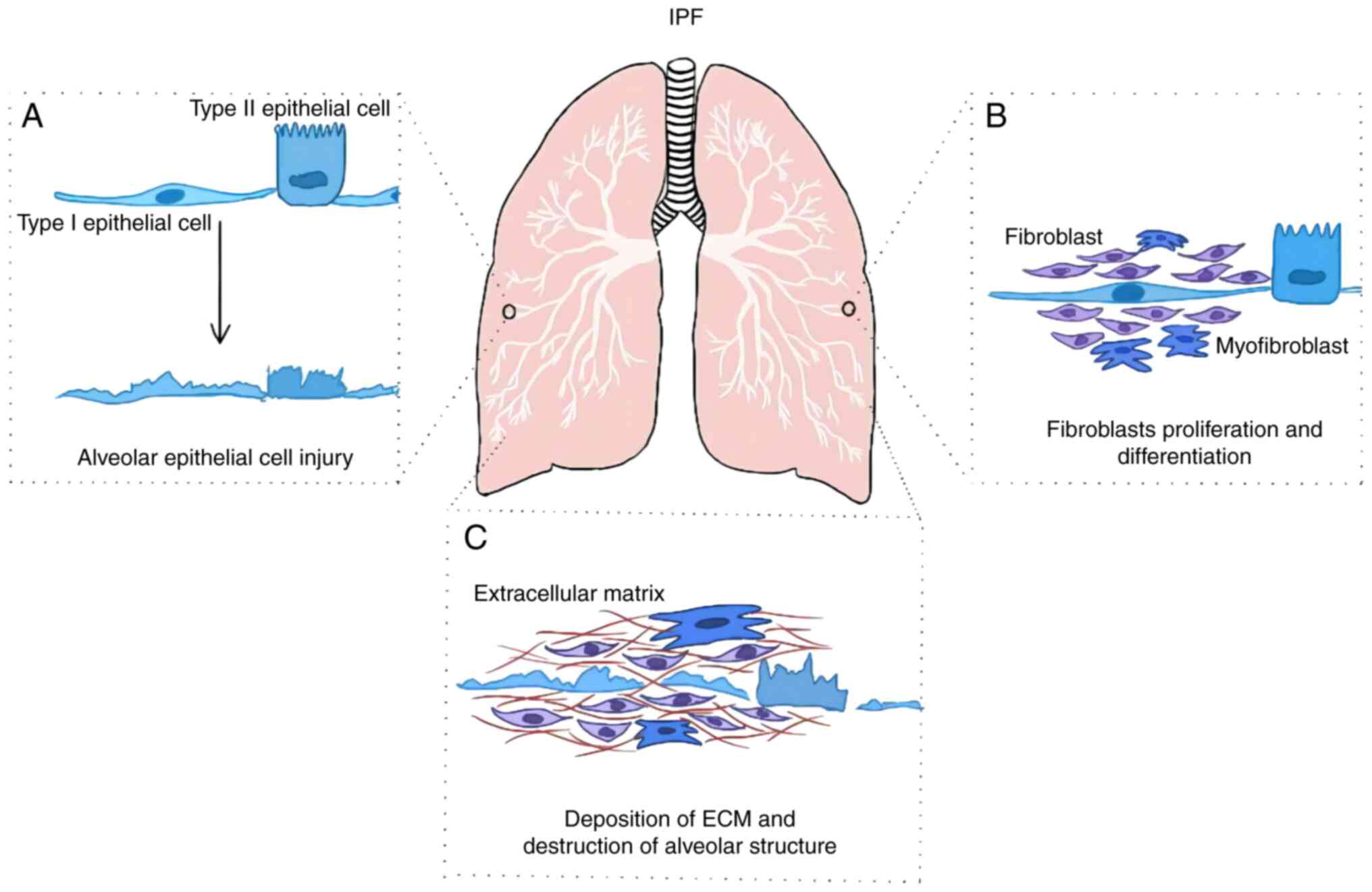
Pulmonary fibrosis (PF) is a respiratory disease
that is characterized by scarring in the lungs and subsequent
breathing difficulties (1). There
are various types of PF including asbestosis, hypersensitivity
pneumonitis and idiopathic pulmonary fibrosis (IPF), with IPF being
one of the most common and severe forms (2). IPF is an interstitial lung disease of
unknown etiology that is chronic, progressive, and irreversible
(3). It is distinguished by
epithelial cell activation and injury, fibroblast proliferation and
differentiation, extracellular matrix (ECM) deposition,
irreversible destruction of the alveolar structure and respiratory
insufficiency (Fig. 1) (4). IPF primarily occurs among the
middle-aged and elderly populations, where it is limited to the
lungs (5,6). The treatment of IPF typically
involves a combination of medications, pulmonary rehabilitation
and, in some cases, lung transplantation (7). IPF is a chronic and progressive
disease, and existing treatments, including antifibrotic
medications, aim to slow down the progression rather than cure the
condition (7). However, the
antifibrotic medications may have side effects, and not all
individuals with IPF can tolerate these drugs (8). The causes and pathogenesis of IPF
remain unclear and the effects conferred by currently available
therapeutic methods are limited (9,10).
The survival rate after IPF diagnosis is typically only 2-5 years,
where the prognosis of which is even worse compared with that of
several types of cancer including uterine, breast and colon cancer
(5).
Epithelial cell dysfunction and senescence has
emerged as a central component of the IPF pathophysiology (11,12).
The alveolar epithelium consists of alveolar epithelial type 1
(AT1) and alveolar epithelial type 2 cells (AT2). The alveolar
surface is mostly covered by AT1 cells, whose thin squamous
morphology and intimate contact with the adjacent capillary plexus
permit efficient gas exchange (13). Although loss of AT1 cells is
considered to be a cardinal feature of the IPF histology,
accumulating evidence has revealed AT2 cells to also serve an
important role in IPF (14,15).
This is in part due to its function in alveolar niche homeostasis
through the production of pulmonary surfactants and as a progenitor
cell for both self-renewal and transdifferentiation into AT1 cells
if needed (13). In particular,
single-cell RNA sequencing has previously identified a cell
population expressing both AT2 (SOX4, SOX9, COL1A1 and FN1) and AT1
(COL1A1, FN1 and SCGB1A1) markers in IPF lungs, suggesting a subset
of epithelial cells transitioning between the AT1 and AT2 phenotype
(16).
In eukaryotic cells, a substantial proportion of
signal transduction activity is facilitated by protein kinases,
which is achieved through phosphorylation of target substrates
(17,18). This process serves pivotal roles in
the regulation of a wide variety of cellular functions, including
proliferation, differentiation, metabolism and programmed cell
death (19,20). In humans, protein kinases can be
categorized into nine groups based on the evolutionary
relationships of their catalytic domains (17,18,21).
One such group is one consisting of cAMP-dependent protein kinase
A, cGMP-dependent protein kinase G and phospholipid-dependent
protein kinase C (AGC), collectively known as AGC kinases (21).
AGC kinases form a highly conserved group of kinases
that are ubiquitously distributed across different orders of
eukaryotic organisms (21).
Members of the AGC kinases group have been reported to regulate
different cellular processes, where their targets may have
therapeutic implications for various human diseases, including but
not limited to cancer, diabetes, obesity, immunological disorders,
inflammation, neurological disorders, viral infections and muscular
dystrophies (21-24).
Therefore, targeting members of the AGC kinases may prove to be a
potential method of treatment for PF.
The present review summarizes the reported
significant effects of AGC kinases on the pathological procession
of PF, before discussing their potential as molecular targets for
the treatment of this disease. In addition, focus will be placed on
the role of different families of AGC kinases in PF.
The AGC kinase group is comprised of 63
serine/threonine protein kinases that are evolutionarily related.
This group includes the protein kinase G and protein kinase C
families of kinases, Akt/protein kinase B, Aurora kinases,
ribosomal protein S6 kinases and the phosphoinositide-dependent
kinases (17,22,24).
In addition, the majority of AGC kinases each have multiple
isoforms and splice variants, increasing the complexity of this
family of kinases (25).
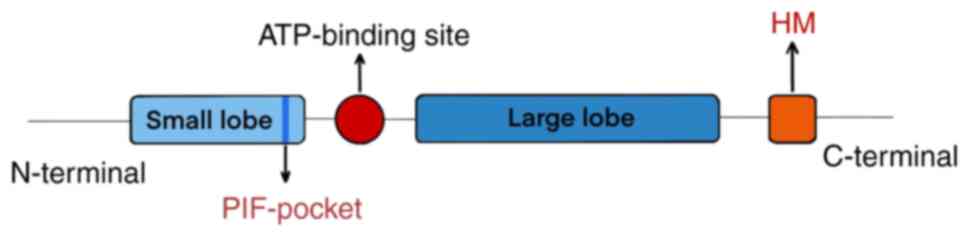
AGC kinases typically exhibit a conserved fold that
is characterized by a catalytic domain consisting of a small
N-terminal lobe and a large C-terminal lobe (21). The predominant secondary structure
of the large C-terminal lobe is α-helical, whereas the small
N-terminal lobe is comprised of a single helix (α-C) and a
5-stranded β-sheet (22). An
ATP-binding site is located between the two lobes (21,26),
where the bound ATP serves as the phosphate donor during
phosphorylation (25). The
activation loop originating from an Aspartate-Phenylalanine-Glycine
motif is also situated amidst the large and the small lobe
(21). In addition. the majority
of AGC kinases contain a conserved catalytic core with a C-terminal
hydrophobic motif (HM) sequence (21). This HM sequence is known to bind to
a co-evolved hydrophobic site in the small lobe of the catalytic
core, which is referred to as the 3-phosphoinositide-dependent
protein kinase-1-interacting fragment (PIF)-pocket (Fig. 2) (27,28).
According to a previous study, the PIF-pocket is proposed to be a
central and common on-off switch in the AGC kinases (22). Apart from the conserved catalytic
domain, the AGC kinases group contains various functional domains.
The AGC kinases can be classified into 14 families and 21
subfamilies based on homology outside the catalytic domain
(17,25).
AGC kinases serve a crucial role in regulating a
multitude of cellular functions, including but not limited to cell
cycle progression, cellular differentiation, cell survival and
apoptosis (21). In both animals
and yeast, AGC kinases have been documented to serve as key
mediators that are capable of transducing signaling cascades
initiated by secondary messengers through substrate phosphorylation
(29,30). In plants, AGC kinases have been
demonstrated to serve indispensable roles in diverse cellular and
developmental processes including growth, immunity, cell death and
defense responses (31-33).
PDK1 is a serine/threonine kinase that was initially
discovered in previous studies on insulin-activated Akt signaling
in the presence of phosphatidylinositol-3,4,5-triphosphate (PIP3)
(34-36).
PDK1 is a conserved protein kinase that is expressed in eukaryotes
(37). PDK1 is mainly located in
the cytoplasm, but under certain conditions it can be induced to
translocate into the nucleus (38). PDK1 was originally considered to be
a regulator of glycolysis in the cytoplasm (39-41).
Subsequent studies have revealed that PDK1 can regulate a number of
physiological processes, such as blood vessel formation, metabolism
and development (42,43). In addition, the pathological
processes of Alzheimer's disease (44), diabetes (45) and cancer (46,47)
have all been reported to be caused at least in part by PDK1
activity (39).
Previous studies have revealed that PDK1 serves a
role in the regulation of PF. The PDK1 gene was previously
shown to be a direct target gene of hypoxia-inducible factor-1
(HIF-1) (48,49). Glycolytic metabolism, which is
mediated by PDK-1, serves a crucial role in the progression of PF
(50-52).
Goodwin et al (53)
previously reported that hypoxia markedly enhanced transforming
growth factor-β (TGF-β)-induced myofibroblast differentiation in
fibrotic lesions via HIF-1α. However, overexpression of PDK1 was
sufficient in activating glycolysis and potentiate myofibroblast
differentiation regardless of the existence of HIF-1α.
Additionally, bleomycin (BLM)-induced PF can be significantly
attenuated by using dichloroacetate, a potent PDK inhibitor
(54,55). Yang et al (56) revealed that PDK1 knockdown can
attenuate PF by inhibiting the NF-κB/p65 signaling pathway.
Mannan-binding lectin (MBL) can interact with and ubiquitinate PDK1
to inhibit epithelial-mesenchymal transition (EMT) in PF by
attenuating store-operated calcium entry (SOCE) signaling (57). However, the specific mechanism of
PDK1 in IPF remains unclear.
ROCK is a downstream target protein of Rho and has
been implicated in a wide range of cell functions, such as
proliferation, migration, adhesion, apoptosis and differentiation
(58-60).
ROCK has two isoforms, namely ROCK-I and ROCK-II, which regulate
cytoskeletal reorganization by phosphorylating myosin phosphatase
to increase the phosphorylation level of myosin light chain
(61). In addition to brain and
muscle tissues, the expression of ROCK-I is widespread, whilst
ROCK-II expression tends to be limited to the brain and muscle,
especially in the smooth muscle (62). However, the functional differences
between ROCK-I and ROCK-II remain unclear (59).
ROCK-II mRNA expression has been previously revealed
to be increased in a murine model of lung fibrosis induced by BLM
(59). The Rho/ROCK signaling
pathway can be inhibited to prevent fibrosis by decreasing the
levels of inflammatory cells (macrophages, neutrophils and
lymphocytes) and cytokine (TGF-β1, connective tissue growth factor
(CTGF) and plasminogen activator inhibitor (PAI)-1 levels (59,63).
Shimizu et al (64)
demonstrated that the expression and activity of ROCK-II was
increased in several types of lung cells in patients with IPF,
including bronchial epithelial cells, airway smooth muscle cells,
vascular smooth muscle cells and fibroblasts (64). The RhoA/ROCK-I signaling pathway
has also been demonstrated to promote the migration of lung
fibroblasts and synthesis of collagen by myofibroblasts, both of
which can exacerbate PF (65).
Rho/ROCK inhibitors, such as Fasudil, have been shown to attenuate
BLM-induced lung fibrosis by suppressing the recruitment of
inflammatory cells such as neutrophils and reducing the production
of TGF-β1, CTGF, α-smooth muscle actin (α-SMA) and PAI-1 in
BLM-induced mouse lungs (63).
Recently, compound 9b, a novel selective inhibitor of ROCK-II, has
demonstrated marked anti-PF effects by suppressing the expression
of α-SMA and collagen I in BLM-induced IPF mice model (66). Notably, dual pharmacological
inhibition of ROCK-I and -II was found to counteract TGF-β-induced
PF in an organoid assay, which included freshly isolated
EpCAM+ mouse lung cells co-cultured with human lung
fibroblasts (67). However, it
should be emphasized that although the main role of ROCK in PF has
been established, the precise regulatory mechanisms mediated by
Rho/ROCK signaling require further clarification.
LATS1 and 2 are important components of the kinase
cascades in the Hippo signal pathway in mammalian cells (68-70).
A number of studies have demonstrated that LATS2 and its downstream
signaling pathway have a vital impact on the proliferation,
migration, differentiation and immunomodulation of mesenchymal stem
cells (MSCs) (71,72). Dong and Li (71) previously revealed that
LATS2-underexpressing bone marrow-derived MSCs (transfected with
LATS2-interfering lentivirus vector) can repair the alveolar
epithelium damaged by lipopolysaccharide in a mouse model of acute
lung injury (ALI).
AKT, also known as PKB, has three isoforms in
mammals, namely AKT1, AKT2 and AKT3. It can regulate numerous
cellular processes, such as cell survival, proliferation,
differentiation and intermediary metabolism (76-81).
Specifically, it has been previously revealed that both AKT1 and
AKT2 can modulate the migration and invasion of cancer cells. AKT1
can stimulate prostate cancer cell motility, whereas AKT2 inhibits
motility and migration in breast cancer and ovarian cancer cells
(82,83). Since AKT3 is primarily expressed in
the brain tissue and has only been reported to serve a role in
neuronal development (84),
research on the role of AKT in PF has mainly concentrated on AKT1
and AKT2(81).
Previous studies have demonstrated that TGF-β1 can
regulate the activation of AKT in myofibroblasts and that
inhibiting the function of AKT can alleviate TGF-β1-induced PF
(85,86). It has been found that AKT1 and AKT2
can mediate significant roles in regulating the function of
alveolar macrophages in IPF (87,88).
Specifically, AKT1 can promote macrophage mitochondrial reactive
oxygen species (ROS) and mitophagy, as well as increase TGF-β1
expression, resulting in the development of fibrosis (88). In addition, the pro-fibrotic
cytokine IL-13, can be upregulated by AKT1 in macrophages in PF
(89). Nie et al (87) revealed that AKT2 phosphorylation is
upregulated in the tissues of patients with PF. AKT2 deficiency
protects against BLM-induced PF and inflammation (87). In conclusion, AKT may serve an
important role in the development of IPF, suggesting that it can be
a potential molecular target for its therapeutic intervention.
PKC is a type of phospholipid-dependent
serine/threonine kinase for which 12 isozymes have been identified
(90). PKCs are classified into
three subfamilies, based on structural and activation
characteristics: conventional or classic PKCs (cPKCs: α, βI, βII
and γ), novel or non-classic PKCs (nPKCs: δ, ε, η and θ), and
atypical PKCs (aPKC: ζ, ι and λ) (91). PKC isozymes participate in signal
transduction by either directly or indirectly activating or
inactivating target proteins through phosphorylation (92). PKC has been documented to mediate
various cellular processes, including proliferation, migration,
apoptosis, adhesion and differentiation (93-96).
The role of PKC-δ in IPF remains controversial,
despite its reported involvement in the progression of PF (97). PKC-δ has been reported to inhibit
NF-κB signaling by enhancing the activity and stability of A20,
which is an endogenous negative regulator of NF-κB (97). In addition, the deficiency of PKC-δ
has been reported to increase the expression of proinflammatory
cytokines to exacerbate inflammation and PF induced by BLM
(97), suggesting that PKC-δ may
serve a protective role in IPF. Previous studies have demonstrated
that the inhibitor of PKC-δ rottlerin can downregulate the
expression of type I and type III collagen gene, and suppress the
type I collagen production in cultured dermal fibroblasts derived
from patients with systemic sclerosis (98). Additionally, Song et al
(99) revealed that thrombin
induces EMT and collagen I secretion by activating
protease-activated receptor (PAR-1), PKC (α/β, δ and ε) and ERK1/2
in A549 cells. A549 is an adenocarcinomic human alveolar basal
epithelial cell line, that is widely used as a model of alveolar
epithelial-like behavior in IPF study (100). Therefore, targeting PAR-1 or
specific PKC isoforms (α, β, δ and ε) may halt the fibrotic process
in human IPF by preventing thrombin-induced EMT. Results reported
by the aforementioned studies suggest that PKC-δ may promote IPF.
However, the possible link between PKC and IPF require further
studies.
RPS6Ks can be divided into two subfamilies, p90
ribosomal S6 kinase (RSK) and p70 ribosomal S6 kinase (p70S6K). The
p70S6K subfamily has two members of the p70S6K (S6K1 and S6K2),
whilst the RSK subfamily has four members (RSK1-4) (101,102). RSK is activated by the ERK
signaling pathway. p70S6K is activated through a complex network of
signaling molecules, and mTOR serine/threonine kinase is necessary
for its full activation (103).
These kinases are involved in various signaling pathways and can
regulate multiple cellular processes, such as cell proliferation,
differentiation, growth, transformation and apoptosis (104-108).
The AGC kinases form a widely conserved family of
protein kinases that have been implicated in various pathologies,
including cancer, metabolic disorders, cardiovascular disease,
immunological disorders and neurological disorders. Accumulating
evidence has shown that AGC kinases can exert important roles in
IPF through distinct mechanistic pathways (Table I). Several inhibitors of AGC
kinases such as RSK-inhibitor peptide (112), dichloroacetate (53), BX795 and BX912(113) have been found to attenuate PF.
However, additional research is required to fully comprehend the
contribution of AGC kinases towards IPF. Identification of AGC
kinases with the potential to serve as therapeutic targets of IPF
may facilitate the discovery of novel drugs for IPF treatment.
At present, to the best of our knowledge, only a
small number of AGC kinases have been found to be involved in
regulating the pathological process of PF. To further explore the
key role of AGC kinases comprehensively in the pathogenesis of PF,
more functions of AGC kinases in PF need to be explored. As
single-cell sequencing and spatial proteomics technology advance,
the distinct functions mediated by AGC kinase in various cell types
during different stages of PF will be elucidated (114-116).
In addition, continuous advancements in organoid technology are
expected to facilitate studies into the microenvironment of lung
tissues in the pathological process of PF in the future, where the
role of AGC kinases should also be investigated. Understanding the
specific substrates and associated signaling pathways by AGC
kinases in the regulation of PF will also be a focus of future
attention. Based on the results and findings of existing studies,
it would be of benefit to screen small molecule inhibitors or
targeted drugs for AGC kinase and conduct relevant clinical trials,
providing more effective treatment options and strategies for
patients with IPF.
Not applicable.
Funding: The present study was supported by grants from the
National Natural Science Foundation of China (grant nos. 82060023
and 82160133) and the Jiangxi Provincial Natural Science Foundation
(grant nos. 20202ACBL206015, 20224BAB206007 and
20212ACB216005).
Not applicable.
YL conceived the study and revised the manuscript.
CM prepared the figures and wrote the manuscript. TC, XH and CX
participated in the writing of the manuscript. SC reviewed the
manuscript. All authors have read and approved the final
manuscript. Data authentication is not applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Noble PW, Barkauskas CE and Jiang D:
Pulmonary fibrosis: Patterns and perpetrators. J Clin Invest.
122:2756–2762. 2012.PubMed/NCBI View
Article : Google Scholar
|
|
2
|
Kreuter M, Ladner UM, Costabel U, Jonigk D
and Heussel CP: The diagnosis and treatment of pulmonary fibrosis.
Dtsch Arztebl Int. 118:152–162. 2021.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Richeldi L, Collard HR and Jones MG:
Idiopathic pulmonary fibrosis. Lancet. 389:1941–1952.
2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Günther A, Korfei M, Mahavadi P, von der
Beck D, Ruppert C and Markart P: Unravelling the progressive
pathophysiology of idiopathic pulmonary fibrosis. Eur Respir Rev.
21:152–160. 2012.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Raghu G, Collard HR, Egan JJ, Martinez FJ,
Behr J, Brown KK, Colby TV, Cordier JF, Flaherty KR, Lasky JA, et
al: An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary
fibrosis: Evidence-based guidelines for diagnosis and management.
Am J Respir Crit Care Med. 183:788–824. 2011.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Lederer DJ and Martinez FJ: Idiopathic
pulmonary fibrosis. N Engl J Med. 378:1811–1823. 2018.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Abuserewa ST, Duff R and Becker G:
Treatment of idiopathic pulmonary fibrosis. Cureus.
13(e15360)2021.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Khor YH: Antifibrotic therapy for
idiopathic pulmonary fibrosis: Combining real world and clinical
trials for totality of evidence. Chest. 160:1589–1591.
2021.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Desai O, Winkler J, Minasyan M and Herzog
EL: The role of immune and inflammatory cells in idiopathic
pulmonary fibrosis. Front Med (Lausanne). 5(43)2018.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Fujimoto H, Kobayashi T and Azuma A:
Idiopathic pulmonary fibrosis: Treatment and prognosis. Clin Med
Insights Circ Respir Pulm Med. 9 (Suppl 1):S179–S185.
2016.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Selman M and Pardo A: The leading role of
epithelial cells in the pathogenesis of idiopathic pulmonary
fibrosis. Cell Signal. 66(109482)2020.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Tu M, Wei T, Jia Y, Wang Y and Wu J:
Molecular mechanisms of alveolar epithelial cell senescence and
idiopathic pulmonary fibrosis: A narrative review. J Thorac Dis.
15:186–203. 2023.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Katzen J and Beers MF: Contributions of
alveolar epithelial cell quality control to pulmonary fibrosis. J
Clin Invest. 130:5088–5099. 2020.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Parimon T, Yao C, Stripp BR, Noble PW and
Chen P: Alveolar epithelial type II cells as drivers of lung
fibrosis in idiopathic pulmonary fibrosis. Int J Mol Sci.
21(2269)2020.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Zhu W, Tan C and Zhang J: Alveolar
epithelial type 2 cell dysfunction in idiopathic pulmonary
fibrosis. Lung. 200:539–547. 2022.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Habermann AC, Gutierrez AJ, Bui LT, Yahn
SL, Winters NI, Calvi CL, Peter L, Chung MI, Taylor CJ, Jetter C,
et al: Single-cell RNA sequencing reveals profibrotic roles of
distinct epithelial and mesenchymal lineages in pulmonary fibrosis.
Sci Adv. 6(eaba1972)2020.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Manning G, Whyte DB, Martinez R, Hunter T
and Sudarsanam S: The protein kinase complement of the human
genome. Science. 298:1912–1934. 2002.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Roskoski R Jr: A historical overview of
protein kinases and their targeted small molecule inhibitors.
Pharmacol Res. 100:1–23. 2015.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Deribe YL, Pawson T and Dikic I:
Post-translational modifications in signal integration. Nat Struct
Mol Biol. 17:666–672. 2010.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Attwood MM, Fabbro D, Sokolov AV, Knapp S
and Schiöth HB: Trends in kinase drug discovery: Targets,
indications and inhibitor design. Nat Rev Drug Discov. 20:839–861.
2021.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Arencibia JM, Pastor-Flores D, Bauer AF,
Schulze JO and Biondi RM: AGC protein kinases: From structural
mechanism of regulation to allosteric drug development for the
treatment of human diseases. Biochim Biophys Acta. 1834:1302–1321.
2013.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Leroux AE, Schulze JO and Biondi RM: AGC
kinases, mechanisms of regulation and innovative drug development.
Semin Cancer Biol. 48:1–17. 2018.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Rath N and Olson MF: Rho-associated
kinases in tumorigenesis: Re-considering ROCK inhibition for cancer
therapy. EMBO Rep. 13:900–908. 2012.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Turnham RE and Scott JD: Protein kinase A
catalytic subunit isoform PRKACA; history, function and physiology.
Gene. 577:101–108. 2016.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Pearce LR, Komander D and Alessi DR: The
nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol.
11:9–22. 2010.PubMed/NCBI View
Article : Google Scholar
|
|
26
|
Zheng J, Knighton DR, ten Eyck LF,
Karlsson R, Xuong N, Taylor SS and Sowadski JM: Crystal structure
of the catalytic subunit of cAMP-dependent protein kinase complexed
with MgATP and peptide inhibitor. Biochemistry. 32:2154–2161.
1993.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Biondi RM, Cheung PC, Casamayor A, Deak M,
Currie RA and Alessi DR: Identification of a pocket in the PDK1
kinase domain that interacts with PIF and the C-terminal residues
of PKA. EMBO J. 19:979–988. 2000.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Biondi RM, Komander D, Thomas CC, Lizcano
JM, Deak M, Alessi DR and van Aalten DM: High resolution crystal
structure of the human PDK1 catalytic domain defines the regulatory
phosphopeptide docking site. EMBO J. 21:4219–4228. 2002.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Zhang Y and McCormick S: AGCVIII kinases:
At the crossroads of cellular signaling. Trends Plant Sci.
14:689–695. 2009.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Sobko A: Systems biology of AGC kinases in
fungi. Sci STKE. 2006(re9)2006.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Lanassa Bassukas AE, Xiao Y and
Schwechheimer C: Phosphorylation control of PIN auxin transporters.
Curr Opin Plant Biol. 65(102146)2022.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Jiang Y, Liu X, Zhou M, Yang J, Ke S and
Li Y: Genome-wide identification of the AGC protein kinase gene
family related to photosynthesis in rice (Oryza sativa). Int J Mol
Sci. 23(12557)2022.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Glanc M, Van Gelderen K, Hoermayer L, Tan
S, Naramoto S, Zhang X, Domjan D, Včelařová L, Hauschild R, Johnson
A, et al: AGC kinases and MAB4/MEL proteins maintain PIN polarity
by limiting lateral diffusion in plant cells. Curr Biol.
31:1918–1930.e5. 2021.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Wick KL and Liu F: A new molecular target
of insulin action: Regulating the pivotal PDK1. Curr Drug Targets
Immune Endocr Metabol Disord. 1:209–221. 2001.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Alessi DR, James SR, Downes CP, Holmes AB,
Gaffney PR, Reese CB and Cohen P: Characterization of a
3-phosphoinositide-dependent protein kinase which phosphorylates
and activates protein kinase Balpha. Curr Biol. 7:261–269.
1997.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Stokoe D, Stephens LR, Copeland T, Gaffney
PR, Reese CB, Painter GF, Holmes AB, McCormick F and Hawkins PT:
Dual role of phosphatidylinositol-3,4,5-trisphosphate in the
activation of protein kinase B. Science. 277:567–570.
1997.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Dittrich ACN and Devarenne TP:
Perspectives in PDK1 evolution: Insights from photosynthetic and
non-photosynthetic organisms. Plant Signal Behav. 7:642–649.
2012.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Scheid MP, Parsons M and Woodgett JR:
Phosphoinositide-dependent phosphorylation of PDK1 regulates
nuclear translocation. Mol Cell Biol. 25:2347–2363. 2005.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Gagliardi PA, di Blasio L and Primo L:
PDK1: A signaling hub for cell migration and tumor invasion.
Biochim Biophys Acta. 1856:178–188. 2015.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Cohen P, Alessi DR and Cross DA: PDK1, one
of the missing links in insulin signal transduction? FEBS Lett.
410:3–10. 1997.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Zhou Y, Guo Y, Ran M, Shan W, Granchi C,
Giovannetti E, Minutolo F, Peters GJ and Tam KY: Combined
inhibition of pyruvate dehydrogenase kinase 1 and lactate
dehydrogenase a induces metabolic and signaling reprogramming and
enhances lung adenocarcinoma cell killing. Cancer Lett.
577(216425)2023.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Feng Q, Di R, Tao F, Chang Z, Lu S, Fan W,
Shan C, Li X and Yang Z: PDK1 regulates vascular remodeling and
promotes epithelial-mesenchymal transition in cardiac development.
Mol Cell Biol. 30:3711–3721. 2010.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Lawlor MA, Mora A, Ashby PR, Williams MR,
Murray-Tait V, Malone L, Prescott AR, Lucocq JM and Alessi DR:
Essential role of PDK1 in regulating cell size and development in
mice. EMBO J. 21:3728–3738. 2002.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Pietri M, Dakowski C, Hannaoui S,
Alleaume-Butaux A, Hernandez-Rapp J, Ragagnin A, Mouillet-Richard
S, Haik S, Bailly Y, Peyrin JM, et al: PDK1 decreases TACE-mediated
α-secretase activity and promotes disease progression in prion and
Alzheimer's diseases. Nat Med. 19:1124–1131. 2013.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Hashimoto N, Kido Y, Uchida T, Asahara S,
Shigeyama Y, Matsuda T, Takeda A, Tsuchihashi D, Nishizawa A, Ogawa
W, et al: Ablation of PDK1 in pancreatic beta cells induces
diabetes as a result of loss of beta cell mass. Nat Genet.
38:589–593. 2006.PubMed/NCBI View
Article : Google Scholar
|
|
46
|
Choucair KA, Guérard KP, Ejdelman J,
Chevalier S, Yoshimoto M, Scarlata E, Fazli L, Sircar K, Squire JA,
Brimo F, et al: The 16p13.3 (PDPK1) genomic gain in prostate
cancer: A potential role in disease progression. Transl Oncol.
5:453–460. 2012.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Maurer M, Su T, Saal LH, Koujak S, Hopkins
BD, Barkley CR, Wu J, Nandula S, Dutta B, Xie Y, et al:
3-Phosphoinositide-dependent kinase 1 potentiates upstream lesions
on the phosphatidylinositol 3-kinase pathway in breast carcinoma.
Cancer Res. 69:6299–6306. 2009.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Kim JW, Tchernyshyov I, Semenza GL and
Dang CV: HIF-1-mediated expression of pyruvate dehydrogenase
kinase: A metabolic switch required for cellular adaptation to
hypoxia. Cell Metab. 3:177–185. 2006.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Papandreou I, Cairns RA, Fontana L, Lim AL
and Denko NC: HIF-1 mediates adaptation to hypoxia by actively
downregulating mitochondrial oxygen consumption. Cell Metab.
3:187–197. 2006.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Li J, Zhai X, Sun X, Cao S, Yuan Q and
Wang J: Metabolic reprogramming of pulmonary fibrosis. Front
Pharmacol. 13(1031890)2022.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Hamanaka RB and Mutlu GM: Metabolic
requirements of pulmonary fibrosis: Role of fibroblast metabolism.
FEBS J. 288:6331–6352. 2021.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Henderson J and O'Reilly S: The emerging
role of metabolism in fibrosis. Trends Endocrinol Metab.
32:639–653. 2021.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Goodwin J, Choi H, Hsieh MH, Neugent ML,
Ahn JM, Hayenga HN, Singh PK, Shackelford DB, Lee IK, Shulaev V, et
al: Targeting hypoxia-inducible factor-1α/pyruvate dehydrogenase
kinase 1 axis by dichloroacetate suppresses bleomycin-induced
pulmonary fibrosis. Am J Respir Cell Mol Biol. 58:216–231.
2018.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Stacpoole PW: Review of the pharmacologic
and therapeutic effects of diisopropylammonium dichloroacetate
(DIPA). J Clin Pharmacol J New Drugs. 9:282–291. 1969.PubMed/NCBI
|
|
55
|
Stacpoole PW, Kurtz TL, Han Z and Langaee
T: Role of dichloroacetate in the treatment of genetic
mitochondrial diseases. Adv Drug Deliv Rev. 60:1478–1487.
2008.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Yang K, Li B and Chen J: Knockdown of
phosphoinositide-dependent kinase 1 (PDK1) inhibits fibrosis and
inflammation in lipopolysaccharide-induced acute lung injury rat
model by attenuating NF-κB/p65 pathway activation. Ann Transl Med.
9(1671)2021.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Liu Y, Xie X, Wang P, Luo J, Chen Y, Xu Q,
Zhou J, Lu X, Zhao J, Chen Z and Zuo D: Mannan-binding lectin
reduces epithelial-mesenchymal transition in pulmonary fibrosis via
inactivating the store-operated calcium entry machinery. J Innate
Immun. 15:37–49. 2023.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Loirand G, Guérin P and Pacaud P: Rho
kinases in cardiovascular physiology and pathophysiology. Circ Res.
98:322–334. 2006.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Shimizu Y, Dobashi K, Iizuka K, Horie T,
Suzuki K, Tukagoshi H, Nakazawa T, Nakazato Y and Mori M:
Contribution of small GTPase Rho and its target protein rock in a
murine model of lung fibrosis. Am J Respir Crit Care Med.
163:210–217. 2001.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Barcelo J, Samain R and Sanz-Moreno V:
Preclinical to clinical utility of ROCK inhibitors in cancer.
Trends Cancer. 9:250–263. 2023.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Shimokawa H and Takeshita A: Rho-kinase is
an important therapeutic target in cardiovascular medicine.
Arterioscler Thromb Vasc Biol. 25:1767–1775. 2005.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Knipe RS, Tager AM and Liao JK: The Rho
kinases: Critical mediators of multiple profibrotic processes and
rational targets for new therapies for pulmonary fibrosis.
Pharmacol Rev. 67:103–117. 2015.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Jiang C, Huang H, Liu J, Wang Y, Lu Z and
Xu Z: Fasudil, a Rho-kinase inhibitor, attenuates bleomycin-induced
pulmonary fibrosis in mice. Int J Mol Sci. 13:8293–8307.
2012.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Shimizu Y, Dobashi K, Sano T and Yamada M:
ROCK activation in lung of idiopathic pulmonary fibrosis with
oxidative stress. Int J Immunopathol Pharmacol. 27:37–44.
2014.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Ghatak S, Hascall VC, Markwald RR,
Feghali-Bostwick C, Artlett CM, Gooz M, Bogatkevich GS,
Atanelishvili I, Silver RM, Wood J, et al: Transforming growth
factor β1 (TGFβ1)-induced CD44V6-NOX4 signaling in pathogenesis of
idiopathic pulmonary fibrosis. J Biol Chem. 292:10490–10519.
2017.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Fu S, Wen Y, Peng B, Tang M, Shi M, Liu J,
Yang Y, Si W, Guo Y, Li X, et al: Discovery of indoline-based
derivatives as effective ROCK2 inhibitors for the potential new
treatment of idiopathic pulmonary fibrosis. Bioorg Chem.
137(106539)2023.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Wu X, Verschut V, Woest ME, Ng-Blichfeldt
JP, Matias A, Villetti G, Accetta A, Facchinetti F, Gosens R and
Kistemaker LEM: Rho-kinase 1/2 inhibition prevents transforming
growth factor-β-induced effects on pulmonary remodeling and repair.
Front Pharmacol. 11(609509)2021.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Hong AW, Meng Z and Guan KL: The Hippo
pathway in intestinal regeneration and disease. Nat Rev
Gastroenterol Hepatol. 13:324–337. 2016.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Wu Z and Guan KL: Hippo signaling in
embryogenesis and development. Trends Biochem Sci. 46:51–63.
2021.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Landry NM, Rattan SG, Filomeno KL, Meier
TW, Meier SC, Foran SJ, Meier CF, Koleini N, Fandrich RR, Kardami
E, et al: SKI activates the Hippo pathway via LIMD1 to inhibit
cardiac fibroblast activation. Basic Res Cardiol.
116(25)2021.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Dong L and Li L: Lats2-underexpressing
bone marrow-derived mesenchymal stem cells ameliorate LPS-induced
acute lung injury in mice. Mediators Inflamm.
2019(4851431)2019.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Antebi B, Walker KP III, Mohammadipoor A,
Rodriguez LA, Montgomery RK, Batchinsky AI and Cancio LC: The
effect of acute respiratory distress syndrome on bone
marrow-derived mesenchymal stem cells. Stem Cell Res Ther.
9(251)2018.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Kuwano K, Miyazaki H, Hagimoto N, Kawasaki
M, Fujita M, Kunitake R, Kaneko Y and Hara N: The involvement of
Fas-Fas ligand pathway in fibrosing lung diseases. Am J Respir Cell
Mol Biol. 20:53–60. 1999.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Cai SX, Liu AR, Chen S, He HL, Chen QH, Xu
JY, Pan C, Yang Y, Guo FM, Huang YZ, et al: The orphan receptor
tyrosine kinase ROR2 facilitates MSCs to repair lung injury in ARDS
animal model. Cell Transplant. 25:1561–1574. 2016.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Han J, Lu X, Zou L, Xu X and Qiu H:
E-prostanoid 2 receptor overexpression promotes mesenchymal stem
cell attenuated lung injury. Hum Gene Ther. 27:621–630.
2016.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Fernández-Hernando C, Ackah E, Yu J,
Suárez Y, Murata T, Iwakiri Y, Prendergast J, Miao RQ, Birnbaum MJ
and Sessa WC: Loss of Akt1 leads to severe atherosclerosis and
occlusive coronary artery disease. Cell Metab. 6:446–457.
2007.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Iliopoulos D, Polytarchou C,
Hatziapostolou M, Kottakis F, Maroulakou IG, Struhl K and Tsichlis
PN: MicroRNAs differentially regulated by Akt isoforms control EMT
and stem cell renewal in cancer cells. Sci Signal.
2(ra62)2009.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Revathidevi S and Munirajan AK: Akt in
cancer: Mediator and more. Semin Cancer Biol. 59:80–91.
2019.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Risso G, Blaustein M, Pozzi B, Mammi P and
Srebrow A: Akt/PKB: One kinase, many modifications. Biochem J.
468:203–214. 2015.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Toker A and Yoeli-Lerner M: Akt signaling
and cancer: Surviving but not moving on. Cancer Res. 66:3963–3966.
2006.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Wang J, Hu K, Cai X, Yang B, He Q, Wang J
and Weng Q: Targeting PI3K/AKT signaling for treatment of
idiopathic pulmonary fibrosis. Acta Pharm Sin B. 12:18–32.
2022.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Virtakoivu R, Pellinen T, Rantala JK,
Perälä M and Ivaska J: Distinct roles of AKT isoforms in regulating
β1-integrin activity, migration, and invasion in prostate cancer.
Mol Biol Cell. 23:3357–3369. 2012.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Arboleda MJ, Lyons JF, Kabbinavar FF, Bray
MR, Snow BE, Ayala R, Danino M, Karlan BY and Slamon DJ:
Overexpression of AKT2/protein kinase Bbeta leads to up-regulation
of beta1 integrins, increased invasion, and metastasis of human
breast and ovarian cancer cells. Cancer Res. 63:196–206.
2003.PubMed/NCBI
|
|
84
|
Baek ST, Copeland B, Yun EJ, Kwon SK,
Guemez-Gamboa A, Schaffer AE, Kim S, Kang HC, Song S, Mathern GW
and Gleeson JG: An AKT3-FOXG1-reelin network underlies defective
migration in human focal malformations of cortical development. Nat
Med. 21:1445–1454. 2015.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Kang HR, Lee CG, Homer RJ and Elias JA:
Semaphorin 7A plays a critical role in TGF-beta1-induced pulmonary
fibrosis. J Exp Med. 204:1083–1093. 2007.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Horowitz JC, Rogers DS, Sharma V, Vittal
R, White ES, Cui Z and Thannickal VJ: Combinatorial activation of
FAK and AKT by transforming growth factor-beta1 confers an
anoikis-resistant phenotype to myofibroblasts. Cell Signal.
19:761–771. 2007.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Nie Y, Sun L, Wu Y, Yang Y, Wang J, He H,
Hu Y, Chang Y, Liang Q, Zhu J, et al: AKT2 regulates pulmonary
inflammation and fibrosis via modulating macrophage activation. J
Immunol. 198:4470–4480. 2017.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Larson-Casey JL, Deshane JS, Ryan AJ,
Thannickal VJ and Carter AB: Macrophage Akt1 kinase-mediated
mitophagy modulates apoptosis resistance and pulmonary fibrosis.
Immunity. 44:582–596. 2016.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Nie Y, Hu Y, Yu K, Zhang D, Shi Y, Li Y,
Sun L and Qian F: Akt1 regulates pulmonary fibrosis via modulating
IL-13 expression in macrophages. Innate Immun. 25:451–461.
2019.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Kazanietz MG and Cooke M: Protein kinase C
signaling ‘in’ and ‘to’ the nucleus: Master kinases in
transcriptional regulation. J Biol Chem: 105692, 2024 (Epub ahead
of print).
|
|
91
|
Silnitsky S, Rubin SJS, Zerihun M and Qvit
N: An update on protein kinases as therapeutic targets-part I:
Protein kinase C activation and its role in cancer and
cardiovascular diseases. Int J Mol Sci. 24(17600)2023.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Kang JH, Toita R, Kim CW and Katayama Y:
Protein kinase C (PKC) isozyme-specific substrates and their
design. Biotechnol Adv. 30:1662–1672. 2012.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Abe MK, Kartha S, Karpova AY, Li J, Liu
PT, Kuo WL and Hershenson MB: Hydrogen peroxide activates
extracellular signal-regulated kinase via protein kinase C, Raf-1,
and MEK1. Am J Respir Cell Mol Biol. 18:562–569. 1998.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Barman SA: Potassium channels modulate
canine pulmonary vasoreactivity to protein kinase C activation. Am
J Physiol. 277:L558–L565. 1999.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Das M, Stenmark KR, Ruff LJ and Dempsey
EC: Selected isozymes of PKC contribute to augmented growth of
fetal and neonatal bovine PA adventitial fibroblasts. Am J Physiol.
273:L1276–L1284. 1997.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Harrington EO, Löffler J, Nelson PR, Kent
KC, Simons M and Ware JA: Enhancement of migration by protein
kinase Calpha and inhibition of proliferation and cell cycle
progression by protein kinase Cdelta in capillary endothelial
cells. J Biol Chem. 272:7390–7397. 1997.PubMed/NCBI View Article : Google Scholar
|
|
97
|
Wang J, Sun L, Nie Y, Duan S, Zhang T,
Wang W, Ye RD, Hou S and Qian F: Protein kinase C δ (PKCδ)
attenuates bleomycin induced pulmonary fibrosis via inhibiting
NF-κB signaling pathway. Front Physiol. 11(367)2020.PubMed/NCBI View Article : Google Scholar
|
|
98
|
Jimenez SA, Gaidarova S, Saitta B,
Sandorfi N, Herrich DJ, Rosenbloom JC, Kucich U, Abrams WR and
Rosenbloom J: Role of protein kinase C-delta in the regulation of
collagen gene expression in scleroderma fibroblasts. J Clin Invest.
108:1395–1403. 2001.PubMed/NCBI View Article : Google Scholar
|
|
99
|
Song JS, Kang CM, Park CK and Yoon HK:
Thrombin induces epithelial-mesenchymal transition via PAR-1, PKC,
and ERK1/2 pathways in A549 cells. Exp Lung Res. 39:336–348.
2013.PubMed/NCBI View Article : Google Scholar
|
|
100
|
Barosova H, Meldrum K, Karakocak BB, Balog
S, Doak SH, Petri-Fink A, Clift MJD and Rothen-Rutishauser B:
Inter-laboratory variability of A549 epithelial cells grown under
submerged and air-liquid interface conditions. Toxicol In Vitro.
75(105178)2021.PubMed/NCBI View Article : Google Scholar
|
|
101
|
McMullen JR, Shioi T, Zhang L, Tarnavski
O, Sherwood MC, Dorfman AL, Longnus S, Pende M, Martin KA, Blenis
J, et al: Deletion of ribosomal S6 kinases does not attenuate
pathological, physiological, or insulin-like growth factor 1
receptor-phosphoinositide 3-kinase-induced cardiac hypertrophy. Mol
Cell Biol. 24:6231–6240. 2004.PubMed/NCBI View Article : Google Scholar
|
|
102
|
Ludwik KA and Lannigan DA: Ribosomal S6
kinase (RSK) modulators: A patent review. Expert Opin Ther Pat.
26:1061–1078. 2016.PubMed/NCBI View Article : Google Scholar
|
|
103
|
Shima H, Pende M, Chen Y, Fumagalli S,
Thomas G and Kozma SC: Disruption of the p70(s6k)/p85(s6k) gene
reveals a small mouse phenotype and a new functional S6 kinase.
EMBO J. 17:6649–6659. 1998.PubMed/NCBI View Article : Google Scholar
|
|
104
|
Magnuson B, Ekim B and Fingar DC:
Regulation and function of ribosomal protein S6 kinase (S6K) within
mTOR signalling networks. Biochem J. 441:1–21. 2012.PubMed/NCBI View Article : Google Scholar
|
|
105
|
Pullen N, Dennis PB, Andjelkovic M, Dufner
A, Kozma SC, Hemmings BA and Thomas G: Phosphorylation and
activation of p70s6k by PDK1. Science. 279:707–710. 1998.PubMed/NCBI View Article : Google Scholar
|
|
106
|
Roux PP, Shahbazian D, Vu H, Holz MK,
Cohen MS, Taunton J, Sonenberg N and Blenis J: RAS/ERK signaling
promotes site-specific ribosomal protein S6 phosphorylation via RSK
and stimulates cap-dependent translation. J Biol Chem.
282:14056–14064. 2007.PubMed/NCBI View Article : Google Scholar
|
|
107
|
Soares HP, Ni Y, Kisfalvi K, Sinnett-Smith
J and Rozengurt E: Different patterns of Akt and ERK feedback
activation in response to rapamycin, active-site mTOR inhibitors
and metformin in pancreatic cancer cells. PLoS One.
8(e57289)2013.PubMed/NCBI View Article : Google Scholar
|
|
108
|
Frödin M and Gammeltoft S: Role and
regulation of 90 kDa ribosomal S6 kinase (RSK) in signal
transduction. Mol Cell Endocrinol. 151:65–77. 1999.PubMed/NCBI View Article : Google Scholar
|
|
109
|
Madala SK, Thomas G, Edukulla R, Davidson
C, Schmidt S, Schehr A and Hardie WD: p70 ribosomal S6 kinase
regulates subpleural fibrosis following transforming growth
factor-α expression in the lung. Am J Physiol Lung Cell Mol
Physiol. 310:L175–L186. 2016.PubMed/NCBI View Article : Google Scholar
|
|
110
|
Han Q, Lin L, Zhao B, Wang N and Liu X:
Inhibition of mTOR ameliorates bleomycin-induced pulmonary fibrosis
by regulating epithelial-mesenchymal transition. Biochem Biophys
Res Commun. 500:839–845. 2018.PubMed/NCBI View Article : Google Scholar
|
|
111
|
Zou W, Zhang X, Zhao M, Zhou Q and Hu X:
Cellular proliferation and differentiation induced by single-layer
molybdenum disulfide and mediation mechanisms of proteins via the
Akt-mTOR-p70S6K signaling pathway. Nanotoxicology. 11:781–793.
2017.PubMed/NCBI View Article : Google Scholar
|
|
112
|
Kim S, Han JH, Kim S, Lee H, Kim JR, Lim
JH and Woo CH: p90RSK inhibition ameliorates TGF-β1 signaling and
pulmonary fibrosis by inhibiting smad3 transcriptional activity.
Cell Physiol Biochem. 54:195–210. 2020.PubMed/NCBI View Article : Google Scholar
|
|
113
|
Jia S, Agarwal M, Yang J, Horowitz JC,
White ES and Kim KK: Discoidin domain receptor 2 signaling
regulates fibroblast apoptosis through PDK1/Akt. Am J Respir Cell
Mol Biol. 59:295–305. 2018.PubMed/NCBI View Article : Google Scholar
|
|
114
|
Wang L, Li Z, Wan R, Pan X, Li B, Zhao H,
Yang J, Zhao W, Wang S, Wang Q, et al: Single-cell RNA sequencing
provides new insights into therapeutic roles of thyroid hormone in
idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol.
69:456–469. 2023.PubMed/NCBI View Article : Google Scholar
|
|
115
|
Adams TS, Schupp JC, Poli S, Ayaub EA,
Neumark N, Ahangari F, Chu SG, Raby BA, DeIuliis G, Januszyk M, et
al: Single-cell RNA-seq reveals ectopic and aberrant lung-resident
cell populations in idiopathic pulmonary fibrosis. Sci Adv.
6(eaba1983)2020.PubMed/NCBI View Article : Google Scholar
|
|
116
|
Yang L, Gilbertsen A, Smith K, Xia H,
Higgins L, Guerrero C and Henke CA: Proteomic analysis of the IPF
mesenchymal progenitor cell nuclear proteome identifies
abnormalities in key nodal proteins that underlie their fibrogenic
phenotype. Proteomics. 22(e2200018)2022.PubMed/NCBI View Article : Google Scholar
|