Introduction
Phenytoin (PHT, an antiepileptic drug), cyclosporin
A (an immunosuppressant) and nifedipine (a calcium channel blocker)
cause gingival overgrowth as side effects (1-3).
Among those drugs, a high probability of gingival overgrowth caused
by PHT is known (approximately 50%) (4). A characteristic of gingival
overgrowth in clinical conditions is an increase in the size of the
gingiva (5). Overgrowth of the
gingiva not only disrupts normal mastication but also results in
unusual facial features that can cause mental distress (3,6-8).
Basic periodontal treatments and surgery are usually performed to
improve gingival overgrowth (9),
however, an effective medication has not been identified at the
present time.
Based on histological observations, PHT-induced
gingival overgrowth has characteristic increases in the
proliferation of fibroblasts and accumulated amounts of collagen in
the gingiva (3,10,11).
Gingival fibroblasts are the primary cell type in gingival
connective tissue and their role is the maintenance and repair of
that tissue (12). The pathogenic
mechanisms responsible for PHT-associated gingival overgrowth have
been determined using an in vitro model and the effects of
PHT on gingival fibroblasts in tissue culture have been
investigated (13-17).
Fibroblast proliferation is observed in periodontal tissues with
PHT-induced gingival overgrowth (18). Furthermore, the interaction of
drugs with inflammation causes the increased growth and reduced
apoptosis of gingival fibroblasts, and consequently the overgrowth
of gingiva proceeds (18,19).
Licorice has long been used as a medicinal herb and
as a sweetener to give sweetness to food products (20,21).
It contains many phytochemicals including more than 300 flavonoids
and 20 triterpenoids (22), and it
also inhibits mild inflammation and heals ulcers. In addition,
licorice inhibits cell proliferation through blocking the cell
cycle in mammalian cells (23),
and it also induces apoptosis (24). 18-alpha-Glycyrrhetinic acid
(18α-GA) is a bioactive compound extracted from licorice that
exhibits many biological and pharmacological effects such as
anti-inflammatory effects (25).
18α-GA is also apoptotic promoter in epithelial cell rests of
Malassez (24). Also, 18α-GA
induces apoptosis in leukemic HL60 cells (26) and in ovarian cancer A2780 cells
(27). These findings suggest that
18α-GA could be used to treat PHT-influenced gingival overgrowth
since it may induce the apoptosis of gingival fibroblasts.
In this research, we investigated the effects of
18α-GA on apoptosis and on apoptotic regulators in gingival
fibroblasts exposed to PHT, to evaluate the therapeutic potential of
18α-GA. The results show that 18α-GA regulates caspase activity in
the death receptor pathway in gingival fibroblasts, which results
in the induction of apoptosis.
Materials and methods
Cell culture
The methods used in this study are based on
previously published reports (3,17,20,28,29).
PHT and 18α-GA were purchased from Sigma-Aldrich, Japan K.K.
(Tokyo, Japan). Four primary cultures of fibroblasts derived from
the gingiva of healthy donors were obtained from ScienCell™
Research Laboratories (cat. no. 2620, https://sciencellonline.com/human-gingival-fibroblasts/,
San Diego, CA, USA). Those cells had been cryopreserved at passage
one and delivered frozen. Cells were cultured in an atmosphere of
5% CO2/95% air maintained at 37˚C in Dulbecco's modified
Eagle medium (High Glucose) with L-Glutamine and Phenol Red (D-MEM,
FUJIFILM Wako Pure Chemical Corporation, Osaka, Japan) supplemented
with 10% foetal bovine serum, 50 units/ml penicillin and 50 µg/ml
streptomycin (Gibco, Thermo Fisher Scientific, Inc., Waltham, MA,
USA) until they reached semi-confluence. Cells were routinely
passaged using 0.05 w/v% Trypsin-0.53 mmol/l EDTA·4Na Solution with
Phenol Red (FUJIFILM Wako Pure Chemical Corporation). Cells were
used between passages 6 and 9 for subsequent experiments (Fig. 1). The concentrations of PHT and
18α-GA used in this study were decided according to the results of
previous studies as follows: 0.25 µM PHT significantly inhibited
the G1 cell cycle arrest and increased the cell
proliferation of gingival fibroblasts compared with the untreated
control (17,29); 10 µM 18α-GA significantly decreased
the proliferation of gingival fibroblasts compared with 0, 0.1, and
1 µM 18α-GA (20).
Apoptosis assay
Apoptosis assays were performed using an
APOPercentage™ Apoptosis Assay Kit (BiocolourLtd., Northern
Ireland, UK). After semiconfluent cells were treated with 0.25 µM
PHT with or without 10 µM 18α-GA in serum-free D-MEM for 24, 48 and
72 h, the apoptotic cells were labelled with APOPercentage Dye in
fresh D-MEM at 37˚C in 5% CO2 for 1 h. The D-MEM
containing the dye was removed, after which the APOPercentage Dye
release reagent was added into the cell culture plates and the
plates were gently shaken for 10 min. The absorbance of the
released dye at 550 nm was then determined. The methods used in
this study are based on previously published reports (3,17,20).
Propidium iodide staining and flow
cytometric analysis
The propidium iodide staining and flow cytometric
analysis were performed using a CycleTEST™ plus DNA Reagent Kit
(Becton Dickinson and Company, Franklin Lakes, NJ, USA; BD). After
semiconfluent cells were treated with 0.25 µM PHT with or without
10 µM 18α-GA in serum-free D-MEM for 48 h, cells were harvested by
trypsinization, washed three times with Buffer Solution, and then
treated with Solution A (trypsin buffer), Solution B (trypsin
inhibitor and RNase buffer) and Solution C (PI stain solution) in
accordance with the manufacturer's instructions. A BD FACSCalibur™
Flow Cytometer (BD Biosciences) acquired 20,000 events for each
sample, and the percentage of cells in the Sub-G1
(apoptotic), G0/G1, S and G2/M
phases of the cell cycle were determined using BD CellQuest Pro
Software (version 3.1, BD Biosciences). The methods used in this
study are based on previously published reports (3,17,20).
RNA isolation and reverse
transcription-quantitative PCR (RT-qPCR)
After semiconfluent cells were treated with 0.25 µM
PHT with or without 10 µM 18α-GA in serum-free D-MEM for 12 h,
total RNA was immediately extracted from the cells using a RNeasy
Mini Kit (QIAGEN, Tokyo, Japan). A standard spectrophotometric
method was used to assess the concentration and purity of each
extracted total RNA. One µg of each total RNA was then
reverse-transcribed using a PrimeScript™ RT reagent Kit (TAKARA BIO
INC., Shiga, Japan). The cDNAs were analyzed by qPCR in an Eco™
Real-Time PCR System (Illumina, Inc., San Diego, CA, USA) using a
KAPA SYBR® FAST qPCR Master Mix Kit (KAPA BIOSYSTEMS
Inc., Wilmington, MA, USA). The following thermocycling conditions
were used for qPCR: Enzyme activation at 95˚C for 30 sec, followed
by 45 cycles of denaturation at 95˚C for 5 sec and annealing and
extension at 60˚C for 20 sec. A Perfect Real Time Support System
(TAKARA BIO INC.) was used to synthesize the following PCR primers:
B-cell CLL/lymphoma 2 (BCL2); baculoviral IAP repeat containing 3
(BIRC3); CASP8 and FADD-like apoptosis regulator (CFLAR); CASP2 and
RIPK1 domain containing adaptor with death domain (CRADD); Fas
(TNFRSF6)-associated via death domain (FADD); receptor
(TNFRSF)-interacting serine-threonine kinase 1 (RIPK1); tumor
necrosis factor receptor superfamily; member 1A (TNFRSF1A); TNF
receptor-associated factor 2 (TRAF2); and
glyceraldehyde-3-phosphate dehydrogenase (GAPDH). These primer
sequences were used according to the method of Takeuchi et
al (3). The PCR primers for
Caspases-2, -3, -8, -9 and -10 were synthesized by Custom DNA
Oligos (Merck KGaA, Darmstadt, Germany) and Primer-BLAST (National
Library of Medicine, Bethesda, MD, USA). Primer sequences used are
listed in Table I. Relative
quantification was calculated using the 2-∆∆Cq method
(30). After normalization to
GAPDH, RNA ratios in treated vs. control cultures were determined.
The methods used in this study are based on previously published
reports (3).
 | Table IPrimers used for reverse
transcription-quantitative PCR. |
Table I
Primers used for reverse
transcription-quantitative PCR.
| Gene symbol | Sequence
(5'-3') | Product size,
bp | GenBank accession
no. |
|---|
| BCL2 | F:
TGGACAACCATGACCTTGGAC | 170 | NM_000633.2 |
| | R:
GTGCTCAGCTTGGTATGCAGAA | | |
| BIRC3 | F:
GGACAGGAGTTCATCCGTCAAG | 176 | NM_001165.3 |
| | R:
GCAGCATTAATCACAGGAGTATTCA | | |
| CFLAR | F:
TGCAGTTCAGTCAAACATTGGAAG | 177 | NM_003879.5 |
| | R:
GGGTTCCAGATGGTCCAGAAATA | | |
| CRADD | F:
CCTAACAGTCAGGATTCCGGTTG | 107 | NM_003805.3 |
| | R:
CGAAGTGAGCGGAGTACTTGTTTG | | |
| FADD | F:
ATGCGCGGGTCCCTTAGTT | 84 | NM_003824.3 |
| | R:
CACTCCGGTGCCTGATTCAC | | |
| RIPK1 | F:
TTTCAAAGCCCACCTGAAACC | 170 | NM_003804.3 |
| | R:
GCCAGATTGACCATCACCACA | | |
| TNFRSF1A | F:
ACAGAACACCGTGTGCACCT | 103 | NM_001065.3 |
| | R:
GCACAACTTCGTGCACTCC | | |
| TRAF2 | F:
GATGGAGGCATCCACCTACGA | 163 | NM_021138.3 |
| | R:
GCCGTTCAGGTAGATACGCAGAC | | |
| CASP2 | F:
CAGCATGTACTCCCACCGTT | 197 | NM_032982.4 |
| | R:
GCCAGCTGGAAGTGTGTTTG | | |
| CASP3 | F:
CCTTGAAATCCCAGGCCGT | 168 | NM_001354777.2 |
| | R:
TCCAGAGTCCATTGATTCGCT | | |
| CASP8 | F:
CTTTCTGGGCACGTGAGGTT | 182 | NM_001080124.2 |
| | R:
CAGGCTCAGGAACTTGAGGG | | |
| CASP9 | F:
AGGCCCCATATGATCGAGGA | 193 | NM_001229.5 |
| | R:
TCGACAACTTTGCTGCTTGC | | |
| CASP10 | F:
TCTTGGAAGCCTTACCGCAG | 78 | NM_032977.4 |
| | R:
GTGCACCATTTGTGGCTCTG | | |
| GAPDH | F:
GCACCGTCAAGGCTGAGAAC | 138 | NM_002046.5 |
| | R:
TGGTGAAGACGCCAGTGGA | | |
Western blot analysis
After semiconfluent cells were treated with 0.25 µM
PHT with or without 10 µM 18α-GA in serum-free D-MEM for 24 h, the
cells were washed with 37˚C PBS and lysed using β-ME Sample
Treatment for Tris SDS (COSMO BIO Co., LTD, Tokyo, Japan). Protein
concentrations were determined using a TaKaRa Bradford Protein
Assay Kit (TAKARA BIO INC.). Equal quantities of protein extracts
(10 µg/lane) were separated via SDS-PAGE with Running Buffer
Solution for SDS-PAGE (NACALAI TESQUE, INC., Kyoto, Japan) after
which the proteins were transferred to PVDF membranes. The
membranes were blocked for 30 min at room temperature with Bullet
Blocking One for Western Blotting (NACALAI TESQUE), after which
they were probed at room temperature for 1 h with primary
antibodies against Caspase-2 (1:500), Caspase-3 (1:1,000),
Caspase-9 (1:1,000) and β-Actin (1:1,000). The membranes were
washed three times with Tris Buffered Saline with 0.05%-Detergent
(NACALAI TESQUE) for 5 min at room temperature, and were then
incubated with the secondary antibody (1:10,000) at room
temperature for 45 min. The primary and secondary antibodies were
diluted using Can Get Signal® Immunoreaction Enhancer
Solution (TOYOBO CO., LTD., Osaka, Japan). After washing, the blots
were detected using Chemi-Lumi One Super (NACALAI TESQUE) and
ChemiDoc™ MP Imaging System (Bio-Rad Laboratories, Inc., Hercules,
CA, USA). The densities of western blot bands were measured using
ImageJ (1.53t; Java 1.8.0_345 [64-bit]). Primary antibodies against
Caspase-3 (cat. no. #9662), Caspase-9 (cat. no. #9502) and β-Actin
(cat. no. #4967), as well as anti-rabbit HRP-conjugated IgG were
purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA).
Rabbit monoclonal anti-Caspase-2 antibody (cat. no. ab32021) was
purchased from Abcam plc. (Cambridge, UK). The secondary antibody
(anti-rabbit IgG, HRP-linked antibody; cat. no. #7074) was
purchased from Cell Signaling Technology. The methods used in this
study are based on previously published reports (3).
Detection of caspase activity
After semiconfluent cells were treated with 0.25 µM
PHT with or without 10 µM 18α-GA in serum-free D-MEM for 24 h,
Caspase-2, -3 and -9 Colorimetric Assay Kits (Medical &
Biological Laboratories Co., Ltd., Nagoya, Japan) and a
spectrophotometer at 405 nm were used according to the
manufacturer's protocols to assess caspase activities. Caspase-2,
caspase-3 and caspase-9 were labelled using the synthetic peptide
substrates VDVAD-p-nitroanilide (pNA),
DEVD-pNA and LEHD-pNA respectively. The methods used
in this study are based on previously published reports (3,20,28).
Statistical analysis
All data are reported as mean ± standard error of
the mean (SEM). Statistical analysis was carried out using Welch's
t-test. P values <0.05 were considered to indicate a
statistically significant difference.
Results
Relative number of apoptotic cells
after treatment of gingival fibroblasts with 18α-GA
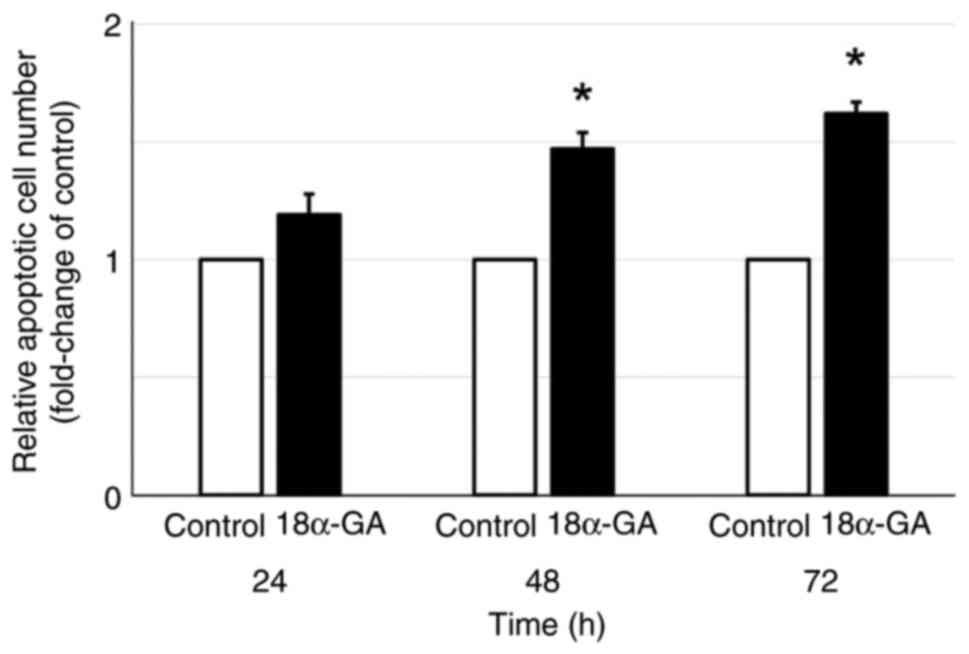
Apoptosis was assessed in gingival fibroblasts after
treatment with or without 18α-GA. As shown in Fig. 2, gingival fibroblasts treated with
18α-GA showed a time-dependent increase in the relative number of
apoptotic cells compared to the untreated control with significant
increases at 48 h (1.5-fold) and at 72 h (1.6-fold).
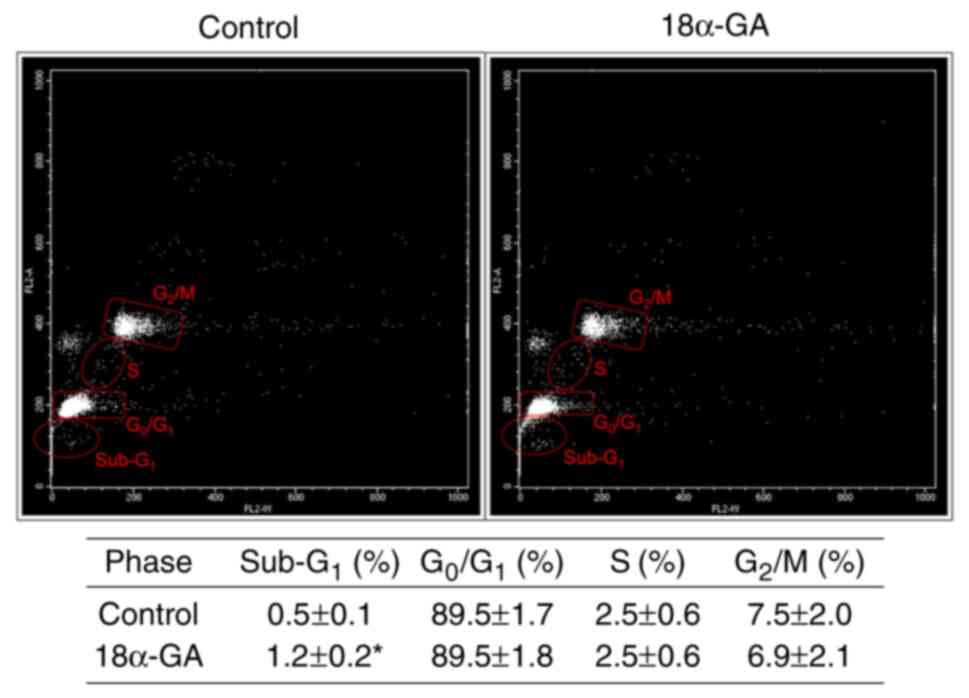
Apoptotic population and cell cycle
dynamics of gingival fibroblasts treated with 18α-GA
We analyzed the apoptotic cell population
(sub-G1) and the distribution of cell cycle phases
(G0/G1, S, and G2/M) in gingival
fibroblasts treated with or without 18α-GA. As shown in Fig. 3, treatment with 18α-GA
significantly increased the number of apoptotic cells, however it
did not change the distribution of cells in the
G0/G1, S and G2/M phase.
mRNA expression levels in gingival
fibroblasts treated with 18α-GA
We analyzed the effects of 18α-GA treatment of
gingival fibroblasts on their mRNA expression levels of apoptotic
factors (BCL2, BIRC3, CFLAR, CRADD, FADD, RIPK1, TNFRSF1A, TRAF2,
CASP2, CASP3, CASP8, CASP9 and CASP10) using qPCR. As shown in
Fig. 4, the treatment of gingival
fibroblasts with 18α-GA significantly reduced the BCL2 (0.5-fold)
mRNA expression level and significantly increased CRADD (1.7-fold),
FADD (7.1-fold), RIPK1 (10.3-fold), TNFRSF1A (7.8-fold) and TRAF2
(13.0-fold) mRNA expression levels. Treatment with 18α-GA also
increased the BIRC3 (1.7-fold) mRNA expression level and decreased
the CFLAR (0.8-fold) mRNA expression level but not significantly.
Treatment of gingival fibroblasts with 18α-GA also significantly
increased CASP2 (2.2-fold), CASP3 (2.6-fold) and CASP9 (1.6-fold)
mRNA expression levels and increased CASP8 (2.9-fold) and CASP10
(2.7-fold) mRNA expression levels but not significantly.
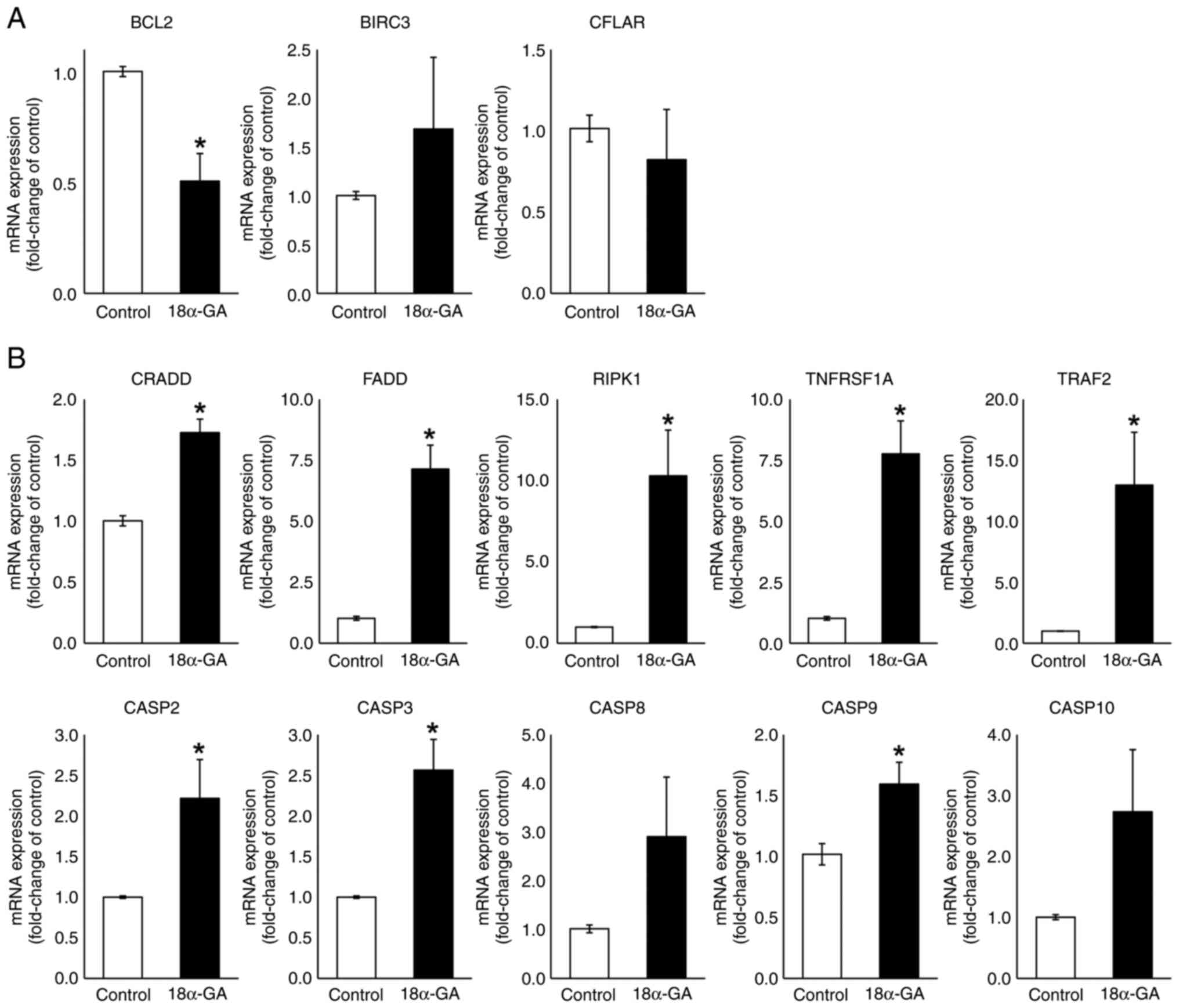 | Figure 4mRNA expression levels of apoptotic
regulators in gingival fibroblasts treated with PHT in the presence
or absence of 18α-GA. Semiconfluent cells were incubated in
serum-free D-MEM containing PHT (0.25 µM) with or without (control)
18α-GA (10 µM) for 12 h, after which reverse
transcription-quantitative PCR analysis was performed. Relative
quantification was performed using the 2-∆∆Cq method.
After normalization to GAPDH, RNA ratios in treated vs. control
cultures were determined. Data are presented as the mean ± SEM.
*P<0.05 compared with the control using Welch's
t-test (n=4). (A) Anti-apoptotic genes. (B) Pro-apoptotic genes.
18α-GA, 18-α-glycyrrhetinic acid; BIRC3, baculoviral IAP repeat
containing 3; CASP, caspase; CFLAR, CASP8 and FADD-like apoptosis
regulator; CRADD, CASP2 and RIPK1 domain containing adaptor with
death domain; FADD, Fas (TNFRSF6)-associated via death domain; PHT,
phenytoin; RIPK1, receptor (TNFRSF)-interacting serine-threonine
kinase 1; TNFRSF1A, tumor necrosis factor receptor superfamily;
member 1A; TRAF2, TNF receptor-associated factor 2. |
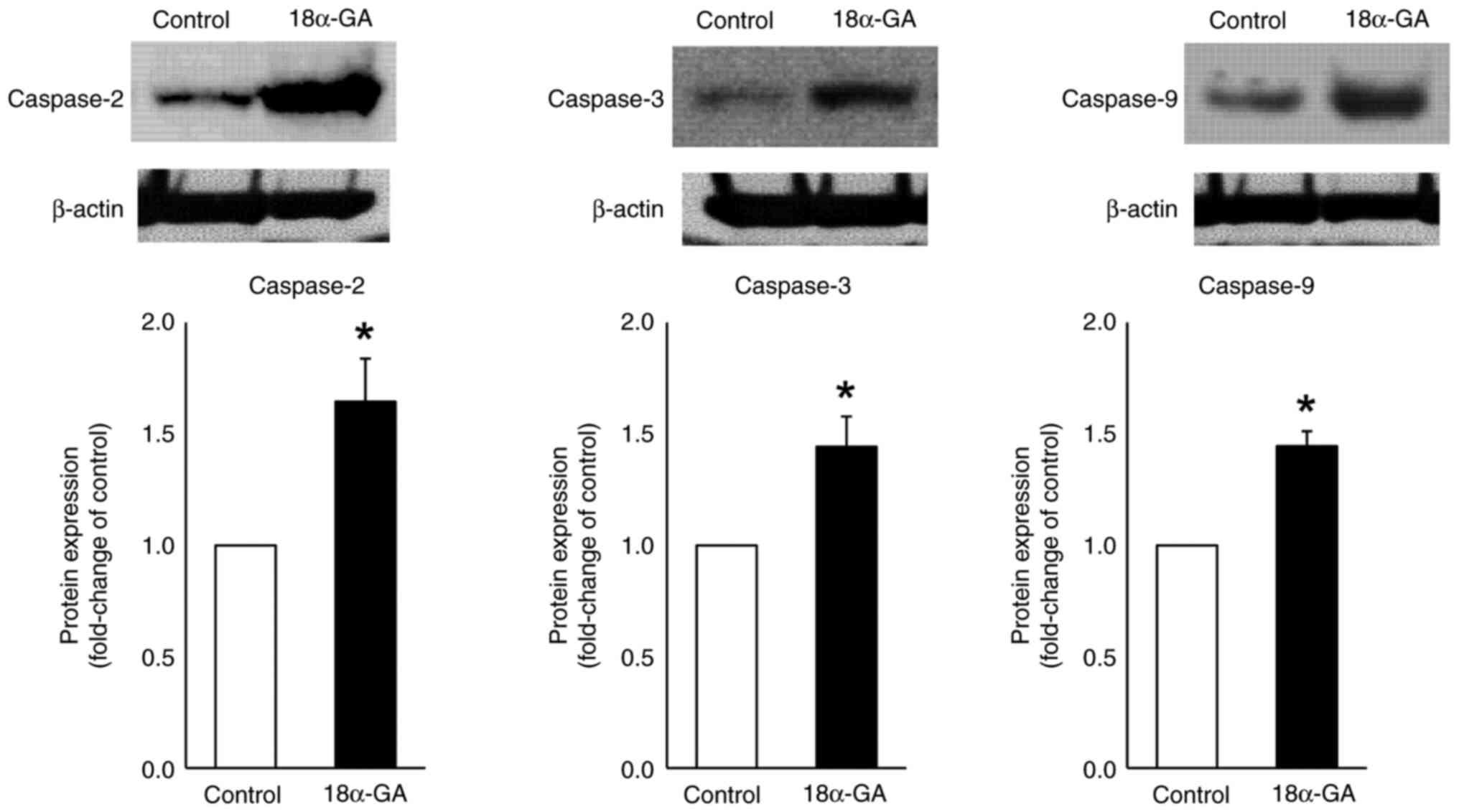
Protein expression in gingival
fibroblasts treated with 18α-GA
We analyzed the effects of 18α-GA treatment of
gingival fibroblasts on the protein expression of caspases- 2, 3
and 9 using western blot analysis. As shown in Fig. 5, treatment of gingival fibroblasts
with 18α-GA significantly increased the protein expression levels
of caspase-2 (2.3-fold), caspase-3 (2.7-fold) and caspase-9
(2.8-fold) compared to the levels observed in control cells.
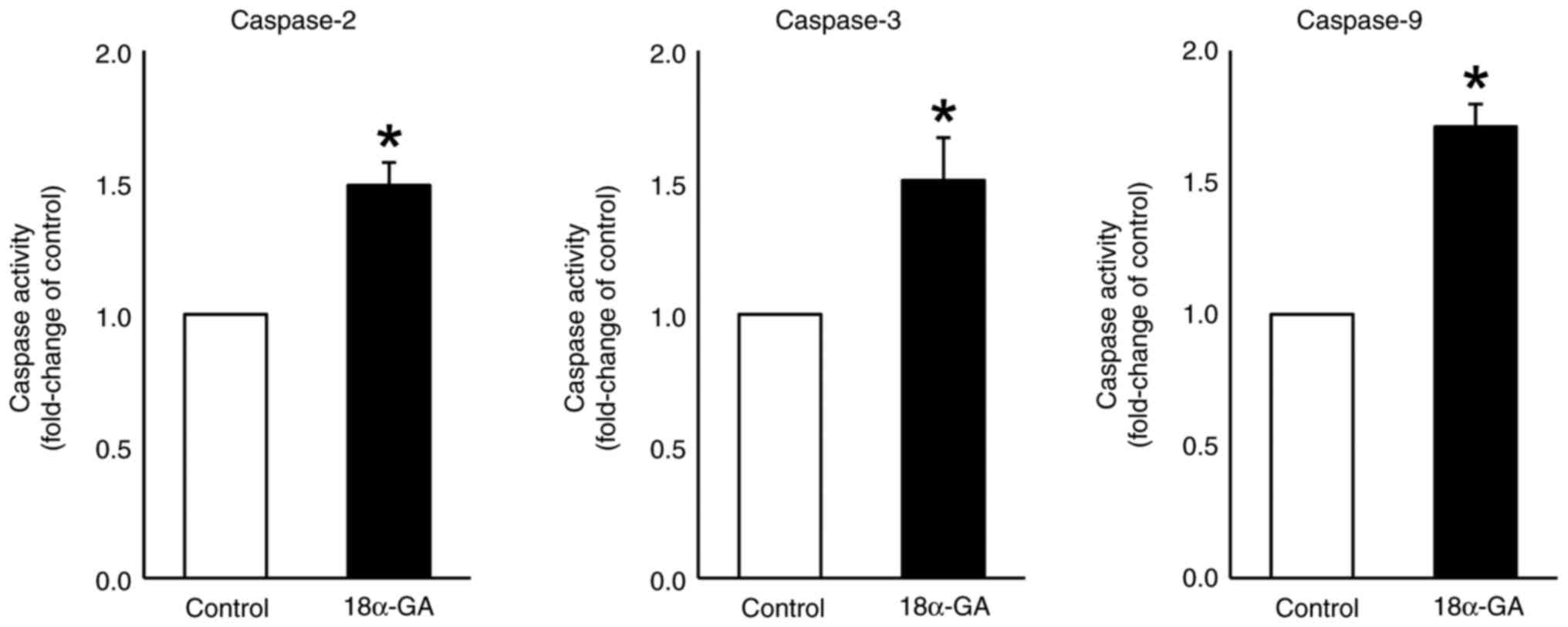
Caspase activity in gingival
fibroblasts treated with 18α-GA
We assessed the effects of treating gingival
fibroblasts with 18α-GA on the activities of caspases- 2, 3 and 9.
As shown in Fig. 6, treatment with
18α-GA significantly up-regulated the activities of caspase-2
(1.5-fold), caspase-3 (1.5-fold) and caspase-9 (1.7-fold) compared
to the levels observed in control cells.
Discussion
In the present study, we determined whether
treatment with 18α-GA affects apoptosis of gingival fibroblasts
exposed to PHT. The purpose of this study was to establish a basis
for the therapeutic application of 18α-GA to treat PHT-induced
gingival overgrowth. We found that 18α-GA induced the apoptosis of
gingival fibroblasts by activating the caspase cascade in the death
receptor pathway.
Gingival overgrowth is caused by the increased
proliferation and the decreased apoptosis of gingival fibroblasts
that are exposed to drugs, such as PHT (3). The pathogenesis of this disease is
also associated with the gingiva including inflammation (31). 18α-GA promotes the apoptosis of
multiple types of cells, including porcine epithelial cell rests of
Malassez cells (24), leukemic
HL60 cells (26), ovarian cancer
A2780 cells (27) and murine
microglial BV2 cells (32). In
this study, we found that 18α-GA induced the apoptosis of gingival
fibroblasts. Conversely, transformed cells with severe DNA damage
are cleared by cellular apoptosis (33). Several studies have proposed that
the pathogenesis of gingival overgrowth involves the inhibition of
apoptosis (3,34,35).
We have also demonstrated that gingival overgrowth is attributed to
reduced apoptosis in gingival fibroblasts derived from patients with
gingival overgrowth (28).
Apoptosis is programmed cell death characterized by
an elaborate sequence of morphological events including nuclear
condensation (pyknosis) and fragmentation (karyorrhexis), along
with blebbing of the plasma membrane, both of which contribute to
the formation of apoptotic bodies (36,37).
The two main pathways of apoptotic cell death are the intrinsic and
extrinsic pathways. The intrinsic pathway is marked by
mitochondrial outer membrane permeabilization, which releases
cytochrome c from the mitochondrial intermembrane space (38). The extrinsic pathway is activated
in response to specific death receptors which are Fas, TNF receptor
1 (TNFR1) or TNF-related apoptosis-inducing ligand receptor. Both
pathways trigger downstream effector caspases such as caspase-3
that lead to apoptotic cell death (36,39).
The expression of caspase-3 protein is attenuated in tissues of
gingiva derived from patients treated with cyclosporin A and/or
nifedipine, and from those with PHT-induced gingival overgrowth
(3,18,35).
PHT treatment of gingival fibroblasts from healthy donors also
reduced the expression and activity of caspase-3(3). In this study, we show that treatment
with 18α-GA enhances the mRNA and protein expression levels of
caspase-3 and increases the activation of caspase-3.
To elucidate the pro-apoptotic mechanism of 18α-GA
in the death receptor pathway of gingival fibroblasts exposed to
PHT, we examined the mRNA expression levels of apoptotic genes,
including inducers (CRADD, FADD, RIPK1, TNFRSF1A and TRAF2), an
effector (CASP3), initiators (CASP2, CASP8, CASP9 and CASP10),
inhibitors (BCL2, BIRC3 and CFLAR) and the protein expression
levels and activities of caspases (2, 3 and 9) in those cells. We
found that treatment with 18α-GA reduced the mRNA expression level
of BCL2, enhanced the mRNA expression levels of CASP2, CASP3,
CASP9, CRADD, FADD, RIPK1, TNFRSF1A and TRAF2, and increased the
protein expression levels and activities of caspase-2, caspase-3
and caspase-9 in gingival fibroblasts treated with PHT.
As mentioned above, the major death receptors are
Fas and TNFR1. After those receptors are activated by extracellular
ligands such as Fas ligand (Fas-L) and TNF ligands, initiator
caspases (2, 8, 9 and 10) activate executioner caspases (3, 6 and
7) in cooperation with various apoptotic mediators, and
consequently, apoptosis is induced (40). When Fas is bound by its ligand
Fas-L, the resulting conformational change in Fas DD (Fas death
domain) allows it to bind to FADD DD. The binding of Fas DD to FADD
DD causes the exposed DED (death effector domain) on FADD (40). The binding of TNF-α (one of the TNF
ligands) to TNF receptor-1, which is encoded by TNFRSF1A, results
in the recruitment of the adapter proteins FADD and TRADD (3,41-43).
FADD then activates caspase-8 and caspase-10. Caspase-8 can cleave
and activate the apoptosis executioner caspase-3(40). The action of c-FLIP, which is
encoded by CFLAR, prevents FADD recruitment (44,45).
On the other hand, when the adapter protein TRAF2 binds to the TNF
receptor-1/TRADD complex, NF-κB is activated and apoptosis is
inhibited (46). c-IAP2, which is
encoded by BIRC3, inhibits the activation of NF-κB (47,48).
A complex consisting of CRADD-activated caspase-2, RIPK1 and TNF
receptor-1 activates caspase-9 and caspase-3, which can induce
apoptosis (49). BCL2 prevents
apoptosis by depressing the activation of caspase-9(50).
Our results show that 18α-GA has the following
effects on gingival fibroblasts exposed to PHT: a decrease in the
BCL2 mRNA expression level; increases in CRADD, FADD, RIPK1,
TNFRSF1A and TRAF2 mRNA expression levels; increases in CASP2,
CASP3 and CASP9 mRNA expression levels and increases in caspase-2,
caspase-3 and caspase-9 protein expression levels and activities.
Based on the above results, the apoptotic mechanism of 18α-GA in
gingival fibroblasts treated with PHT may be as follows: 18α-GA
modulates the TNF pathway by upregulating TNFRSF1A, TRAF2, RIPK1
and CRADD, which induce apoptosis via the activation of caspase-2,
caspase-9 and caspase-3; 18α-GA affects the Fas pathway by
upregulating FADD and induces apoptosis by downregulating BCL2
(shown schematically in Fig. 7).
The release of cytochrome c to the cytoplasm from mitochondria also
activates caspase-9(3). Thus,
18α-GA may affect apoptosis via the mitochondrial pathway in
gingival fibroblasts.
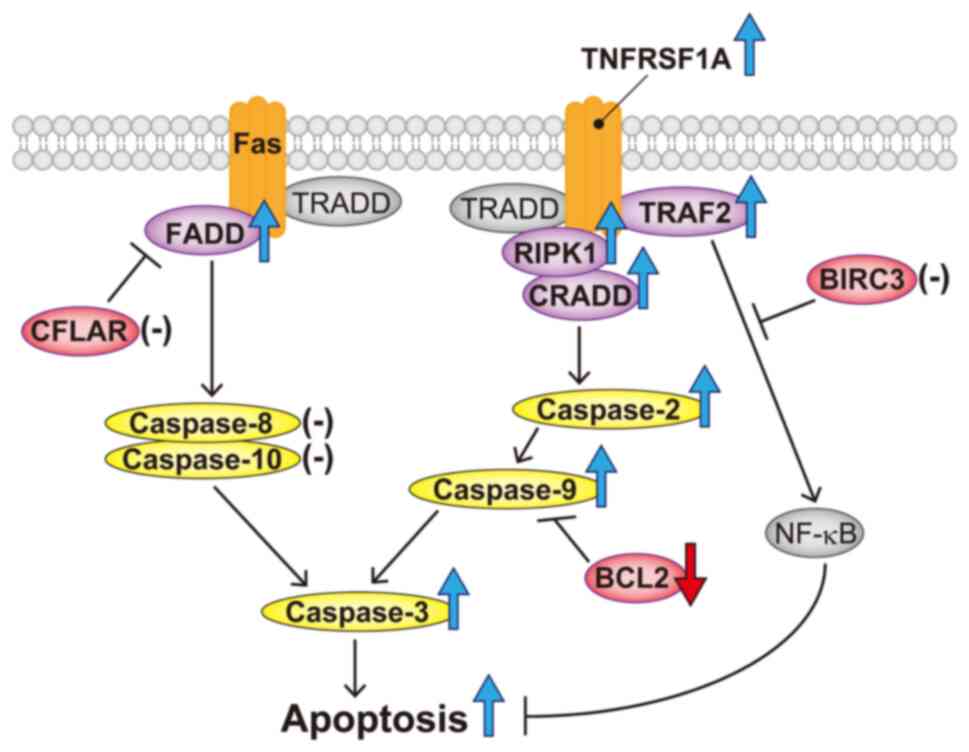 | Figure 7Schematic representation of apoptosis
accelerated by 18α-GA in gingival fibroblasts treated with
phenytoin. 18α-GA induced the upregulation of FADD and caspase-3,
leading to an increase in the apoptotic Fas pathway. 18α-GA also
induced the upregulation of RIPK1, CRADD, caspase-2, caspase-9 and
caspase-3 in the TNF pathway, which resulted in apoptosis
acceleration. Furthermore, 18α-GA decreased BCL2, which increased
caspase-9. Purple (components of death-inducing signaling complex),
red (antiapoptotic factors) and yellow (caspases) ellipses denote
the molecules analyzed in the present study. The blue or red large
arrows denote upregulation or downregulation, respectively,
following 18α-GA treatment. Hyphens denote the molecules that are
unaffected by 18α-GA treatment. 18α-GA, 18-α-glycyrrhetinic acid;
BIRC3, baculoviral IAP repeat containing 3; CFLAR, CASP8 and
FADD-like apoptosis regulator; CRADD, CASP2 and RIPK1 domain
containing adaptor with death domain; FADD, Fas
(TNFRSF6)-associated via death domain; RIPK1, receptor
(TNFRSF)-interacting serine-threonine kinase 1; TNFRSF1A, tumor
necrosis factor receptor superfamily; member 1A; TRADD, TNFRSF1A
associated via death domain; TRAF2, TNF receptor-associated factor
2. |
This study demonstrates that 18α-GA induces
apoptosis through activating the pathways of Fas and TNF in the
death receptor signaling pathway of gingival fibroblasts treated
with PHT. In conclusion, 18α-GA has a therapeutic potential for the
treatment of PHT-induced gingival overgrowth. Future studies should
investigate the alterations of the mitochondrial pathway in
gingival fibroblasts caused by 18α-GA treatment. The mechanism of
gingival overgrowth induced by PHT is related to the accumulation
of collagen by its enhanced production in numerous gingival
fibroblasts (3) or by the impaired
metabolism caused by TNF-α and PHT together (7). The fact that TNF-α activates NF-κB
may also be related to the accumulation of collagen in the gingiva
(3). Thus, future studies should
aim to clarify whether 18α-GA affects collagen
production/metabolism in gingival fibroblasts exposed to PHT.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by JSPS KAKENHI (grant
no. 23K09514).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Author's contributions
RT, TNomo and KH designed the study. RT, TNomu, MY,
CT, IS, HM and YO carried out the experiments. RT, TNomu and MY
performed the apoptosis assay and the flow cytometric analysis. RT,
CT and IS performed the reverse transcription-quantitative PCR. RT,
TNomu and YO performed the western blot analysis. RT and HM
detected the caspase activity. RT, HS and KA analyzed the data. RT
and MY confirmed the authenticity of all raw data. RT drafted the
original manuscript. All authors reviewed the manuscript draft and
revised it critically for intellectual content. All authors have
read and approved the final manuscript.
Ethics approval and consent to
participate
For the use of primary human cell lines, ScienCell
Research Laboratories strictly comply with the policies provided on
website: https://sciencellonline.com/technical-support/ethical-statement.html.
This study was approved by the Ethics Review Committee of Nihon
University School of Dentistry at Matsudo (approval no.
EC23-18).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Takeuchi R: The effect of basic fibroblast
growth factor on cell cycle in human gingival fibroblasts from
nifedipine responder and non-responder. J Oral Sci. 46:37–44.
2004.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Takeuchi R, Matsumoto H, Okada H, Hori M,
Gunji A, Hakozaki K, Akimoto Y and Fujii A: Differences of cell
growth and cell cycle regulators induced by basic fibroblast growth
factor between nifedipine responders and non-responders. J
Pharmacol Sci. 103:168–174. 2007.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Takeuchi R, Matsumoto H, Arikawa K,
Taguchi C, Nakayama R, Nasu I and Hiratsuka K: Phenytoin-induced
gingival overgrowth caused by death receptor pathway malfunction.
Oral Dis. 23:653–659. 2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Nakib N and Ashrafi SS: Drug-induced
gingival overgrowth. Dis Mon. 57:225–230. 2011.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Corrêa JD, Queiroz-Junior CM, Costa JE,
Teixeira AL and Silva TA: Phenytoin-induced gingival overgrowth: A
review of the molecular, immune, and inflammatory features. ISRN
Dent. 2011(497850)2011.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Hassell TM and Hefti AF: Drug-induced
gingival overgrowth: Old problem, new problem. Crit Rev Oral Biol
Med. 2:103–137. 1991.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Kato T, Okahashi N, Ohno T, Inaba H, Kawai
S and Amano A: Effect of phenytoin on collagen accumulation by
human gingival fibroblasts exposed to TNF-alpha in vitro. Oral Dis.
12:156–162. 2006.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Bajkovec L, Mrzljak A, Likic R and Alajbeg
I: Drug-induced gingival overgrowth in cardiovascular patients.
World J Cardiol. 13:68–75. 2021.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Morikawa S, Nasu M, Miyashita Y and
Nakagawa T: Treatment of calcium channel blocker-induced gingival
overgrowth without modifying medication. BMJ Case Rep.
14(e238872)2021.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Hallmon WW and Rossmann JA: The role of
drugs in the pathogenesis of gingival overgrowth. A collective
review of current concepts. Periodontol 2000. 21:176–196.
1999.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Naruishi K: Biological roles of
fibroblasts in periodontal diseases. Cells. 11(3345)2022.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Alikhani M, Alikhani Z and Graves DT:
Apoptotic effects of LPS on fibroblasts are indirectly mediated
through TNFR1. J Dent Res. 83:671–676. 2004.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Shafer WG: Effect of dilantin sodium
analogues on cell proliferation in tissue culture. Proc Soc Exp
Biol Med. 106:205–207. 1961.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Smith QT and Hinrichs JE: Phenytoin and
5-(p-hydroxyphenyl)-5-phenylhydantoin do not alter the effects of
bacterial and amplified plaque extracts on cultures of fibroblasts
from normal and overgrown gingivae. J Dent Res. 66:1393–1398.
1987.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Vernillo AT and Schwartz NB: The effects
of phenytoin (5,5-diphenylhydantoin) on human gingival fibroblasts
in culture. J Periodontal Res. 22:307–312. 1987.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Vijayasingham SM, Dykes PJ and Marks R:
Phenytoin has little effect on in-vitro models of wound healing. Br
J Dermatol. 125:136–139. 1991.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Takeuchi R, Matsumoto H, Akimoto Y and
Fujii A: Inhibition of G1 cell cycle arrest in human gingival
fibroblasts exposed to phenytoin. Fundam Clin Pharmacol.
28:114–119. 2014.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Kantarci A, Augustin P, Firatli E, Sheff
MC, Hasturk H, Graves DT and Trackman PC: Apoptosis in gingival
overgrowth tissues. J Dent Res. 86:888–892. 2007.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Jung JY, Jeong YJ, Jeong TS, Chung HJ and
Kim WJ: Inhibition of apoptotic signals in overgrowth of human
gingival fibroblasts by cyclosporin A treatment. Arch Oral Biol.
53:1042–1049. 2008.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Takeuchi R, Hiratsuka K, Arikawa K, Ono M,
Komiya M, Akimoto Y, Fujii A and Matsumoto H: Possible
pharmacotherapy for nifedipine-induced gingival overgrowth:
18a-glycyrrhetinic acid inhibits human gingival fibroblast growth.
Br J Pharmacol. 173:913–924. 2016.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Ding Y, Brand E, Wang W and Zhao Z:
Licorice: Resources, applications in ancient and modern times. J
Ethnopharmacol. 298(115594)2022.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Moustafa GO, Shalaby A, Naglah AM, Mounier
MM, El-Sayed H, Anwar MM and Nossier ES: Synthesis,
characterization, in vitro anticancer potentiality, and
antimicrobial activities of novel peptide-glycyrrhetinic-acid-based
derivatives. Molecules. 26(4573)2021.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Chu XT, de la Cruz J, Hwang SG and Hong H:
Tumorigenic effects of endocrine-disrupting chemicals are
alleviated by licorice (Glycyrrhiza glabra) root extract through
suppression of AhR expression in mammalian cells. Asian Pac J
Cancer Prev. 15:4809–4813. 2014.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Haku K, Muramatsu T, Hara A, Kikuchi A,
Hashimoto S, Inoue T and Shimono M: Epithelial cell rests of
Malassez modulate cell proliferation, differentiation and apoptosis
via gap junctional communication under mechanical stretching in
vitro. Bull Tokyo Dent Coll. 52:173–182. 2011.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Tiboni M, Benedetti S, Skouras A, Curzi G,
Perinelli DR, Palmieri GF and Casettari L: 3D-printed microfluidic
chip for the preparation of glycyrrhetinic acid-loaded ethanolic
liposomes. Int J Pharm. 584(119436)2020.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Pirzadeh S, Fakhari S, Jalili A, Mirzai S,
Ghaderi B and Haghshenas V: Glycyrrhetinic acid induces apoptosis
in Leukemic HL60 cells through upregulating of CD95/CD178. Int J
Mol Cell Med. 3:272–278. 2014.PubMed/NCBI
|
|
27
|
Haghshenas V, Fakhari S, Mirzaie S,
Rahmani M, Farhadifar F, Pirzadeh S and Jalili A: Glycyrrhetinic
acid inhibits cell growth and induces apoptosis in ovarian cancer
a2780 cells. Adv Pharm Bull. 4 (Suppl 1):S437–S441. 2014.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Takeuchi R, Matsumoto H, Akimoto Y and
Fujii A: Reduction in lipopolysaccharide-induced apoptosis of
fibroblasts obtained from a patient with gingival overgrowth during
nifedipine-treatment. Arch Oral Biol. 56:1073–1080. 2011.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Sano M, Ohuchi N, Inoue T, Tono K,
Tachikawa T, Kizawa Y and Murakami H: Proliferative response to
phenytoin and nifedipine in gingival fibroblasts cultured from
humans with gingival fibromatosis. Fundam Clin Pharmacol.
18:465–470. 2004.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Mitic K, Popovska M, Pandilova M,
Jovanovic R, Spasovski G and Nikolov V: The role of inflammation
and apoptosis in cyclosporine A-induced gingival overgrowth. Bosn J
Basic Med Sci. 13:14–20. 2013.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Ma Y, Cao W, Wang L, Jiang J, Nie H, Wang
B, Wei X and Ying W: Basal CD38/cyclic ADP-ribose-dependent
signaling mediates ATP release and survival of microglia by
modulating connexin 43 hemichannels. Glia. 62:943–955.
2014.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Gottlieb TM and Oren M: p53 in growth
control and neoplasia. Biochim Biophys Acta. 1287:77–102.
1996.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Bulut S, Ozdemir BH, Alaaddinoĝlu EE,
Oduncuoĝlu FB, Bulut OE and Demirhan B: Effect of cyclosporin A on
apoptosis and expression of p53 and bcl-2 proteins in the gingiva
of renal transplant patients. J Periodontol. 76:691–695.
2005.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Bulut S and Ozdemir BH: Apoptosis and
expression of caspase-3 in cyclosporin-induced gingival overgrowth.
J Periodontol. 78:2364–2368. 2007.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Das S, Shukla N, Singh SS, Kushwaha S and
Shrivastava R: Mechanism of interaction between autophagy and
apoptosis in cancer. Apoptosis. 26:512–533. 2021.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Feng S, Zha Z, Wang Z, Yang P, Wu J, Li X,
Xu Q and Liu Y: Anticancer activity of oleiferoside B involving
autophagy and apoptosis through increasing ROS release in MCF-7
cells and SMMC-7721 cells. Nat Prod Res. 35:4865–4869.
2021.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Peña-Blanco A and García-Sáez AJ: Bax, Bak
and beyond-mitochondrial performance in apoptosis. FEBS J.
285:416–431. 2018.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Füllsack S, Rosenthal A, Wajant H and
Siegmund D: Redundant and receptor-specific activities of TRADD,
RIPK1 and FADD in death receptor signaling. Cell Death Dis.
10(122)2019.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Green DR: The death receptor pathway of
apoptosis. Cold Spring Harb Perspect Biol.
14(a041053)2022.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Hsu H, Xiong J and Goeddel DV: The TNF
receptor 1-associated protein TRADD signals cell death and NF-kappa
B activation. Cell. 81:495–504. 1995.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Grimm S, Stanger BZ and Leder P: RIP and
FADD: Two ‘death domain’-containing proteins can induce apoptosis
by convergent, but dissociable, pathways. Proc Natl Acad Sci USA.
93:10923–10927. 1996.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Wajant H: The Fas signaling pathway: More
than a paradigm. Science. 296:1635–1636. 2002.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Nikoletopoulou V, Markaki M, Palikaras K
and Tavernarakis N: Crosstalk between apoptosis, necrosis and
autophagy. Biochim Biophys Acta. 1833:3448–3459. 2013.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Irmler M, Thome M, Hahne M, Schneider P,
Hofmann K, Steiner V, Bodmer JL, Schröter M, Burns K, Mattmann C,
et al: Inhibition of death receptor signals by cellular FLIP.
Nature. 388:190–195. 1997.PubMed/NCBI View
Article : Google Scholar
|
|
46
|
Palladino MA, Bahjat FR, Theodorakis EA
and Moldawer LL: Anti-TNF-alpha therapies: The next generation. Nat
Rev Drug Discov. 2:736–46. 2003.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Maas C, Tromp JM, van Laar J, Thijssen R,
Elias JA, Malara A, Krippner-Heidenreich A, Silke J, van Oers MH
and Eldering E: CLL cells are resistant to smac mimetics because of
an inability to form a ripoptosome complex. Cell Death Dis.
4(e782)2013.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Gray CM, McCorkell KA, Chunduru SK,
McKinlay MA and May MJ: Negative feedback regulation of
NF-κB-inducing kinase is proteasome-dependent but does not require
cellular inhibitors of apoptosis. Biochem Biophys Res Commun.
450:341–346. 2014.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Krumschnabel G, Sohm B, Bock F, Manzl C
and Villunger A: The enigma of caspase-2: The laymen's view. Cell
Death Differ. 16:195–207. 2009.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Werner AB, de Vries E, Tait SW, Bontjer I
and Borst J: Bcl-2 family member Bfl-1/A1 sequesters truncated bid
to inhibit its collaboration with pro-apoptotic Bak or Bax. J Biol
Chem. 277:22781–22788. 2002.PubMed/NCBI View Article : Google Scholar
|