U
and dos Santos JL: Immunolocalization of VEGF A and its receptors,
VEGFR1 and VEGFR2, in the liver from patients with biliary atresia.
Appl Immunohistochem Mol Morphol. 19:360–368. 2011.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Bolha L, Ravnik-Glavač M and Glavač D:
Circular RNAs: Biogenesis, function, and a role as possible cancer
biomarkers. Int J Genomics. 2017(6218353)2017.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Zeng X, Lin W, Guo M and Zou Q: A
comprehensive overview and evaluation of circular RNA detection
tools. PLoS Comput Biol. 13(e1005420)2017.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Verduci L, Strano S, Yarden Y and Blandino
G: The circRNA-microRNA code: Emerging implications for cancer
diagnosis and treatment. Mol Oncol. 13:669–680. 2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Hansen TB, Wiklund ED, Bramsen JB,
Villadsen SB, Statham AL, Clark SJ and Kjems J: miRNA-dependent
gene silencing involving Ago2-mediated cleavage of a circular
antisense RNA. EMBO J. 30:4414–4422. 2011.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Abdelmohsen K, Panda AC, Munk R,
Grammatikakis I, Dudekula DB, De S, Kim J, Noh JH, Kim KM,
Martindale JL and Gorospe M: Identification of HuR target circular
RNAs uncovers suppression of PABPN1 translation by CircPABPN1. RNA
Biol. 14:361–369. 2017.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Miao Q, Zhong Z, Jiang Z, Lin Y, Ni B,
Yang W and Tang J: RNA-seq of circular RNAs identified circPTPN22
as a potential new activity indicator in systemic lupus
erythematosus. Lupus. 28:520–528. 2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Li LJ, Zhu ZW, Zhao W, Tao SS, Li BZ, Xu
SZ, Wang JB, Zhang MY, Wu J, Leng RX, et al: Circular RNA
expression profile and potential function of hsa_circ_0045272 in
systemic lupus erythematosus. Immunology. 155:137–149.
2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Wang L, Shen C, Wang Y, Zou T, Zhu H, Lu
X, Li L, Yang B, Chen J, Chen S, et al: Identification of circular
RNA Hsa_circ_0001879 and Hsa_circ_0004104 as novel biomarkers for
coronary artery disease. Atherosclerosis. 286:88–96.
2019.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Lei M, Zheng G, Ning Q, Zheng J and Dong
D: Translation and functional roles of circular RNAs in human
cancer. Mol Cancer. 19(30)2020.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Floris G, Zhang L, Follesa P and Sun T:
Regulatory role of circular RNAs and neurological disorders. Mol
Neurobiol. 54:5156–5165. 2017.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Zhang X, Lu N, Wang L, Wang Y, Li M, Zhou
Y, Yan H, Cui M, Zhang M and Zhang L: Circular RNAs and esophageal
cancer. Cancer Cell Int. 20(362)2020.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Chen X, Zhu S, Li HD, Wang JN, Sun LJ, Xu
JJ, Hui YR, Li XF, Li LY, Zhao YX, et al:
N6-methyladenosine-modified circIRF2, identified by
YTHDF2, suppresses liver fibrosis via facilitating FOXO3 nuclear
translocation. Int J Biol Macromol. 248(125811)2023.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Wang Q, Long Z, Zhu F, Li H, Xiang Z,
Liang H, Wu Y, Dai X and Zhu Z: Integrated analysis of
lncRNA/circRNA-miRNA-mRNA in the proliferative phase of liver
regeneration in mice with liver fibrosis. BMC Genomics.
24(417)2023.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Bolger AM, Lohse M and Usadel B:
Trimmomatic: A flexible trimmer for Illumina sequence data.
Bioinformatics. 30:2114–2120. 2014.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Kozomara A and Griffiths-Jones S: miRBase:
Annotating high confidence microRNAs using deep sequencing data.
Nucleic Acids Res. 42 (Database Issue):D68–D73. 2014.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Friedländer MR, Mackowiak SD, Li N, Chen W
and Rajewsky N: miRDeep2 accurately identifies known and hundreds
of novel microRNA genes in seven animal clades. Nucleic Acids Res.
40:37–52. 2012.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Robinson MD, McCarthy DJ and Smyth GK:
edgeR: A Bioconductor package for differential expression analysis
of digital gene expression data. Bioinformatics. 26:139–140.
2010.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Li JH, Liu S, Zhou H, Qu LH and Yang JH:
starBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA
interaction networks from large-scale CLIP-Seq data. Nucleic Acids
Res. 42 (Database Issue):D92–D97. 2014.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Szklarczyk D, Gable AL, Lyon D, Junge A,
Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork
P, et al: STRING v11: Protein-protein association networks with
increased coverage, supporting functional discovery in genome-wide
experimental datasets. Nucleic Acids Res. 47 (D1):D607–D613.
2019.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Robin X, Turck N, Hainard A, Tiberti N,
Lisacek F, Sanchez JC and Müller M: pROC: An open-source package
for R and S+ to analyze and compare ROC curves. BMC Bioinformatics.
12(77)2011.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Xu X, Song B, Zhang Q, Qi W and Xu Y:
Hsa_circ_0022383 promote non-small cell lung cancer tumorigenesis
through regulating the miR-495-3p/KPNA2 axis. Cancer Cell Int.
23(282)2023.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Zhang C and He W: Circ_0020014 mediates
CTSB expression and participates in IL-1β-prompted chondrocyte
injury via interacting with miR-24-3p. J Orthop Surg Res.
18(877)2023.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Karreth FA, Reschke M, Ruocco A, Ng C,
Chapuy B, Léopold V, Sjoberg M, Keane TM, Verma A, Ala U, et al:
The BRAF pseudogene functions as a competitive endogenous RNA and
induces lymphoma in vivo. Cell. 161:319–332. 2015.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Lakshminarayanan B and Davenport M:
Biliary atresia: A comprehensive review. J Autoimmun. 73:1–9.
2016.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Girard M and Panasyuk G: Genetics in
biliary atresia. Curr Opin Gastroenterol. 35:73–81. 2019.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Debray D, Corvol H and Housset C: Modifier
genes in cystic fibrosis-related liver disease. Curr Opin
Gastroenterol. 35:88–92. 2019.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Costa FF: Non-coding RNAs, epigenetics and
complexity. Gene. 410:9–17. 2008.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Salmena L, Poliseno L, Tay Y, Kats L and
Pandolfi PP: A ceRNA hypothesis: The Rosetta Stone of a hidden RNA
language? Cell. 146:353–358. 2011.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Calvopina DA, Coleman MA, Lewindon PJ and
Ramm GA: Function and regulation of microRNAs and their potential
as biomarkers in paediatric liver disease. Int J Mol Sci.
17(1795)2016.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Vicens Q and Westhof E: Biogenesis of
circular RNAs. Cell. 159:13–14. 2014.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Li H, Zheng X, Gao J, Leung KS, Wong MH,
Yang S, Liu Y, Dong M, Bai H, Ye X and Cheng L: Whole transcriptome
analysis reveals non-coding RNA's competing endogenous gene pairs
as novel form of motifs in serous ovarian cancer. Comput Biol Med.
148(105881)2022.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Wu S, Wu Y, Deng S, Lei X and Yang X:
Emerging roles of noncoding RNAs in human cancers. Discov Oncol.
14(128)2023.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Ramljak S, Schmitz M, Repond C, Zerr I and
Pellerin L: Altered mRNA and protein expression of monocarboxylate
transporter MCT1 in the cerebral cortex and cerebellum of prion
protein knockout mice. Int J Mol Sci. 22(1566)2021.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Acalovschi M: Gallstones in patients with
liver cirrhosis: Incidence, etiology, clinical and therapeutical
aspects. World J Gastroenterol. 20:7277–7285. 2014.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Konyn P, Alshuwaykh O, Dennis BB,
Cholankeril G, Ahmed A and Kim D: Gallstone disease and its
association with nonalcoholic fatty liver disease, all-cause and
cause-specific mortality. Clin Gastroenterol Hepatol.
21:940–948.e2. 2023.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Li M, Xiao Y, Liu M, Ning Q, Xiang Z,
Zheng X, Tang S and Mo Z: MiR-26a-5p regulates proliferation,
apoptosis, migration and invasion via inhibiting hydroxysteroid
dehydrogenase like-2 in cervical cancer cell. BMC Cancer.
22(876)2022.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Xing X, Guo S, Zhang G, Liu Y, Bi S, Wang
X and Lu Q: miR-26a-5p protects against myocardial
ischemia/reperfusion injury by regulating the PTEN/PI3K/AKT
signaling pathway. Braz J Med Biol Res. 53(e9106)2020.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Ye Y, Li Z, Feng Q, Chen Z, Wu Z, Wang J,
Ye X, Zhang D, Liu L, Gao W, et al: Downregulation of microRNA-145
may contribute to liver fibrosis in biliary atresia by targeting
ADD3. PLoS One. 12(e0180896)2017.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Zhang C, Zhou H, Yuan K, Xie R and Chen C:
Overexpression of hsa_circ_0136666 predicts poor prognosis and
initiates osteosarcoma tumorigenesis through miR-593-3p/ZEB2
pathway. Aging (Albany NY). 12:10488–10496. 2020.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Abd El-Aziz A, El-Desouky MA, Shafei A,
Elnakib M and Abdelmoniem AM: Influence of pentoxifylline on gene
expression of PAG1/miR-1206/SNHG14 in ischemic heart disease.
Biochem Biophys Rep. 25(100911)2021.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Zhang Y, Wang D, Zhu T, Yu J, Wu X, Lin W,
Zhu M, Dai Y and Zhu J: CircPUM1 promotes hepatocellular carcinoma
progression through the miR-1208/MAP3K2 axis. J Cell Mol Med.
25:600–612. 2021.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Roskoski R Jr: Small molecule inhibitors
targeting the EGFR/ErbB family of protein-tyrosine kinases in human
cancers. Pharmacol Res. 139:395–411. 2019.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Tzavlaki K and Moustakas A: TGF-β
signaling. Biomolecules. 10(487)2020.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Chung-Davidson YW, Ren J, Yeh CY, Bussy U,
Huerta B, Davidson PJ, Whyard S and Li W: TGF-β signaling plays a
pivotal role during developmental biliary atresia in sea lamprey
(petromyzon marinus). Hepatol Commun. 4:219–234. 2019.PubMed/NCBI View Article : Google Scholar
|