Introduction
Numerous rearrangements in the distal part of the
short arm of chromosome 8 have previously been reported in the
literature. This part of chromosome 8 is prone to a variety of
rearrangements due to the existence of two olfactory receptor gene
clusters, REPD (distal repeat) and REPP (proximal repeat), that
flank a ~5 Mb region of 8p23.1. These rearrangements include, among
others, the deletion or duplication of 8p23.1 between the two
clusters, the 8p interstitial inverted duplication with associated
terminal deletion (inv dup del(8p)) and the isolated terminal 8p23
deletion (1,2). There is a wide range of
characteristics that are associated with such rearrangements. More
specifically, deletions in 8p23 are associated with motor
impairment, microcephaly, dysmorphism, epilepsy, growth and
psychomotor delay, cardiac defects, hypotonia, facial anomalies,
speech delay, attention deficit hyperactivity disorder,
intellectual disability and seizures (2-6).
However, the exact genotype-phenotype correlation regarding the
8p23 deletions still remains unknown. Deletions in 8p23.1 and gene
GATA4 in particular have been identified to be responsible
for heart defects in patients with 8p23 deletion (3,7,8).
Additionally, it has been observed that the 8p23.2-p23.3 deletion
usually leads to a milder phenotype (2,9).
Herein, we describe a 30-year-old man that carries a de novo
deletion in 8p23.2-p23.3 and is clinically asymptomatic.
Case report
The patient is a phenotypically normal 30-year-old
man. His wife and him were referred to our lab due to the couple's
two miscarriages. Physical examination as well as blood test were
normal. Neurobehavioral testing and academic performance was
normal. His height was 182 cm, his weight was 84 kg and his body
mass index was 25.4 kg/m2.
Conventional karyotyping of the patient's blood
T-lymphocytes was performed using high-resolution banding
techniques. Twenty metaphases were analyzed following GTG banding.
Conventional cytogenetic analysis of the patient's blood showed a
deletion in 8p [karyotype according to ISCN was 46,XY,del(8)(p)]. Next, conventional cytogenetic
analysis of the patient's parents followed to establish whether the
deletion was inherited or if it occurred de novo. The
parents had normal karyotypes (results not shown) indicating that
the deletion occurred de novo in the patient. Lastly,
cytogenetic analysis was performed in the patient's wife because of
the couple's miscarriages. Her karyotype was also normal (data not
shown).
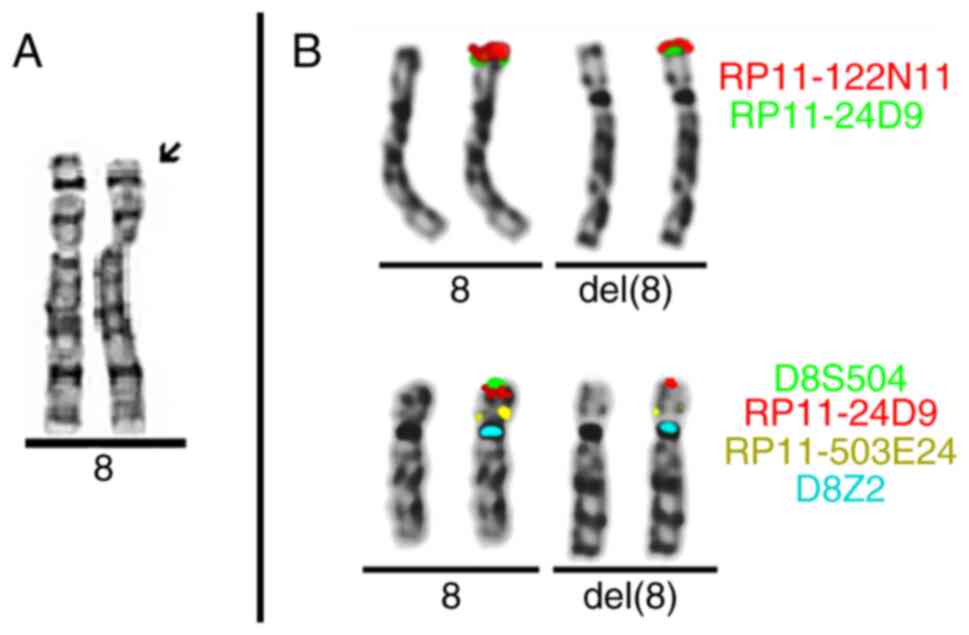
Chromosome analysis on fixed blood cell suspension
was performed (15 metaphases) using fluorescence in situ
hybridization (FISH). Probes specific to genomic locations 8p23.1
(RP11-122N11, RP11-24D9, wcp 8), 8p23.3 (D8S504), 8p11.21
(RP11-503E24) and 8p11.1q11.1 (cep 8=D8Z2) were used. FISH analysis
results suggested a terminal deletion in 8p23.3 [ish del(8)(p23.3)(wcp8+,D8S504-,RP11-122N11+,RP11-24D9+,RP11-503E24+,D8Z2+)]
(Fig. 1).
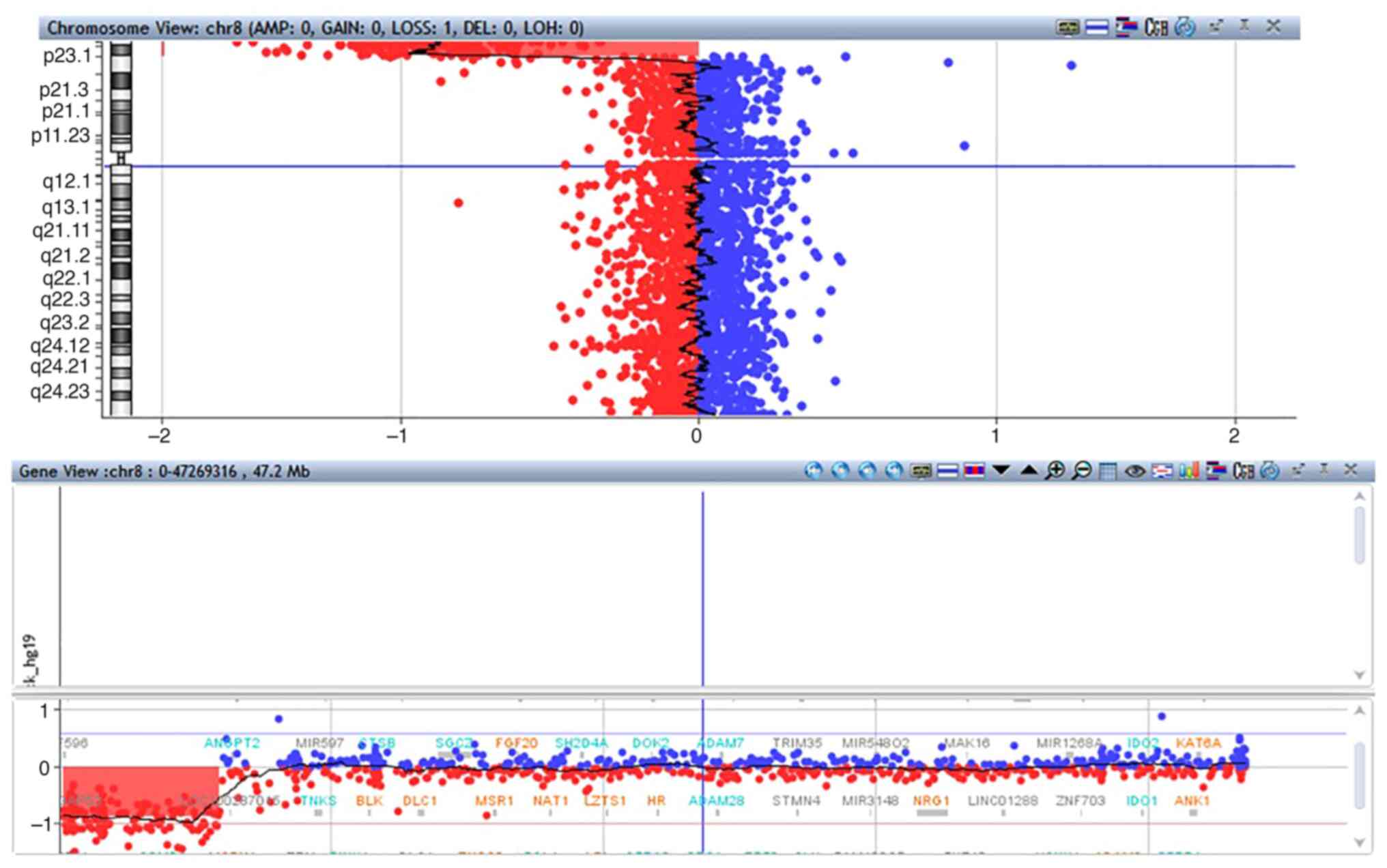
High resolution molecular karyotyping was performed
using an Array Comparative Genomic Hybridization (aCGH) platform of
60,000 oligonucleotides (Agilent technologies). DNA was extracted
from the patient's whole blood cells and from embryonic tissue from
the products of conception (POC) of the couple's second miscarriage
using Promega Maxwell 16 and was hybridized with the human
reference DNA of the same gender (Promega Biotech). The statistical
test that was used as a parameter to estimate the number of copies
was ADM-2 (provided by Agilent Technologies DNA analytics software)
with a window of 0.5 Mb and a threshold of 6. Only those copy
number changes that affected at least 5 consecutive probes with
identically oriented change were considered as copy number
variations. For most of the genome, the average analysis resolution
was 200 kb. Molecular karyotyping of the patient's blood revealed a
5.721 Mb deletion in the 8p23.2-p23.3 region: arr[hg19]
8p23.3p23.2(191,530-5,912,296)x1 (Fig.
2). The deletion involves 25 genes, of which 10 are coding
genes. More specifically, the coding genes located in the deleted
area are ARHGEF10, CLN8, CSMD1, DLGAP2,
ERICH1, FBXO25, KBTBD11, MYOM2,
TDRP and ZNF596. All genes except for CSMD1
are located in 8p23.3 while CSMD1 is located in 8p23.2
(Fig. 3). All genes except for
KBTBD11, MYOM2, and ZNF596 have a high
probability of exhibiting haploinsufficiency (pHaplo scores are
0.80-0.96). Lastly, molecular karyotyping in the POC from the
miscarriage indicated a normal male genomic profile of the fetus
(data not shown).
Discussion
Numerous rearrangements in the distal part of
chromosome 8p have been reported, including deletions in 8p23.1 and
8p23.2-pter. In this case report, we present the case of a man that
is carrying a 5.72 Mb de novo deletion in 8p23.2-p23.3 that
involves 10 coding genes, namely ZNF596, FBXO25,
TDRP, ERICH1, DLGAP2, CLN8,
ARHGEF10, KBTBD11, MYOM2 and CSMD1. The
patient was diagnosed using conventional and molecular karyotyping
as well as FISH analysis.
Rearrangements in this chromosomal area have been
linked to a wide range of phenotypic characteristics that affect,
among others, one's intellectual ability, motor skills,
development, etc. The 8p23.2-p23.3 deletion has been associated
with milder phenotypic characteristics (2,9).
Interestingly, our patient remains completely asymptomatic. His
clinical examination showed a normal male phenotype. Molecular
karyotyping of the POC of the couple's miscarriage indicated a
normal male genotype, suggesting that the miscarriage cannot be
attributed to the 8p deletion our patient carries.
Due to the variability in phenotypic
characteristics, we decided to compare reported cases of deletions
similar to our patient's. In this comparison, we decided to only
include patients that did not have any additional chromosomal
abnormalities, since those could have an additive effect on the
patients' phenotype. Searching in DECIPHER and the literature, we
discovered patients that have similar deletions to our patient in
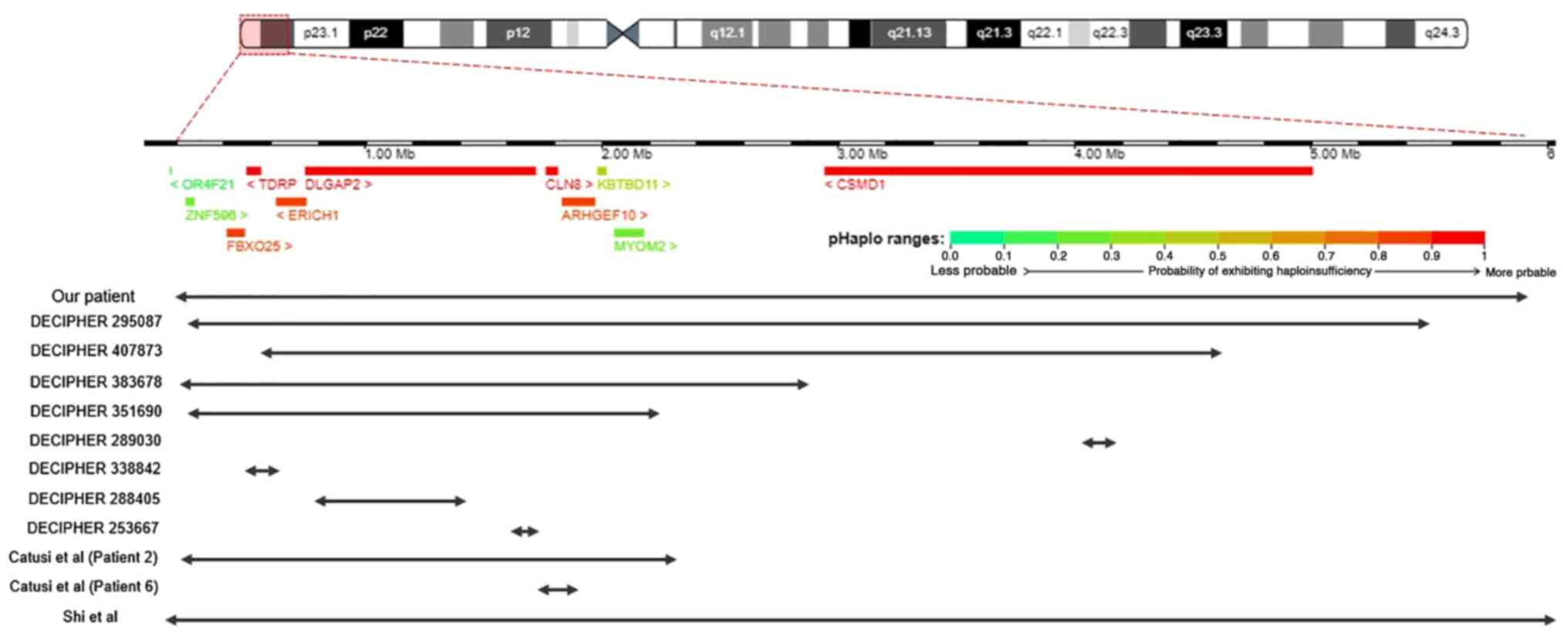
the 8p23.2-p23.3 area that are summarized in Table I (Fig.
3). Patient 295087 and Patient 2 from Catusi et al
(4) carry deletions (5.25 and 2.12
Mb respectively) that involve the same coding genes as our patient.
Both these patients have an abnormal phenotype, with the first one
having moderate intellectual disability and the second one
presenting with a variety of symptoms including developmental delay
and mobility issues. It is important to note that Patient 2 has
inherited the deletion from his mother who is asymptomatic. Both
Patient 2 and his mother also carry a microdeletion in 5p15.2.
According to Catusi et al, this 5p microdeletion would
probably be classified as likely benign but it could have an
additive effect on the pathogenic phenotype (4). Another case of a similar deletion is
DECIPHER patient 407873. He has a 4.37 Mb deletion in 8p23.2-23.3
that involves the same coding genes as our patient, except for
ZNF596. His symptoms include abnormalities of the immune and
nervous system. Another similar case is the patient from Shi et
al who has a 5.85 Mb deletion in 8p23.2-8p23.3 that involves
gene OR4F21 in addition to all other coding genes of our
patient's deletion. This patient has mildly dysmorphic features,
psychomotor delay, poor language and motor skills, attention
deficit and others (2). The
OR4F21 gene is an olfactory receptor gene and its deletion
is most likely not responsible for the patient's symptoms (has a
low probability of exhibiting haploinsufficiency). Next, DECIPHER
patients 383678 and 351690 carry deletions (sizes 2.59 and 1.98 Mb
respectively) that involve the same coding genes as our patient,
except for CSMD1 which is located in 8p23.2. For patient
383678 no phenotype is included in DECIPHER, however, the deletion
was classified as pathogenic. According to DECIPHER, patient 351690
has intellectual disability.
 | Table IData regarding our patient, 8 DECIPHER
patients, 2 patients from Catusi et al (4) and the patient from Shi et al
(2). |
Table I
Data regarding our patient, 8 DECIPHER
patients, 2 patients from Catusi et al (4) and the patient from Shi et al
(2).
| |
Patient |
|---|
| Variable | Our patient | 295087 | 407873 | 383678 | 351690 | 289030 | 338842 | 288405 | 253667 | Patient 2 (Catusi
et al, 2021) | Patient 6 (Catusi,
et al, 2021) | Patient from Shi
et al, 2017 |
|---|
| Deletion size | 5.72 Mb | 5.25 Mb | 4.37 Mb | 2.59 Mb | 1.98 Mb | 124.35 kb | 28.72 kb | 632.65 kb | 67.7 kb | 2.12 Mb | 123 kb | 5.85 Mb |
| Deleted genes | ARHGEF10,
CLN8, CSMD1, DLGAP2, ERICH1,
FBXO25, KBTBD11, MYOM2, TDRP,
ZNF596 | ARHGEF10,
CLN8, CSMD1, DLGAP2, ERICH1,
FBXO25, KBTBD11, MYOM2, TDRP,
ZNF596 | ARHGEF10,
CLN8, CSMD1, DLGAP2, ERICH1,
FBXO25, KBTBD11, MYOM2, TDRP | ARHGEF10,
CLN8, DLGAP2, ERICH1, FBXO25,
KBTBD11, MYOM2, TDRP, ZNF596 | ARHGEF10,
CLN8, DLGAP2, ERICH1, FBXO25,
KBTBD11, MYOM2, TDRP, ZNF596 | CSMD1 | TDRP | DGALP2
MYOM2, ORF4F21, | DGALP2 | ARHGEF10,
CLN8, DLGAP2, ERICH1, FBXO25,
KBTBD11, KBTBD11, MYOM2, TDRP,
ZNF596 | ARHGEF10,
CLN8 | ARHGEF10,
CLN8, CSMD1, DLGAP2, ERICH1,
FBXO25, OR4F21, TDRP, ZNF596 |
| Pathogenicity
(based on DECIPHER, if available) | N/A | Pathogenic | Pathogenic | Pathogenic | Uncertain | Likely benign | Likely benign | Likely benign | N/A | N/A | N/A | N/A |
| Phenotype | Normal | Moderate ID | Thin upper lip
vermilion, recurrent otitis media, abnormal emotion/ affect
behavior, DSLD, low frustration tolerance, poor fine motor
coordination | | ID | Autism | DSLD | Autism, ID,
seizure | Epicanthus, spotty
hyperpigmentation, clinodactyly of 5th finger, microcephaly,
ID | DD, motor
instability, balance/ coordination problems, limb hypotonia,
hyperkinetic behavior, irritability, xerosis cutis, skin anomalies,
mild hepatomegaly, cerebral parenchymal anomalies | DD, fine and gross
coordination problems, epilepsy, scoliosis | Mildly dysmorphic
facial features, growth delay, psychomotor delay, poor language
& motor skills, poor balance & coordination, attention
deficit, hyperactivity, impulsivity |
| Additional
CNVs | | | | | | | | | | 5p15.2
microdeletion | | |
Patients with smaller deletions in the 8p23.2-p23.3
region also present with symptoms. For instance, Patient 6 from
Catusi et al has a 123 Kb deletion that only involves genes
ARHGEF10 and CLN8. His symptoms involve epilepsy and
scoliosis. Interestingly, his asymptomatic brother also carried the
same deletion. Moreover, DECIPHER patient 289030, who has autism,
has a 124.35 kb deletion in 8p23.2 that only involves the
CSMD1 gene. Patient 338842 has a 28.72 kb deletion in 8p23.3
that only involves the TDRP gene and has abnormalities of
the nervous system. Lastly, two DECIPHER patients, 288405 and
253667 have a 632.65 and 67.7 kb deletion respectively that only
involve the DLGAP2 gene. Patient's 288405 symptoms include
autism and intellectual disability while patient's 253667 symptoms
include microcephaly and intellectual disability.
The exact genotype-phenotype correlation still
remains unknown (2). Wu et
al (10) suggested a critical
region (CR) 2.05 Mb in size in 8p23.3 responsible for developmental
delay, intellectual disability, microcephaly and neurobehavioral
problems. This CR comprises of genes that have a role in neural
differentiation and neural function (4,10).
Genes ARHGEF10 (OMIM 608236), CLN8 (OMIM 607837),
DLGAP2 (OMIM 605438) and CSMD1 (OMIM 608397) are
associated to the central nervous system. ARHGEF10 (Rho
guanine nucleotide exchange factor 10) encodes a Rho guanine
nucleotide exchange factor that is thought to have a role in neural
morphogenesis and connectivity. The ARHGEF10 protein has a role in
developmental myelination of peripheral nerves (11). CLN8 (Ceroid lipofuscinosis,
neuronal, 8) encodes a transmembrane protein that plays a role in
lipid synthesis, transport and sensing. Mutations in this gene
cause progressive epilepsy with intellectual disabilities (12,13).
DLGAP2 (Discs large-associated protein 2) encodes a protein
that is said to be involved in synapse scaling and is important for
postsynaptic density. It is linked to a variety of neurological
disorders including schizophrenia and the autism spectrum disease
(14). CSMD1 (CUB and Sushi
multiple domains 1) encodes a protein that is involved in brain
circuits' development, signaling, neurotransmission etc. It is
thought to be a regulator of complement activation and inflammation
in the developing central nervous system and may play a role in
nerve growth cone function (15,16).
As mentioned above, there is not a clear
genotype-phenotype relationship established so far. Motor
impairment has been linked to genes CLN8 and ARHGEF10
(4). Moreover, Shi et al
hypothesized that the candidate genes responsible for developmental
delay, intellectual disability, microcephaly and neurobehavioral
disorders are DLGAP2, CLN8, ARHGEF10 and
CSMD1 (2). In a recent
genotype analysis performed by Catusi et al in order to
narrow down the CR, the strongest candidate gene responsible for
neurodevelopmental/behavioral phenotypes was DLGAP2
(4). Additionally, because of the
asymptomatic mother of Patient 2 and brother of Patient 6, Catusi
et al suggested that reduced penetrance should be further
investigated and that more cases are needed in order to strengthen
the hypothesis that incomplete penetrance of 8p23.2-pter deletions
exists (4). Our case is in further
support of the incomplete penetrance hypothesis of 8p23.2-p23.3
deletions.
Our patient, a carrier of a 5.72 Mb de novo
deletion in 8p23.2-p23.3 that involves all genes that are in the
critical region that is hypothesized to be responsible for the
abnormal phenotypic traits in patients with such deletions, is a
phenotypically normal male with no clinical symptoms. Other
patients with similar deletions exhibit a variety of symptoms. This
supports the hypothesis that there is incomplete penetrance of
8p23.2-p23.3 deletions.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets generated and/or analyzed during the
current study are available in the ArrayExpress repository under
accession no. E-MTAB-13528 (https://www.ebi.ac.uk/biostudies/arrayexpress/studies/E-MTAB-13528).
Authors' contributions
CK substantially contributed to the design of the
work and prepared the manuscript. EM was in charge of overall
patient management and project supervision, and critically revised
the manuscript. IP performed and analyzed the molecular karyotype
results and critically revised the manuscript. EPap was responsible
for conventional cytogenetic analysis. TL performed the FISH
experiment and interpreted the results. MBP significantly
contributed to the design of the work and revised the manuscript.
EPav, KK and AG were responsible for medical treatment and
assessment of the patient and his wife. EM and IP confirm the
authenticity of all the raw data. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Written informed consent was obtained from the
patient for publication of this case report and any accompanying
images. Any information revealing the patient's identity was not
included. All procedures followed were conducted according to The
Declaration of Helsinki 1975, as revised in 2008.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Yu S, Fiedler S, Stegner A and Graf WD:
Genomic profile of copy number variants on the short arm of human
chromosome 8. Eur J Hum Genet. 18:1114–1120. 2010.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Shi S, Lin S, Chen B and Zhou Y: Isolated
chromosome 8p23.2-pter deletion: Novel evidence for developmental
delay, intellectual disability, microcephaly and neurobehavioral
disorders. Mol Med Rep. 16:6837–6845. 2017.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Páez MT, Yamamoto T, Hayashi K, Yasuda T,
Harada N, Matsumoto N, Kurosawa K, Furutani Y, Asakawa S, Shimizu N
and Matsuoka R: Two patients with atypical interstitial deletions
of 8p23.1: Mapping of phenotypical traits. Am J Med Genet A.
146A:1158–1165. 2008.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Catusi I, Garzo M, Capra AP, Briuglia S,
Baldo C, Canevini MP, Cantone R, Elia F, Forzano F, Galesi O, et
al: 8p23.2-pter microdeletions: Seven new cases narrowing the
candidate region and review of the literature. Genes (Basel).
12(652)2021.PubMed/NCBI View Article : Google Scholar
|
|
5
|
de Die-Smulders CE, Engelen JJ,
Schrander-Stumpel CT, Govaerts LC, de Vries B, Vles JS, Wagemans A,
Schijns-Fleuren S, Gillessen-Kaesbach G and Fryns JP: Inversion
duplication of the short arm of chromosome 8: Clinical data on
seven patients and review of the literature. Am J Med Genet.
59:369–374. 1995.PubMed/NCBI View Article : Google Scholar
|
|
6
|
García-Santiago FA, Martínez-Glez V,
Santos F, García-Miñaur S, Mansilla E, Meneses AG, Rosell J,
Granero ÁP, Vallespín E, Fernández L, et al: Analysis of
invdupdel(8p) rearrangement: Clinical, cytogenetic and molecular
characterization. Am J Med Genet A. 167A:1018–1025. 2015.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Wat MJ, Shchelochkov OA, Holder AM, Breman
AM, Dagli A, Bacino C, Scaglia F, Zori RT, Cheung SW, Scott DA and
Kang SH: Chromosome 8p23.1 deletions as a cause of complex
congenital heart defects and diaphragmatic hernia. Am J Med Genet
A. 149A:1661–1677. 2009.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Ballarati L, Cereda A, Caselli R,
Selicorni A, Recalcati MP, Maitz S, Finelli P, Larizza L and
Giardino D: Genotype-phenotype correlations in a new case of 8p23.1
deletion and review of the literature. Eur J Med Genet. 54:55–59.
2011.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Burnside RD, Pappas JG, Sacharow S,
Applegate C, Hamosh A, Gadi IK, Jaswaney V, Keitges E, Phillips KK,
Potluri VR, et al: Three cases of isolated terminal deletion of
chromosome 8p without heart defects presenting with a mild
phenotype. Am J Med Genet A. 161A:822–828. 2013.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Wu Y, Ji T, Wang J, Xiao J, Wang H, Li J,
Gao Z, Yang Y, Cai B, Wang L, et al: Submicroscopic subtelomeric
aberrations in Chinese patients with unexplained developmental
delay/mental retardation. BMC Med Genet. 11(72)2010.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Verhoeven K, De Jonghe P, Van de Putte T,
Nelis E, Zwijsen A, Verpoorten N, De Vriendt E, Jacobs A, Van
Gerwen V, Francis A, et al: Slowed conduction and thin myelination
of peripheral nerves associated with mutant rho guanine-nucleotide
exchange factor 10. Am J Hum Genet. 73:926–932. 2003.PubMed/NCBI View
Article : Google Scholar
|
|
12
|
Mole SE, Williams RE and Goebel HH:
Correlations between genotype, ultrastructural morphology and
clinical phenotype in the neuronal ceroid lipofuscinoses.
Neurogenetics. 6:107–126. 2005.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Passantino R, Cascio C, Deidda I, Galizzi
G, Russo D, Spedale G and Guarneri P: Identifying protein partners
of CLN8, an ER-resident protein involved in neuronal ceroid
lipofuscinosis. Biochim Biophys Acta. 1833:529–540. 2013.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Rasmussen AH, Rasmussen HB and
Silahtaroglu A: The DLGAP family: Neuronal expression, function and
role in brain disorders. Mol Brain. 10(43)2017.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Kraus DM, Elliott GS, Chute H, Horan T,
Pfenninger KH, Sanford SD, Foster S, Scully S, Welcher AA and
Holers VM: CSMD1 is a novel multiple domain complement-regulatory
protein highly expressed in the central nervous system and
epithelial tissues. J Immunol. 176:4419–4430. 2006.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Gayed EMAE, Rizk MS, Ramadan AN and Bayomy
NR: mRNA expression of the CUB and sushi multiple domains 1 (CSMD1)
and its serum protein level as predictors for psychosis in the
familial high-risk children and young adults. ACS Omega.
6(24128)2021.PubMed/NCBI View Article : Google Scholar
|