Introduction
Growing evidence suggests that disruptions in
mitochondrial energy metabolism can contribute to the development
of various cardiac diseases (1-5).
Among the fuel sources utilized by the myocardium, increased fatty
acid oxidation plays a pivotal role in driving the pathogenesis of
cardiomyopathy (1-7).
The inhibition of fatty acid oxidation has been demonstrated to
alleviate cardiac dysfunction (2,4,6,8).
Acetyl-CoA carboxylases (ACCs) are key enzymes responsible for
catalyzing the carboxylation of acetyl-CoA into malonyl-CoA
(9). Mammalian cells contain two
isoforms of ACC, namely ACC1 and ACC2. ACC1 serves as the
rate-limiting enzyme responsible for fatty acid biosynthesis in the
cytoplasm of adipocytes and hepatocytes. ACC2 is located in the
outer mitochondrial membrane of cardiomyocytes and governs
mitochondrial fatty acid oxidation (9,10).
Therapeutics targeting ACC2 have the potential to regulate
mitochondrial fatty acid utilization, reduce oxidative stress and
confer cardioprotective benefits.
For the preceding five decades, lithium has served
as the first-line medication for treating bipolar disorder
(11). Human and preclinical
studies have both demonstrated that in addition to its
mood-stabilizing effects, lithium can enhance the activity of
mitochondrial complex I while hindering the formation of free
radicals and reducing lipid peroxidation and DNA damage (12-14).
Furthermore, low-dose lithium has been shown to promote longevity
(15,16) and reduce the risk of heart failure
(17,18). These findings suggest that low-dose
lithium may possess cytoprotective properties and thus may play an
essential role in regulating metabolic stress. However, whether
lithium exerts cardioprotective effects through the modulation of
cardiac metabolism remains unclear. Laboratory evidence has
indicated that lithium directly targets glycogen synthase kinase-3
beta (GSK-3β) (19), a crucial
protein kinase that inhibits the activity of ACC (20,21).
The present study investigated whether lithium can regulate the
activity of ACC2 and modulate mitochondrial fatty acid oxidation in
cardiomyocytes, with a focus on the potential of lithium for
mitigating metabolic stress. The results revealed that at a low
physiological concentration (0.3 mM), LiCl upregulated the
expression of phosphorylated (p-)GSK-3β and downregulated the level
of p-ACC2 in H9c2 cardiomyocytes. Additionally, when used in
combination with the GSK-3β inhibitor TWS119, LiCl (0.3 mM)
downregulated the expression of p-ACC2, an effect comparable to
treatment with TWS119 alone. Furthermore, LiCl (0.3 mM) inhibited
mitochondrial fatty acid oxidation, enhanced coupling efficiency
and the cellular respiratory control ratio, suppressed reactive
oxygen species (ROS) production and proton leakage and restored
mitochondrial membrane potential in glucose transporter type 4
(GLUT4)-knockdown H9c2 cardiomyocytes. Taken together, these
findings suggest that at a low physiological concentration, lithium
can inhibit mitochondrial fatty acid utilization and mitigate
oxidative stress in cardiomyocytes, potentially through its
upregulation of p-GSK-3β and downregulation of p-ACC2.
Materials and methods
Cell culture and treatment
The H9c2 cell line (cat. no. 60096) was purchased
from the Bioresource Collection and Research Center and cultured in
Dulbecco's modified Eagle's medium (DMEM; Sigma-Aldrich; Merck
KGaA) supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich;
Merck KGaA) at 37˚C in a humidified atmosphere with 5%
CO2. To retain the differentiation capacity and
mitochondrial respiratory activity, a subculture was performed when
the cells reached 80% confluence. The culture medium was changed
every 2-3 days. H9c2 cells were treated with LiCl (Sigma-Aldrich;
Merck KGaA) at concentrations of 0.1 mM (i.e., subtherapeutic), 0.3
mM (i.e., low therapeutic), or 1.0 mM (i.e., high therapeutic) for
48 h. Additionally, these cells were cotreated with LiCl and a
GSK-3β inhibitor, namely TWS119 (Sigma-Aldrich; Merck KGaA), at a
concentration of 8 µM for 48 h to evaluate whether lithium
downregulated p-ACC2 by modulating GSK-3β activity. Each experiment
was performed at least three times.
GLUT4-knockdown cellular model
To simulate insulin resistance and induce metabolic
stress in H9c2 cardiomyocytes, a GLUT4-knockdown cellular model was
established. H9c2 cardiomyocytes were seeded at a density of
2x105 cells per well on a 6-well plate and transfected
when reaching ~80% confluence. Transfection was performed with
either 50 nM GLUT4 small interfering (si)RNA (sense
5'-GCUGUUUUCUACUAUUCAAtt-3', antisense 5'-UUGAAUAGUAGAAAACAGCat-3';
cat. no. s73928; Thermo Fisher Scientific, Inc.) or 50 nM negative
control siRNA (sense 5'-UAACGACGCGACGACGUAAtt-3', antisense
5'-UUACGUCGUCGCGUCGUUAtt-3'; cat. no. 4390843; Thermo Fisher
Scientific, Inc.) using Lipofectamine® RNAiMax
Transfection Reagent (Thermo Fisher Scientific, Inc.) for 48 h at
37˚C. The protein knockdown efficiency of GLUT4 and protein
expression levels of carnitine palmitoyl transferase 1, the
mitochondrial enzyme responsible for the translocation of fatty
acids from the cytosol to the mitochondrial matrix, were assessed
48 h after siRNA transfection (Fig.
S1). LiCl was administered 24 h post-initiation of
transfection. For experiments in which GLUT4-knockdown was combined
with LiCl administration, cells were incubated for an additional 48
h. In groups without LiCl treatment, cells were incubated for an
equivalent 48 h period, matching the incubation time of the
LiCl-treated groups.
Measurement of mitochondrial
bioenergetic function
Mitochondrial bioenergetic function was assessed
using the XFe24 Extracellular Flux Analyzer (Seahorse Bioscience)
and Seahorse XF Cell Mito Stress Test kit (Seahorse Bioscience).
H9c2 cells were initially seeded at a density of 7,000 cells per
well on a Seahorse XFe24 culture plate and cultured with DMEM
supplemented with 10% FBS for 48 h. On the day of the assay, the
culture medium was substituted with Seahorse assay medium
supplemented with 25 mM glucose, 1 mM pyruvate and 2 mM glutamine.
Subsequently, a series of injections was administered, including
1.5 µM oligomycin, 3 µM carbonyl cyanide p-trifluoromethoxy
phenylhydrazone (FCCP) and 0.5 µM rotenone/antimycin A. Basal
respiration (last rate measurement before oligomycin
injection-minimum rate measurement after rotenone/antimycin A
injection), adenosine triphosphate (ATP) production (last rate
measurement before oligomycin injection-minimum rate measurement
after oligomycin injection), proton leakage (minimum rate
measurement after oligomycin injection-minimum rate measurement
after rotenone/antimycin A injection) and maximal respiration
(maximum rate measurement after FCCP injection-minimum rate
measurement after rotenone/antimycin A injection) were determined
using our previously described methods (22). In addition, coupling efficiency and
the cell respiratory control ratio were analyzed as these measures
are internally normalized bioenergetic parameters used to assess
the proportion of mitochondrial respiratory activity contributing
to ATP generation (i.e., coupling efficiency) and the degree of
change in mitochondrial respiratory activity attributable to proton
leakage (i.e., the cell respiratory control ratio) (23,24).
Coupling efficiency was calculated by dividing ATP production by
basal respiration. The cell respiratory control ratio was
calculated by dividing maximal respiration by proton leakage.
Measurement of mitochondrial fatty
acid oxidation
Mitochondrial fatty acid oxidation was assessed
using the XFe24 Extracellular Flux Analyzer (Seahorse Bioscience)
and Seahorse XF substrate oxidation stress test kit (Seahorse
Bioscience). H9c2 cells were initially seeded at a density of 7,000
cells per well on a Seahorse XFe24 culture plate and cultured with
DMEM supplemented with 10% FBS for 48 h. On the day of the assay,
the culture medium was substituted with Seahorse assay medium
containing 25 mM glucose, 1 mM pyruvate and 2 mM glutamine. During
the substrate oxidation stress test, etomoxir was injected at a
concentration of 40 µM. Mitochondrial fatty acid oxidation was
estimated by monitoring the change in the oxygen consumption rate
following etomoxir injection.
Measurement of mitochondrial ROS
Mitochondrial ROS levels were assessed using MitoSox
Red dye (Invitrogen; Thermo Fisher Scientific, Inc.) and a
fluorescence microscopy system (EVOS M5000 Imaging System; Thermo
Fisher Scientific, Inc.) in accordance with our previously
described methods (22). In brief,
H9c2 cells were seeded at a density of 5,000 cells per well on a
96-well plate and cultured with DMEM supplemented with 10% FBS for
48 h. Prior to fluorescence microscopy, the H9c2 cells were loaded
with MitoSox Red at a concentration of 5 µM and Hoechst 33423
(Sigma-Aldrich; Merck KGaA) at a concentration of 1 µg/ml for a
30-min incubation period at 37˚C. Fluorescence intensity was then
measured in four randomly selected fields in each well and
quantified using ImageJ 1.52a software (National Institutes of
Health).
Measurement of mitochondrial membrane
potential
Mitochondrial membrane potential was assessed using
the TMRE Mitochondrial Membrane Potential Assay Kit (Cayman
Chemical Company) and fluorescence microscopy (EVOS M5000 Imaging
System; Thermo Fisher Scientific, Inc.). In brief, H9c2 cells were
seeded at a density of 5,000 cells per well on a 96-well plate and
cultured with DMEM supplemented with 10% FBS for 48 h. Prior to
fluorescence microscopy, the H9c2 cells were loaded with
tetramethylrhodamine ethyl ester at a concentration of 125 nM and
HOECHST 33423 at a concentration of 1 µg/ml for a 30-min incubation
period at 37˚C in accordance with the manufacturer's instructions.
Fluorescence intensity was measured in four randomly selected
fields within each well and quantified using ImageJ 1.52a software
(National Institutes of Health).
Western blot analysis
Western blotting was performed as previously
described (25). Briefly, H9c2
cells were lysed using protein extraction reagent (cat. no. 78501;
Thermo Fisher Scientific, Inc.). Protein concentrations were
determined using Qubit™ Protein Assay Kits (Thermo Fisher
Scientific, Inc.). Subsequently, 30 µg protein/lane was separated
using 8% sodium dodecyl sulfate-polyacrylamide gel electrophoresis.
This process was followed by the electrophoretic transfer of the
separated proteins onto equilibrated polyvinylidene difluoride
membranes. These membranes were then blocked with 5% skimmed milk
for 1 h at room temperature. Following this blocking procedure, the
membranes were incubated overnight at 4˚C with specific antibodies
against total ACC2 (1:2,000; monoclonal; cat. no. ab45174; Abcam),
p-(p-)ACC2 (1:500; polyclonal; cat. no. 07303; Millipore), total
AMP-activated protein kinase (AMPK; 1:500; monoclonal; cat. no.
5831; Cell Signaling), p-AMPK (1:1,000; polyclonal; cat. no. 07681;
Millipore), calcineurin (1:10,000; monoclonal; cat. no. ab109412;
Abcam), total GSK-3β (1:1,000; monoclonal; cat. no. 9315; Cell
Signaling), p-GSK-3β (1:1,000; polyclonal; cat. no. 9336; Cell
Signaling) and GLUT4 (1:500; monoclonal; cat. no. sc-53566; Santa
Cruz). After washing with PBS containing Tween 20 (0.1%) for 15 min
at room temperature, a peroxidase-conjugated secondary antibody
(anti-rabbit IgG; 1:1,000; cat. no. HAF008; or anti-mouse IgG;
1:500; cat. no. HAF007; R&D Systems, Inc.) was added for
incubation for 1 h at room temperature. Bound antibodies were
detected using an enhanced chemiluminescence detection system
(MilliporeSigma) and the results were analyzed using AlphaEaseFC
4.0.0.34 software (ProteinSimple). Targeted bands were normalized
to glyceraldehyde 3-phosphate dehydrogenase (1:50,000; monoclonal;
cat. no. M171-1; MBL) or β-actin (1:10,000; polyclonal; cat. no.
ab6274; Abcam) to confirm equal protein loading.
Statistical analysis
Quantitative data are presented as mean ± standard
error of the mean. Statistical significance in H9c2 cells exposed
to various conditions was determined using a one-way analysis of
variance followed by Tukey's post hoc test. Statistical analysis
was performed using SigmaPlot 12.3 software (Systat Software,
Inc.). P<0.05 was considered to indicate a statistically
significant difference.
Results
Effects of lithium on ACC2 in H9c2
cardiomyocytes
In H9c2 cardiomyocytes treated with 0.3 mM LiCl for
48 h, the expression of p-ACC2 was significantly downregulated
compared with the control (Fig.
1A). However, LiCl at concentrations of 0.1 and 1.0 mM had only
a minimal effect on the expression of p-ACC2. Notably, no
significant differences were observed in the expression of total
ACC2 between the control group and H9c2 cardiomyocytes treated with
LiCl at concentrations of 0.1, 0.3 and 1 mM.
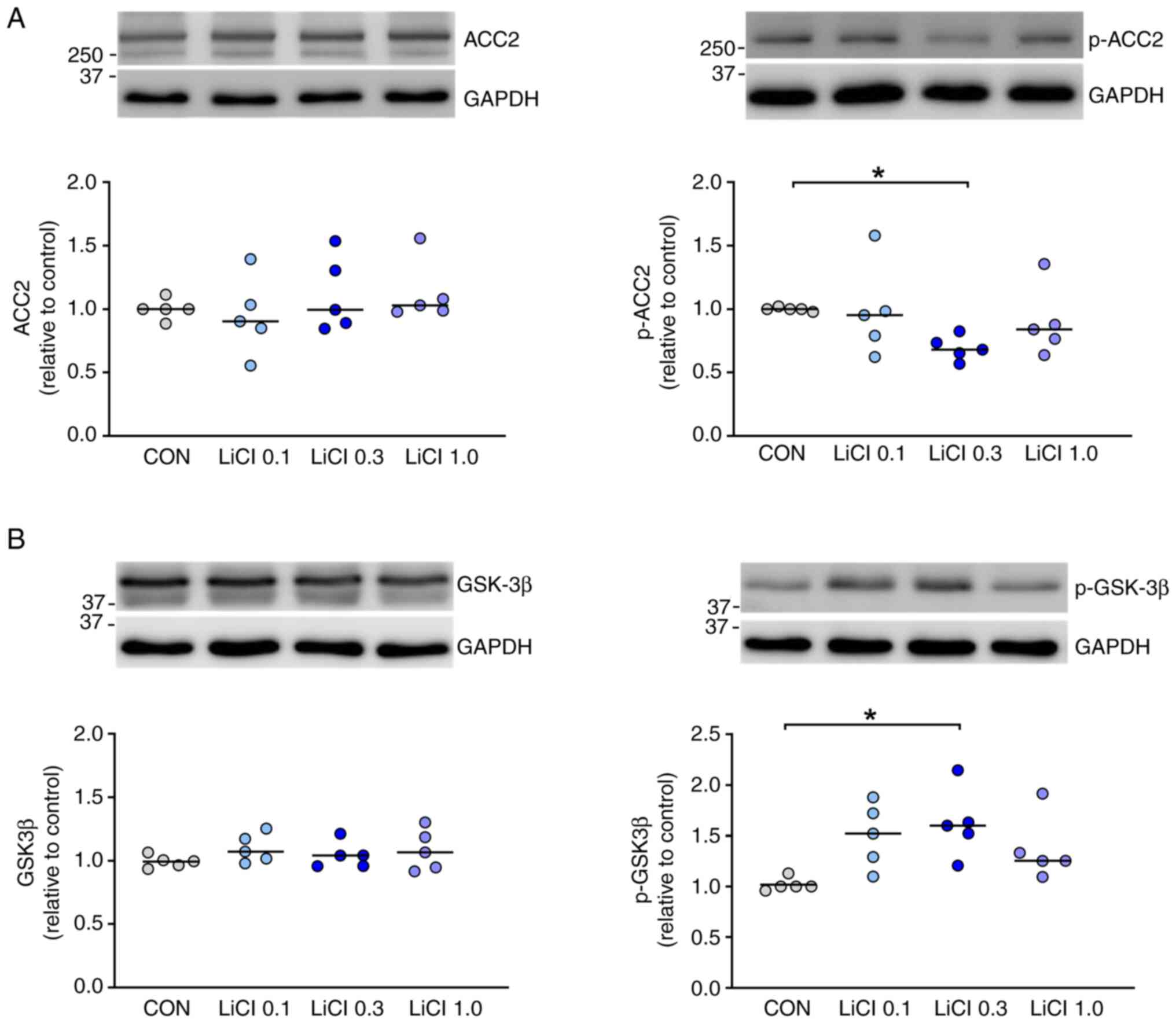 | Figure 1Expression ACC2 and GSK-3β in H9c2
cardiomyocytes treated with LiCl. (A) Compared with control cells
(n=5), H9c2 cells treated with LiCl at 0.3 mM (n=5) for 48 h
exhibited downregulated expression of p-ACC2. However, LiCl at 0.1
(n=5) or 1.0 mM (n=5) had no significant effect on the expression
of p-ACC2. Additionally, the expression of total ACC2 did not
significantly differ between the control cells and those treated
with LiCl at concentrations of 0.1, 0.3 and 1 mM. (B) H9c2 cells
treated with LiCl at 0.3 mM (n=5) for 48 h exhibited upregulated
expression of p-GSK-3β relative to the control cells (n=5).
However, LiCl at 0.1 (n=5) and 1.0 mM (n=5) did not significantly
affect the expression of p-GSK-3β. The expression of total GSK-3β
did not significantly differ between the control cells and those
treated with LiCl at concentrations of 0.1, 0.3 and 1 mM.
*P<0.05. ACC2, acetyl-CoA carboxylase 2; GSK-3β,
glycogen synthase kinase-3 beta; p-, phosphorylated; CON,
control. |
Effects of lithium on GSK-3β in H9c2
cardiomyocytes
As GSK-3β is a potential regulator of ACC2, it was
investigated whether lithium regulated GSK-3β activity in H9c2
cardiomyocytes. Compared with the control, LiCl at a concentration
of 0.3 mM upregulated the expression of p-GSK-3β by 55.4% (Fig. 1B). By contrast, LiCl at
concentrations of 0.1 and 1.0 mM did not significantly affect the
expression of p-GSK-3β in H9c2 cardiomyocytes. No significant
differences in the expression of total GSK-3β were observed between
the control group and H9c2 cardiomyocytes treated with LiCl at
concentrations of 0.1, 0.3 and 1 mM. To confirm whether lithium
downregulated the expression of p-ACC2 by modulating GSK-3β
activity, H9c2 cardiomyocytes were treated with TWS119, a GSK-3β
inhibitor. The expression of p-ACC2 in H9c2 cardiomyocytes was
downregulated to a similar extent in cells subjected to combined
treatment with TWS119 (8 µM) and LiCl (0.3 mM) and in those treated
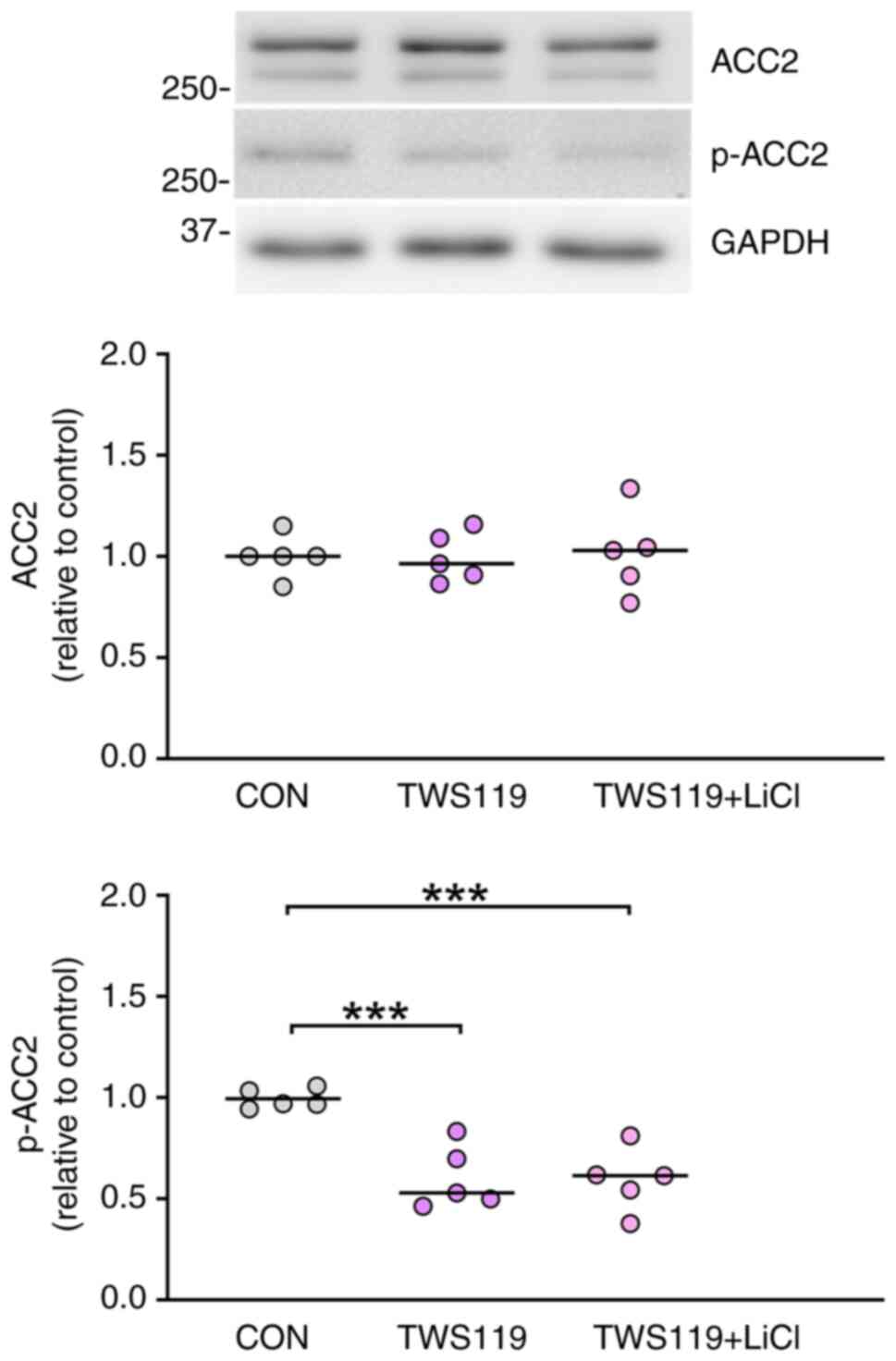
with TWS119 (8 µM) alone (Fig. 2).
These findings suggest that lithium downregulated the expression of
p-ACC2 through the modulation of GSK-3β activity. As the activities
of ACC1 and ACC2 are regulated by numerous protein kinases and
phosphatases, the present study also examined the effects of
lithium on AMPK and calcineurin, the two principal protein kinases
involved in the regulation of ACC2 activity in H9c2 cardiomyocytes.
It was observed that LiCl did not significantly affect the
expression level of total or p-AMPK or calcineurin in H9c2
cardiomyocytes (Fig. S2).
Effects of lithium on mitochondrial
bioenergetic function in GLUT4-knockdown H9c2 cardiomyocytes
To explore the therapeutic potential of lithium for
mitigating metabolic stress, mitochondrial bioenergetic function
was evaluated in GLUT4-knockdown H9c2 cardiomyocytes treated with
lithium. Compared with the control cells, the GLUT4-knockdown H9c2
cardiomyocytes exhibited greater proton leakage (Fig. 3). Additionally, the GLUT4-knockdown
H9c2 cardiomyocytes exhibited greater reductions in coupling
efficiency and the cell respiratory control ratio compared with the
control cells. Furthermore, GLUT4-knockdown H9c2 cardiomyocytes
treated with LiCl (0.3 mM) exhibited a reduction in proton leakage
and improvement in coupling efficiency and cell respiratory control
ratio compared with cells not treated with LiCl. These findings
indicated that LiCl at a concentration of 0.3 mM enhanced
mitochondrial bioenergetic function in GLUT4-knockdown H9c2
cardiomyocytes.
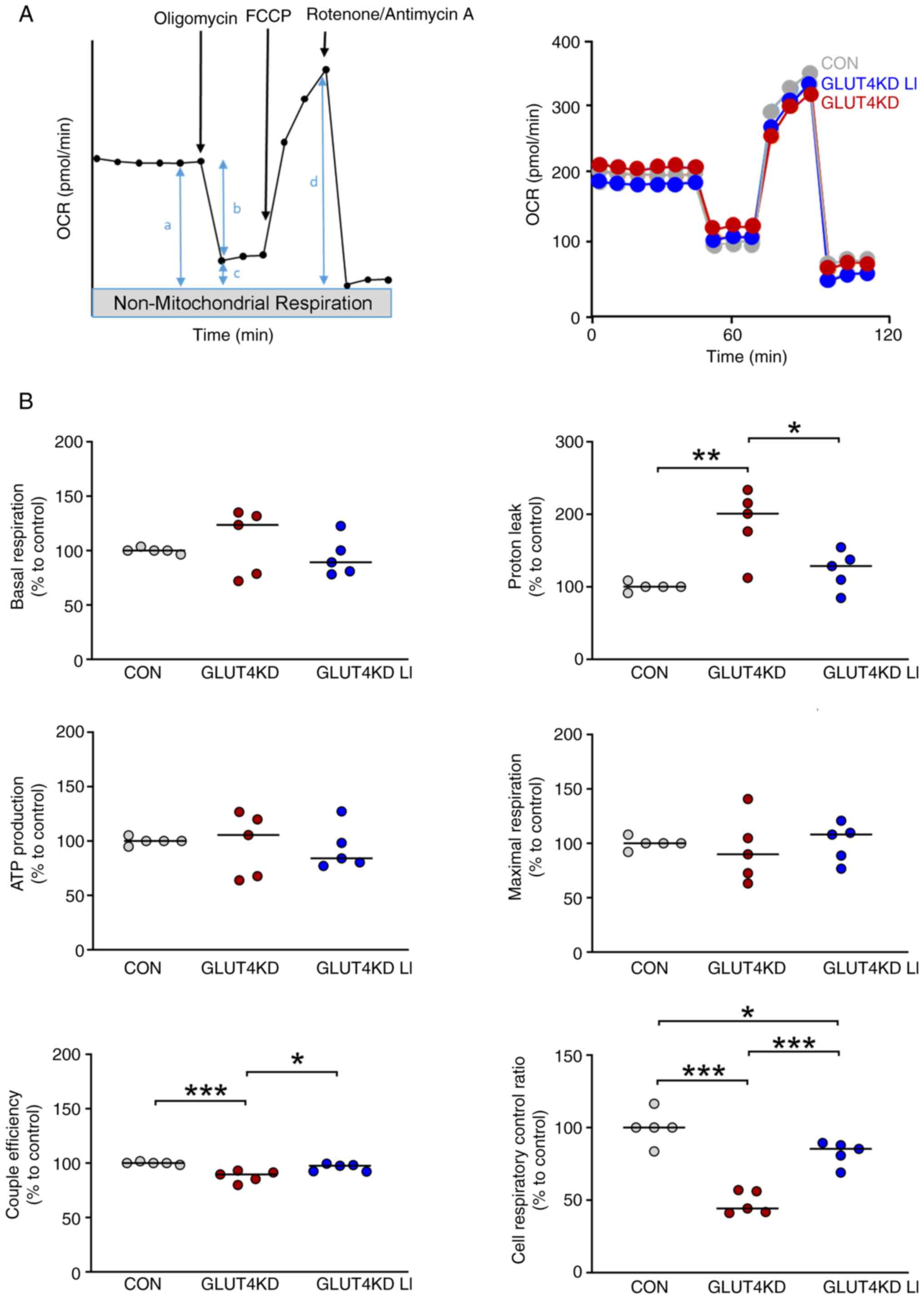 | Figure 3Mitochondrial bioenergetic function
in GLUT4-knockdown H9c2 cardiomyocytes treated with LiCl. (A)
Schematic and representative tracing of oxygen consumption rates,
following the sequential injection of oligomycin (1.5 µM), carbonyl
cyanide p-trifluoromethoxy phenylhydrazone (3 µM) and
rotenone/antimycin A (0.5 µM). The derived bioenergetic parameters
are indicated as a, basal respiration; b, ATP production; c, proton
leakage; d, maximal respiration; b/a: coupling efficiency; d/c:
cell respiratory control ratio. (B) The knockdown of GLUT4 with
small interfering RNA at 50 nM in H9c2 cardiomyocytes for 48 h
(n=5) resulted in elevated proton leakage, impaired mitochondrial
coupling efficiency and a reduced cell respiratory control ratio
compared with the control cells (n=5). Treatment with LiCl at a
concentration of 0.3 mM (n=5) attenuated proton leakage and
enhanced mitochondrial coupling efficiency and the cell respiratory
control ratio in GLUT4-knockdown H9c2 cardiomyocytes.
*P<0.05, **P<0.01,
***P<0.001. GLUT4, glucose transporter type 4; KD,
knockdown; CON, control. |
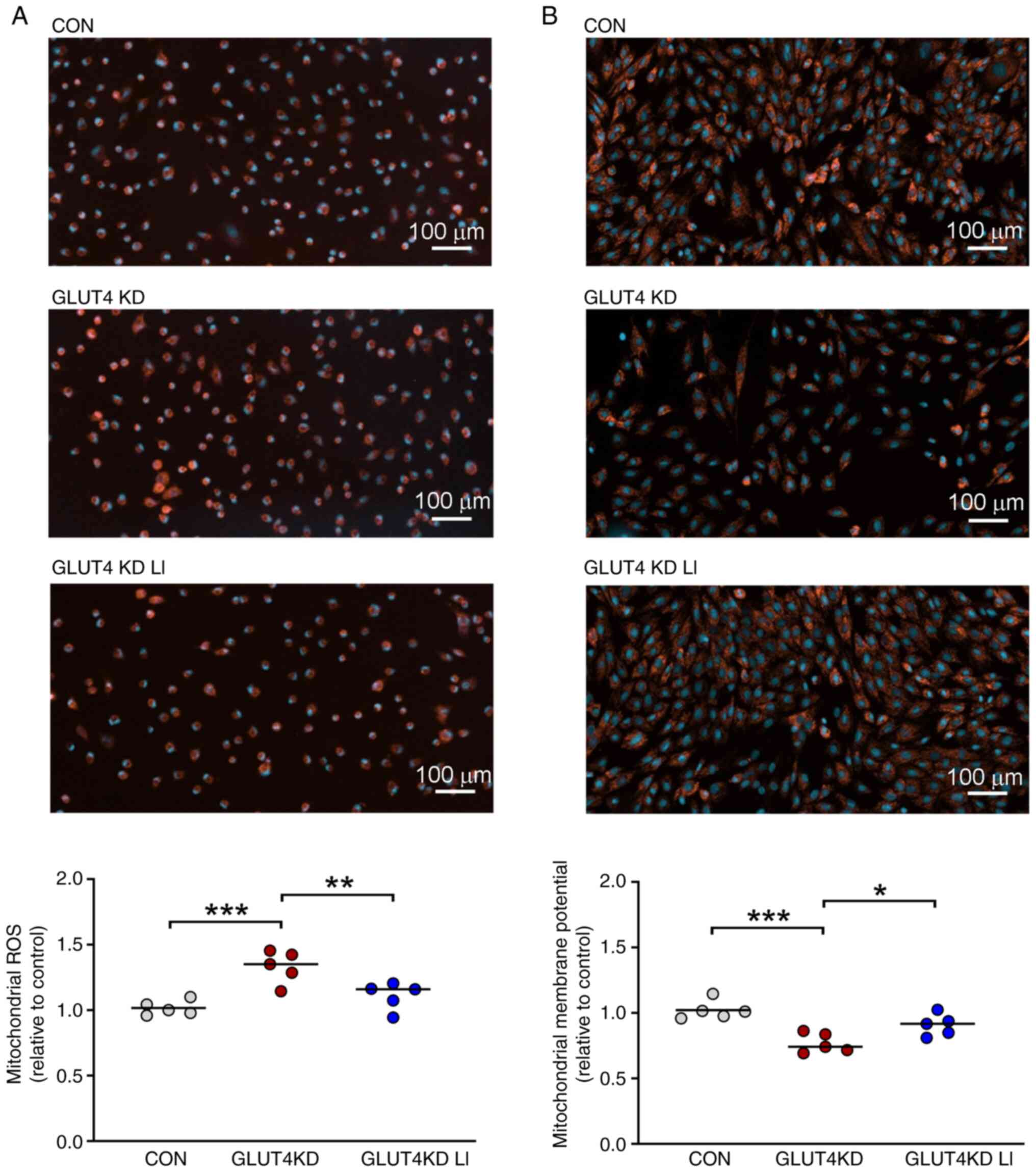
Effects of lithium on mitochondrial
ROS and membrane potential in GLUT4-knockdown H9c2
cardiomyocytes
The effects of lithium on mitochondrial ROS and
membrane potential in GLUT4-knockdown H9c2 cardiomyocytes are
illustrated in Fig. 4. Compared
with the control cells, the GLUT4-knockdown H9c2 cardiomyocytes
exhibited higher levels of mitochondrial ROS. By contrast, the
GLUT4-knockdown cells treated with LiCl (0.3 mM) had mitochondrial
ROS levels similar to those in the control cells. Furthermore, the
GLUT4-knockdown H9c2 cardiomyocytes exhibited reduced mitochondrial
membrane potential in contrast to both the control cells and the
LiCl (0.3 mM)-treated GLUT4-knockdown cells. These findings
suggested that LiCl at a concentration of 0.3 mM attenuated
mitochondrial ROS and restored the mitochondrial membrane potential
in GLUT4-knockdown H9c2 cardiomyocytes.
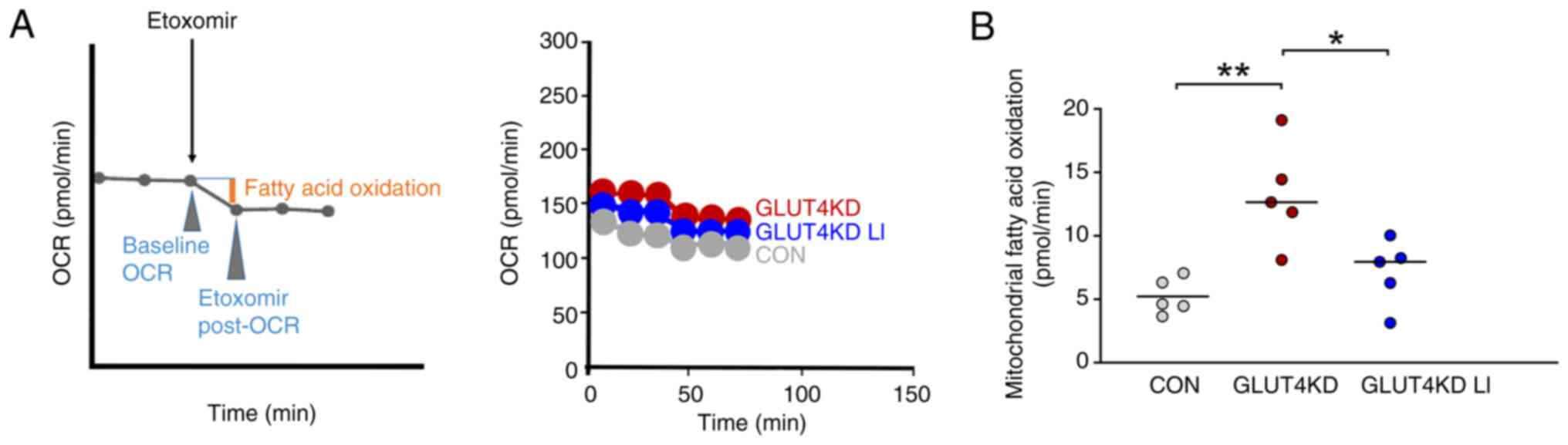
Effects of lithium on mitochondrial
fatty acid oxidation in GLUT4-knockdown H9c2 cardiomyocytes
The effects of lithium on mitochondrial fatty acid
oxidation in GLUT4-knockdown H9c2 cardiomyocytes are depicted in
Fig. 5. Compared with the control
cells, the GLUT4-knockdown H9c2 cardiomyocytes exhibited a greater
elevation in mitochondrial fatty acid oxidation. Additionally, when
GLUT4-knockdown H9c2 cardiomyocytes were treated with LiCl (0.3
mM), they exhibited a greater reduction in mitochondrial fatty acid
oxidation compared with GLUT4-knockdown H9c2 cardiomyocytes not
treated with LiCl. These findings suggested that LiCl at a
concentration of 0.3 mM attenuated the increase in mitochondrial
fatty acid oxidation in GLUT4-knockdown H9c2 cardiomyocytes.
Discussion
Studies have suggested that ACC2 can be inactivated
through phosphorylation (9,10).
In the present study, for the first time to the best of the
authors' knowledge, LiCl at a concentration of 0.3 mM, representing
a low therapeutic level but not a supraphysiological level, was
observed to downregulate the expression of p-ACC2 in H9c2
cardiomyocytes but not to affect the expression of total ACC2. The
downregulation of p-ACC2 induced by lithium may activate ACC2 and
subsequently elevate the level of malonyl-CoA (9). Such an increase in malonyl-CoA levels
can inhibit carnitine palmitoyltransferase 1, which is crucial for
the transport of long-chain fatty acyl-CoAs into the mitochondria
for β-oxidation (26,27). Additionally, the present study also
revealed that LiCl at a low therapeutic level attenuated
mitochondrial fatty acid oxidation in H9c2 cardiomyocytes.
The inhibition of fatty acid oxidation has been
demonstrated to have beneficial effects related to heart failure
associated with diabetes mellitus (2,4,8,28,29).
In patients with diabetes, heart failure often presents a shift in
cardiac fuel substrate utilization toward an increased reliance on
more mitochondrial fatty acid oxidation, a change driven by insulin
resistance (1,6). Such an increase in fatty acid
oxidation can overwhelm mitochondria, resulting in oxidative stress
(6). The subsequent generation of
ROS from lipid-overburdened mitochondria may worsen insulin
resistance and accelerate the progression of heart failure
(6). Thus, inhibiting fatty acid
oxidation in patients with diabetic cardiomyopathy may offer
cardioprotection by reducing oxidative stress, promoting glucose
utilization and enhancing cardiac efficiency (6,30-32).
Supporting this hypothesis, a previous study found that a low dose
of lithium (0.36±0.03 mM) mitigated cardiac dysfunction in an
experimental animal model to investigate sleep deprivation
(33). The current study
corroborated the cardioprotective potential of lithium in a
GLUT4-knockdown cellular model designed to simulate insulin
resistance (34-36).
This finding agrees with previous findings on the benefits of
lithium on cardiac metabolism (12-14,37).
Nonetheless, failing hearts show varying mitochondrial fatty acid
oxidation patterns, which could increase or decrease depending on
the heart failure type (4). For
example, in heart failure associated with conditions such as
hypertension and ischemia, myocardial fatty acid oxidation tends to
decline. Hence, the implications of the present findings might not
extend to all types of heart failure given the variability in
underlying disease processes.
ROS-induced proton leakage is mediated by adenine
nucleotide translocase (38,39),
a protein that is a central component of the mitochondrial
permeability transition pore (40). The opening of the mitochondrial
permeability transition pore, particularly under pathological
conditions, leads to the dissipation of mitochondrial membrane
potential, which in turn triggers the mitochondrial pathway of
apoptosis (41). Studies have
demonstrated that lithium exerts a cardioprotective effect by
enhancing the threshold at which the mitochondrial permeability
transition pore is activated by ROS (42,43).
The present study demonstrated that low-dose lithium hinders ROS
generation and proton leakage while restoring mitochondrial
membrane potential in GLUT4-knockdown H9c2 cardiomyocytes.
GSK-3β signaling plays a pivotal role in the
regulation of multiple mitochondrial functions, including energy
bioenergetics, biogenesis and apoptosis (44). The inhibition of GSK-3β has been
shown to reduce the apoptosis of cardiomyocytes and alleviate
cardiac dysfunction (45-48).
In the present study, LiCl at a concentration of 0.3 mM inhibited
GSK-3β and activated ACC2 in H9c2 cardiomyocytes. Although studies
have reported that lithium may regulate the activity of AMPK and
calcineurin (49-52),
the present study revealed that lithium at a concentration of 0.3
mM had no significant effect on the expression of p-AMPK or that of
calcineurin in H9c2 cardiomyocytes. These findings suggested that
the biological effects of lithium may be concentration dependent.
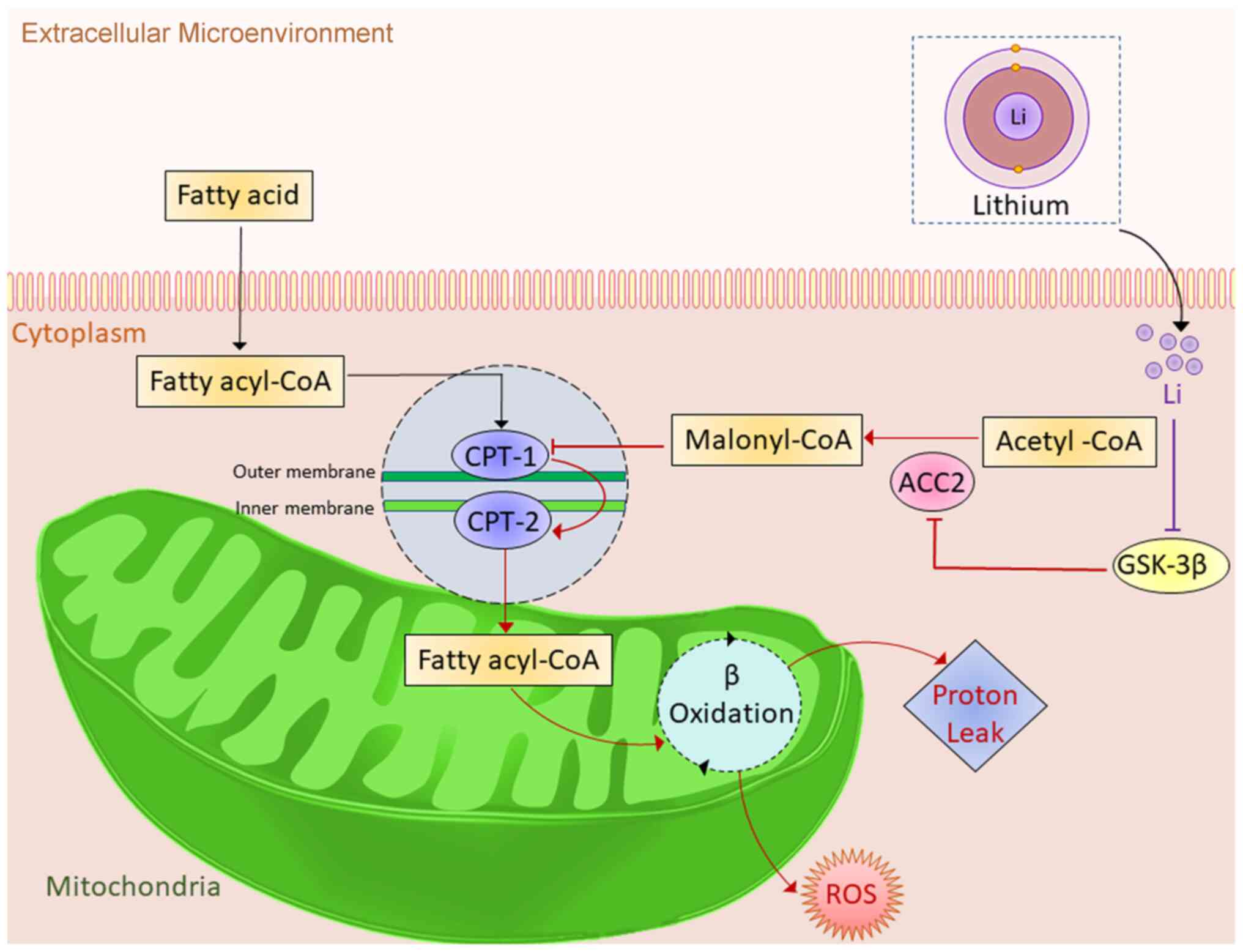
As summarized in Fig. 6, the
results of the present study suggested that at a low therapeutic
concentration, lithium inhibits mitochondrial fatty acid
utilization and mitigates oxidative stress in cardiomyocytes. These
effects are probably achieved through the inhibition of GSK-3β and
the activation of ACC2.
In the present study, lithium had a biphasic
dose-response effect on ACC2 and GSK-3β activities. This phenomenon
may stem from interactions among multiple signaling pathways
targeted by lithium (53),
resulting in dose-response curves that significantly differ from
the monotonic curves typically seen in pharmaceuticals acting on
specific receptors (54). Numerous
studies have identified the biphasic dose-response effects of
lithium on a wide range of signaling pathways in multiple cell
types (55-58).
To gain a comprehensive understanding of the mechanisms underlying
the effects of lithium on mitochondrial energy metabolism in
cardiomyocytes at the system level, future research involving
large-scale multiomics data is warranted.
In conclusion, the findings of the present study
indicate that lithium directly regulated mitochondrial fatty acid
utilization and mitigated oxidative stress in cardiomyocytes at low
therapeutic concentrations. These findings suggest that lithium
possessed cardioprotective properties and may attenuate metabolic
stress in the myocardium. Considering the inherent limitations of
in vitro research, future in vivo studies are
necessary to confirm the cardioprotective effects of lithium in
metabolic stress.
Supplementary Material
Expression of GLUT4 and CPT1 in H9c2
cardiomyocytes after transfection with either negative control or
GLUT4 siRNA. Relative to transfection with negative control, GLUT4
expression was downregulated by 43.1% after transfection with GLUT4
siRNA at 50 nM for 48 h. Additionally, the expression of CPT1, the
mitochondrial enzyme responsible for the translocation of fatty
acids from the cytosol to the mitochondrial matrix, where fatty
acid oxidation occurs, was upregulated by 11.6% after transfection
with GLUT4 siRNA at 50 nM. A paired t-test was conducted to compare
the protein expression in H9c2 cardiomyocytes after transfection
with negative control with that after transfection with GLUT4
siRNA. GLUT4, glucose transporter type 4; CPT1, carnitine
palmitoyltransferase 1; siRNA, small interfering RNA; CON, control;
p-, phosphorylated.
Expression of AMPK and calcineurin in
H9c2 cardiomyocytes treated with LiCl. The expression of
calcineurin and the total and p-forms of AMPK did not significantly
differ between control cells (n=5) and H9c2 cardiomyocytes treated
with 0.1 mM (n=5), 0.3 mM (n=5) and 1.0 mM (n=5) of LiCl. A one-way
repeated measures analysis of variance was performed to compare
protein expression under multiple treatment conditions. AMPK.
adenosine monophosphate-activated protein kinase; CON, control; p-,
phosphorylated.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by research grants from
the National Science and Technology Council of Taiwan (grant nos.
NSTC 111-2314-B-038-061 and NSTC 112-2314-B-038-050), Taipei
Medical University (grant no. TMU110-AE1-B19) and Wan Fang Hospital
(grant no. 108-wf-swf-06). The National Science and Technology
Council of Taiwan, Taipei Medical University and Wan Fang Hospital
had no involvement in the study design; collection, analysis and
interpretation of data; composition of the report; or decisions
regarding article publication.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
PHC, YHY, YHK and YJC conceived and designed the
study. PHC, YHK and YJC conducted the experiments and analyzed the
data. PHC, YHK and YJC confirm the authenticity of all the raw
data. PHC and CCC acquired funding. PHC, TWL, SHL, TVH and CCC
contributed to the data interpretation and prepared the manuscript.
TVH assisted with data visualization and created graphs. PHC
composed the first draft of the manuscript. YHK, YHY and YJC
reviewed and edited the manuscript. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bertero E and Maack C: Metabolic
remodelling in heart failure. Nat Rev Cardiol. 15:457–470.
2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Fillmore N, Mori J and Lopaschuk GD:
Mitochondrial fatty acid oxidation alterations in heart failure,
ischaemic heart disease and diabetic cardiomyopathy. Br J
Pharmacol. 171:2080–2090. 2014.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Lesnefsky EJ, Chen Q and Hoppel CL:
Mitochondrial metabolism in aging heart. Circ Res. 118:1593–611.
2016.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Lopaschuk GD, Karwi QG, Tian R, Wende AR
and Abel ED: Cardiac energy metabolism in heart failure. Circ Res.
128:1487–1513. 2021.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Marin W, Marin D, Ao X and Liu Y:
Mitochondria as a therapeutic target for cardiac
ischemia-reperfusion injury (Review). Int J Mol Med. 47:485–499.
2021.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Fukushima A and Lopaschuk GD: Cardiac
fatty acid oxidation in heart failure associated with obesity and
diabetes. Biochim Biophys Acta. 1861:1525–1534. 2016.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Lkhagva B, Lee TW, Lin YK, Chen YC, Chung
CC, Higa S and Chen YJ: Disturbed cardiac metabolism triggers
atrial arrhythmogenesis in diabetes mellitus: Energy substrate
alternate as a potential therapeutic intervention. Cells.
11(2915)2022.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Zuurbier CJ, Bertrand L, Beauloye CR,
Andreadou I, Ruiz-Meana M, Jespersen NR, Kula-Alwar D, Prag HA,
Eric Botker H, Dambrova M, et al: Cardiac metabolism as a driver
and therapeutic target of myocardial infarction. J Cell Mol Med.
24:5937–5954. 2020.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Tong L: Structure and function of
biotin-dependent carboxylases. Cell Mol Life Sci. 70:863–891.
2013.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Brownsey RW, Boone AN, Elliott JE, Kulpa
JE and Lee WM: Regulation of acetyl-CoA carboxylase. Biochem Soc
Trans. 34(Pt 2):223–227. 2006.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Malhi GS, Gessler D and Outhred T: The use
of lithium for the treatment of bipolar disorder: Recommendations
from clinical practice guidelines. J Affect Disord. 217:266–280.
2017.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Machado-Vieira R: Lithium, stress and
resilience in bipolar disorder: Deciphering this key homeostatic
synaptic plasticity regulator. J Affect Disord. 233:92–99.
2018.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Ochoa ELM: Lithium as a neuroprotective
agent for bipolar disorder: An overview. Cell Mol Neurobiol.
42:85–97. 2022.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Singulani MP, De Paula VJR and Forlenza
OV: Mitochondrial dysfunction in Alzheimer's disease: Therapeutic
implications of lithium. Neurosci Lett. 760(136078)2021.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Castillo-Quan JI, Li L, Kinghorn KJ,
Ivanov DK, Tain LS, Slack C, Kerr F, Nespital T, Thornton J, Hardy
J, et al: Lithium promotes longevity through GSK3/NRF2-dependent
hormesis. Cell Rep. 15:638–650. 2016.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Zarse K, Terao T, Tian J, Iwata N, Ishii N
and Ristow M: Low-dose lithium uptake promotes longevity in humans
and metazoans. Eur J Nutr. 50:387–389. 2011.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Chen PH, Hsiao CY, Chiang SJ, Shen RS, Lin
YK, Chung KH and Tsai SY: Cardioprotective potential of lithium and
role of fractalkine in euthymic patients with bipolar disorder.
Aust N Z J Psychiatry. 57:104–114. 2023.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Chen PH, Hsiao CY, Chiang SJ and Tsai SY:
Association of lithium treatment with reduced left ventricular
concentricity in patients with bipolar disorder. Psychiatry Clin
Neurosci. 77:59–61. 2023.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Snitow ME, Bhansali RS and Klein PS:
Lithium and therapeutic targeting of GSK-3. Cells.
10(255)2011.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Domoto T, Pyko IV, Furuta T, Miyashita K,
Uehara M, Shimasaki T, Nakada M and Minamoto T: Glycogen synthase
kinase-3β is a pivotal mediator of cancer invasion and resistance
to therapy. Cancer Sci. 170:1363–1372. 2016.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Terrand J, Bruban V, Zhou L, Gong W, El
Asmar Z, May P, Zurhove K, Haffner P, Philippe C, Woldt E, et al:
LRP1 controls intracellular cholesterol storage and fatty acid
synthesis through modulation of Wnt signaling. J Biol Chem.
284:381–388. 2009.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Lkhagva B, Kao YH, Lee TI, Lee TW, Cheng
WL and Chen YJ: Activation of Class I histone deacetylases
contributes to mitochondrial dysfunction in cardiomyocytes with
altered complex activities. Epigenetics. 13:376–385.
2018.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Brand MD and Nicholls DG: Assessing
mitochondrial dysfunction in cells. Biochem J. 435:297–312.
2011.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Nisr RB and Affourtit C: Insulin acutely
improves mitochondrial function of rat and human skeletal muscle by
increasing coupling efficiency of oxidative phosphorylation.
Biochim Biophys Acta. 1837:270–276. 2014.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Chen PH, Chung CC, Lin YF, Kao YH and Chen
YJ: Lithium reduces migration and collagen synthesis activity in
human cardiac fibroblasts by inhibiting store-operated
Ca2+ entry. Int J Mol Sci. 22(842)2021.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Eaton S: Control of mitochondrial
beta-oxidation flux. Prog Lipid Res. 41:197–239. 2022.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Schlaepfer IR and Joshi M: CPT1A-mediated
fat oxidation, mechanisms and therapeutic potential. Endocrinology.
161(bqz046)2020.PubMed/NCBI View Article : Google Scholar
|
|
28
|
McCarthy CP, Mullins KV and Kerins DM: The
role of trimetazidine in cardiovascular disease: Beyond an
anti-anginal agent. Eur Heart J Cardiovasc Pharmacother. 2:266–272.
2016.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Zhao C, Jin C, He X and Xiang M: The
efficacy of trimetazidine in non-ischemic heart failure patients: A
meta-analysis of randomized controlled trials. Rev Cardiovasc Med.
22:1451–1459. 2021.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Karwi QG, Sun Q and Lopaschuk GD: The
contribution of cardiac fatty acid oxidation to diabetic
cardiomyopathy severity. Cells. 10(3259)2021.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Shu H, Peng Y, Hang W, Zhou N and Wang DW:
Trimetazidine in heart failure. Front Pharmacol.
11(569132)2021.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Sung MM, Hamza SM and Dyck JR: Myocardial
metabolism in diabetic cardiomyopathy: Potential therapeutic
targets. Antioxid Redox Signal. 22:1606–1630. 2015.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Chen PH, Chung CC, Liu SH, Kao YH and Chen
YJ: Lithium treatment improves cardiac dysfunction in rats deprived
of rapid eye movement sleep. Int J Mol Sci.
23(11226)2022.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Jia G, DeMarco VG and Sowers JR: Insulin
resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat
Rev Endocrinol. 12:144–153. 2016.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Riehle C and Abel ED: Insulin signaling
and heart failure. Circ Res. 118:1151–1169. 2016.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Saotome M, Ikoma T, Hasan P and Maekawa Y:
Cardiac insulin resistance in heart failure: The role of
mitochondrial dynamics. Int J Mol Sci. 20(3552)2019.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Chen PH, Chao TF, Kao YH and Chen YJ:
Lithium interacts with cardiac remodeling: The fundamental value in
the pharmacotherapy of bipolar disorder. Prog Neuropsychopharmacol
Biol Psychiatry. 88:208–214. 2019.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Cheng J, Nanayakkara G, Shao Y, Cueto R,
Wang L, Yang WY, Tian Y, Wang H and Yang X: Mitochondrial proton
leak plays a critical role in pathogenesis of cardiovascular
diseases. Adv Exp Med Biol. 982:359–370. 2017.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Nadtochiy SM, Tompkins AJ and Brookes PS:
Different mechanisms of mitochondrial proton leak in
ischaemia/reperfusion injury and preconditioning: Implications for
pathology and cardioprotection. Biochem J. 395:611–618.
2006.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Carrer A, Laquatra C, Tommasin L and
Carraro M: Modulation and pharmacology of the mitochondrial
permeability transition: A journey from F-ATP synthase to ANT.
Molecules. 26(6463)2021.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Bonora M, Giorgi C and Pinton P: Molecular
mechanisms and consequences of mitochondrial permeability
transition. Nat Rev Mol Cell Biol. 23:266–285. 2022.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Juhaszova M, Zorov DB, Kim SH, Pepe S, Fu
Q, Fishbein KW, Ziman BD, Wang S, Ytrehus K, Antos CL, et al:
Glycogen synthase kinase-3beta mediates convergence of protection
signaling to inhibit the mitochondrial permeability transition
pore. J Clin Invest. 113:1535–1549. 2004.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Juhaszova M, Zorov DB, Yaniv Y, Nuss HB,
Wang S and Sollott SJ: Role of glycogen synthase kinase-3beta in
cardioprotection. Circ Res. 104:1240–1252. 2009.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Yang K, Chen Z, Gao J, Shi W, Li L, Jiang
S, Hu H, Liu Z, Xu D and Wu L: The key roles of GSK-3β in
regulating mitochondrial activity. Cell Physiol Biochem.
44:1445–1459. 2017.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Hirotani S, Zhai P, Tomita H, Galeotti J,
Marquez JP, Gao S, Hong C, Yatani A, Avila J and Sadoshima J:
Inhibition of glycogen synthase kinase 3beta during heart failure
is protective. Circ Res. 101:1164–1174. 2007.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Murphy E and Steenbergen C: Inhibition of
GSK-3beta as a target for cardioprotection: The importance of
timing, location, duration and degree of inhibition. Expert Opin
Ther Targets. 9:447–456. 2005.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Sharma AK, Bhatia S, Al-Harrasi A, Nandave
M and Hagar H: Crosstalk between GSK-3β-actuated molecular cascades
and myocardial physiology. Heart Fail Rev. 26:1495–1504.
2021.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Sharma AK, Thanikachalam PV and Bhatia S:
The signaling interplay of GSK-3β in myocardial disorders. Drug
Discov Today. 25:633–641. 2020.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Bao H, Zhang Q, Liu X, Song Y, Li X, Wang
Z, Li C, Peng A and Gong R: Lithium targeting of AMPK protects
against cisplatin-induced acute kidney injury by enhancing
autophagy in renal proximal tubular epithelial cells. FASEB J.
33:14370–14381. 2019.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Lee Y, Kim SM, Jung EH, Park J, Lee JW and
Han IO: Lithium chloride promotes lipid accumulation through
increased reactive oxygen species generation. Biochim Biophys Acta
Mol Cell Biol Lipids. 1865(158552)2020.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Liu P, Zhang Z, Wang Q, Guo R and Mei W:
Lithium chloride facilitates autophagy following spinal cord injury
via ERK-dependent pathway. Neurotox Res. 32:535–543.
2017.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Qu Z, Sun D and Young W: Lithium promotes
neural precursor cell proliferation: Evidence for the involvement
of the non-canonical GSK-3β-NF-AT signaling. Cell Biosci.
1(18)2011.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Jakobsson E, Argüello-Miranda O, Chiu SW,
Fazal Z, Kruczek J, Nunez-Corrales S, Pandit S and Pritchet L:
Towards a unified understanding of lithium action in basic biology
and its significance for applied biology. J Membr Biol.
250:587–604. 2017.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Parris GE: A hypothesis concerning the
biphasic dose-response of tumors to angiostatin and endostatin.
Dose Response 13: dose-response. 14-020. Parris, 2015.
|
|
55
|
Ge W and Jakobsson E: Systems biology
understanding of the effects of lithium on cancer. Front Oncol.
9(296)2019.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Erguven M, Oktem G, Kara AN and Bilir A:
Lithium chloride has a biphasic effect on prostate cancer stem
cells and a proportional effect on midkine levels. Oncol Lett.
12:2948–2955. 2016.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Suganthi M, Sangeetha G, Gayathri G and
Ravi Sankar B: Biphasic dose-dependent effect of lithium chloride
on survival of human hormone-dependent breast cancer cells (MCF-7).
Biol Trace Elem Res. 150:477–486. 2012.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Gao XM, Fukamauchi F and Chuang DM:
Long-term biphasic effects of lithium treatment on phospholipase
C-coupled M3-muscarinic acetylcholine receptors in cultured
cerebellar granule cells. Neurochem Int. 22:395–403.
1993.PubMed/NCBI View Article : Google Scholar
|