Introduction
Osteonecrosis of the femoral head (ONFH) is a common
orthopedic disease caused by a decrease in blood supply to the
femoral head, with frequently reported features of osteocyte
necrosis, trabecular bone fracture and articular surface collapse
(1). It is estimated that there
are ~8.12 million individuals over the age of 15 years with ONFH in
China and the total number of patients with ONFH worldwide will
reach 20 million in the next 10 years (2). ONFH can be categorized into traumatic
ONFH and non-traumatic ONFH, with the latter being further
categorized into steroid- or corticosteroid-induced ONFH (SONFH)
and alcohol-induced ONFH (AONFH) (3). Excessive alcohol consumption is
recognized to be a major risk factor for AONFH (4). Aberrant alcohol metabolism may
contribute to femoral head tissue damage through the production of
a number of toxic byproducts, such as acetaldehyde, free radicals
and acetaldehyde adducts. In addition, alcohol metabolic
impairments can adversely affect intravascular coagulation and the
clotting cascade (4,5). However, the pathogenic mechanism of
AONFH remains poorly understood.
The gut microbiota (GM) has been reported to be an
important symbiotic partner in the regulation of human physiology
(6). A number of studies have
previously reported that gut microbiome composition and
corresponding metabolic activity can participate in the regulation
of bone homeostasis to exert pivotal effects on the development of
osteochondral or bone diseases (6,7).
These include estrogen deprivation-induced bone loss (8) and bisphosphonate-related
osteonecrosis of the jaw (9). In
addition, alcohol consumption can alter the GM composition and is
related to overall health, and cause diseases such as inflammatory
bowel disease, gastrointestinal cancers and liver injury (10). However, to the best of our
knowledge, the interaction among alcohol, the GM and GM
metabolites, in addition to their roles in the development of ONFH,
has not been reported to date.
On the basis of the previously reported regulatory
effects of GM on bone (6,7), it may be hypothesized that
alcohol-induced gut dysbiosis may participate in the development of
AONFH. Therefore, in the present study, fecal integrated omics
analysis was performed, including 16S rDNA gene sequencing,
metagenomic and metabolomic analyses, to define the gut metabolome
and metabolic profiles in patients with AONFH.
Materials and methods
Sample collection and ethical
approval
The present study enrolled 98 Chinese men, including
48 healthy adults [negative control (NC); age, 41.75±10.50 years]
and 50 patients with AONFH (age, 43.98±11.40 years), from June 2021
to June 2022 at the Luoyang Orthopedic-Traumatological Hospital of
Henan Province (Luoyang, China). The selected participants were Han
Chinese from similar geographic areas, experienced the same
environmental factors, with similar hygiene status and diet (except
alcohol consumption).
The patients with AONFH were required to meet the
following inclusion criteria: i) Aged 18-80 years; ii) history of
any type of alcoholic beverage intake of >320 ml/week for >6
months (11); iii) AONFH diagnosis
within 1 year of alcohol consumption at the aforementioned levels;
iv) AONFH diagnosed by clinical examination, X-ray, CT and MRI; and
v) no history of other osteoarticular diseases (such as injury,
osteoarthritis, rheumatoid arthritis, gout or skeletal fluorosis),
chronic diseases (hypertension, diabetes or coronary heart disease)
or bowel diseases (inflammatory bowel disease, irritable bowel
syndrome or colorectal cancer), for which they received treatment
in the past 6 months. The exclusion criteria of healthy controls
were as follows: i) Musculoskeletal pathologies or recent injuries;
and ii) use of antibiotics, probiotics, prebiotics or symbiotics in
the previous 2 months.
The general clinical data of patients were recorded,
including age, educational background, height, weight and BMI. The
present study was approved by the ethics committee of Luoyang
Orthopedic-Traumatological Hospital of Henan Province (approval no.
KY2021-007-01). All participants provided written informed consent
for participation into the present study and the study protocols
followed the ethical guidelines of The Declaration of Helsinki.
Fecal samples were collected by the participants and immediately
transported to the laboratory, where they were divided into three
portions per sample, packed into three freezer tubes, frozen in
liquid nitrogen overnight and preserved at -80˚C for further
testing.
DNA extraction and 16S rDNA gene
sequencing
A total of 48 NC samples and 50 AONFH samples were
subjected to 16S rDNA gene sequencing analysis. DNA was extracted
using the E.Z.N.A.® Stool DNA Kit (cat. no. D4015; Omega
Bio-Tek, Inc.) according to the manufacturer's protocols.
Nuclease-free water was used as the negative control. Total DNA
from each sample was eluted in 50 µl elution buffer and stored at
-80˚C until PCR was performed.
The V3-V4 region of the prokaryotic 16S rDNA gene
was amplified using the following primers: 341 forward,
5'-CCTACGGGNGGCWGCAG-3'; and 805 reverse,
5'-GACTACHVGGGTATCTAATCC-3' (N, A+C+G+T; H, A+C+T; V, A+C+G; W,
A+T) (12). The 5' ends of the
primers were tagged with specific barcodes for each sample, which
were sequenced using universal primers (forward,
5'-GTGCCAGCMGCCGCGGTAA-3'; reverse, 5'-GGACTACHVGGGTWTCTAAT-3').
PCR amplification was performed in a reaction mixture with a total
volume of 25 µl, containing 25 ng template DNA, 12.5 µl PCR premix,
2.5 µl of each primer and PCR-grade water to adjust to the final
volume, 1 µl of KOD DNA polymerase (2.5 U/µl; Toyobo). The PCR
conditions used to amplify the prokaryotic 16S fragments were as
follows: Initial denaturation at 98˚C for 30 sec; followed by 32
cycles of 98˚C for 10 sec, 54˚C for 30 sec and 72˚C for 45 sec and
a final extension at 72˚C for 10 min. PCR product amplification was
confirmed using 2% agarose gel electrophoresis (Genecolour™;
GBY-II; Beijing Jinboyi Biotechnology Co., Ltd). Throughout the DNA
extraction process, ultrapure water was used instead of sample
solution as a negative control to exclude the possibility of
false-positive PCR results. The PCR products were purified using
AMPure XT beads (Beckman Coulter, Inc.) and quantified using Qubit
3.0 fluorometer (Invitrogen; Thermo Fisher Scientific, Inc.). The
final library DNA concentration was 10 ng/µl. The amplicon pools
were prepared for sequencing and the size and quantity of the
amplicon library were assessed using an Agilent 2100 Bioanalyzer
(Agilent Technologies, Inc.) and the Library Quantification Kit for
Illumina (Kapa Biosystems; Roche Diagnostics), respectively. The
libraries were sequenced using the NovaSeq PE250 platform according
to the manufacturer's instructions (Illumina, Inc.).
The raw 150 bp paired-end reads were assigned to
samples based on their unique barcodes and truncated by cutting off
the barcode and primer sequence. Paired-end reads were merged using
FLASH (version 1.2.8; http://ccb.jhu.edu/software/FLASH/). Quality filtering
of the raw reads was performed to obtain high-quality clean tags
using ‘fqtrim’ (version 0.94, http://ccb.jhu.edu/software/fqtrim/). Chimeric
sequences were filtered using Vsearch software (version 2.3.4;
https://github.com/torognes/vsearch).
Dereplication with DADA2(13)
generated a feature table and feature sequence. Alpha diversity and
beta diversity were calculated using QIIME2 (version 2019.7;
https://qiime2.org/), for which the same number of
sequences were extracted randomly by reducing the number of
sequences to the minimum for certain samples, and the relative
abundance was used to determine the bacterial taxonomy. Alpha
diversity and beta diversity figures were produced using the
ggplot2 (version 3.2.0) toolbox implemented in R software. Blast
(http://www.ncbi.nlm.nih.gov/BLAST)
was used for sequence alignment and each representative feature
sequence was annotated using the SILVA database (version 138.1,
http://www.arb-silva.de) (14).
Fecal metagenomics analysis
As one sample from the NC group was missed in the
fecal metagenomics analysis, a total of 47 NC and 50 AONFH
fecal samples were subjected to metagenomics analysis. The DNA
library was constructed using a TruSeq Nano DNA LT Library
Preparation Kit (cat. no. FC-121-4001; Illumina, Inc.). DNA was
fragmented using dsDNA Fragmentase (cat. no. M0348S; New England
BioLabs, Inc.) and incubated at 37˚C for 30 min, before the
sequencing library was constructed from the fragmented cDNA.
Blunt-end DNA fragments were generated using a combination of
fill-in reactions and exonuclease activity, and size selection was
performed with the provided sample purification beads. An A-base
was then added to the blunt ends of each strand, preparing them for
ligation to the indexed adapters. Each adapter contained a T-base
overhang for ligating the adapter to the A-tailed fragmented DNA.
These adapters contained the full complement of sequencing primer
hybridization sites for single, paired-end and indexed reads.
Single- or dual-index adapters were ligated to the fragments and
the ligated products were amplified by PCR using the following
thermocycling conditions: Initial denaturation at 95˚C for 3 min;
followed by 8 cycles of 98˚C for 15 sec, 60˚C for 15 sec and 72˚C
for 30 sec and a final extension at 72˚C for 5 min.
Raw sequencing reads were processed to obtain valid
reads for further analysis. First, sequencing adapters were removed
from sequencing reads using ‘cutadapt’ (version 1.9, http://cutadapt.readthedocs.org/en/stable/guide.html).
The low-quality reads were then trimmed by ‘fqtrim’ (version 0.94,
http://ccb.jhu.edu/software/fqtrim/)
using a sliding-window algorithm. The reads were next aligned to
the host genome using ‘bowtie2’ (version 2.2.0) to remove host DNA
contamination (15). Once
quality-filtered reads were obtained, they were de novo
assembled to construct the metagenome for each sample using IDBA-UD
(version 1.1.1) (16). All coding
regions (CDS) within the metagenomic contigs were predicted using
‘MetaGeneMark’ (version 3.26) (17). CDS sequences from all samples were
clustered using CD-HIT (version 4.6.1) to obtain unigenes (18). Unigene abundance for individual
samples were estimated by transcripts per million based on the
number of aligned reads using bowtie2 (version 2.2.0). The lowest
common taxonomic ancestors of the unigenes were obtained by
aligning them against the National Center for Biotechnology
Information Non-Redundant Protein Sequence database using DIAMOND
(version 0.9.14) (19). Similarly,
the functional annotation of unigenes were obtained using Gene
Ontology (GO) database (version go_2018.12.21, http://www.geneontology.org/) Kyoto Encyclopedia of
Genes and Genomes (KEGG-release_87.1, http://www.genome.jp/kegg/).
A random forest model was constructed using the
random forest package in R software (version 3.4.4). Receiver
operating characteristic (ROC) curves were generated and the area
under the curve (AUC) values were computed using pROC in R
software. Functional annotation of the unigenes was also performed
using Blast (http://www.ncbi.nlm.nih.gov/BLAST). Finally,
differentially expressed unigenes were identified at the taxonomic,
functional or gene level by Fisher's exact test based on the
taxonomic annotation, functional annotation and abundance profiles,
respectively.
Metabolomics and data analysis
One sample from the AONFH group was missed in the
metabolomics analysis, so a total of 48 NC samples and 49 AONFH
samples were subjected to metabolomics analysis. The metabolites
were extracted from fecal samples with 50% methanol buffer and
incubated at 24˚C for 10 min. The extraction mixture was stored
overnight at -20˚C. After centrifugation at 4,000 x g for 20 min at
room temperature, the supernatants were transferred into 96-well
plates and stored at -80˚C prior to being subjected to liquid
chromatography-mass spectrometry (LC-MS) analysis to identify the
metabolites. Pooled quality control (QC) samples were prepared by
combining 10 µl each extraction mixture. Chromatographic separation
was performed using the UltiMate 3000 high-performance LC system
(Thermo Fisher Scientific, Inc.). No internal standard was used
(20). An ACQUITY UPLC BEH C18
column (size, 100x2.1 mm; 1.8 µm; Waters Corporation) was used for
the reversed phase separation. The column temperature was
maintained at 35˚C. The flow rate was 0.4 ml/min, and the mobile
phase consisted of solvent A (water, 0.1% formic acid) and solvent
B (acetonitrile, 0.1% formic acid). The gradient elution conditions
were as follows: 0-0.5 min, 5% solvent B; 0.5-7 min, 5-100% solvent
B; 7-8 min, 100% solvent B; 8-8.1 min, 100-5% solvent B; 8.1-10
min, 5% solvent B. The injection volume for each sample was 4
µl.
A high-resolution triple time-of-flight (TOF) 5600
Plus tandem mass spectrometer (SCIEX) was operated in both positive
ionization mode and negative ionization mode for detecting
metabolites eluted from the column. The curtain gas was set to 30
psi, ion source gas1 was set to 60 psi, ion source gas2 was set to
60 psi and the interface heater temperature was set to 650˚C. For
positive ion mode, the Ionspray voltage was set at 5,000 V. For
negative ion mode, the Ionspray voltage was set at -4,500 V. The
mass spectrometry data were acquired in information-dependent
acquisition mode. The TOF mass range was in the 60-1,200-Da range.
The survey scans were acquired in 150 msec, and ≥12 product ion
scans were collected if they reached the threshold of >100
counts/sec with a 1+ charge-state. The total cycle time was fixed
at 0.56 sec. A total of four-time bins were summed for each scan at
a pulser frequency value of 11 kHz through monitoring the 40 GHz
multichannel thermal conductivity detector with four-anode/channel
detection. Dynamic exclusion was set at 4 sec. During acquisition,
the mass accuracy was calibrated every 20 samples. Furthermore, to
evaluate the stability of the LC-MS procedure throughout
acquisition, a QC sample (pooled sample) was processed after every
10 samples. The following multiple reaction monitoring transitions
were selected: m/z 1861.3→70.02 (positive), m/z 1889.0→72.02
(negative).
The acquired MS data were pretreated by peak
picking, peak grouping, retention time correction, second peak
grouping and annotation of isotopes and adducts using the XCMS
software. LC-MS raw data files were converted into the ‘mzXML’
format and then processed using XCMS, CAMERA and the metaX toolbox
implemented with the R software (version 3.5.3 R Core Team, 2019;
https://www.R-project.org/). Each ion
was identified by combining the retention time and m/z data. The
intensity of each peak was recorded and a 3D matrix containing
arbitrarily assigned peak indices (retention time-m/z pairs),
sample names (observations) and ion intensity information
(variables) was generated.
The KEGG and Human Metabolome Database (HMDB 5.0,
http://www.hmdb.ca) databases were used to annotate
the metabolites by matching the exact molecular mass data (m/z) of
samples with those from the databases. If the mass difference
between the observed and database values was <10 parts per
million, the metabolite would be annotated, and the molecular
formula of the metabolite would be further identified and validated
by isotopic distribution measurements. An in-house metabolite
fragment spectrum library was used to validate the identified
metabolites (21).
Peak intensity data were further preprocessed using
metaX. Features that were detected in <50% of QC samples or 80%
of biological samples were removed and the remaining peaks with
missing values were imputed using the ‘k-nearest neighbor’
algorithm to further improve data quality (22). Principal component (PC) analysis
was performed for outlier detection and batch effect evaluation
using the pre-processed dataset. QC-based robust locally weighted
scatter-plot smoother signal correction was then fitted to the QC
data with respect to the order of injection to minimize signal
intensity drift over time. In addition, the relative standard
deviations of the metabolic features were calculated across all QC
samples and any that were >30% were removed.
Unparied Student's t-tests were conducted to detect
differences in metabolite concentrations between the two groups.
The P-value was adjusted for multiple tests with a false-discovery
rate (FDR) using the Benjamini-Hochberg method. A median
FDR<0.05 was used as a cutoff value. Supervised partial least
squares discriminant analysis (PLS-DA) was conducted using metaX to
discriminate between the different variables whereas the XCMS
software was used to pretreat the acquired MS data. The raw LC-MS
raw data files were processed with metaX using the ‘XCMS’ package
for peak detection and the ‘CAMERA’ package for peak annotation in
R. The variable importance in projection (VIP) was calculated,
where a VIP cut-off value of 1.0 was used to select important
features.
Statistical analysis
Continuous variables were presented as the mean ±
standard deviation (SD) and Student's t-test or the Wilcoxon
signed-rank test was used to compare their differences. Pearson's
chi-squared test or Fisher's exact test was used to compare
differences for count data. Spearman's correlation analysis was
conducted to calculate the correlation between species and
metabolites. P<0.05 was considered to indicate a statistically
significant difference. All data were analyzed using GraphPad Prism
software (version 6; Dotmatics), R software and Microsoft Excel
(version 3.3.2.13, Microsoft Corp.).
Results
Characteristics of the study
population
A total of 50 patients with AONFH and 48 healthy
adults were included in the present study. The demographic
characteristics of the two groups did not show any statistically
significant difference (Table I),
suggesting that there were no confounding factors that could have
influenced the results.
 | Table ICharacteristics of participants in
the present study. |
Table I
Characteristics of participants in
the present study.
|
Characteristica | Patients with
alcohol-induced osteonecrosis of the femoral head (n=50) | Negative controls
(n=48) | P-value |
|---|
| Age, years | 43.98±11.40 | 41.75±10.50 | 0.32 |
| Height, cm | 172.64±5.30 | 174.58±5.76 | 0.09 |
| Weight, kg | 72.46±11.74 | 71.19±11.48 | 0.59 |
| BMI,
kg/m2 | 24.31±3.79 | 23.35±3.54 | 0.20 |
| Alcohol
consumption | 50(100) | 0 (0) | <0.001 |
| Smoking | 4(8) | 0 (0) | 0.12 |
GM changes in patients with AONFH
To identify changes in the gut microbiome in
patients with AONFH, 16S rRNA metagenomics analysis was conducted
on 98 fecal samples, including 48 samples from the NC group and 50
samples from the AONFH group. After QC, >21 million valid bases
were obtained for each sample (Table
SI).
The rRNA sequences were then grouped into
operational taxonomic units (OTUs) based on sequence similarity to
classify the microbial diversity in terms of bacterial strains.
Performing a 97% similarity cluster analysis identified 4,966 OTUs
in the NC group and 4,248 OTUs in the AONFH group, where 1,697 OTUs
were shared between the two groups (Fig. 1A).
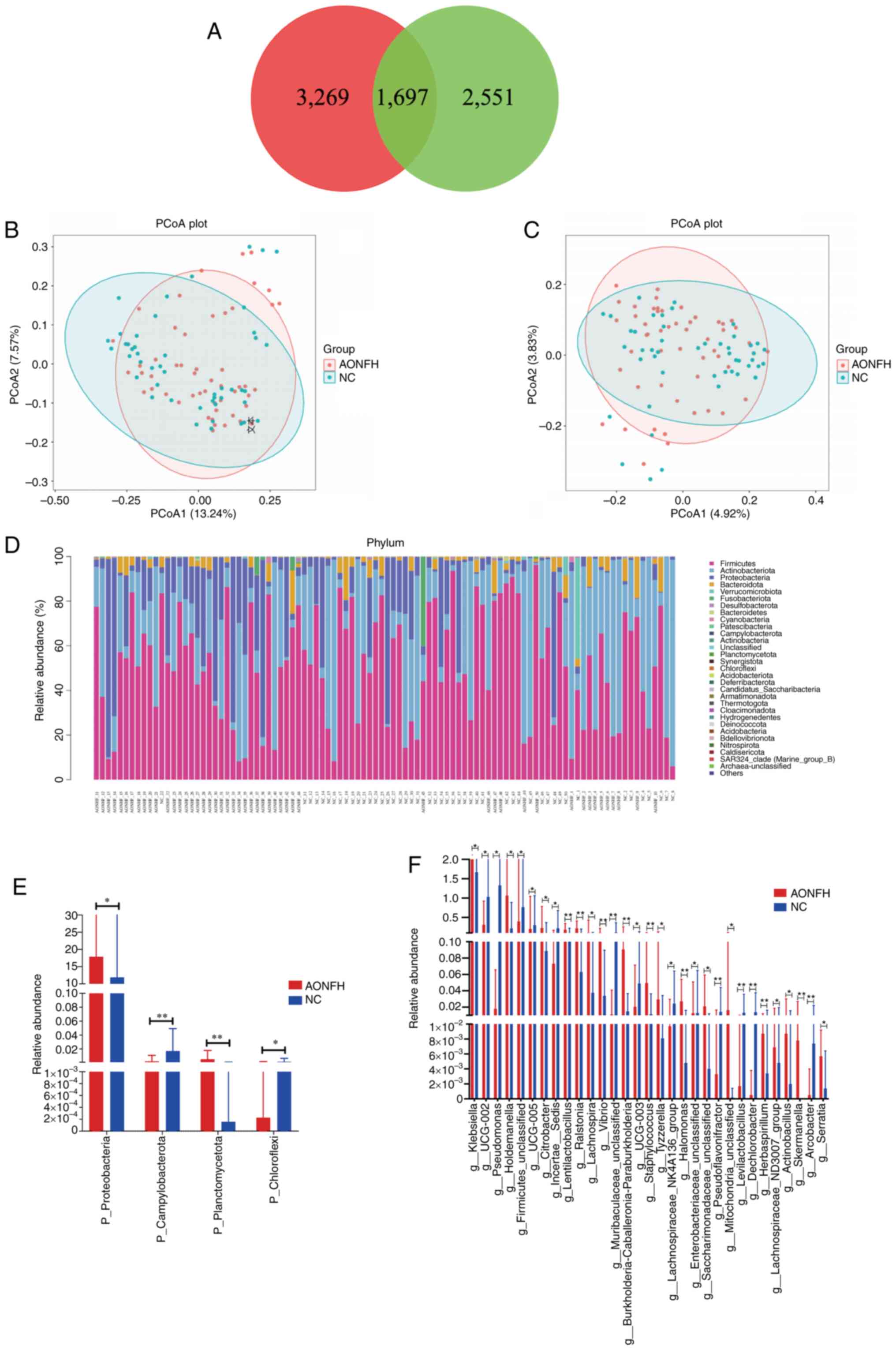 | Figure 1Gut microbiome diversity and
structure analysis of patients with AONFH and NCs. (A) Venn diagram
of the observed features (amplicon sequence variants) in the AONFH
and NC groups. PCoA of the microbiota based on (B) unweighted
UniFrac (ANOSIM, R=0.058, P=0.005) and (C) Jaccard (ANOSIM,
R=-0.060, P=0.003) distance matrices for the AONFH and NC groups.
(D) Heatmap was generated at phylum level based on the relative
abundance values. Statistically significant differences in
bacterial abundance at the (E) phylum and (F) genus level between
the AONFH and NC groups (mean ± SD). *P<0.05,
**P<0.01. AONFH, alcohol-induced osteonecrosis of the
femoral head; NC, negative control; PCoA, principal coordinate
analysis; ANOSIM, analysis of similarities; p_, at phylum level,
g_, at genus level. |
Alpha diversity analysis was then used to analyze
the complexity of species diversity in each sample using several
indices, including the observed OTUs, Chao1, Shannon and Simpson
indices. The richness and diversity rarefaction curve in the two
groups tended to be flat or reach a plateau, suggesting
satisfactory sequencing depth (Fig.
S1). Alpha diversity analysis demonstrated no significant
differences in observed OTUs, Chao1, Shannon and Simpson indices
between the NC group and the AONFH group (Fig. S2), which indicated that the
complexity of species diversity was similar.
Principle coordinate analysis (PCoA) and analysis of
similarities (ANOSIM) testing for beta diversity demonstrated a
significant difference in GM composition and abundance between the
two groups (unweighted Unifrac P=0.005 and Jaccard P=0.003;
Fig. 1B and C).
AONFH-related changes in gut
microbiome composition
Taxon-dependent analysis (Fig. 1D) identified 31 phyla present in
both groups, with Firmicutes, Actinobacteriota, Proteobacteria,
Bacteroidota and Verrucomicrobiota being the most dominant phyla.
Firmicutes was the most predominant phylum, accounting for 55.23
and 48.19% of the GM in the NC group and the AONFH group,
respectively. Further analyses demonstrated that, at the phylum
level, Campylobacterota (P=0.0048) and Chloroflexi (P=0.0392) were
significantly more abundant in the NC group compared with those in
the AONFH group, whereas Planctomycetota (P=0.0099) and
Proteobacteria (P=0.0438) were significantly more abundant in the
AONFH group compared with those in the NC group (Fig. 1E).
At the genus level, the abundance of 58 genera was
significantly different between the NC group and the AONFH group.
Among them, UCG-002 (P=0.0241), Pseudomonas
(P=0.0342), UCG-005 (P=0.0436) and Incertae sedis
(P=0.0208) were significantly more abundant in the NC group
compared with those in the AONFH group, whereas Klebsiella
(P=0.0232), Holdemanella (P=0.0229), Citrobacter
(P=0.0468) and Lentilactobacillus (P=0.0093) were
significantly more abundant in the AONFH group compared with the NC
group (Fig. 1F).
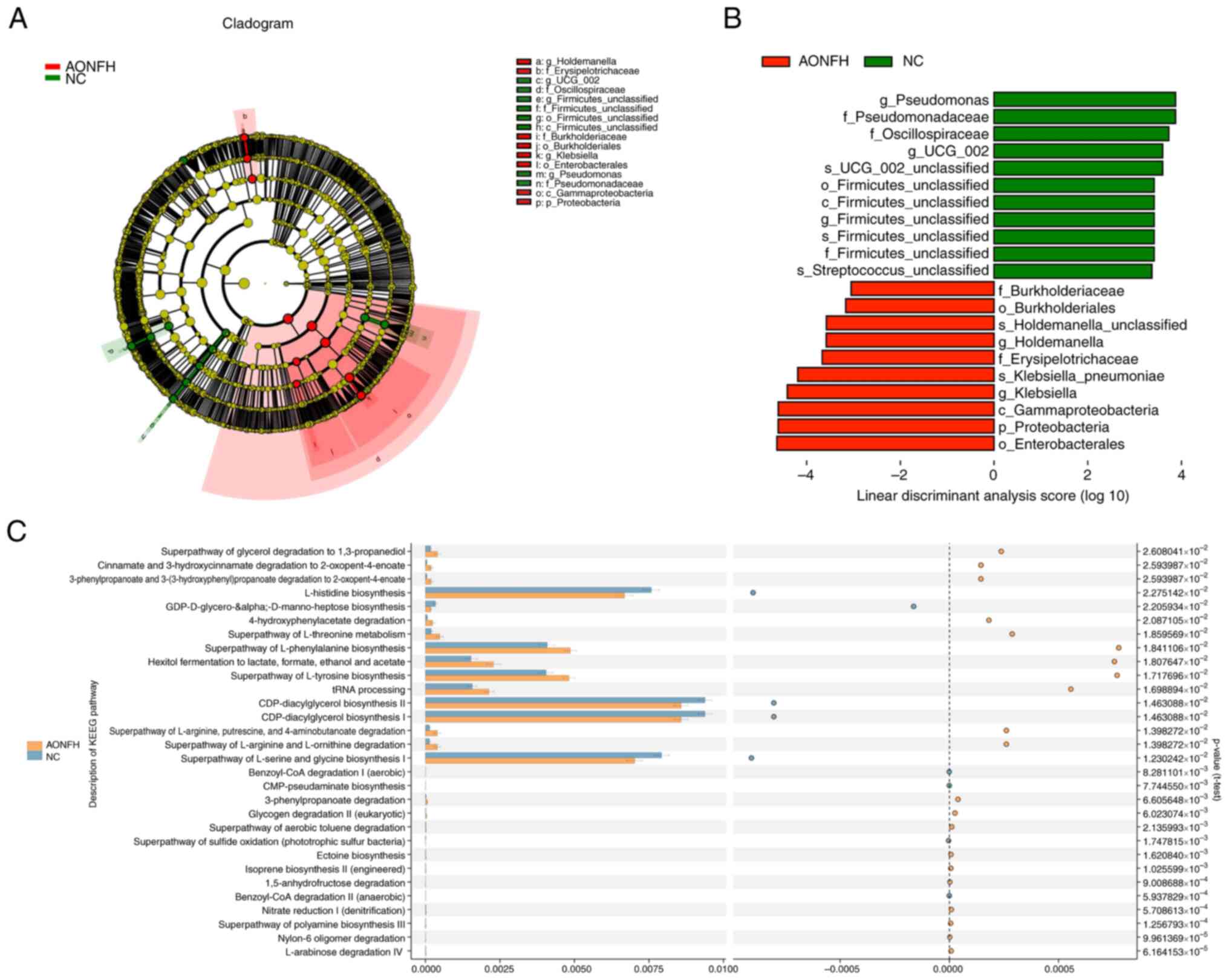
Next, linear discriminant analysis (LDA) was
performed and integrated with effect size to generate a cladogram
to identify the specific bacteria species that dominate in the GM
of patients with AONFH (Fig. 2A).
Significant differences were demonstrated in 21 OTUs (LDA>3),
including Pseudomonas, Pseudomonadaceae, Oscillospiraceae,
UCG-002, Firmicutes and Streptococcus, which were
more abundant in the NC group compared with those in the AONFH
group. By contrast, Burkholderiaceae, Buekholderiales,
Holdemanella, Erysipelotrichaceae, Klebsiella
pneumoniae, Klebsiella, Gammaproteobacteria,
Proteobacteria and Enterobacterales were more abundant in the AONFH
group compared with those in the NC group (Fig. 2B).
Prediction of gene function in the
GM
Next, Phylogenetic Investigation of Communities by
Reconstruction of Unobserved States (PICRUSt 2.2.0b http://huttenhower.sph.harvard.edu/galaxy/root?tool_id=PICRUSt_normalize)
was used to compare gut microbial gene functions across Clusters of
Orthologous Genes (COGs), KEGG and KEGG orthology functional
orthologues between the AONFH and NC groups (Fig. S3, Fig. S4 and Fig. S5). KEGG pathway analysis
identified important functions, such as ‘CDP-diacylglycerol
biosynthesis I/II’, ‘L-histidine biosynthesis’ and ‘superpathway of
L-serine and glycine biosynthesis I’ (Fig. 2C), whereas COG database analysis
highlighted ‘Flp pilus assembly protein TadG’, ‘lipid-binding SYLF
domain’ and ‘CBS-domain-containing membrane proteins’ in the AONFH
group were higher when compared with the NC group (Fig. S3). KEGG analysis indicated that
gut microbial genes were upregulated in AONFH group, such as those
participating in ‘propanoate metabolism’, ‘fructose and mannose
metabolism’ and ‘phosphotransferase system’. However, those
participating in RNA degradation and transcription machinery were
downregulated (Fig. S4). KEGG
orthology analysis indicated that ‘AgrD protein’, ‘formate
dehydrogenase’, ‘diapolycopene oxygenase’, ‘kumamolisin’ and
‘carotenoid cleavage dioxygenase’ were enhanced in the AONFH group
(Fig. S5).
Metagenomic sequencing demonstrates
significant differences between the AONFH and NC groups
Metagenomic sequencing was performed on fecal
samples from 50 patients with AONFH and 47 healthy individuals. A
total of 317,243 genes were identified. The samples from the NC
group contained 3,177 specific genes that were not detected in the
AONFH samples. Compared with the NC group, 20,823 unigenes were
found to be differentially expressed in the AONFH group (10,171
upregulated and 10,652 downregulated).
The alpha diversity was lower in the AONFH group
compared with that in the NC group, as measured by the observed
species and Chao1 indices, Whilst the Shannon and Simpson indices
did not demonstrate any significant difference in alpha diversity
between the groups (Fig. S6). The
results indicated that the complexities of species diversity of the
two groups were similar, although the species richness was lower in
the AONFH group.
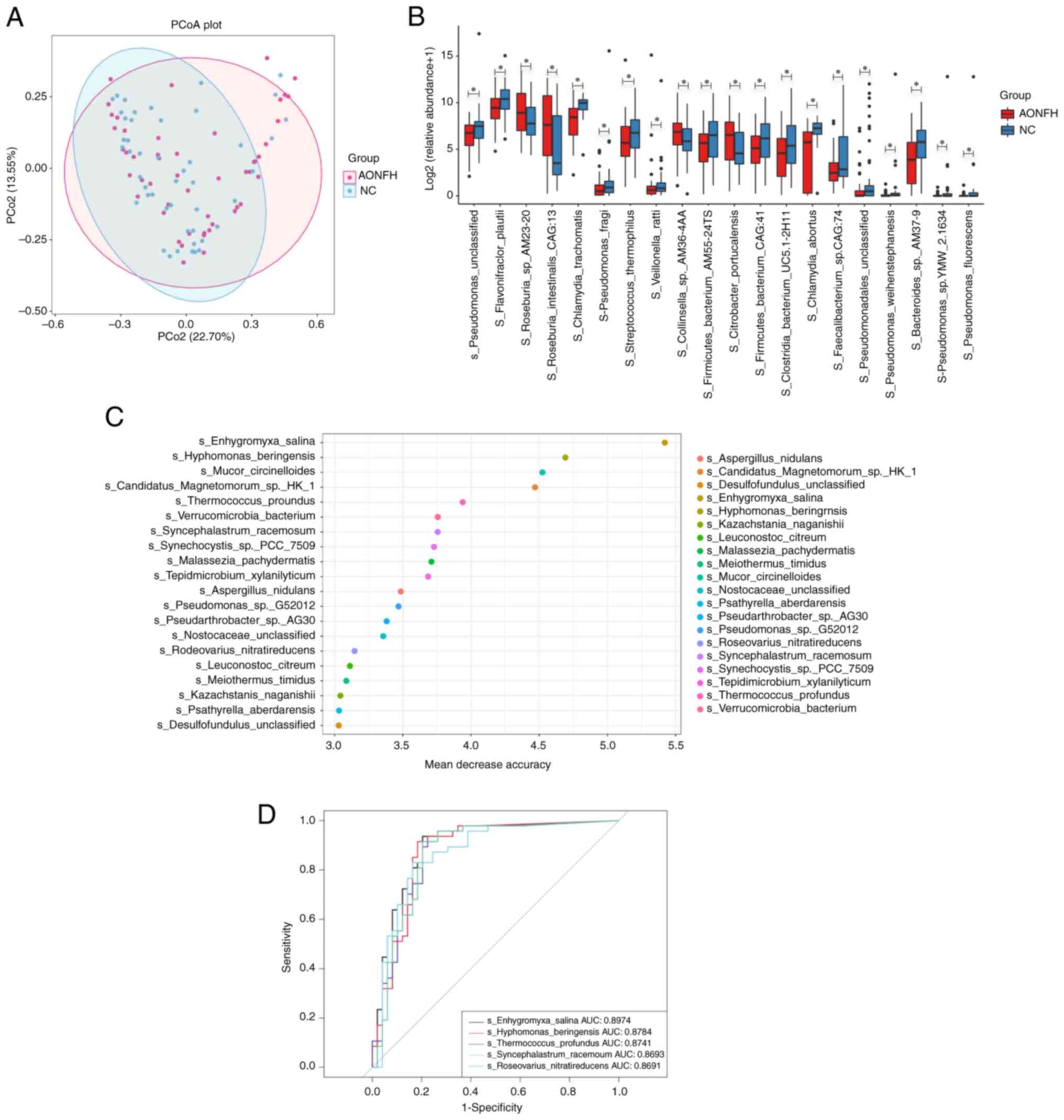
PCoA and ANOSIM testing for beta diversity
demonstrated no significant difference in microbial composition
between the AONFH and NC groups at the species level (Bray-Curtis
Unifrac P=0.06; Fig. 3A).
Comparing the profiles of the AONFH and NC groups demonstrated that
Pseudomonas, Pseudomonas fluorescens, Pseudomonas
sp. TMW-2.1634, Pseudomonas weihenstephanensis and
Pseudomonas fragi were significantly less abundant in the
AONFH group compared with those in the NC group (Fig. 3B and Table SII).
Potential role of GM biomarkers in
AONFH risk assessment
A random forest model was constructed based on the
genera that demonstrated significantly different abundances to
identify potential diagnostic biomarkers that could be used to
predict AONFH. The optimal model that provided the best
discriminatory power utilized 20 genera (Fig. 3C). According to the aforementioned
analysis, there were significant differences in the composition of
the microbial community between AONFH and NC groups. Therefore, to
determine the ability of the identified bacterial biomarkers to
discriminate between the two groups, receiver operating
characteristic curves were produced and the AUC was calculated. The
top five AUC values were for Enhygromyxa salina (89.47%),
Hyphomonas beringensis (87.84%), Thermococcus
profundus (87.41%), Syncephalastrum racemosum (86.93%)
and Roseovarius nitratireducens (86.91%; Fig. 3D).
Functional analysis of differentially
expressed genes in the AONFH group identified by metagenomic
sequencing
The top 10 GO items for the three types of gene
classifications provided by the GO database were selected (Fig. S7A). Next, GO enrichment analysis
of the differentially expressed unigenes between the two groups was
performed and the top 20 GO terms were analyzed (Fig. S7B). KEGG analysis of the
differentially expressed unigenes was then performed to identify
the metabolic pathways that differed most significantly between the
two groups. The expression of genes related to ‘starch and sucrose
metabolism’, ‘RNA degradation’, ‘pentose and glucuronate
interconversions’, ‘glutathione metabolism’, ‘flagellar assembly’
and ‘bacterial chemotaxis’ differed significantly between the AONFH
and NC groups (P≤0.01; unigene number >50; Fig. S7C and Table SIII).
Metabolomic analysis reveals abnormal
metabolic alterations in patients with AONFH
To identify changes in the gut microbiome in
patients with AONFH, metabolomic analysis was performed on 97 fecal
samples, including 48 samples from the NC group and 49 samples from
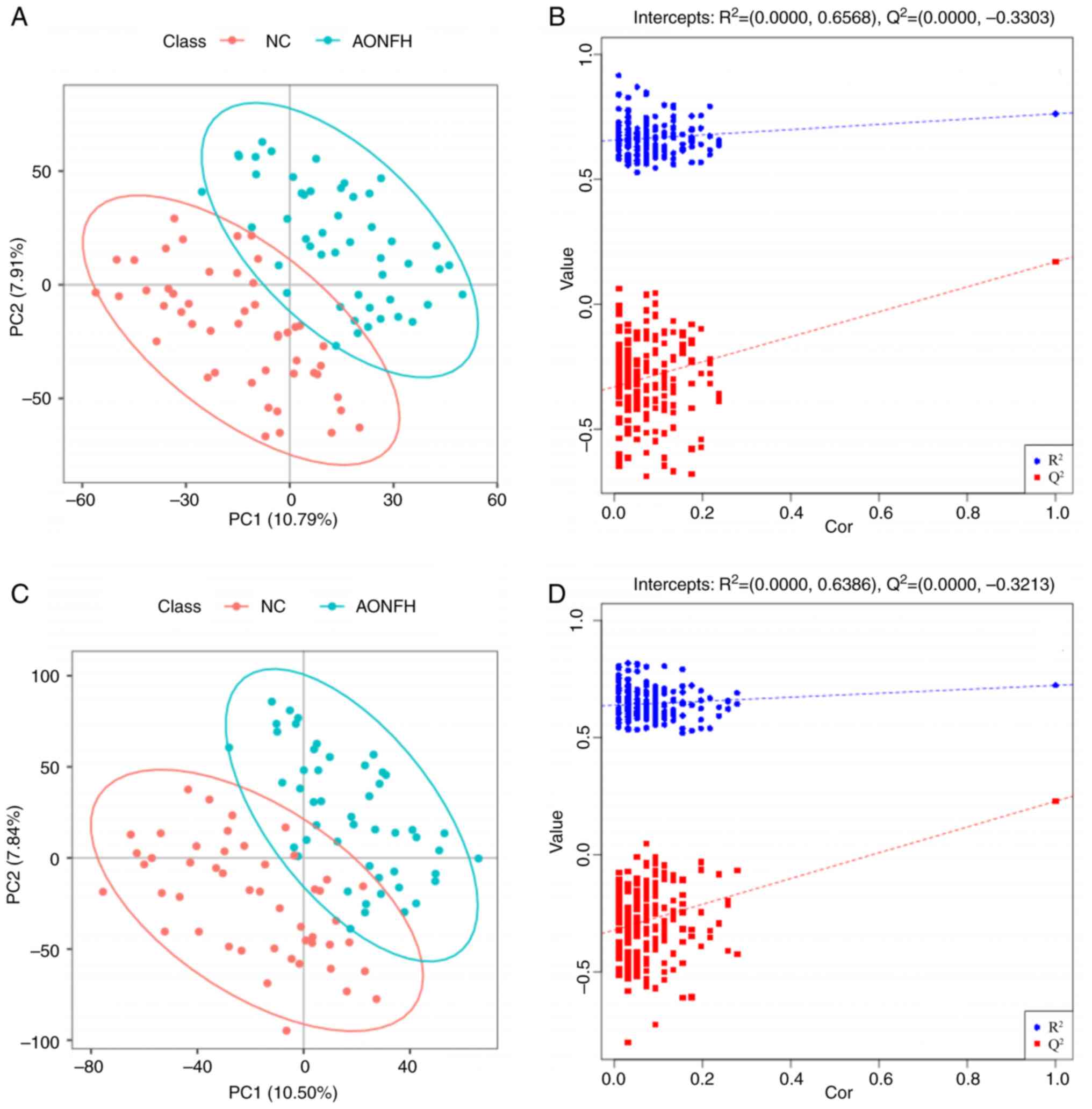
the AONFH group. PLS-DA was performed to identify discriminant
metabolites in the fecal samples from these two groups. Using
negative ion mode (NIM), there was an apparent trend towards the
separation of metabolic features in the fecal samples from patients
with AONFH and NCs (Fig. 4A). The
combined explained variance of PC1 and PC2 (Fig. 4B) was 18.84%, R2=(PC1,
0.0; PC2, 0.6568) and Q2=(PC1, 0.0; PC2, -0.3303). In
positive ion mode (PIM), there was also an apparent trend toward
separation between the metabolic features of the fecal samples from
patients with AONFH and NCs (Fig.
4C). The combined explained variance of PC1 and PC2 was 18.41%
(Fig. 4D), R2=(PC1,
0.0; PC2, 0.6386) and Q2=(PC1, 0.0; PC2-0.3213).
Fecal metabolomic changes in patients
with AONFH
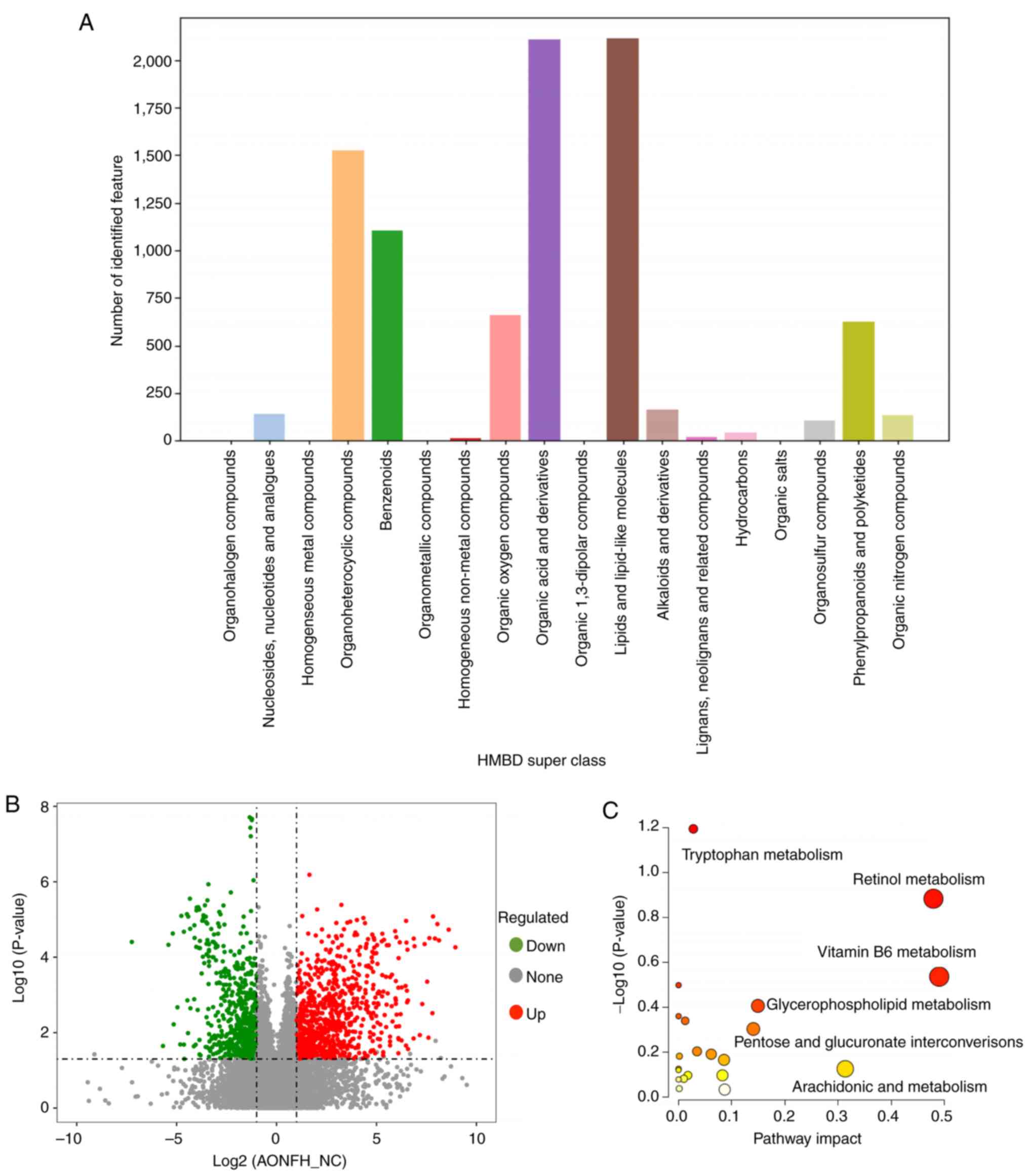
Metabolomic analysis identified 21,486 features and
11,723 metabolites in PIM and 14,155 and 7,576 metabolites in NIM.
The obtained data were used as the batch query against the HMDB for
single-stage mass spectrometry analysis, which annotated 5,098 and
3,689 individual samples with the features identified in PIM and
NIM, respectively. In the HMDB superclass analysis, the most
abundant metabolites were ‘lipids and lipid-like molecules’ and
‘organic acid and derivatives’ (Fig.
5A).
Comparative metabolomic analysis demonstrated clear
differences in fecal metabolite profiles between patients with
AONFH and NCs (Fig. 5B). A total
of 483 significantly upregulated and 396 significantly
downregulated metabolites in PIM, whereas 358 significantly
upregulated and 296 significantly downregulated metabolites in NIM
were identified, in the AONFH group compared with those the control
group (Table SIV). These
metabolites can potentially be regarded as part of various
signaling pathway networks underlying AONFH occurrence.
Metabolite profiling and AONFH-related
pathways
Pathway analysis demonstrated the detailed impact of
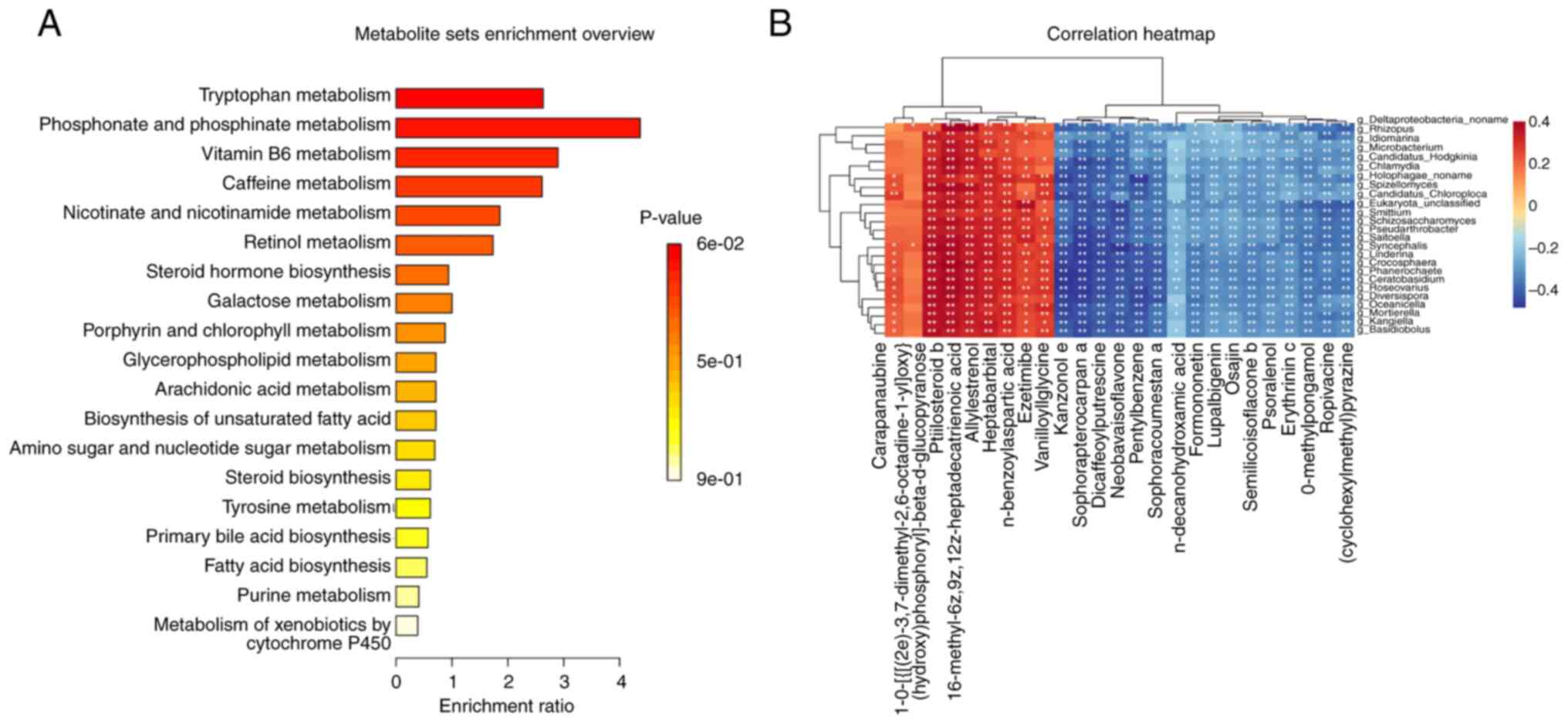
AONFH-related alterations in metabolic networks (Fig. 5C). The most influential metabolic
pathway had a pathway impact of >0.05 and
log10(P-value)>0.3. A total of four metabolic
pathways were identified as being disturbed in the fecal profiles
of patients with AONFH, which included ‘vitamin B6 metabolism’,
‘retinol metabolism’, ‘pentose and glucuronate interconversions’
and ‘glycerophospholipid metabolism’. KEGG enrichment analysis
identified the most abundant metabolic pathways in the two groups
(Fig. 6A). Based on P-values,
pathway impact and enrichment ratios, the top two differentially
expressed metabolic pathways were deemed to be ‘vitamin B6
metabolism’ and ‘retinol metabolism’.
A correlation matrix was next created using Spearman
correlation and correlation network analyses to explore the
potential relationships between changes in the GM and changes in
metabolic product concentrations (Fig.
6B and Table SV). The levels
of ptilosteroid b, 16-methyl-6z,9z,12z-heptadecatrienoic acid,
allylestrenol, heptabarbital, n-benzoylaspartic acid, ezetimibe and
vanilloylglycine were found to be positively correlated with the
abundance of various genera, such as Schizosaccharomyces,
Oceanicella, Basidiobolus, Mortierella and
Roseovarius. The levels of kanzonol E, sophoracoumestan A,
dicaffeoylputrescine, neobavaisoflavone, pentylbenzene,
sophorapterocarpan A, pyrazine, ropivacaine, o-methylpongamol,
erythrinin C and semilicoisoflavone B were negatively correlated
with the abundance of various genera, such as Phanerochaete,
Ceratobasidium, Candidatus Hodgkinia,
Smittium, Schizosaccharomyces and
Syncephalis.
Discussion
GM is an important symbiotic partner that
facilitates the maintenance of physiology in animals and humans. In
addition, it can regulate several aspects of host physiology, such
as nutritional metabolism (5). The
GM has also been reported to be implicated in a number of
conditions, including neurodegenerative diseases (23), cancer (24), obesity (25) and Kashin-Beck disease (26). As the prime pathogenic factor
contributing to AONFH, alcohol is largely metabolized within the
gastrointestinal tract (27).
Alcohol can alter GM composition, impact the gut immune system and
lead to downstream systemic effects on immune system communication
with other organs (28). High
levels of alcohol intake can result in nutrient malabsorption and
deficiencies, including vitamin D, alter the gut microbiome and gut
metabolites, affect the expression of bone metabolism-regulating
hormones, induce osteoclast activation and influence GM composition
(28). In addition,
glucocorticoids can induce the loss of Lactobacillus
animalis and its extracellular vesicles from the gut, which is
associated with the pathogenesis of glucocorticoid-induced ONFH
(29). Therefore, it is possible
that the GM can serve a role in ONFH. However, to the best of our
knowledge, no prior study has systematically investigated the role
of GM and associated gut metabolites in the development of ONFH to
date.
The results from the present study demonstrated that
gut dysbiosis occurred in patients with AONFH, suggesting that
alcohol may participate in AONFH pathogenesis by altering the GM
composition. The 16S rDNA gene sequencing results demonstrated that
Pseudomonas, Pseudomonadaceae, Oscillospiraceae, Firmicutes
and Streptococcus were more abundant in the NC group
compared with those in the AONFH group. By contrast,
Burkholderiaceae, Buekholderiales, Holdemanella,
Erysipelotrichaceae, Klebsiella pneumoniae,
Klebsiella, Proteobacteria and Enterobacterales were more
abundant in the AONFH group compared with those in the NC group.
The metagenomics analysis results demonstrated that
Pseudomonas was significantly less abundant in the AONFH
group compared with those in the NC group.
In previous studies of clinical alcohol use disorder
(AUD), the associated dysbiosis was characterized by a lower
abundance of Bacteroidetes and Akkermansia muciniphila
(30). In animal models of
high-dose alcohol consumption, a decrease in bacterial diversity
was observed, exemplified by fewer Bacteroidetes and Firmicutes
(31,32), coupled with increased
Proteobacter, Proteobacteria and Actinobacter
(31-33).
In particular, Proteobacteria is one of the most abundant phyla in
the human GM and is frequently overrepresented in diseases,
especially in those associated with an inflammatory phenotype, such
as inflammatory bowel disease, asthma and chronic obstructive
pulmonary disease (34). The
present study demonstrated that Proteobacteria were more abundant
in patients with AONFH, suggesting this species to be attributable
to alcohol consumption. Pseudomonas is a member of the
Proteobacteria phylum that has been reported to be associated with
alcohol-related diseases and ONFH. This species is particularly
prevalent in the intestine of rats with alcohol-related liver
injury, where their abundance could be reduced by transplantation
with fecal filtrate from a healthy rat (35). Liu et al (36) previously reported that
Pseudomonas aeruginosa and Pseudomonas putida may be
pathogens in patients with ONFH. However, the present study found
decreased Pseudomonas abundance in patients with AONFH,
suggesting that Pseudomonas abundance was decreased by
alcohol consumption. The discrepancy between this study and Liu
et al (36) may be due to
all kinds of ONFH patients being included in their study, while
only patients with AONFH were included in the present study.
Another reason is that they reported the result at the species
level, while the present study reported them at the genus level.
Klebsiella and Streptococcus form another two members
of the Proteobacteria phylum family. Individuals who consume
excessive amounts of alcohol have previously been found to exhibit
increased susceptibility to lung infection by Streptococcus
pneumoniae and Klebsiella pneumoniae (37). Yuan et al (38) reported that ≤60% of individuals
with non-alcoholic fatty liver disease in a Chinese cohort were
infected with Klebsiella pneumoniae, a bacterial strain that
produces alcohol as a byproduct of glucose. Taken together, these
aforementioned studies suggest that alcohol consumption can
increase susceptibility to Klebsiella pneumoniae infection
and the subsequent excessive endogenous alcohol production due to
GM alteration. Since Klebsiella pneumoniae and
Klebsiella were found to be more abundant in the AONFH group
in the present study, alcohol consumption may likely increase
Klebsiella pneumoniae abundance in this group, resulting in
the excessive production of endogenous alcohol and aggravation of
AONFH pathogenesis.
Firmicutes and Bacteroidetes are two major phyla in
the healthy human GM. They are documented to be involved in colonic
metabolism through a complex metabolic energy-harvesting mechanism
based on cross-feeding and co-metabolism (39). The Firmicutes/Bacteroidetes ratio
has been implicated in the predisposition to several disease states
(40). Wang et al (26) previously reported that patients
with Kashin-Beck disease were characterized by decreased Firmicutes
levels and a significantly decreased Firmicutes/Bacteroidetes
ratio. In addition, Firmicutes abundance was found to be positively
associated with calcium absorption (41), which was significantly decreased in
the presence of alcohol (42), a
finding that was verified by Cheng et al (28). In the present study, it was
demonstrated that Firmicutes levels were decreased in patients with
AONFH, suggesting that alcohol consumption may contribute to AONFH
pathogenesis by decreasing Firmicutes abundance.
ONFH develops because of varying degrees of necrosis
in the local microenvironment (5).
This can in turn lead to a number of pathological changes caused by
erroneous metabolic processes, including intravascular fat
embolism, endovascular coagulation, lipid metabolism, apoptosis and
inhibition of angiogenesis (5). It
can therefore be possible that alterations in certain metabolic
molecular markers are evident in the bloodstream during the early
stages of the ONFH pathological process. In the present study,
several important gut microbial gene functions were identified,
such as CDP-diacylglycerol biosynthesis I/II, L-histidine
biosynthesis and the L-serine and glycine biosynthesis I
superpathway. CDP-diacylglycerol is a critical intermediate in
lipid metabolism, taking part in the synthesis of
phosphatidylglycerol, cardiolipin and phosphatidylinositol
(42). CDP-diacylglycerol synthase
(CDS) produces CDP-diacylglycerol from phosphatidic acid and
cytidine triphosphate (43). CDS
has been reported to serve an important role in various processes,
including mitochondrial function, signal transduction, membrane
trafficking, secretion and cytoskeletal rearrangements (44). In the present study,
CDP-diacylglycerol biosynthesis was identified as one of the most
important gut microbial gene functions altered in patients with
AONFH. Therefore, CDP-diacylglycerol and CDS may serve key roles in
AONFH pathogenesis, but further studies are needed to investigate
the underlying mechanism of this process.
A previous bioinformatics analysis performed by Yang
et al (1) revealed that
histidine, cysteine and methionine metabolism were associated with
ONFH pathogenesis, where subsequent metabolic pathway analysis
found that L-histidine is a key molecule in histidine metabolism
and L-serine has a central role in cysteine and methionine
metabolism. Histidine is an essential amino acid in mammals that
can regulate gene expression, biological activity of proteins and
signal transduction (45). By
contrast, L-serine has been reported to promote osteoclast
formation and therefore induce bone resorption (46). These aforementioned previous
findings support the microbial gene function prediction results of
the present study, suggesting that L-histidine and L-serine may
serve regulatory roles in the pathology of AONFH.
In the present study, it was found that glycine
biosynthesis may participate in the pathological process of ONFH.
Betaine is a trimethyl derivative of glycine and an important
nutrient for humans, which regulates a series of vital biological
processes, including oxidative stress, inflammatory responses,
osteoblast differentiation and apoptosis (47-49).
Yang et al (50) previously
reported that betaine is a potential pharmacotherapy option for
alcohol-induced ONFH in vivo, since it was observed to exert
a protective role against ethanol-induced suppression of
osteogenesis and mineralization of human bone marrow mesenchymal
stem cells.
Vitamin A is vital for a variety of bodily
functions, including gene expression, reproduction, embryonic
development and immune function (51). Whilst insufficient vitamin A intake
can cause a number of adverse effects, including low bone density,
excessive vitamin A consumption can also cause bone loss and
increase the risk of fracture, leaving a narrow range of optimal
dosage (52). As the biologically
active form of vitamin A, retinol can enhance osteoblast
proliferation and hinder osteoclast resorption (53). It has previously been reported that
chronic alcohol consumption can mediate adverse effects on vitamin
A metabolism, which can be directly associated with the development
of alcohol-induced disease (54).
In the present study, retinol metabolism was identified to be one
of the most important AONFH-related pathways. Therefore, alcohol
may disturb retinol metabolism in this disease, forming a part of
AONFH pathogenesis. Vitamin B6 deficiency is common in individuals
with alcoholism (55). This
condition was previously found to be a potential risk factor for
osteoporosis and bone fracture (56). Additionally, serum vitamin B6
concentration was observed to significantly associate inversely
with the concentration of the bone resorption marker parathyroid
hormone, whilst significantly associating positively with the
concentration of 25-hydroxyvitamin D (57). Vitamin B6 deficiency may affect the
mechanical property of the bone due to reduced cortical thickness,
trabecular osteoid and coarse trabeculation (57). Therefore, alcohol may potentially
induce vitamin B6 deficiency, which can disturb the balance between
bone resorption and bone reconstruction in AONFH.
Dysfunction of lipid metabolism is reported to serve
a crucial role in ONFH. A previous study reported different serum
lipidomic profiles between patients with SONFH and healthy
controls, where glycerophospholipids occupied a large part of this
difference (58). In addition, it
was reported that glycerophospholipids were distinguished in the
bone trabecula and plasma of patients with ONFH compared with those
in healthy controls, most glycerophospholipids were upregulated in
patients with ONFH (59,60). Mei et al (61) previously reported that
glycerophospholipid metabolism was the metabolic pathway that was
the most significantly altered in rabbits with SONFH, suggesting it
to be a potential pathway for the targeted intervention against
SONFH. In the future, the detailed mechanism of how
glycerophospholipid metabolism impacts AONFH requires further
study.
In conclusion, the present study demonstrated that
gut dysbiosis occurred in patients with AONFH, possibly causing
alterations in gut metabolites. This altered GM profile and
metabolites may serve as potential diagnostic markers for AONFH,
even if the GM metabolites are from host cells or food in the
intestinal tract. In particular, analysis of the interactions among
alcohol, GM, metabolites and AONFH discussed in the present study
may enhance the understanding of the mechanisms underlying AONFH
pathogenesis. The findings from the present study regarding GM and
metabolite changes may also facilitate the discovery of novel
therapeutic targets. However, several research directions remain
that require further elucidation. Detection of additional
biochemical indicators associated with bone metabolism is required
to understand the relationship between the GM and biochemical
indicators. This may in turn deepen the understanding into the role
of GM in AONFH pathogenesis. In addition, since probiotics,
prebiotics or symbiotics can regulate the GM (26), future studies are required to
investigate the relationship among such substances, gut dysbiosis
and AONFH.
Supplementary Material
Complexity of species diversity of
each group was analyzed by (A) Chao1, (B) observed OTUs, (C)
Shannon and (D) Simpson indices. AONFH, alcohol-induced
osteonecrosis of the femoral head; NC, negative control; OTU,
operational taxonomic unit.
Species diversity differences between
the AONFH and NC groups were estimated by (A) Chao1, (B) observed
OTUs, (C) Shannon and (D) Simpson indices. AONFH, alcohol-induced
osteonecrosis of the femoral head; NC, negative control; OTU,
operational taxonomic unit.
Functional analysis of dysregulated
gut microbial gene in patients with AONFH compared with NCs by
Clusters of Orthologous Genes analysis. AONFH, alcohol-induced
osteonecrosis of the femoral head; NC, negative control.
Functional analysis of dysregulated
gut microbial gene in patients with AONFH compared with NCs by
Kyoto Encyclopedia of Genes and Genomes analysis. AONFH,
alcohol-induced osteonecrosis of the femoral head; NC, negative
control.
Functional analysis of dysregulated
gut microbial gene in patients with AONFH and NCs by Kyoto
Encyclopedia of Genes and Genomes orthology analysis. AONFH,
alcohol-induced osteonecrosis of the femoral head; NC, negative
control.
Species diversity differences between
the AONFH and NC groups were estimated by (A) Chao1, (B) observed
species, (C) Shannon and (D) Simpson indices between the NC group
and the AONFH group. ***P<0.005. AONFH,
alcohol-induced osteonecrosis of the femoral head; NC, negative
control.
Differentially expressed unigenes
between the alcohol-induced osteonecrosis of the femoral head and
negative control groups were detected by (A) GO functional
classification analysis, (B) GO enrichment analysis and (C) Kyoto
Encyclopedia of Genes and Genomes analysis. AONFH, alcohol-induced
osteonecrosis of the femoral head; NC, negative control; GO, Gene
Ontology.
Clean 16S rDNA data of all
samples.
Different abundance in microbial
composition between the AONFH and NC groups at the species
level.
Metabolic pathways and unigene number
between the patients with alcohol-induced osteonecrosis of the
femoral head and negative controls according to Kyoto Encyclopedia
of Genes and Genomes analysis.
Differentially regulated metabolites
between patients with AAONFH and NCs in positive-ion mode and
negative-ion mode.
Potential relationships between
changes in the gut microbiome and changes in metabolic product
concentrations.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by the National Natural
Science Foundation of China (grant no. 82074472), the Zhejiang
Provincial Natural Science Foundation (grant no. LQ22H060003) and
the Heluo Youth Talent Promotion Project (grant no.
2022HLTJ15).
Availability of data and materials
The data generated in the present study may be
found in the China National Centre for Bioinformation repository
for metagenomic sequencing (accession no. PRJCA021196; https://bigd.big.ac.cn/gsa/browse/CRA014109), for 16S
rDNA analysis (accession no. PRJCA022064; https://bigd.big.ac.cn/gsa/browse/CRA014110) and
metabolomics (accession no. PRJCA022518; https://ngdc.cncb.ac.cn/omix/release/OMIX005517).
Authors' contributions
CY, YL and BX contributed to the study conception
and design. MM, YY and JG performed the experiments. Data
collection and analysis were performed by CY, HL and YL. CY and BX
confirm the authenticity of all the raw data. The manuscript was
drafted by CY and BX and all authors commented on previous versions
of the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was approved by the ethics
committee of Luoyang Orthopedic-Traumatological Hospital of Henan
Province (approval no. KY2021-007-01) and the study was performed
in accordance with The Declaration of Helsinki. All participants
provided written informed consent for participation into the
present study and the study protocols followed the ethical
guidelines of The Declaration of Helsinki.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Yang G, Zhao G, Zhang J, Gao S, Chen T,
Ding S and Zhu Y: Global urinary metabolic profiling of the
osteonecrosis of the femoral head based on UPLC-QTOF/MS.
Metabolomics. 15(26)2019.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Tan B, Li W, Zeng P, Guo H, Huang Z, Fu F,
Gao H, Wang R and Chen W: Epidemiological study based on china
osteonecrosis of the femoral head database. Orthop Surg.
13:153–160. 2021.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Yan Y, Wang J, Huang D, Lv J, Li H, An J,
Cui X and Zhao H: Plasma lipidomics analysis reveals altered lipids
signature in patients with osteonecrosis of the femoral head.
Metabolomics. 18(14)2022.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Seamon J, Keller T, Saleh J and Cui Q: The
pathogenesis of nontraumatic osteonecrosis. Arthritis.
2012(601763)2012.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Pouya F and Kerachian MA: Avascular
necrosis of the femoral head: Are any genes involved? Arch Bone Jt
Surg. 3:149–55. 2015.PubMed/NCBI
|
|
6
|
Ticinesi A, Lauretani F, Milani C,
Nouvenne A, Tana C, Del Rio D, Maggio M, Ventura M and Meschi T:
Aging gut microbiota at the Cross-Road between nutrition, physical
frailty, and sarcopenia: Is there a gut-muscle axis? Nutrients.
9(1303)2017.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Lucas S, Omata Y, Hofmann J, Böttcher M,
Iljazovic A, Sarter K, Albrecht O, Schulz O, Krishnacoumar B,
Krönke G, et al: Short-chain fatty acids regulate systemic bone
mass and protect from pathological bone loss. Nat Commun.
9(55)2018.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Li JY, Chassaing B, Tyagi AM, Vaccaro C,
Luo T, Adams J, Darby TM, Weitzmann MN, Mulle JG, Gewirtz AT, et
al: Sex steroid deficiency-associated bone loss is microbiota
dependent and prevented by probiotics. J Clin Invest.
126:2049–2063. 2016.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Wei X, Pushalkar S, Estilo C, Wong C,
Farooki A, Fornier M, Bohle G, Huryn J, Li Y, Doty S and Saxena D:
Molecular profiling of oral microbiota in jawbone samples of
bisphosphonate-related osteonecrosis of the jaw. Oral Dis.
18:602–612. 2012.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Wang SC, Chen YC, Chen SJ, Lee CH and
Cheng CM: Alcohol addiction, gut microbiota, and alcoholism
treatment: A review. Int J Mol Sci. 21(6413)2020.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Gabriel S, Ziaugra L and Tabbaa D: SNP
genotyping using the Sequenom MassARRAY iPLEX platform. Curr Protoc
Hum Genet: Chapter 2: Unit 2.12, 2009.
|
|
12
|
Logue JB, Stedmon CA, Kellerman AM,
Nielsen NJ, Andersson AF, Laudon H, Lindström ES and Kritzberg ES:
Experimental insights into the importance of aquatic bacterial
community composition to the degradation of dissolved organic
matter. ISME J. 10:533–545. 2015.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Callahan BJ, McMurdie PJ, Rosen MJ, Han
AW, Johnson AJ and Holmes SP: DADA2: High-resolution sample
inference from Illumina amplicon data. Nat Methods. 13:581–583.
2016.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Quast C, Pruesse E, Yilmaz P, Gerken J,
Schweer T, Yarza P, Peplies J and Glöckner FO: The SILVA ribosomal
RNA gene database project: Improved data processing and web-based
tools. Nucleic Acids Res. 41 (Database Issue):D590–D596.
2013.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Langmead B and Salzberg SL: Fast
gapped-read alignment with Bowtie 2. Nat Methods. 9:357–359.
2012.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Peng Y, Leung HC, Yiu SM and Chin FY:
IDBA-UD: A de novo assembler for single-cell and metagenomic
sequencing data with highly uneven depth. Bioinformatics.
28:1420–1428. 2012.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Zhu W, Lomsadze A and Borodovsky M: Ab
initio gene identification in metagenomic sequences. Nucleic Acids
Res. 38(e132)2010.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Li W and Godzik A: Cd-hit: A fast program
for clustering and comparing large sets of protein or nucleotide
sequences. Bioinformatics. 22:1658–1659. 2006.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Buchfink B, Reuter K and Drost HG:
Sensitive protein alignments at tree-of-life scale using DIAMOND.
Nat Methods. 18:366–368. 2021.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Delinsky DC, Hill KT, White CA and
Bartlett MG: Quantitation of the large polypeptide glucagon by
protein precipitation and LC/MS. Biomed Chromatogr. 18:700–705.
2004.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Zheng JB, Xie YZ, Li F, Zhou Y, Qi LQ, Liu
LB and Chen Z: Lactoferrin improves cognitive function and
attenuates brain senescence in aged mice. J Functional Foods.
65(103736)2019.
|
|
22
|
Long NP, Heo D, Kim HY, Kim TH, Shin JG,
Lee A and Kim DH: Metabolomics-guided global pathway analysis
reveals better insights into the metabolic alterations of breast
cancer. J Pharm Biomed Anal. 202(114134)2021.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Zhang H, Chen Y, Wang Z, Xie G, Liu M,
Yuan B, Chai H, Wang W and Cheng P: Implications of gut microbiota
in neurodegenerative diseases. Front Immunol.
13(785644)2022.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Vivarelli S, Falzone L, Leonardi GC,
Salmeri M and Libra M: Novel insights on gut microbiota
manipulation and immune checkpoint inhibition in cancer (Review).
Int J Oncol. 59(75)2021.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Li R, Huang X, Liang X, Su M, Lai KP and
Chen J: Integrated omics analysis reveals the alteration of gut
microbe-metabolites in obese adults. Brief Bioinform.
22(bbaa165)2021.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Wang X, Ning Y, Li C, Gong Y, Huang R, Hu
M, Poulet B, Xu K, Zhao G, Zhou R, et al: Alterations in the gut
microbiota and metabolite profiles of patients with Kashin-Beck
disease, an endemic osteoarthritis in China. Cell Death Dis.
12(1015)2021.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Bishehsari F, Magno E, Swanson G, Desai V,
Voigt RM, Forsyth CB and Keshavarzian A: Alcohol and Gut-Derived
inflammation. Alcohol Res. 38:163–171. 2017.PubMed/NCBI
|
|
28
|
Cheng M, Tan B, Wu X, Liao F, Wang F and
Huang Z: Gut microbiota is involved in alcohol-induced osteoporosis
in young and old rats through immune regulation. Front Cell Infect
Microbiol. 11(636231)2021.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Chen CY, Rao SS, Yue T, Tan YJ, Yin H,
Chen LJ, Luo MJ, Wang Z, Wang YY, Hong CG, et al:
Glucocorticoid-induced loss of beneficial gut bacterial
extracellular vesicles is associated with the pathogenesis of
osteonecrosis. Sci Adv. 8(eabg8335)2022.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Caslin B, Mohler K, Thiagarajan S and
Melamed E: Alcohol as friend or foe in autoimmune diseases: A role
for gut microbiome? Gut Microbes. 13(1916278)2021.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Mutlu EA, Gillevet PM, Rangwala H,
Sikaroodi M, Naqvi A, Engen PA, Kwasny M, Lau CK and Keshavarzian
A: Colonic microbiome is altered in alcoholism. Am J Physiol
Gastrointest Liver Physiol. 302:G966–G978. 2012.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Dubinkina VB, Tyakht AV, Odintsova VY,
Yarygin KS, Kovarsky BA, Pavlenko AV, Ischenko DS, Popenko AS,
Alexeev DG, Taraskina AY, et al: Links of gut microbiota
composition with alcohol dependence syndrome and alcoholic liver
disease. Microbiome. 5(141)2017.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Yan AW and Schnabl B: Bacterial
translocation and changes in the intestinal microbiome associated
with alcoholic liver disease. World J Hepatol. 4:110–118.
2012.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Rizzatti G, Lopetuso LR, Gibiino G, Binda
C and Gasbarrini A: Proteobacteria: A common factor in human
diseases. Biomed Res Int. 2017(9351507)2017.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Yu L, Wang L, Yi H and Wu X: Beneficial
effects of LRP6-CRISPR on prevention of alcohol-related liver
injury surpassed fecal microbiota transplant in a rat model. Gut
Microbes. 11:1015–1029. 2020.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Liu C, Li W, Zhang C, Pang F and Wang DW:
Previously unexplored etiology for femoral head necrosis:
Metagenomics detects no pathogens in necrotic femoral head tissue.
World J Clin Cases. 10:2138–2146. 2022.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Raju SV, Painter RG, Bagby GJ, Nelson S
and Wang G: Response of differentiated human airway epithelia to
alcohol exposure and klebsiella pneumoniae challenge. Med Sci
(Basel). 1:2–19. 2013.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Yuan J, Chen C, Cui J, Lu J, Yan C, Wei X,
Zhao X, Li N, Li S, Xue G, et al: Fatty liver disease caused by
high-alcohol-producing klebsiella pneumoniae. Cell Metab.
30:675–688.e7. 2019.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Cheng L, Chen Y, Zhang X, Zheng X, Cao J,
Wu Z, Qin W and Cheng K: A metagenomic analysis of the modulatory
effect of Cyclocarya paliurus flavonoids on the intestinal
microbiome in a high-fat diet-induced obesity mouse model. J Sci
Food Agric. 99:3967–3975. 2019.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Jandhyala SM, Talukdar R, Subramanyam C,
Vuyyuru H, Sasikala M and Nageshwar Reddy D: Role of the normal gut
microbiota. World J Gastroenterol. 21:8787–8803. 2015.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Whisner CM, Martin BR, Nakatsu CH, Story
JA, MacDonald-Clarke CJ, McCabe LD, McCabe GP and Weaver CM:
Soluble corn fiber increases calcium absorption associated with
shifts in the gut microbiome: A randomized Dose-Response trial in
Free-Living pubertal females. J Nutr. 146:1298–1306.
2016.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Qamar N, Castano D, Patt C, Chu T,
Cottrell J and Chang SL: Meta-analysis of alcohol induced gut
dysbiosis and the resulting behavioral impact. Behav Brain Res.
376(112196)2019.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Jennings W and Epand RM:
CDP-diacylglycerol, a critical intermediate in lipid metabolism.
Chem Phys Lipids. 230(104914)2020.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Wang X, Devaiah SP, Zhang W and Welti R:
Signaling functions of phosphatidic acid. Prog Lipid Res.
45:250–278. 2006.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Klumpp S and Krieglstein J:
Phosphorylation and dephosphorylation of histidine residues in
proteins. Eur J Biochem. 269:1067–1071. 2002.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Ogawa T, Ishida-Kitagawa N, Tanaka A,
Matsumoto T, Hirouchi T, Akimaru M, Tanihara M, Yogo K and Takeya
T: A novel role of L-serine (L-Ser) for the expression of nuclear
factor of activated T cells (NFAT)2 in receptor activator of
nuclear factor kappa B ligand (RANKL)-induced osteoclastogenesis in
vitro. J Bone Miner Metab. 24:373–379. 2006.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Veskovic M, Mladenovic D, Milenkovic M,
Tosic J, Borozan S, Gopcevic K, Labudovic-Borovic M, Dragutinovic
V, Vucevic D, Jorgacevic B, et al: Betaine modulates oxidative
stress, inflammation, apoptosis, autophagy, and Akt/mTOR signaling
in methionine-choline deficiency-induced fatty liver disease. Eur J
Pharmacol. 848:39–48. 2019.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Shi H, Wang XL, Quan HF, Yan L, Pei XY,
Wang R and Peng XD: Effects of betaine on LPS-Stimulated activation
of microglial M1/M2 phenotypes by suppressing TLR4/NF-κB pathways
in N9 cells. Molecules. 24(367)2019.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Li C, Wang Y, Li L, Han Z, Mao S and Wang
G: Betaine protects against heat exposure-induced oxidative stress
and apoptosis in bovine mammary epithelial cells via regulation of
ROS production. Cell Stress Chaperones. 24:453–460. 2019.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Yang Q, Yin W, Chen Y, Zhu D, Yin J, Zhang
C and Gao Y: Betaine alleviates alcohol-induced osteonecrosis of
the femoral head via mTOR signaling pathway regulation. Biomed
Pharmacother. 120(109486)2019.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Conaway HH, Henning P and Lerner UH:
Vitamin a metabolism, action, and role in skeletal homeostasis.
Endocr Rev. 34:766–797. 2013.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Yee MMF, Chin KY, Ima-Nirwana S and Wong
SK: Vitamin A and Bone Health: A review on current evidence.
Molecules. 26(1757)2021.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Vu AA, Kushram P and Bose S: Effects of
Vitamin A (Retinol) release from calcium phosphate matrices and
Porous 3D printed scaffolds on bone cell proliferation and
maturation. ACS Appl Bio Mater. 5:1120–1129. 2022.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Clugston RD and Blaner WS: The adverse
effects of alcohol on vitamin A metabolism. Nutrients. 4:356–371.
2012.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Tobias SL, van der Westhuyzen J, Davis RE,
Icke GC and Atkinson PM: Alcohol intakes and deficiencies in
thiamine and vitamin B6 in black patients with cardiac failure. S
Afr Med J. 76:299–302. 1989.PubMed/NCBI
|
|
56
|
Wang J, Chen L, Zhang Y, Li CG, Zhang H,
Wang Q, Qi X, Qiao L, Da WW, Cui XJ, et al: Association between
serum vitamin B6 concentration and risk of osteoporosis in the
middle-aged and older people in China: A cross-sectional study. BMJ
Open. 9(e028129)2019.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Masse PG, Pritzker KP, Mendes MG, Boskey
AL and Weiser H: Vitamin B6 deficiency experimentally-induced bone
and joint disorder: Microscopic, radiographic and biochemical
evidence. Br J Nutr. 71:919–932. 1994.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Wang XY, Zhang LL, Jiang C, Hua BX, Ji ZF,
Fan WS, Gong LJ, Zhu L, Wang XD and Yan ZQ: Altered lipidomic
profiles in patients with and without osteonecrosis of the femoral
head after 1-month glucocorticoid treatment. Clin Transl Med.
11(e298)2021.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Zhu W, Chen T, Ding S, Gang Y, Xu Z, Xu K,
Zhang S, Ma T and Zhang J: Metabolomic study of the bone trabecula
of osteonecrosis femoral head patients based on UPLC-MS/MS.
Metabolomics. 12(48)2016.
|
|
60
|
Liu X, Li Q, Sheng J, Hu B, Zhu Z, Zhou S,
Yin J, Gong Q, Wang Y and Zhang C: Unique plasma metabolomic
signature of osteonecrosis of the femoral head. J Orthop Res.
34:1158–1167. 2016.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Mei R, Chen D, Zhong D, Li G, Lin S, Zhang
G, Chen K and Yu X: Metabolic profiling analysis of the effect and
mechanism of gushiling capsule in rabbits with
Glucocorticoid-Induced osteonecrosis of the femoral head. Front
Pharmacol. 13(845856)2022.PubMed/NCBI View Article : Google Scholar
|