Introduction
Osteoarthritis (OA) is a degenerative joint disease
characterized by articular surface lesions, cartilage degeneration,
chronic inflammation, subchondral bone alterations and osteophyte
formation (1,2). Chronic inflammation, excessive
mechanical loading, aging, obesity and trauma are all risk factors
for OA (3). However, the
pathological development of OA may be a result of numerous factors,
including those at the cellular, ultra-structural and tissue levels
(4). Current treatment options for
OA in clinical practice include non-steroidal anti-inflammatory
drugs and analgesics, which are the first-line treatment option for
alleviating the symptoms of pain associated with OA (5). However, the aforementioned drugs do
not have an effect on the progression of OA, and may lead to
gastrointestinal and cardiovascular adverse events (6). Thus, novel therapeutic strategies
that effectively inhibit the pathogenesis of OA are required.
OA may result due to imbalances in joint cartilage
homeostasis, and a loss of articular cartilage may be facilitated
by the hypertrophy-like phenotype switch of chondrocytes (7). Hypertrophic chondrocytes act as
markers for numerous degenerative disorders, including OA (8). In hypertrophic chondrocytes, the
expression of collagen X and runt-related transcription factor 2
(Runx2) is significantly increased (9). Collagen X is a protein marker for
hypertrophic chondrocytes, and the presence of collagen X in the
extracellular matrix (ECM) may be an indicator of progressive
alterations in the growth plate during endochondral ossification
(10). Runx2, a transcription
factor expressed in pre-hypertrophic and hypertrophic chondrocytes,
promotes the differentiation of proliferative chondrocytes into
hypertrophic chondrocytes (11).
Inactivation of Runx2 leads to decreased chondrocyte hypertrophy
and reduced collagen X expression (12). This suggests that collagen X and
Runx2 play key roles in chondrocyte hypertrophy and bone diseases.
Moreover, the increased expression levels of collagen X and Runx2
are associated with cartilage destruction during OA development
(13).
Bone morphogenetic protein (BMP)/Smads signaling may
induce chondrocyte hypertrophy (14). Following treatment with BMP4, human
chondrocytes exhibit an increase in the nuclear translocation of
phosphorylated (p-)Smad1/5/8, resulting in the increased expression
of collagen X (15). Runx2 is also
a downstream factor of the BMP4/Smad1/5/8 signaling pathway.
Results of a previous study revealed that BMP4 promotes
endochondral bone formation by increasing Runx2 expression
(16). Inhibition of the
BMP4/Smad/Runx2 pathway is associated with the suppression of
osteogenic differentiation in bone marrow mesenchymal stem cells
(BMSCs) (17). BMP-binding
endothelial regulator (BMPER), a secreted glycoprotein, directly
interacts with BMP4 to modulate its functions (18). BMPER acts as an agonist of BMP4 and
mediates angiogenesis in endothelial cells (19). However, whether BMPER mediates
chondrocyte hypertrophy and endochondral ossification remains
unknown.
Forkhead box O (FOXO), a member of the FOX family,
is a transcription factor with a highly conserved DNA-binding
domain. There are four elements in the FOXO sub-family: Namely,
FOXO1, FOXO3a, FOXO4 and FOXO6. FOXO3a activity is regulated by
post-translational modifications, such as acetylation,
phosphorylation and ubiquitination (20). The results of a previous study
revealed that histone deacetylases, such as Sirt1 and Sirt3,
deacetylate FOXO3a and mediate its transcriptional activity
(21).
5,7,3',4'-tetramethoxyflavone (TMF) is a natural
flavonoid derived from Murraya exotica L. The results of our
previous study revealed that TMF exerts protective effects against
the development of OA through activation of the Sirt1/FOXO3a
signaling pathway (22). However,
the molecular mechanism underlying TMF in the inhibition of OA
development remains to be fully elucidated. Notably, BMPER is a
downstream factor of FOXO3a (19).
Thus, the present study aimed to determine whether TMF inhibited
chondrocyte hypertrophy and OA progression by mediating
FOXO3a/BMPER signaling.
Materials and methods
Animals
The present study (approval no. 2020365) was
approved by The Institutional Animal Care and Use Committee of
Gannan Medical University (Ganzhou, China), according to the
principles of the Declaration of Helsinki. A total of 24 male rats
(age, 8 weeks; weight, 220±20 g), provided by the Experimental
Animal Center of Gannan Medical University (Ganzou, China), were
housed in an SPF-grade room with a 12/12 h light/dark cycle, a
temperature of 21-23˚C and a humidity of 45-55%. All animals had
free access to food and water, and were left to acclimate to the
conditions for 7 days.
Establishment of rat OA models
Rats were anesthetized with 3% sodium pentobarbitone
(40 mg/kg) via an intraperitoneal injection (i.p.). After routine
shaving and disinfection in the right knee, 10 µl of sterile saline
with 1 mg of monosodium iodoacetate (MIA; Sigma-Aldrich; Merck
KGaA) was intra-articularly injected (23). Rats in the negative control group
received the same volume of vehicle. Rats were divided into four
groups (n=6) as follows: Negative control group, MIA-induced model
group, model group intragastrically treated with 25 mg/kg TMF and
model group intragastrically treated with 100 mg/kg TMF (24). Signs in rats, such as excessive fur
grooming, 20% loss of body weight and difficulty breathing during
the experiment were considered humane endpoints, requiring
immediate intervention. However, no cases required euthanasia due
to observation of a humane endpoint. After 8 weeks, rats were
sacrificed via cervical dislocation after euthanasia by sodium
pentobarbitone (40 mg/kg; i.p.). Cartilage tissues were obtained
from the joints for further examination.
Hematoxylin and eosin (H&E)
staining and immunohistochemistry (IHC)
Cartilage tissues were fixed in 4% paraformaldehyde
(Beyotime Institute of Biotechnology) for 48 h at 4˚C and
subsequently decalcified in 10% EDTA solution for 25 days at 4˚C.
Decalcified cartilage was embedded in paraffin and samples were cut
into 4-µm slices. Subsequently, all samples were pre-heated for 20
min at 60˚C. The samples were then deparaffinized in xylene-I for
20 min at 60˚C and then in xylene-II for 10 min at room
temperature, and rehydrated with 100, 95, 80 and 75% ethanol.
H&E (Beijing Solarbio Science & Technology Co., Ltd.)
staining was performed and histological evaluation was carried out.
Briefly, sections were stained with hematoxylin for 5 min at 35˚C.
Sections were then rinsed with running water, immersed in 1%
hydrochloric acid alcohol for 5 sec, rinsed with running water,
immersed in 1% aqueous ammonia for 5 sec, and rinsed with running
water again. After soaking in 80, 90 and 95% alcohol for 5 min
each, sections were stained with eosin for 5 min at 35˚C. Next, the
stained sections were dehydrated using ascending ethanol solutions
before sealing. The sections were analyzed using a confocal laser
scanning microscope (LSM880; Carl Zeiss AG).
For immunohistochemical analysis, deparaffinized
slices were eluted in 3% H2O2 to eliminate
endogenous peroxidase activity. Samples were treated with goat
serum (10%) for 30 min at room temperature, and subsequently
incubated with the following primary antibodies: Anti-collagen X
(1:200; cat. no. DF13214; Affinity Biosciences, Ltd.), anti-Runx2
(1:200; cat. no. AF5186; Affinity Biosciences, Ltd.), anti-BMPER
(1:200; cat. no. AV52569; Sigma-Aldrich; Merck KGaA) or anti-FOXO3a
(1:200; cat. no. AF6020; Affinity Biosciences, Ltd.) overnight at
4˚C. Following primary incubation, samples were incubated with the
goat anti-rabbit horseradish peroxidase-conjugated secondary
antibody (1:2,000; cat. no. BA1039; Boster Biological Technology)
for 30 min at 37˚C. Samples were subsequently stained with DAB at
room temperature for 10 min and analyzed using a confocal laser
scanning microscope (LSM880; Carl Zeiss AG) and ImageJ software
(version 1.51r; National Institutes of Health).
Cell culture
Human immortal C28/I2 cells (Procell Life Science
& Technology Co., Ltd.) were cultured in low-glucose DMEM
(Thermo Fisher Scientific, Inc.) supplemented with penicillin,
streptomycin and 10% fetal bovine serum at 37˚C in an incubator
with 5% CO2. Recombinant human interleukin 1β (IL-1β;
cat. no. IL038; Sigma-Aldrich; Merck KGaA) was used to establish
OA-like chondrocyte models at 37˚C for 24 h. Samples were treated
with TMF at concentrations of 5 and 20 µg/ml at 37˚C for 24 h
(22,24,25).
Lentivirus (LV) infection
LV-BMPER and LV-short hairpin RNA (sh/shRNA)-FOXO3a
lentiviral particles were constructed, screened and verified by
OBiO Technology Corp. Ltd. (Shanghai, China). The coding sequence
of BMPER was cloned into the pLV-CMV-MCS-EF1-ZsGreen1-T2A-Puro
vector. The FOXO3a short hairpin RNA (shRNA) was cloned into the
pSLenti-U6-shRNA-CMV-EGFP-F2A-Puro-WPRE vector. The 2nd generation
system was used to package the lentivirus. The lentiviral particles
were prepared by transfecting 293T cells (Cell Bank of Type Culture
Collection of The Chinese Academy of Science) with 12 µg of either
a plasmid containing the coding sequence or an shRNA plasmid along
with lentiviral packaging plasmid psPAX2 and the envelope plasmid
pMD2.G at a ratio of 4:3:1 at 37˚C for 72 h. The 293T cells were
cultured with DMEM containing 10% FBS, penicillin (100 U/ml) and
streptomycin (100 U/ml) at 37˚C for 48 h. The supernatant was
collected by centrifugation (3,000 x g) at 4˚C for 10 min. The
lentiviral particles were harvested after centrifugation (100,000 x
g) at 4˚C for 120 min. Chondrocytes (5x104 cells/ml)
were incubated into 6-well plates at 37˚C for 12-24 h to 20-30%
confluency. Then, cells were infected with LV-BMPER and
LV-sh-FOXO3a, according to the instructions recommended by
manufacturer (OBiO Technology Corp. Ltd.). Simply, cells were
transferred to serum-free media containing 5 mg/ml polybrene and
lentiviral particles at a multiplicity of infection (MOI) of 10.0.
The cells were cultured at 37˚C for 12 h, and the medium was
replaced with a fresh complete medium. After 72 h of infection,
cells were selected using puromycin-containing medium (1.5 µg/ml
puromycin; Sigma-Aldrich; Merck KGaA) for 14 days, and 0.625 µg/ml
puromycin was maintained for the subsequent experiments. The cells
were observed under a fluorescence microscope. The infection
efficiency was >90%.
Western blot analysis
Total protein was extracted from harvested C28/I2
cells using RIPA lysis buffer (Beyotime Institute of
Biotechnology). Protein concentration was determined using a BCA
kit (Beyotime Institute of Biotechnology), and 25 µg/lane of each
sample was separated by SDS-PAGE on a 10% gel. The separated
proteins were subsequently transferred onto a PVDF membrane and
blocked for 1 h at 37˚C with 10% BSA Tris-buffered saline (TBS)
containing 5% non-fat milk, followed by washing three times with
TBS-Tween-20 (containing 0.05% Tween-20). Membranes were incubated
with the following primary antibodies: Anti-collagen X (1:1,000;
cat. no. DF13214; Affinity Biosciences, Ltd.), anti-Runx2 (1:1,000;
cat. no. AF5186; Affinity Biosciences, Ltd.), anti-BMP4 (1:1,000;
cat. no. AF5175; Affinity Biosciences, Ltd.), anti-Smad1 (1:1,000;
cat. no. AF0614; Affinity Biosciences, Ltd.), anti-p-Smad1
(1:1,000; cat. no. AF8313; Affinity Biosciences, Ltd.), anti-FOXO3a
(1:1,000; cat. no. AF6020; Affinity Biosciences, Ltd.), anti-BMPER
(1:1,000; cat. no. AV52569; Sigma-Aldrich; Merck KGaA) and
anti-β-actin (1:1,000; cat. no. AF7018; Affinity Biosciences, Ltd.)
overnight at room temperature. Following primary incubation,
membranes were incubated with the HRP-labeled goat anti-rabbit
secondary antibody (1:5,000; cat. no. A0208; Beyotime Institute of
Biotechnology) at room temperature for 1 h. Protein expression was
analyzed using enhanced chemiluminescence Omni-ECL™
(cat. no. SQ101L; Epizyme; Ipsen Biopharmaceuticals, Inc.) and
ImageJ software (version, 1.51r; National Institutes of
Health).
Immunofluorescence assay
C28/I2 cells on glass coverslips were fixed with 4%
paraformaldehyde at 37˚C for 30 min, and subsequently treated with
0.1% Triton X-100 for membrane infiltration at 37˚C for 10 min.
Following blocking with 5% BSA at 37˚C for 30 min, C28/I2 cells
were incubated with anti-collagen X (1:200; cat. no. DF13214;
Affinity Biosciences, Ltd.) and anti-Runx2 (1:200; cat. no. AF5186;
Affinity Biosciences, Ltd.) overnight at 4˚C. Following primary
incubation, cells were incubated with goat anti-rabbit secondary
IgG antibody (1:500; cat. no. S0001; Affinity Biosciences, Ltd.)
for 1 h at room temperature. DAPI was added to cells at 37˚C for 20
min, and immunofluorescent intensity was analyzed using a confocal
laser scanning microscope (Zeiss GmbH) and ImageJ software
(National Institutes of Health).
Statistical analysis
Experiments were independently performed three
times, and data are expressed as the mean ± standard deviation.
GraphPad Prism 8 (GraphPad Software, Inc.) was used for statistical
analysis. Comparisons between multiple groups were carried out
using one-way ANOVA followed by Tukey's post hoc test. P<0.05
was considered to indicate a statistically significant
difference.
Results
TMF protects against chondrocyte
hypertrophy in rat OA models
To explore the effects of TMF on chondrocyte
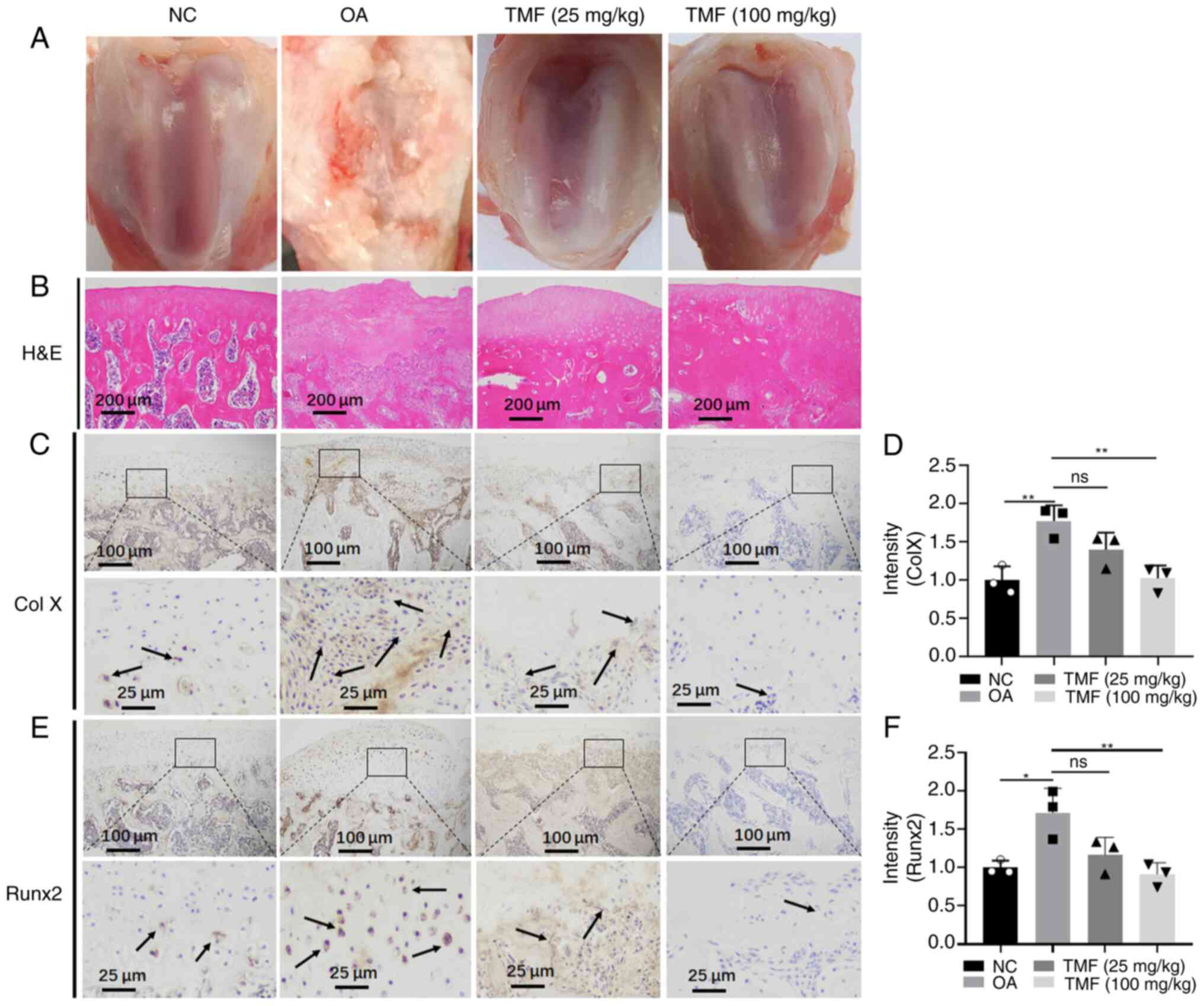
hypertrophy, rat OA models were established (Fig. 1A). Results of the present study
demonstrated that the cartilage in the model group was markedly
damaged (Fig. 1A and B) and TMF exerted protective effects. In
addition, the expression levels of collagen X (Fig. 1C and D) and Runx2 (Fig. 1E and F) were significantly increased in the
cartilage of the OA group compared with those in the NC group.
Consistently, TMF suppressed the expression of collagen X and
Runx2, and these are biomarkers of chondrocyte hypertrophy. Thus,
TMF may protect against chondrocyte hypertrophy in OA rats.
TMF inhibits chondrocyte hypertrophy
in IL-1β-treated C28/I2 cells
To investigate the effects of TMF on chondrocyte
hypertrophy in vitro, C28/I2 cells were treated with IL-1β
(10 ng/ml) to establish OA-like chondrocytes. Results of the
present study demonstrated that, compared with the NC group,
treatment with IL-1β significantly increased the expression of
collagen X (Fig. 2A and B) and Runx2 (Fig. 2A and C) in C28/I2 cells, indicating the
induction of the hypertrophy-like phenotype of chondrocytes.
However, treatment with TMF reversed the effects of IL-1β on C28/I2
cells. In addition, BMPER, BMP4 and p-Smad1/Smad1 ratio protein
expression levels were significantly reduced following treatment
with TMF, indicating that IL-1β-induced BMPER/BMP4/Smad1 signaling
was suppressed in C28/I2 cells (Fig.
2A and D-F). Results of the
immunofluorescent assay also demonstrated that TMF reduced the
expression of collagen X and Runx2 in a concentration-dependent
manner in IL-1β-treated C28/I2 cells (Fig. 2G and H). Thus, the inhibitory activity of TMF
against chondrocyte hypertrophy may be associated with suppression
of the BMPER/BMP4/Smad1 signaling pathway in C28/I2 cells. TMF at
20 µg/ml exhibited higher activity in protecting against
IL-1β-induced chondrocyte hypertrophy than that at 5 µg/ml. To
better understand the protective effects of TMF, the 20 µg/ml
concentration was selected for use in subsequent experiments
instead of 5 µg/ml.
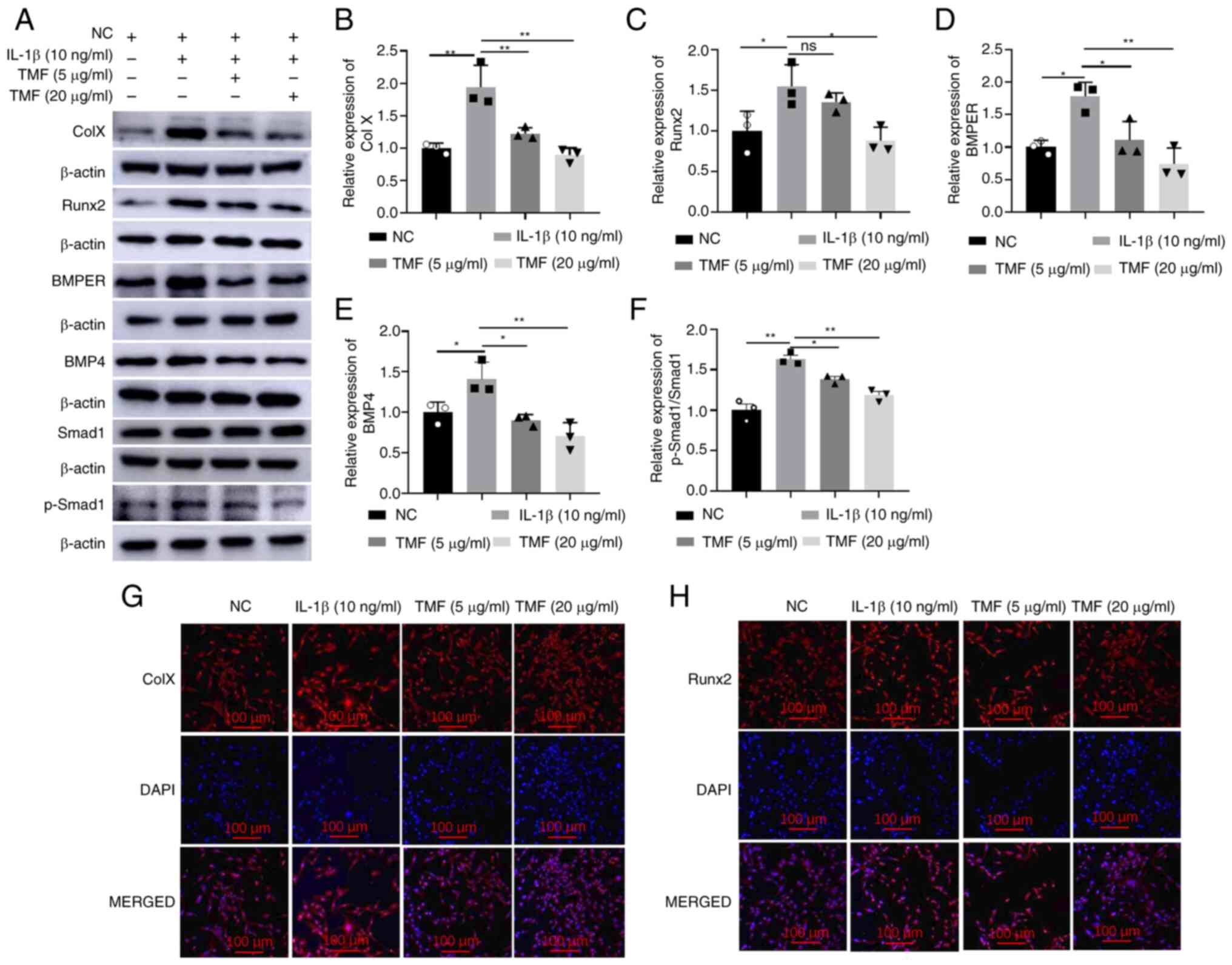 | Figure 2Effects of TMF on chondrocyte
hypertrophy in vitro. Protein expression levels of (A and B)
Col X, (A and C) Runx2, (A and D) BMPER, (A and E) BMP4 and (A and
F) p-Smad1/Smad1. Immunofluorescent intensity of (G) Col X and (H)
Runx2. *P<0.05 and **P<0.01. TMF,
5,7,3',4'-tetramethoxyflavone; BMPER, BMP-binding endothelial
regulator; BMP4, bone morphogenetic protein 4; p-, phosphorylated;
Runx2, runt-related transcription factor 2; Col X, collagen X; NC,
negative control. |
TMF inhibits chondrocyte hypertrophy
by suppressing BMPER/BMP4 signaling
Expression levels of BMPER were significantly
increased in rat OA models. However, TMF exhibited inhibitory
activity against BMPER expression in the cartilage of rat OA models
(Fig. 3A and B). Similar results are also displayed in
Fig. 2A and D. To explore whether TMF inhibited
chondrocyte hypertrophy through suppression of BMPER/BMP4
signaling, BMPER was overexpressed in C28/I2 cells. The
significantly increased expression of BMPER detected by western
blot analysis indicated successful infection (Fig. 3C and D). Results of the present study
demonstrated that BMPER overexpression may reverse the effects of
TMF on chondrocyte hypertrophy, indicated by the increased protein
expression of collagen X (Fig. 3E
and F) and Runx2 (Fig. 3E and G) in the BMPER-OE group compared with the
TMF (20 µg/ml) group. In addition, BMPER overexpression
significantly increased BMPER (Fig.
3E and H), BMP4 (Fig. 3E and I) and p-Smad1/Smad1 (Fig. 3E and J) expression levels in C28/I2 cells
compared with the TMF (20 µg/ml) group. Results of the
immunofluorescent assay also indicated that BMPER overexpression
reversed the effects of TMF on collagen X and Runx2 expression
(Fig. 3K and L). Thus, TMF inhibited chondrocyte
hypertrophy through the suppression of BMPER/BMP4 signaling in
C28/I2 cells.
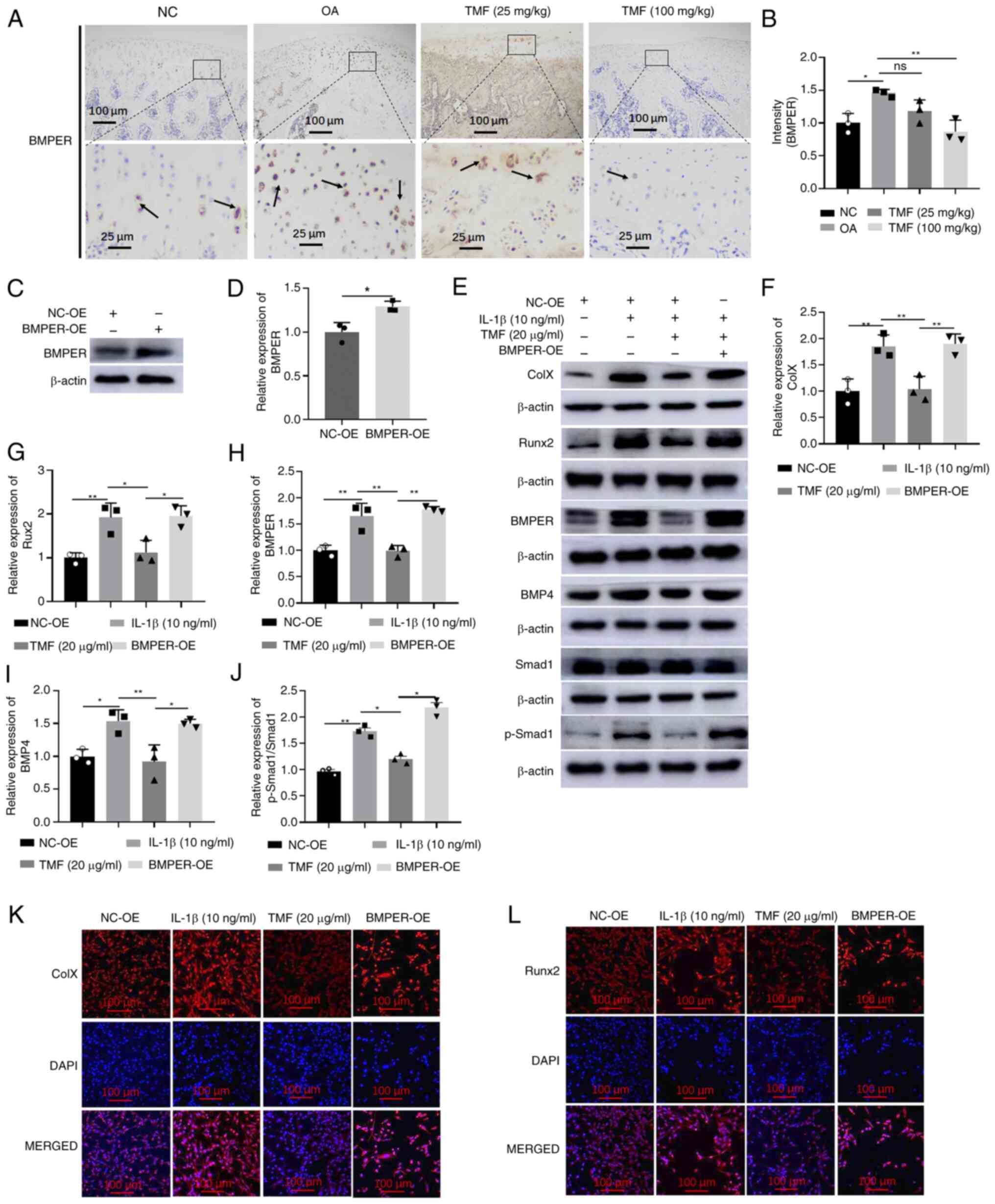 | Figure 3TMF inhibits chondrocyte hypertrophy
by suppressing BMPER/BMP4 signaling. (A and B) Immunohistochemical
analysis of BMPER in rat OA cartilage. The arrows indicate positive
staining. (C and D) The BMPER protein expression was detected by
western blot in LV-BMPER-infected C28/I2 cells. Protein expression
levels of (E and F) collagen X, (E and G) Runx2, (E and H) BMPER,
(E and I) BMP4 and (E and J) p-Smad1/Smad1. Immunofluorescent
intensity of (K) collagen X and (L) Runx2. *P<0.05
and **P<0.01. TMF, 5,7,3',4'-tetramethoxyflavone;
BMPER, BMP binding endothelial regulator; BMP4, bone morphogenetic
protein 4; OA, osteoarthritis; p-, phosphorylated; NC, negative
control; OE, overexpression; LV, lentivirus; Runx2, runt-related
transcription factor 2. |
TMF inhibits BMPER/BMP4
signaling-mediated chondrocyte hypertrophy through increasing
FOXO3a expression
FOXO3a expression levels were reduced in the
cartilage of rat OA models, and treatment with TMF increased FOXO3a
expression (Fig. 4A and B). To further investigate the mechanisms
underlying TMF-mediated inhibition of the BMPER/BMP4 signaling
pathway, FOXO3a expression was silenced following the transfection
of LV-sh-FOXO3a into C28/I2 cells. The significantly decreased
expression of FOXO3a detected by western blot analysis indicated
successful infection (Fig. 4C and
D). Results of the present study
demonstrated that collagen X (Fig.
4E and F) and Runx2 (Fig. 4E and G) expression levels were significantly
increased following FOXO3a knockdown compared with those in the TMF
(20 µg/ml) group, highlighting that FOXO3a knockdown reversed the
protective effects of TMF on chondrocyte hypertrophy in C28/I2
cells. In addition, FOXO3a knockdown also significantly increased
BMPER, BMP4 and p-Smad1/Smad1 expression levels in
LV-sh-FOXO3a-transfected C28/I2 cells compared with the TMF (20
µg/ml) group (Fig. 4E and H-K). Results of the immunofluorescent
assay also indicated that FOXO3a knockdown reversed the effects of
TMF on collagen X and Runx2 expression (Fig. 4L and M). Thus, TMF inhibited BMPER/BMP4
signaling-mediated chondrocyte hypertrophy through increasing
FOXO3a expression.
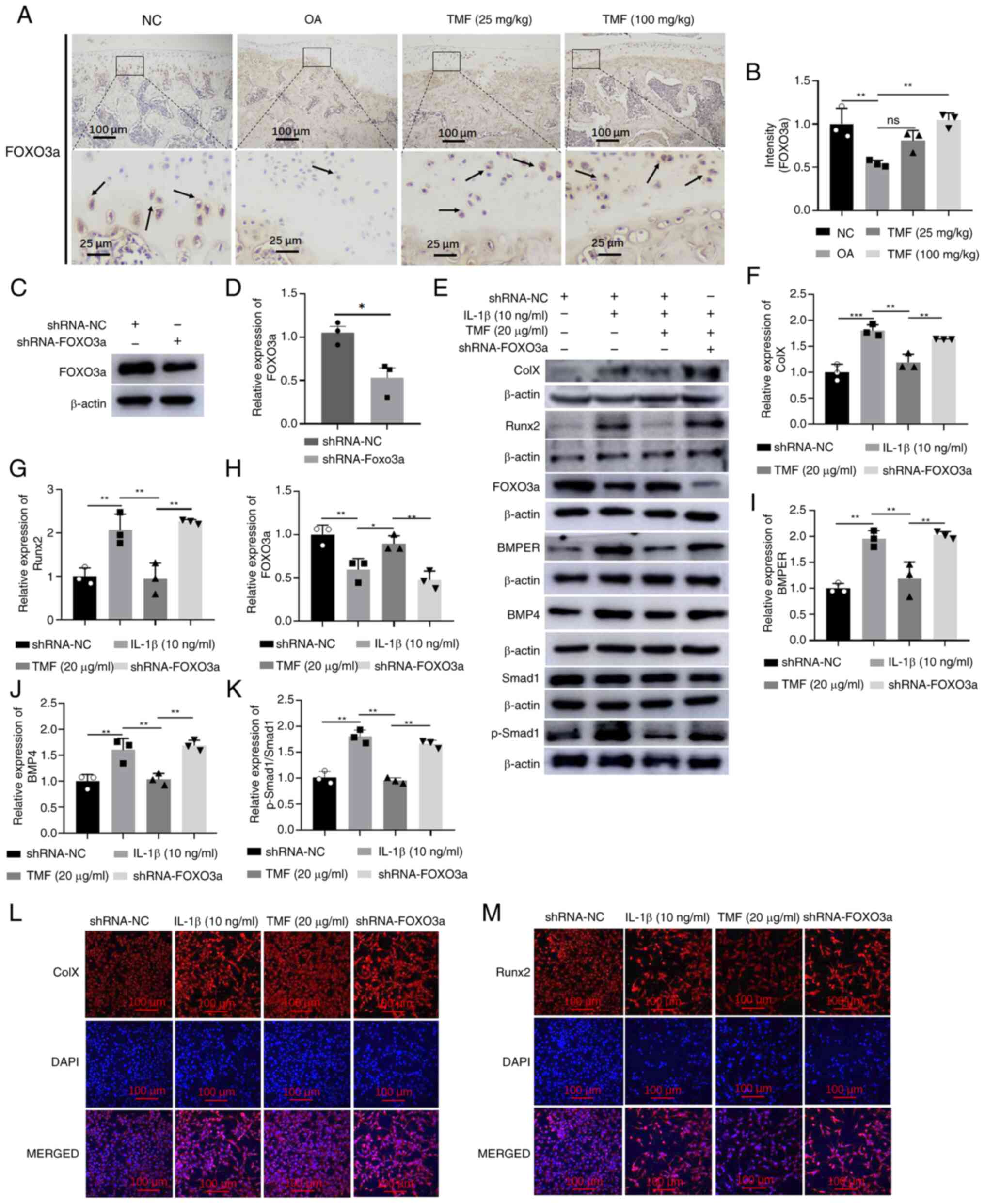 | Figure 4FOXO3a knockdown decreases the
inhibitory activity of TMF against BMPER/BMP4 signaling-mediated
chondrocyte hypertrophy. (A and B) Immunohistochemical analysis of
FOXO3a in the cartilage of rat OA models. The arrows indicate
positive staining. (C and D) The FOXO3a protein expression was
detected by western blot in shRNA-FOXO3a-infected C28/I2 cells.
Protein expression levels of (E and F) collagen X, (E and G) Runx2,
(E and H) FOXO3a, (E and I) BMPER, (E and J) BMP4 and (E and K)
p-Smad1/Smad1. Immunofluorescent intensity of (L) collagen X and
(M) Runx2. *P<0.05, **P<0.01 and
***P<0.001. FOXO3a, forkhead box O 3a; TMF,
5,7,3',4'-tetramethoxyflavone; BMPER, BMP binding endothelial
regulator; BMP4, bone morphogenetic protein 4; OA, osteoarthritis;
p-, phosphorylated; shRNA, short hairpin RNA; Runx2, runt-related
transcription factor 2. |
Discussion
Due to the aberrant terminal hypertrophic
differentiation of quiescent articular chondrocytes, chondrocyte
hypertrophy is a key feature of OA (26). The development of the hypertrophic
phenotype is associated with pathological ECM remodeling and
cartilage destruction (27). The
results of the present study revealed that MIA may severely damage
rat knee joint cartilage and promote the expression of collagen X
and Runx2, which are biomarkers of chondrocyte hypertrophy. In
vitro, the BMPER/BMP4/Smad1 signaling pathway was activated,
and treatment with TMF (20 µg/ml) inhibited chondrocyte hypertrophy
by suppressing the BMPER/BMP4/Smad1 signaling pathway. Notably,
BMPER overexpression or FOXO3a knockdown reversed the protective
effects of TMF on chondrocyte hypertrophy in IL-1β-treated C28/I2
cells.
In addition to the inflammatory responses and
degeneration in joint cartilage, hypertrophic phenotype alterations
of chondrocytes in cartilage are key hallmarks of OA (28). Hypertrophic chondrocytes secrete
catabolic enzymes, such as matrix metalloproteinase 13 (MMP-13) and
a disintegrin and metalloproteinase with thrombospondin 4/5
(ADAMTS4/5), which promote cartilage destruction and OA development
(29). Chondrocytes in the
non-calcified zones of joint cartilage do not undergo hypertrophic
phenotype alterations under normal conditions. However,
OA-associated hypertrophic phenotype alterations in joint cartilage
enhance pathological damage, such as calcification and
vascularization (30,31). Inhibition of chondrocyte
hypertrophy in joint cartilage exhibits potential as a therapeutic
strategy for OA management (31).
At present, numerous studies on the pathogenesis of OA are focused
on inflammatory responses. Thus, further investigations are
required to understand the mechanisms underlying chondrocyte
hypertrophy in OA pathogenesis.
Healthy levels of chondrocyte hypertrophy are
essential for musculoskeletal development. However, OA-associated
chondrocyte hypertrophy may be facilitated by the aberrant
expression of BMP (32),
TGFβ1(33) and Indian Hedgehog
(34) signaling pathways. Collagen
X is minimally expressed in healthy joint cartilage, and increased
collagen X expression in hypertrophic chondrocytes indicates the
initiation of endochondral bone formation (35). Runx2 expression is aberrantly
increased in OA chondrocytes, and this promotes OA pathogenesis
through increasing several target factors, such as COL10A1
(encoding collagen X), MMPs, alkaline phosphatase (ALP),
osteopontin and vascular endothelial growth factor (VEGF) (36). Runx2 is mediated by various
signaling pathways, such as BMP, TGFβ1 and Indian Hedgehog
(37,38). Runx2 plays an important role in
chondrocyte hypertrophy (38).
Results of the present study revealed that collagen X and Runx2
expression levels were significantly increased in vivo and
in vitro, highlighting the hypertrophic alterations of OA
chondrocytes.
The BMP signaling pathway is an important mediator
of chondrocyte hypertrophy and endochondral ossification.
Activation of BMP signaling may result in phosphorylation of Smad
proteins, such as Smad-1, -5 and -8, which are transcriptional
factors that mediate the expression of downstream factors (39). Exogenous BMP4 increases the
chondrocyte differentiation of bone marrow stem cells and promotes
endochondral bone formation (40).
Exogenous BMP4 promotes cartilage maturation and induces
chondrocyte hypertrophy (41).
Results of a previous study have revealed that exogenous BMP4
decreases the expression of collagen II, and increases the
expression of collagen X and ALP in juvenile idiopathic arthritis
synovial fibroblasts (42).
Results of another previous study have revealed that treatment with
the antagonist Noggin inhibits BMP4, which ultimately inhibits the
late differentiation of mesenchymal stem cells and chondrocyte
hypertrophy (43). Results of the
present study revealed that IL-1β increased the expression of the
BMP4/Smad1 signaling pathway and its downstream factors, such as
collagen X and Runx2. Collectively, these results indicated that
activation of the BMP4/Smad1 signaling pathway was associated with
hypertrophic phenotype alterations of OA chondrocytes.
BMPER, also known as the vertebrate homolog of
Drosophila cross-veinless 2, was originally identified in a
screening assay for differentially expressed proteins in embryonic
endothelial precursor cells (44).
BMPER interacts with BMP-2, -4, -6, -7, -9 and BMP receptor Ia/b
(45,46), and activates the BMP signaling
pathway in a concentration-dependent manner (47). Results of a previous study have
revealed that exogenous BMPER at low molar concentrations enhances
Smad activation (47). However, at
higher molar concentrations, BMPER may act as an inhibitor of
BMP4(47). BMPER overexpression
increases BMP-2-induced osteogenic differentiation, VEGF expression
and vascularization (48). BMPER
knockdown inhibits the osteogenic differentiation of cyclic tensile
strain-induced ossification in posterior longitudinal ligament
cells (49). In human vascular
smooth muscle cells, BMPER knockdown significantly decreases the
expression of ALP and Runx-2, both of which are regulated by
BMP2/Smad1/5/8 signaling (50).
However, results of a previous study revealed that the absence of
BMPER in the atrioventricular cushions increases BMP2/Smad1/5/8
signaling and SOX9 expression (51). In addition, in the presence of
twisted gastrulation, full-length BMPER inhibits BMP-signaling in
C2C12 cells (52). The results of
a previous study have also demonstrated that BMPER exerts positive
and negative effects on BMP activity, and these effects are
context-dependent (53). At
present, the specific association between BMPER and BMP4 remains
controversial. The results of the present study revealed that the
expression of BMPER in OA chondrocytes was increased. In addition,
BMPER overexpression activated the BMP4/Smad1 signaling pathway and
increased the expression of collagen X and Runx2. These results
suggested that activation of the BMPER/BMP4 signaling pathway
promoted chondrocyte hypertrophy in OA cartilage.
Results of a previous study demonstrated that
increased FOXO3a expression or inhibited FOXO3a degradation may
inhibit cell apoptosis (54).
DL-3-n-butylphthalide inhibits PI3K/AKT signaling and increases
FOXO3a expression, leading to the inhibition of human OA
chondrocyte apoptosis and increased ECM synthesis (55). The results of a previous study have
revealed that FOXO3a and peroxisome proliferators-activated
receptor γ coactivator 1α (PGC-1α) increase AMPK activity and
inhibit catabolic responses in chondrocytes by suppressing
oxidative stress (56). Notably,
the results of a previous study have revealed the positive role of
FOXO3a in OA chondrocytes (57).
FOXO3a interacts with BMPER and negatively modulates its activity,
leading to suppression of BMP4/Smad1/5 signaling (19). In addition, deletion of FOXO3a
results in increased BMPER/BMP4 signaling (19). The results of a previous study have
revealed that BMP4 activates FOXO3a by regulating PI3K/AKT
signaling in glioma stem cells (58). In the present study, FOXO3a
knockdown increased activation of BMPER/BMP4/Smad1 signaling and
promoted chondrocyte hypertrophy.
Moreover, the results of previous studies have
demonstrated that Sirt1 acts as a positive upstream factor of
FOXO3a (59,60). Results of our previous study
revealed that activation of Sirt1 increases the expression of
FOXO3a (22). TMF is a main
bioactive component derived from M. exotica, which exerts
multiple pharmacological activities, including anti-inflammatory,
antioxidant, analgesic, anti-diabetes and antinociception
properties (61,62). TMF may exhibit potential as an
activator of Sirt1, enhancing the expression of FOXO3a and
mediating cholesterol metabolism in OA chondrocytes (22). TMF also decreases the production of
pro-inflammatory cytokines and exhibits chondroprotective activity
by inhibiting the EP/cAMP/PKA and the β-catenin signaling pathways
in OA chondrocytes (24,63). In the present study, TMF activated
FOXO3a expression, inhibited BMPER/BMP4/Smad1 signaling and
suppressed chondrocyte hypertrophy in OA chondrocytes.
The present study has a number of limitations. In
the IHC assays, three samples (six in total) were randomly selected
for analysis. Further studies on the total samples are still
needed. It is essential to explore the dose response and
time-dependence of TMF on chondrocyte hypertrophy in OA cartilage,
and these were not determined in the present study. In vitro
study, two concentrations of TMF were studied to inhibit the
expression of ColX and Runx-2, which are the biomarkers of OA
chondrocyte hypertrophy (64).
However, the absence of more concentrations to validate the
concentration-dependent TMF inhibition of OA chondrocyte
hypertrophy was also a limitation of the present study. In
addition, the association between FOXO3a and BMPER was not
verified, and the cellular distribution of FOXO3a and BMPER was
also not detected. The effects of BMPER overexpression and FOXO3a
knockdown on OA chondrocytes were investigated; however, further
investigations into the effects of BMPER knockdown and FOXO3a
overexpression on OA chondrocytes are required. The study on BMPER
gene knockout mice will be explored in our future plans. In
addition, the effects of exogenous BMPER and BMP4 on TMF-protected
chondrocyte hypertrophy requires further validation. The nuclear
translocation of p-Smad1 and the mechanisms underlying FOXO3a
mediation of BMP2/4 were also not determined in the present study,
highlighting the requirement for further investigations. The impact
of TMF on the activity of BMP receptors remains unknown, and the
protective effects of TMF on chondrocytes derived from patients
with OA require further validation.
In conclusion, the results of the present study
revealed that collagen X and Runx2 expression levels and the
BMPER/BMP4/Smad1 signaling pathway were increased in the cartilage
of rat OA models. TMF exhibited protective activities against
chondrocyte hypertrophy by activating FOXO3 expression and
inhibiting the BMPER/BMP4/Smad1 signaling pathway. The results of
the present study also demonstrated that BMPER overexpression and
FOXO3a knockdown reversed the protective effects of TMF. Thus, TMF
inhibited chondrocyte hypertrophy by mediating the
FOXO3a/BMPER/BMP4/Smad1 signaling pathway.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by Jiangxi Provincial
Natural Science (grant no. 20212ACB206002).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
YY and LW designed and conceptualized the study. JH,
QR, LJ, SN, CL and JZ conducted the experiments and revised the
manuscript. JH and YY confirm the authenticity of all the raw data.
All authors have read and approved the final manuscript.
Ethics approval and consent to
participate
The present study (approval no. 2020365) was
approved by The Institutional Animal Care and Use Committee of
Gannan Medical University (Ganzhou, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wang CJ, Cheng JH, Chou WY, Hsu SL, Chen
JH and Huang CY: Changes of articular cartilage and subchondral
bone after extracorporeal shockwave therapy in osteoarthritis of
the knee. Int J Med Sci. 14:213–223. 2017.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Li X, Chen W, Liu D, Chen P, Wang S, Li F,
Chen Q, Lv S, Li F, Chen C, et al: Pathological progression of
osteoarthritis: A perspective on subchondral bone. Front Med: Apr
15, 2024 (Epub ahead of print).
|
|
3
|
Wojdasiewicz P, Poniatowski ŁA and
Szukiewicz D: The role of inflammatory and anti-inflammatory
cytokines in the pathogenesis of osteoarthritis. Mediators Inflamm.
2014(561459)2014.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Goldring MB and Otero M: Inflammation in
osteoarthritis. Curr Opin Rheumatol. 23:471–478. 2011.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Kong H, Han JJ, Dmitrii G and Zhang XA:
Phytochemicals against osteoarthritis by inhibiting apoptosis.
Molecules. 29(1487)2024.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Pelletier JP, Martel-Pelletier J, Rannou F
and Cooper C: Efficacy and safety of oral NSAIDs and analgesics in
the management of osteoarthritis: Evidence from real-life setting
trials and surveys. Semin Arthritis Rheum. 45 (4 Suppl):S22–S27.
2016.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Thielen NGM, Neefjes M, Vitters EL, van
Beuningen HM, Blom AB, Koenders MI, van Lent PLEM, van de Loo FAJ,
Blaney Davidson EN, van Caam APM and van der Kraan PM:
Identification of transcription factors responsible for a
transforming growth factor-β-driven hypertrophy-like phenotype in
human osteoarthritic chondrocytes. Cells. 11(1232)2022.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Park S, Bello A, Arai Y, Ahn J, Kim D, Cha
KY, Baek I, Park H and Lee SH: Functional duality of chondrocyte
hypertrophy and biomedical application trends in osteoarthritis.
Pharmaceutics. 13(1139)2021.PubMed/NCBI View Article : Google Scholar
|
|
9
|
van der Kraan PM and van den Berg WB:
Chondrocyte hypertrophy and osteoarthritis: Role in initiation and
progression of cartilage degeneration? Osteoarthritis Cartilage.
20:223–232. 2012.PubMed/NCBI View Article : Google Scholar
|
|
10
|
von der Mark K, Kirsch T, Nerlich A, Kuss
A, Weseloh G, Glückert K and Stöss H: Type X collagen synthesis in
human osteoarthritic cartilage. Indication of chondrocyte
hypertrophy. Arthritis Rheum. 35:806–811. 1992.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Ferrao Blanco MN, Bastiaansen-Jenniskens
YM, Chambers MG, Pitsillides AA, Narcisi R and van Osch GJVM:
Effect of inflammatory signaling on human articular chondrocyte
hypertrophy: Potential involvement of tissue repair macrophages.
Cartilage. 13 (2 Suppl):168S–174S. 2021.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Yoshida CA, Yamamoto H, Fujita T, Furuichi
T, Ito K, Inoue K, Yamana K, Zanma A, Takada K, Ito Y and Komori T:
Runx2 and Runx3 are essential for chondrocyte maturation, and Runx2
regulates limb growth through induction of Indian hedgehog. Genes
Dev. 18:952–963. 2004.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Chen N, Wu RWH, Lam Y, Chan WCW and Chan
D: Hypertrophic chondrocytes at the junction of musculoskeletal
structures. Bone Rep. 19(101698)2023.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Kawashima K, Ogawa H, Komura S, Ishihara
T, Yamaguchi Y, Akiyama H and Matsumoto K: Heparan sulfate
deficiency leads to hypertrophic chondrocytes by increasing bone
morphogenetic protein signaling. Osteoarthritis Cartilage.
28:1459–1470. 2020.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Saitta B, Elphingstone J, Limfat S,
Shkhyan R and Evseenko D: CaMKII inhibition in human primary and
pluripotent stem cell-derived chondrocytes modulates effects of
TGFβ and BMP through SMAD signaling. Osteoarthritis Cartilage.
27:158–171. 2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Li G, Peng H, Corsi K, Usas A, Olshanski A
and Huard J: Differential effect of BMP4 on NIH/3T3 and C2C12
cells: Implications for endochondral bone formation. J Bone Miner
Res. 20:1611–1623. 2005.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Liang C, Sun R, Xu Y, Geng W and Li J:
Effect of the abnormal expression of BMP-4 in the blood of diabetic
patients on the osteogenic differentiation potential of alveolar
BMSCs and the rescue effect of metformin: A bioinformatics-based
study. Biomed Res Int. 2020(7626215)2020.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Helbing T, Wiltgen G, Hornstein A, Brauers
EZ, Arnold L, Bauer A, Esser JS, Diehl P, Grundmann S, Fink K, et
al: Bone morphogenetic protein-modulator BMPER regulates
endothelial barrier function. Inflammation. 40:442–453.
2017.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Heinke J, Wehofsits L, Zhou Q, Zoeller C,
Baar KM, Helbing T, Laib A, Augustin H, Bode C, Patterson C and
Moser M: BMPER is an endothelial cell regulator and controls bone
morphogenetic protein-4-dependent angiogenesis. Circ Res.
103:804–812. 2008.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Jin H, Zhang L, He J, Wu M, Jia L and Guo
J: Role of FOXO3a transcription factor in the regulation of liver
oxidative injury. Antioxidants (Basel). 11(2478)2022.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Zhao X, Liu Y, Zhu G, Liang Y, Liu B, Wu
Y, Han M, Sun W, Han Y, Chen G and Jiang J: SIRT1 downregulation
mediated Manganese-induced neuronal apoptosis through activation of
FOXO3a-Bim/PUMA axis. Sci Total Environ. 646:1047–1055.
2019.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Peng F, Huang X, Shi W, Xiao Y, Jin Q, Li
L, Xu D and Wu L: 5,7,3',4'-Tetramethoxyflavone ameliorates
cholesterol dysregulation by mediating SIRT1/FOXO3a/ABCA1 signaling
in osteoarthritis chondrocytes. Future Med Chem. 13:2153–2166.
2021.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Sakamoto J, Miyahara S, Motokawa S,
Takahashi A, Sasaki R, Honda Y and Okita M: Regular walking
exercise prior to knee osteoarthritis reduces joint pain in an
animal model. PLoS One. 18(e0289765)2023.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Wu L, Liu H, Li L, Liu H, Yang K, Liu Z
and Huang H: 5,7,3',4'-Tetramethoxyflavone exhibits
chondroprotective activity by targeting β-catenin signaling in vivo
and in vitro. Biochem Biophys Res Commun. 452:682–688.
2014.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Yang J, Liu H, Li L, Liu H, Shi W and Wu
L: The chondroprotective role of TMF in PGE2-induced apoptosis
associating with endoplasmic reticulum stress. Evid Based
Complement Alternat Med. 2015(297423)2015.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Horváth E, Sólyom Á, Székely J, Nagy EE
and Popoviciu H: Inflammatory and metabolic signaling interfaces of
the hypertrophic and senescent chondrocyte phenotypes associated
with osteoarthritis. Int J Mol Sci. 24(16468)2023.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Abou-Jaoude A, Courtes M, Badique L, Elhaj
Mahmoud D, Abboud C, Mlih M, Justiniano H, Milbach M, Lambert M,
Lemle A, et al: ShcA promotes chondrocyte hypertrophic commitment
and osteoarthritis in mice through RunX2 nuclear translocation and
YAP1 inactivation. Osteoarthritis Cartilage. 30:1365–1375.
2022.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Yoon DS, Kim EJ, Cho S, Jung S, Lee KM,
Park KH, Lee JW and Kim SH: RUNX2 stabilization by long non-coding
RNAs contributes to hypertrophic changes in human chondrocytes. Int
J Biol Sci. 19:13–33. 2023.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Hu Q and Ecker M: Overview of MMP-13 as a
promising target for the treatment of osteoarthritis. Int J Mol
Sci. 22(1742)2021.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Dreier R: Hypertrophic differentiation of
chondrocytes in osteoarthritis: The developmental aspect of
degenerative joint disorders. Arthritis Res Ther.
12(216)2010.PubMed/NCBI View
Article : Google Scholar
|
|
31
|
Shigley C, Trivedi J, Meghani O, Owens BD
and Jayasuriya CT: Suppressing chondrocyte hypertrophy to build
better cartilage. Bioengineering (Basel). 10(741)2023.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Dicks AR, Maksaev GI, Harissa Z,
Savadipour A, Tang R, Steward N, Liedtke W, Nichols CG, Wu CL and
Guilak F: Skeletal dysplasia-causing TRPV4 mutations suppress the
hypertrophic differentiation of human iPSC-derived chondrocytes.
Elife. 12(e71154)2023.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Lian C, Tao T, Su P, Liao Z, Wang X, Lei
Y, Zhao P and Liu L: Targeting miR-18a sensitizes chondrocytes to
anticytokine therapy to prevent osteoarthritis progression. Cell
Death Dis. 11(947)2020.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Cong L, Jiang P, Wang H, Huang L, Wu G,
Che X, Wang C and Li P, Duan Q, Guo X and Li P: MiR-1 is a critical
regulator of chondrocyte proliferation and hypertrophy by
inhibiting Indian hedgehog pathway during postnatal endochondral
ossification in miR-1 overexpression transgenic mice. Bone.
165(116566)2022.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Hoyland JA, Thomas JT, Donn R, Marriott A,
Ayad S, Boot-Handford RP, Grant ME and Freemont AJ: Distribution of
type X collagen mRNA in normal and osteoarthritic human cartilage.
Bone Miner. 15:151–163. 1991.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Chawla S, Mainardi A, Majumder N, Dönges
L, Kumar B, Occhetta P, Martin I, Egloff C, Ghosh S, Bandyopadhyay
A and Barbero A: Chondrocyte hypertrophy in osteoarthritis:
Mechanistic studies and models for the identification of new
therapeutic strategies. Cells. 11(4034)2022.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Bae SC, Lee KS, Zhang YW and Ito Y:
Intimate relationship between TGF-beta/BMP signaling and runt
domain transcription factor, PEBP2/CBF. J Bone Joint Surg Am. 83-A
(Suppl 1):S48–S55. 2001.PubMed/NCBI
|
|
38
|
Nishimura R, Hata K, Takahata Y, Murakami
T, Nakamura E and Yagi H: Regulation of cartilage development and
diseases by transcription factors. J Bone Metab. 24:147–153.
2017.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Nordin K and LaBonne C: Sox5 is a
DNA-binding cofactor for BMP R-Smads that directs target
specificity during patterning of the early ectoderm. Dev Cell.
31:374–382. 2014.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Simonds MM, Schlefman AR, McCahan SM,
Sullivan KE, Rose CD and Brescia AMC: The culture microenvironment
of juvenile idiopathic arthritis synovial fibroblasts is favorable
for endochondral bone formation through BMP4 and repressed by
chondrocytes. Pediatr Rheumatol Online J. 19(72)2021.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Shum L, Wang X, Kane AA and Nuckolls GH:
BMP4 promotes chondrocyte proliferation and hypertrophy in the
endochondral cranial base. Int J Dev Biol. 47:423–431.
2003.PubMed/NCBI
|
|
42
|
Simonds MM, Schlefman AR, McCahan SM,
Sullivan KE, Rose CD and Brescia AC: Juvenile idiopathic arthritis
fibroblast-like synoviocytes influence chondrocytes to alter BMP
antagonist expression demonstrating an interaction between the two
prominent cell types involved in endochondral bone formation.
Pediatr Rheumatol Online J. 18(89)2020.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Karl A, Olbrich N, Pfeifer C, Berner A,
Zellner J, Kujat R, Angele P, Nerlich M and Mueller MB: Thyroid
hormone-induced hypertrophy in mesenchymal stem cell chondrogenesis
is mediated by bone morphogenetic protein-4. Tissue Eng Part A.
20:178–188. 2014.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Moser M, Binder O, Wu Y, Aitsebaomo J, Ren
R, Bode C, Bautch VL, Conlon FL and Patterson C: BMPER, a novel
endothelial cell precursor-derived protein, antagonizes bone
morphogenetic protein signaling and endothelial cell
differentiation. Mol Cell Biol. 23:5664–5679. 2003.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Yao Y, Jumabay M, Ly A, Radparvar M, Wang
AH, Abdmaulen R and Boström KI: Crossveinless 2 regulates bone
morphogenetic protein 9 in human and mouse vascular endothelium.
Blood. 119:5037–5047. 2012.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Pankratz F, Maksudova A, Goesele R, Meier
L, Proelss K, Marenne K, Thut AK, Sengle G, Correns A, Begelspacher
J, et al: BMPER improves vascular remodeling and the contractile
vascular SMC phenotype. Int J Mol Sci. 24(4950)2023.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Kelley R, Ren R, Pi X, Wu Y, Moreno I,
Willis M, Moser M, Ross M, Podkowa M, Attisano L and Patterson C: A
concentration-dependent endocytic trap and sink mechanism converts
Bmper from an activator to an inhibitor of Bmp signaling. J Cell
Biol. 184:597–609. 2009.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Xiao F, Wang C, Wang C, Gao Y, Zhang X and
Chen X: BMPER enhances bone formation by promoting the
osteogenesis-angiogenesis coupling process in mesenchymal stem
cells. Cell Physiol Biochem. 45:1927–1939. 2018.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Ji N and Yu Z: IL-6/Stat3 suppresses
osteogenic differentiation in ossification of the posterior
longitudinal ligament via miR-135b-mediated BMPER reduction. Cell
Tissue Res. 391:145–157. 2023.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Satomi-Kobayashi S, Kinugasa M, Kobayashi
R, Hatakeyama K, Kurogane Y, Ishida T, Emoto N, Asada Y, Takai Y,
Hirata K and Rikitake Y: Osteoblast-like differentiation of
cultured human coronary artery smooth muscle cells by bone
morphogenetic protein endothelial cell precursor-derived regulator
(BMPER). J Biol Chem. 287:30336–30345. 2012.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Dyer L, Lockyer P, Wu Y, Saha A, Cyr C,
Moser M, Pi X and Patterson C: BMPER promotes
epithelial-mesenchymal transition in the developing cardiac
cushions. PLoS One. 10(e0139209)2015.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Lockhart-Cairns MP, Lim KTW, Zuk A, Godwin
ARF, Cain SA, Sengle G and Baldock C: Internal cleavage and synergy
with twisted gastrulation enhance BMP inhibition by BMPER. Matrix
Biol. 77:73–86. 2019.PubMed/NCBI View Article : Google Scholar
|
|
53
|
He 何 璇 XA, Berenson A, Bernard M, Weber C,
Cook L, Visel A, Fuxman Bass JI and Fisher S: Identification of
conserved skeletal enhancers associated with craniosynostosis risk
genes. Hum Mol Genet. (ddad182)2023.PubMed/NCBI View Article : Google Scholar : (Epub ahead of
print).
|
|
54
|
Chen L, Li S, Zhu J, You A, Huang X, Yi X
and Xue M: Mangiferin prevents myocardial infarction-induced
apoptosis and heart failure in mice by activating the Sirt1/FoxO3a
pathway. J Cell Mol Med. 25:2944–2955. 2021.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Zhang Y, Dai J, Yan L, Lin Q, Miao H, Wang
X, Wang J and Sun Y: DL-3-N-butylphthalide promotes cartilage
extracellular matrix synthesis and inhibits osteoarthritis
development by regulating FoxO3a. Oxid Med Cell Longev.
2022(9468040)2022.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Zhao X, Petursson F, Viollet B, Lotz M,
Terkeltaub R and Liu-Bryan R: Peroxisome proliferator-activated
receptor γ coactivator 1α and FoxO3A mediate chondroprotection by
AMP-activated protein kinase. Arthritis Rheumatol. 66:3073–3082.
2014.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Jiang A, Xu P, Yang Z, Zhao Z, Tan Q, Li
W, Song C, Dai H and Leng H: Increased Sparc release from
subchondral osteoblasts promotes articular chondrocyte degeneration
under estrogen withdrawal. Osteoarthritis Cartilage. 31:26–38.
2023.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Ciechomska IA, Gielniewski B, Wojtas B,
Kaminska B and Mieczkowski J: EGFR/FOXO3a/BIM signaling pathway
determines chemosensitivity of BMP4-differentiated glioma stem
cells to temozolomide. Exp Mol Med. 52:1326–1340. 2020.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Wan Q, Tang L, Jin K, Chen X, Li Y and Xu
X: Quercetin and tanshinone prevent mitochondria from oxidation and
autophagy to inhibit KGN cell apoptosis through the
SIRT1/SIRT3-FOXO3a axis. Cell Mol Biol (Noisy-le-grand).
70:257–263. 2024.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Qiu CW, Chen B, Zhu HF, Liang YL and Mao
LS: Gastrodin alleviates cisplatin nephrotoxicity by inhibiting
ferroptosis via the SIRT1/FOXO3A/GPX4 signaling pathway. J
Ethnopharmacol. 319(117282)2024.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Wu L, Li P, Wang X, Zhuang Z, Farzaneh F
and Xu R: Evaluation of anti-inflammatory and antinociceptive
activities of Murraya exotica. Pharm Biol. 48:1344–1353.
2010.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Rehman R, Anila Muzaffar R, Arshad F,
Hussain R and Altaf AA: Diversity in phytochemical composition and
medicinal value of Murraya paniculata. Chem Biodivers.
20(e202200396)2023.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Wu L, Liu H, Zhang R, Li L, Li J, Hu H and
Huang H: Chondroprotective activity of Murraya exotica
through inhibiting β-catenin signaling pathway. Evid Based
Complement Alternat Med. 2013(752150)2013.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Liu W, Feng M and Xu P: From regeneration
to osteoarthritis in the knee joint: The role shift of
cartilage-derived progenitor cells. Front Cell Dev Biol.
10(1010818)2022.PubMed/NCBI View Article : Google Scholar
|