Introduction
Ischemic heart disease (IHD) is a major cause of
global mortality and is a substantial burden on individuals and
healthcare systems (1).
Conventional clinical strategies for treating acute myocardial
infarction include coronary artery intervention and other
reperfusion therapies to minimize the ischemic time (2). Although timely reperfusion
significantly reduces acute mortality in patients with ST-segment
elevation myocardial infarction (3), partial cell damage occurs during the
reperfusion phase, known as ischemia/reperfusion (I/R) injury,
which has garnered significant attention (4). The mechanisms underlying myocardial
I/R injury are intricate, involving multiple cellular events such
as oxidative stress, intracellular calcium overload cell apoptosis
(5). Notably, the excess
generation of reactive oxygen species (ROS) by mitochondria during
I/R is a crucial trigger of the aforementioned cellular events
(6). The mitochondrial electron
transport chain (ETC), which is damaged during ischemia, is
associated with mitochondrial dysfunction and excessive production
of ROS. During reperfusion, damaged mitochondria undergo further
injury, activating programmed cell death pathways in cardiomyocytes
(7,8).
Translocator protein (TSPO) is an 18 kDa protein
located on the outer mitochondrial membrane (OMM) (9). It is found in the cardiovascular
system and is associated with both myocardial injury and protection
(10,11). TSPO was first discovered in human
cortical tissues (12), and its
primary function had been revealed to involve the transport of
cholesterol from the OMM to the inner mitochondrial membrane
(13). TSPO actively participates
in ATP and ROS production in mitochondria and triggers cell
apoptosis (14). Gatliff et
al (15) and Meng et al
(16) reported that TSPO
overexpression reduces mitochondrial coupling and promotes the
overproduction of ROS in canine mammary gland epithelia. TSPO has
also been shown to inhibit mitophagy, preventing the clearance of
damaged mitochondria (17). It is
apparent that a severe consequence of myocardial I/R is oxidative
injury, which leads to mitochondrial dysfunction (18). Based on the aforementioned
observations, it has been hypothesized that TSPO may play a vital
role in myocardial I/R injury by regulating mitochondrial
homeostasis.
In the present study, an in vitro model of
anoxia/reoxygenation (A/R) injury was established using H9c2
cardiomyocytes to simulate myocardial I/R injury. It was observed
that TSPO expression was significantly increased following A/R
injury. This study aims to explore the function and mechanisms of
TSPO in myocardial ischemia-reperfusion injury. Taken together,
targeting TSPO could be a potential strategy for alleviating
myocardial I/R injury.
Materials and methods
H9c2 cardiomyocyte culture
H9c2 cardiomyocytes were cultured following the
methodology described by Pooja et al (19). Briefly, H9c2 cardiomyocytes were
obtained from The Cell Bank of Type Culture Collection of The
Chinese Academy of Sciences and cultured in DMEM supplemented with
10% FBS (Gibco; Thermo Fisher Scientific, Inc.) and 1%
penicillin/streptomycin solution (Gibco; Thermo Fisher Scientific,
Inc.). The cells were maintained in a humidified incubator at 37˚C
supplied with 5% CO2 air (Thermo Fisher Scientific,
Inc.), and the culture medium was replaced every 2 days.
RNA interference
In the present study, three small interfering
(si)RNA constructs were obtained from Shanghai GenePharma
Biotechnology Co., Ltd. and transfected using siRNA-Mate (Shanghai
GenePharma Biotechnology Co., Ltd.). After confirmation of
knockdown using western blotting, the si-RNA3-TSPO sequence, which
exhibited the best knockdown efficiency, was selected for further
experiments. Briefly, for si-TSPO and A/R-si-TSPO group, 3 µl siRNA
and 4 µl siRNA-mate were diluted in 200 µl Opti-MEM (Gibco; Thermo
Fisher Scientific, Inc.). For si-NC and A/R-si-NC group, 3 µl
siRNA-negative control and 4 µl siRNA-mate were diluted in 200 µl
Opti-MEM (Gibco; Thermo Fisher Scientific, Inc.), incubated at room
temperature for 5 min, mixed thoroughly and incubated for 15 min.
H9c2 cardiomyocytes were treated with the mixture and incubated at
37˚C for 72 h. After incubation, cells in the si-TSPO group and
si-NC group were replaced with complete culture medium and placed
in a culture incubator for cultivation. Cells in the A/R-si-TSPO
group and A/R-si-NC group were immediately subjected to
anoxia/reoxygenation stimulation and were then used together for
subsequent experiments. The sequences of these siRNA fragments are
shown in Table I, and the
knockdown efficiency is shown in Fig.
S1.
 | Table ISequences of three pairs of TSPO gene
interference fragments and negative control. |
Table I
Sequences of three pairs of TSPO gene
interference fragments and negative control.
| siRNA | Sequence
(5'-3') |
|---|
|
siRNA1-TSPO-sense |
GCUCCUACAUAAUCUGGAATT |
|
siRNA1-TSPO-anti-sense |
UUCCAGAUUAUGUAGGAGCTT |
|
siRNA2-TSPO-sense |
CCAUGCUCAACUACUAUGUTT |
|
siRNA2-TSPO-anti-sense |
ACAUAGUAGUUGAGCAUGGTT |
|
siRNA3-TSPO-sense |
GGGCCUUUAAAGCUAAAUATT |
|
siRNA3-TSPO-anti-sense |
UAUUUAGCUUUAAAGGCCCTT |
| siRNA-negative
control-sense |
UUCUCCGAACGUGUCACGUTT |
| siRNA-negative
control-anti-sense |
ACGUGACACGUUCGGAGAATT |
Establishment of the in vitro
anoxia/reoxygenation (A/R) model
The A/R model was established following a procedure
described by Tong et al (20) with modifications. Briefly, cells in
the A/R group were subjected to anoxic treatment for 3 h in
glucose-free DMEM (cat. no. 11966-025; Gibco; Thermo Fisher
Scientific, Inc.) under oxygen-depleted conditions. For Hypoxia,
the cells were placed in an anoxic chamber with a deoxygenation bag
(AnaeroPack™, Mitsubishi Gas Chemical Company, Inc.) and
then reoxygenated for 2 h with complete medium in a humidified
incubator at 37˚C supplied with 5% CO2 air. After 2 h of
reoxygenation treatment, the cells were harvested and the medium
was collected immediately for subsequent analysis.
Flow cytometry
According to the instructions of the reagent kit
(cat. no. WLA001a; Wanleibio Co., Ltd.), flow cytometry was used to
detect cell apoptosis. After specific treatment, the cells were
digested with 0.25% trypsin-EDTA (1x) (Gibco; Thermo Fisher
Scientific, Inc.) and collected by centrifugation at 100 x g for 5
min at room temperature. Subsequently, the cells were resuspended
in 500 µl binding buffer (cat. no. WLA001a; Wanleibio Co., Ltd.).
Propidium iodide and annexin V conjugated with fluorescein
isothiocyanate (cat. no. WLA001a; Wanleibio Co., Ltd.) were added
to the cells and incubated in the dark at room temperature for 15
min. A flow cytometer (model no. B73613, DxFLEX; Beckman Coulter,
Inc.) was then used for analysis, The flow cytometry results were
analyzed using FlowJo™ Software (version 10.8.1; BD Life
Sciences); three independent experiments were performed, and
samples were assessed in triplicate.
Detection of mitochondrial membrane
potential
A Mitochondrial Membrane Potential Assay Kit with
JC-1 (cat. no. C2006; Beyotime Institute of Biotechnology) was used
according to the manufacturer's protocol. JC-1 working solution was
added to H9c2 cells at a dilution of 1:1,000, and the cells were
incubated at 37˚C for 20 min. Cells were subsequently washed three
times with the JC-1 buffer and then observed using a fluorescence
microscope.
ROS staining
Intracellular ROS levels and mitochondrial ROS
(mtROS) generation were detected using DCFH-DA (cat. no. S0033S;
Beyotime Institute of Biotechnology) and MitoSOX, respectively
(cat. no. M36008; Invitrogen; Thermo Fisher Scientific, Inc.).
Cells on slides were treated with 10 µM DCFH-DA for 20 min at 37˚C
in the dark in a humid chamber. Subsequently, they were treated
with MitoSOX red mitochondrial superoxide indicator at a dilution
of 1:1,000 in a dark and humid room at 37˚C for 10 min. The number
of ROS-positive cells in four randomly selected fields of view was
observed using a fluorescence microscope.
Cell viability assay
After specific treatments, the cells were digested
as above, counted, and then transferred to a 96-well plate. A total
of 10 µl CCK8 reagent (cat. no. K1018; APExBIO Technology LLC) was
added to each well, and the plate was incubated at 37˚C for 1 h.
The enzymatic activity was then measured using a microplate reader
at a wavelength of 450 nm.
Determination of LDH, SOD, MDA, and
ATP levels
For determination of lactate dehydrogenase (LDH)
levels, 0.1 ml culture medium was collected from each experimental
group immediately following A/R treatment and analyzed according to
the manufacturer's protocol (cat. no. A020-2; Nanjing Jiancheng
Bioengineering Institute). Subsequently, the cells were washed
twice with PBS and, after complete removal of PBS, lysed with RIPA
lysis buffer (cat. no. P0013D; Beyotime Institute of
Biotechnology). The lysates were then incubated in an ice bath for
10 min, followed by centrifugation at 13,400 g for an additional 10
min at 4˚C to obtain the supernatant. The resulting supernatant was
used to measure the levels of superoxide dismutase (SOD; cat. no.
A001-3-2; Nanjing Jiancheng Bioengineering Institute),
malondialdehyde (MDA; cat. no. A003-1-1; Nanjing Jiancheng
Bioengineering Institute), and ATP (cat. no. A095-1-1; Nanjing
Jiancheng Bioengineering Institute) according to the manufacturer's
instructions.
Immunofluorescence (IF) staining
Slides with cells were fixed with 4% neutral
formaldehyde for 20 min at 4˚C. After washing with 1x PBS, the cell
slides were treated with 0.1% Triton X-100 and 3% goat serum (cat.
no. ZLI-9022; OriGene Technologies, Inc.) for 5 min at 4˚C. All
antibodies were diluted using an antibody dilution buffer (cat. no.
ZLI-9030; OriGene Technologies, Inc.) at a ratio of 1:200. The
cells were then incubated with antibodies against ATP synthase F1 β
subunit (ATP5B; cat. no. CL594-6660; ProteinTech Group, Inc.),
activating transcription factor 6 (ATF6; cat. no. 66563-1-Ig;
ProteinTech Group, Inc.), TSPO (cat. no. ab109497; Abcam) and LC3B
(cat. no. ab51520; Abcam) overnight at 4˚C. Subsequently, the
slides were incubated with chicken anti-rabbit IgG (H+L)
cross-adsorbed secondary antibody (Alexa Fluor™ 488;
cat. no. A21441; Invitrogen, Thermo Fisher Scientific, Inc.) or
F(ab')2-goat anti-mouse IgG (H+L) cross-adsorbed secondary antibody
(Alexa Fluor™ 546; cat. no. A11018; Invitrogen Thermo
Fisher Scientific, Inc.) at room temperature for 1 h. Finally, the
coverslip was sealed with DAPI Fluoromount-G™ (cat. no.
36308ES20, Shanghai Yeasen Biotechnology Co., Ltd.), and the cells
were observed using a fluorescence microscope.
Co-immunoprecipitation (Co-IP)
Co-IP was performed using a rProtein A/G Magnetic
IP/Co-IP Kit (cat. no. AM001-01; ACE Biotechnology) according to
the manufacturer's instructions. ATF6 (cat. no. 24169-1-AP;
ProteinTech Group, Inc.), TSPO (cat. no. ab109497; Abcam), rabbit
IgG control polyclonal antibody (cat. no. 30000-0-AP; ProteinTech
Group, Inc.), and 20 µl magnetic beads each groups were mixed at
4˚C for 10 h. The mixture was then placed on a magnetic stand, and
after separation, the supernatant was discarded. The magnetic beads
were washed with 500 µl lysis buffer. After subjecting H9c2
cardiomyocytes to A/R, a lysis buffer was used to extract total
protein. The protein concentration was determined using a BCA
protein assay kit (cat. no. KGP902; Nanjing KeyGen Biotech Co.,
Ltd.). A portion of the extracted protein, approximately 100 µg,
was used as the input group. Divide the remaining protein into
groups of 400 µg, add them to EP tubes containing magnetic beads
incubated with antibodies, and incubate them in a mixed spin at 4˚C
for 6 h. The supernatant was then discarded, and the magnetic beads
were washed twice with 500 µl lysis buffer. Finally, 5x loading
buffer (cat. no. P0015; Beyotime Institute of Biotechnology) was
diluted to 1x with lysis buffer, and the magnetic beads were heated
in 1x loading buffer at 100˚C for 5 min. Western blotting was then
performed for detection.
Reverse transcription-quantitative PCR
(RT-qPCR)
RT-qPCR analysis for TSPO gene expression: After
specific treatments, samples were collected using Trizol reagent
(cat. no. 10296010CN; Thermo Fisher Scientific, Inc.) to quantify
the TSPO gene expression. The following primers were used for TSPO:
Forward primer (5'-3'), CTGCCCGCTTGCTGTATCCTTAC; and the reverse
primer (5'-3'), CGACCAGAGTTATCACGCCATAC. GAPDH was used as an
internal control: Forward primer (5'-3'), ACAGCAACAGGGTGGTGGAC; and
the reverse primer (5'-3'), TTTGAGGGTGCAGCGAACTT. For the isolation
of total RNA the RNA GeneJET RNA Purification kit (cat. no. K0731;
Thermo Fisher Scientific, Inc.) was used, Dilute RNA to 200 ng/µl.
And cDNA synthesis was used cDNA Synthesis SuperMix (cat. no.
11141ES60; Shanghai Yeasen Biotechnology Co., Ltd.). Add 5 µl of
diluted RNA, 2 µl of RNase free H2O, 2 µl of 5x gDNA Eraser buffer,
and 1 µl of gDNA Eraser; Keep at 42˚C for 2 min to remove genomic
DNA and then cool to 4˚C for cDNA synthesis; Then add 4 µl of
RNA-free H2O, 4 µl of 5x PrimeScript Buffer 2, 1 µl of PrimeScript
RT Enzyme Mix, and 1 µl of RT Primer Mix to the EP tube. Incubate
at 37˚C for 15 min, followed by 85˚C for 5 sec, and finally cool to
4˚C for storage to proceed with cDNA amplification. cDNA
amplification using SYBR Green Master Mix (cat. no. 11200ES08;
Shanghai Yeasen Biotechnology Co., Ltd.). Dilute the above cDNA
from 20 µl to 300 µl using RNase free H2O, take 9.2 µl cDNA, 10 µl
Hieff UNICEF Universal Blue qPCR SYBR Green Master Mix, 0.4 µl
Forward Primer (10 µM), 0.4 µl Reverse Primer (10 µM) into EP
tubes, and wait for amplification. The quantitative PCR thermal
cycling program for 40 cycles was: 1 cycle of enzyme activation at
95˚C for 15 min, denaturation at 95˚C for 30 sec, annealing at 60˚C
for 30 sec and extension at 72˚C for 30 sec. Relative quantification
was calculated using the 2-∆∆Ct method (21).
Western blotting
H9c2 cardiomyocytes were lysed with RIPA buffer on
ice, and the protein concentration was determined using a BCA
protein assay kit (cat. no. KGP902; Nanjing KeyGen Biotech Co.,
Ltd.). Equal amounts of protein (20 µg per lane) were loaded using
8-12% SDS-PAGE and then transferred to a PVDF membrane. Membranes
were blocked with 5% skimmed milk at room temperature for 1 h, then
incubated with the following primary antibodies overnight at 4˚C:
GAPDH (cat. no. HRP-60004; ProteinTech Group, Inc.),
cleaved-caspase-3 (cat. no. 9661S; Cell Signaling Technology,
Inc.), caspase-3 (cat. no. 66470-1-Ig; ProteinTech Group, Inc.),
Bax (cat. no. 2772S; Cell Signaling Technology, Inc.), Bcl-2 (cat.
no. 26593-1-AP; ProteinTech Group, Inc.), TSPO (cat. no. ab109497;
Abcam), LC3B (cat. no. ab51520; Abcam), ATG5 (cat. no. 12994S; Cell
Signaling Technology, Inc.), Beclin 1 (cat. no. 66665-1-Ig;
ProteinTech Group, Inc.), P62 (23214S; ProteinTech Group, Inc.),
PINK1 (cat. no. 23274-1-AP; ProteinTech Group, Inc.), Parkin (cat.
no. 66674-1-Ig; ProteinTech Group, Inc.), Mfn2 (cat. no.
12186-1-AP; ProteinTech Group, Inc.), Drp1 (cat. no. 12957-1-AP;
ProteinTech Group, Inc.), ATP5B (cat. no. 17247-1-AP; ProteinTech
Group, Inc.), PI3K (cat. no. 4257S; Cell Signaling Technology,
Inc.), phosphorylated (p)-PI3K (cat. no. 4228S; Cell Signaling
Technology, Inc.), Akt (cat. no. 9272S, Cell Signaling Technology,
Inc.), p-Akt (cat. no. 4060S; Cell Signaling Technology, Inc.),
mTOR (cat. no. 2983S; Cell Signaling Technology, Inc.), p-mTOR
(cat. no. 2971S; Cell Signaling Technology, Inc.), PKR-like ER
kinase (PERK; cat. no. ab229912; Abcam), p-PERK (cat. no. 3179S;
Cell Signaling Technology, Inc.), ATF6 (cat. no. 24169-1-AP;
ProteinTech Group, Inc.) and inositol-requiring enzyme 1 (IRE1;
cat. no. bs16696R, BIOSS). All primary antibodies were diluted
using an antibody dilution buffer (cat. no. P0023A-500ml; Beyotime
Institute of Biotechnology) at a ratio of 1:1,000. After incubation
with the primary antibody, the membrane was washed three times with
Tris-buffered saline and 0.1% Tween 20 (cat. no. 1247ML500;
Biofroxx, Inc.) and then according to the primary antibody species
incubated with rabbit secondary antibodies (cat. no. 7074P2; Cell
Signaling Technology, Inc.) or mouse secondary antibodies (cat. no.
7076S; Cell Signaling Technology, Inc.) at room temperature for 1
h. All secondary antibodies were diluted using an antibody dilution
buffer (cat. no. P0023A-500ml; Beyotime Institute of Biotechnology)
at a ratio of 1:5,000. Signals were visualized using Immobilon
western chemiluminescence HRP substrate (cat. no. WBKLS0500,
MilliporeSigma) and imaged using a ChemiScope (Clinx Science
Instruments). Densitometry analysis was performed using chemical
analysis software (version 2017.12.6.0; Clinx Science
Instruments).
Statistical analysis
Data are presented as the mean ± the standard error
of the mean. The distribution of data was assessed using a
Shapiro-Wilk test. For comparisons between two groups, a
independent sample t-test was used. Comparisons between multiple
groups were assessed using a one-way ANOVA followed by a post hoc
Tukey's test. All statistical analysis was performed using GraphPad
Prism version 9.0 (GraphPad Software, Inc.; Dotmatics). P<0.05
was considered to indicate a statistically significant
difference.
Results
TSPO knockdown alleviates myocardial
apoptosis and damage following A/R stimulation
To investigate the impact of TSPO on myocardial I/R,
an A/R model was established using the H9c2 myocardial cells. The
western blotting results showed a significant increase in the
protein expression levels of TSPO following anoxia. Knockdown of
TSPO using siRNA led to a significant reduction in TSPO
levels following A/R stimulation compared with the control group
(Fig. 1A and B). Moreover, the results showed that the
levels of proapoptotic molecules Bax and
cleaved-caspase-3/caspase-3 were significantly increased in H9c2
si-NC-transfected cardiomyocytes following A/R stimulation compared
with the si-NC-transfected control cells; subsequently, the levels
of proapoptotic molecules Bax and cleaved-caspase-3/caspase-3 were
significantly decreased following TSPO knockdown. By
contrast, in H9c2 si-TSPO-transfected cardiomyocytes following A/R
stimulation compared with si-NC-transfected cardiomyocytes
following A/R stimulation the show that TSPO knockdown
resulted in a slight upregulation of the antiapoptotic molecule
Bcl-2 (Fig. 1A and B). Consistently, FACS-based apoptosis
analysis revealed that the knockdown of TSPO decreased the
levels of apoptosis induced by A/R (Fig. 1C and D). Furthermore, LDH levels were assessed
as a marker of cell damage and quantified using an enzymatic
activity assay. The results showed that cell damage was
significantly increased following A/R stimulation and was reduced
following TSPO knockdown (Fig.
1E). In agreement with the aforementioned observations, CCK-8
analysis revealed a significant decrease in myocardial cell
activity in response to A/R stimulation, but this decrease was
attenuated following TSPO knockdown (Fig. 1F). The aforementioned results
suggest that inhibiting TSPO may be a potential strategy for
mitigating myocardial I/R injury by decreasing cell apoptosis and
damage.
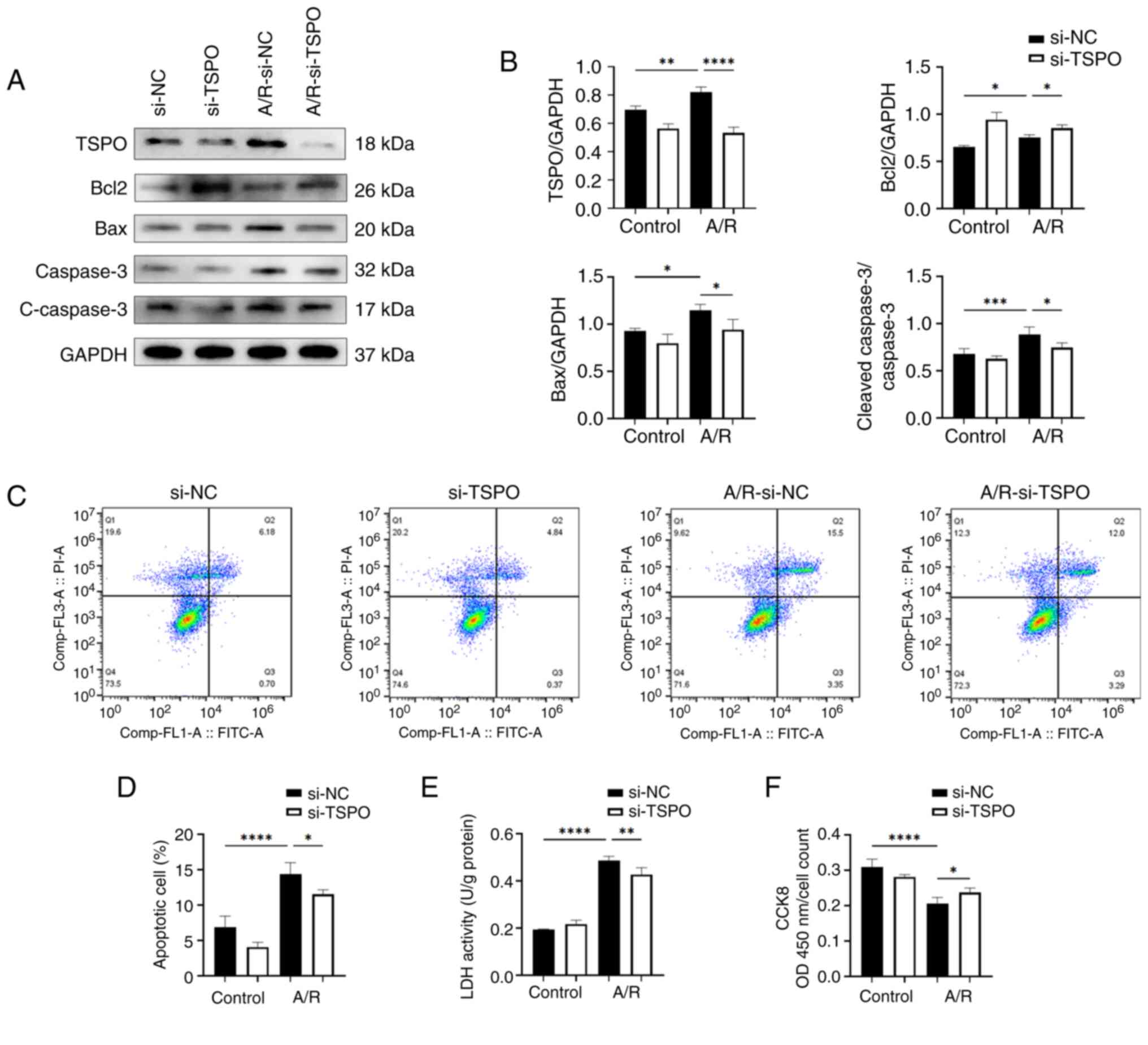 | Figure 1Targeting TSPO alleviates myocardial
apoptosis and damage in an in vitro A/R model. (A and B)
Western blotting and densitometry analysis of TSPO, Bcl2, Bax and
cleaved-caspase-3/caspase-3 expression levels in si-NC and si-TSPO
H9c2 cells with or without A/R stimulation. (C) Representative flow
cytometry results showing the apoptosis of H9c2 cells determined
using Annexin V-FITC/PI double-staining assay. (D) Quantification
of the percentage of late apoptotic H9c2 cells. (E) Cell damage was
measured using an LDH assay. (F) Cell viability was measured using
a Cell Counting Kit-8 assay. n=4 per group. *P<0.05,
**P<0.01, ***P<0.001,
****P<0.0001. TSPO, translocator protein; A/R,
anoxia/reoxygenation; si, small interfering; NC, negative control;
LDH, lactate dehydrogenase; FITC, fluorescein isothiocyanate; PI,
propidium iodide. |
Targeting TSPO improves mitochondrial
dysfunction in myocardial cells following A/R stimulation
Considering the cellular location and previously
reported functions of TSPO, the interplay between TSPO and
mitochondrial dysfunction was next explored. A JC-1 assay kit was
used to detect the mitochondrial membrane potential, as indicated
by the red/green fluorescence ratio. The results revealed a
significant decrease in the membrane potential of myocardial cells
following A/R stimulation, which was significantly attenuated
following TSPO knockdown (Fig.
2A and B). Notably, ATP
production can reflect mitochondrial function. The results further
showed that after TSPO knockdown, ATP production was
significantly increased (Fig. 2C).
Consistent with these findings, ATP5B, a crucial subunit of
mitochondrial ATP synthase, was upregulated following the knockdown
of TSPO and A/R stimulation (Figs. 2D, E and S2).
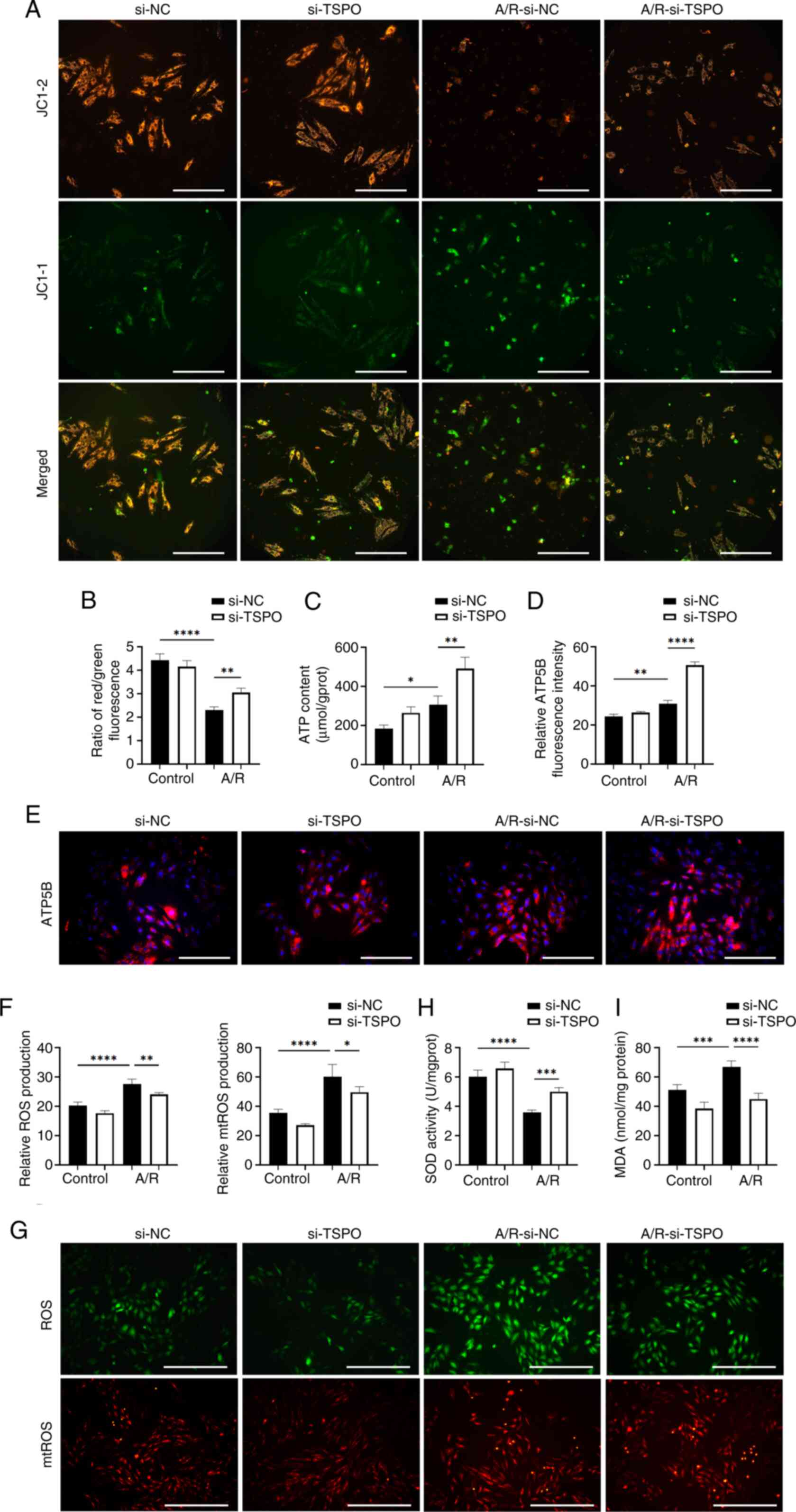 | Figure 2TSPO knockdown reduces
mitochondrial dysfunction in myocardial cells following A/R
stimulation. (A and B) Representative fluorescence images and
quantitative analysis of JC-1 staining in si-NC and si-TSPO H9c2
cells with or without A/R stimulation. Scale bar, 20 µm. (C)
Quantitative analysis of the ATP content in the different groups.
(D and E) Representative fluorescence images and quantitative
analysis of ATP5B expression. Scale bar, 20 µm. (F) Quantitative
analysis and (G) representative images and of ROS and mtROS
staining. Scale bar, 50 µm. (H) Quantitative analysis of SOD
activity and (I) MDA levels. *P<0.05,
**P<0.01, ***P<0.001,
****P<0.0001. TSPO, translocator protein; A/R,
anoxia/reoxygenation; si, small interfering; NC, negative control;
ROS, reactive oxygen species; mtROS, mitochondrial ROS; SOD,
superoxide dismutase; MDA, malondialdehyde; ATP5B, ATP synthase F1
β subunit. |
Subsequently, the effect of TSPO on the oxidative
stress levels of myocardial cells following A/R stimulation.
Notably, a significant increase in ROS and mtROS levels was
observed in si-NC-transfected myocardial cells following A/R
stimulation, and these changes were significantly decreased
following TSPO knockdown (Fig.
2F and G). Moreover, the
levels of SOD, a marker of antioxidant enzymes, significantly
decreased in si-NC-transfected myocardial cells in response to A/R
stimulation and increased after TSPO knockdown (Fig. 2H). By contrast, the levels of MDA,
a marker of cell membrane lipid oxidation, were significantly
increased in si-NC-transfected myocardial cells following A/R
stimulation and decreased following TSPO knockdown (Fig. 2I). These results indicate that
targeting TSPO could efficiently improve mitochondrial function and
concomitantly alleviate oxidative stress.
ER stress and mitophagy may mediate
TSPO-driven mitochondrial dysfunction following A/R
stimulation
To investigate how TSPO knockdown protected
against mitochondrial dysfunction induced by A/R stimulation, the
impact of TSPO on ER stress was explored. ER stress is closely
associated with mitochondrial dysfunction by transferring stress
signals from the ER to the mitochondria (22). Western blotting confirmed that A/R
stimulation resulted in a significant increase in the expression of
key markers (p-PERK/PERK, ATF6 and IRE1) involved in the adaptive
unfolded protein response (UPR), accompanied by downstream
signaling during ER stress (23).
However, TSPO knockdown further increased the expression of
the aforementioned markers compared with the si-NC A/R group
(Fig. 3A and B). Subsequently, Co-IP results show that
there is a protein interaction between TSPO and ATF6 (Fig. 3C). In addition, immunofluorescence
co-staining showed an increase in ATF6 expression in the nucleus
following A/R stimulation. The localization of ATF6 in the
cytoplasm overlapped with TSPO (Fig.
3D). These results indicate that TSPO may play a role in ER
stress by interacting with ATF6 during myocardial I/R injury.
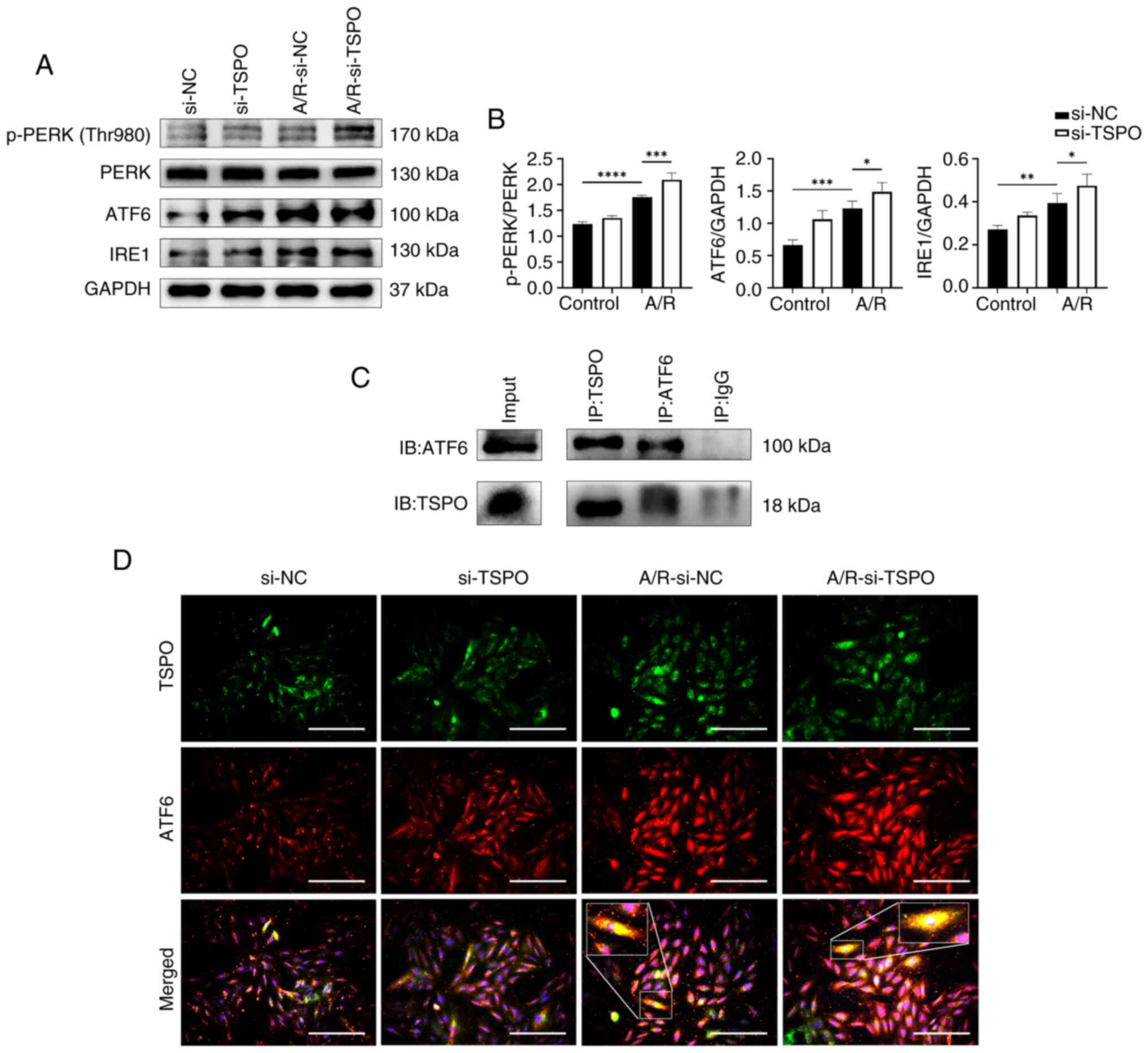 | Figure 3TSPO interacts with ATF6 and promotes
endoplasmic reticulum stress following A/R stimulation. (A) Western
blotting and (B) densitometry analysis of p-PERK, PERK, ATF6 and
IRE1 protein expression levels in si-NC and si-TSPO H9c2 cells with
or without A/R stimulation. (C) Co-immunoprecipitation of TSPO and
ATF6. (D) Multiplex immunocytochemistry analysis of TSPO and ATF6.
4 times magnification for the zoomed in box. Scale bar, 20 µm.
*P<0.05, **P<0.01,
***P<0.001, ****P<0.0001. TSPO,
translocator protein; A/R, anoxia/reoxygenation; si, small
interfering; NC, negative control. IRE1, inositol-requiring enzyme
1. |
ER stress is also a potent trigger for autophagy,
which typically serves an adaptive protective function during
myocardial I/R injury (24).
Paradoxically, the results showed that TSPO knockdown
reduced the expression of autophagy-related markers following A/R
stimulation compared with si-NC A/R cells (Fig. 4A-C). Notably, the PI3K-Akt-mTOR
pathway participates in inhibiting the activation of autophagy
(25). In the present study,
following TSPO knockdown, the ratios of p-PI3K/PI3K,
p-Akt/Akt and p-mTOR/mTOR were significantly increased compared
with si-NC A/R cells (Fig. 4D and
E). These results suggest that
targeting TSPO may inhibit myocardial cell autophagy by promoting
the activation of the PI3K-Akt-mTOR signaling pathway.
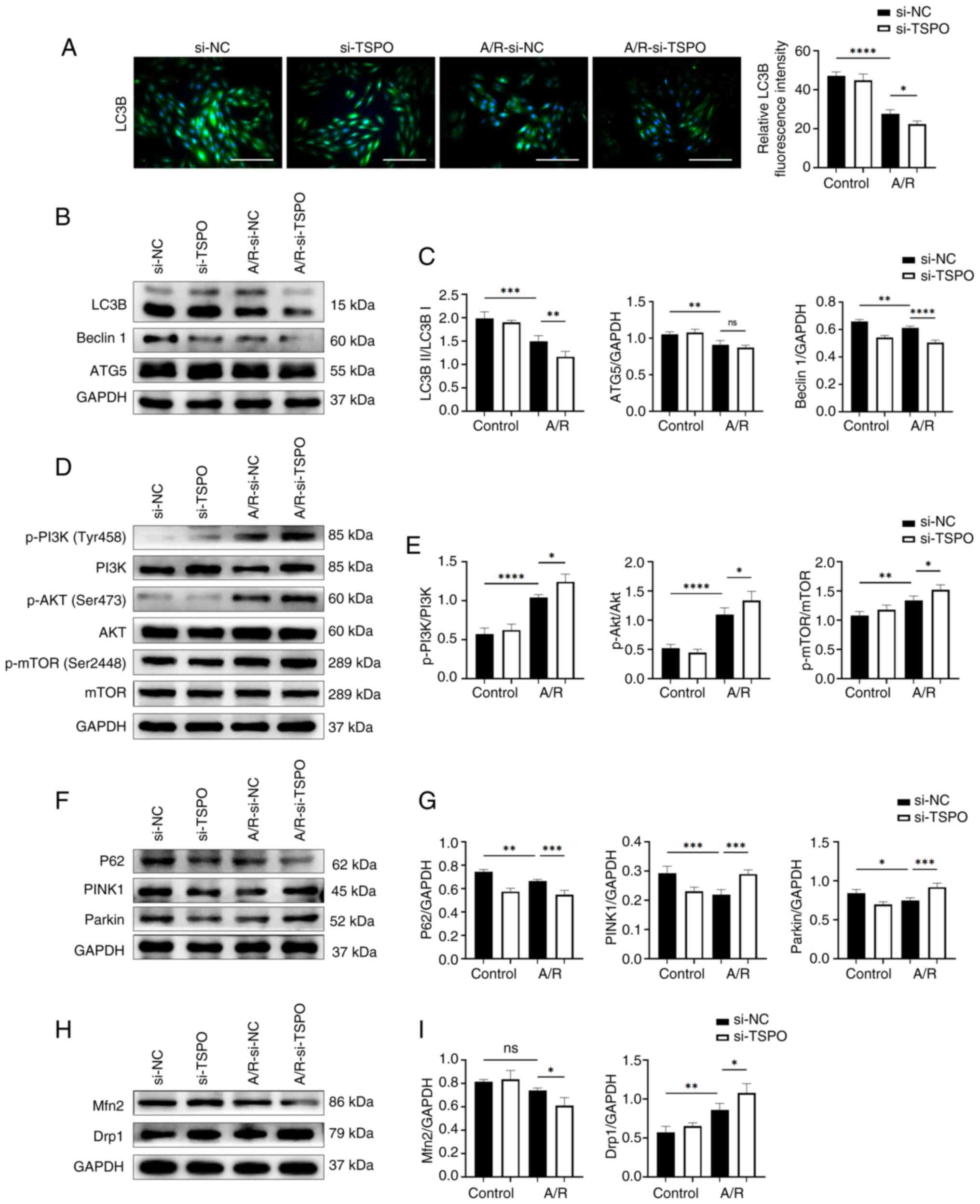 | Figure 4TSPO regulates autophagy and
mitophagy via related pathways following A/R stimulation. (A)
Representative fluorescence images and quantitative analysis of
LC3B expression. Scale bar, 20 µm. (B) Western blotting and (C)
densitometry analysis of LC3B, Beclin1 and ATG5 expression levels
in si-NC and si-TSPO H9c2 cells with or without A/R stimulation.
(D) Western blotting and (E) densitometry analysis of p-PI3K, PI3K,
p-Akt, Akt, p-mTOR and mTOR expression levels. (F) Western blotting
and (G) densitometry analysis of P62, PINK1 and Parkin expression
levels. (H) Western blotting and (I) densitometry analysis of Mfn2
and Drp1 protein expression levels. *P<0.05,
**P<0.01, ***P<0.001,
****P<0.0001. TSPO, translocator protein; A/R,
anoxia/reoxygenation; si, small interfering; NC, negative control;
p-, phosporyalated. |
Subsequently, the expression of mitophagy-related
proteins (P62, PINK1 and Parkin) were assessed. A/R stimulation
induced a significant decrease in mitophagy-related markers, while
TSPO knockdown following A/R treatment upregulated the levels of
these mitophagy markers, as indicated by further decreases in P62
expression and increases in Parkin and PINK1 expression compared
with si-NC A/R controls (Fig. 4F
and G). Furthermore, TSPO
knockdown resulted in a significant decrease in the expression of
the mitochondrial fusion protein Mfn2 following A/R stimulation
compared with si-NC A/R controls. However, the protein levels of
Drp1, a typical marker of mitochondrial fission, significantly
increased following TSPO knockdown compared with si-NC A/R
controls (Fig. 4H and I). These results suggest that inhibiting
TSPO may decrease the fusion of damaged and healthy mitochondria in
myocardial cells, promote mitochondrial fission, and facilitate the
clearance of damaged mitochondria through the Parkin/PINK1-mediated
mitophagy pathway following A/R stimulation. This process may help
alleviate mitochondrial dysfunction during myocardial I/R
injury.
Discussion
Myocardial I/R injury is a significant complication
that occurs following a myocardial infarction (26). Despite significant research efforts
to identify therapeutic targets to address this condition,
advancements in the clinical management of this disease have proven
to be challenging. In the present study, it was demonstrated that
TSPO was significantly induced following A/R injury. Knockdown of
TSPO led to reduced apoptosis and a simultaneous reduction
in mitochondrial dysfunction following A/R injury. Furthermore, it
was found that ER stress persisted in cells subjected to A/R and
was intensified further in the TSPO knockdown cells. The
interaction between TSPO and the ER stress-related protein ATF6
suggested that ER stress may play a role in mediating TSPO-driven
mitochondrial dysfunction following A/R stimulation, highlighting
the need for further investigations. Taken together, these results
suggest that targeting TSPO may be a promising strategy for
alleviating myocardial I/R injury. TSPO may thus function as a
potential mediator of the relationship between ER stress and
mitochondrial dysfunction.
Emerging evidence has confirmed that apoptosis in
cardiomyocytes may result from mitochondrial injury, autophagy,
oxidative stress and ER stress (27). However, the underlying link between
the aforementioned cellular events remains elusive. Since TSPO is
an OMM protein expressed in the heart (11), and the TSPO ligand
4'-chlorodiazepam has been shown to reduce infarct size and enhance
mitochondrial function post-ischemia-reperfusion (28), a focus was placed on assessing the
impact of TSPO on myocardial I/R injury. The results of the present
study revealed that following A/R stimulation, TSPO expression
significantly increased, as did cardiomyocyte apoptosis, which was
notably reduced following TSPO knockdown.
The role of mtROS production and related pathways in
myocardial I/R injury has been extensively studied (7). Myocardial ischemia hinders the
mitochondrial ETC due to oxygen deprivation, leading to excessive
ROS production and consequent cellular damage during reperfusion,
ultimately resulting in apoptosis (6). However, the exact mechanism of
myocardial I/R injury has not been fully determined, and relevant
clinical trials have proven to be ineffective (5). These findings support previous
research indicating that TSPO knockdown alleviates oxidative
stress and consequent apoptosis in cardiomyocytes during A/R.
To further clarify the underlying mechanisms of TSPO
in myocardial I/R injury, its regulatory effect on mitochondrial
function was investigated. Liu et al (14) demonstrated the involvement of TSPO
in the regulation of ATP synthesis. The authors noted that
overexpression of TSPO in Jurkat cells increases
mitochondrial ATP production and cellular excitability.
Paradoxically, Gatliff et al (15) reported that TSPO overexpression, in
conjunction with its interaction with voltage-dependent anion
channel 1, enhances mtROS synthesis while inhibiting mitophagy and
ATP production in canine mammary epithelial cells. In the present
study, it was confirmed that targeting TSPO facilitated ATP
synthesis in cardiomyocytes following A/R stimulation. This effect
was likely attributed to reduced ROS production and efficient
clearance of damaged mitochondria (29). Moreover, the decrease in the
mitochondrial membrane potential was mitigated following
TSPO knockdown. Thus, TSPO may participate in myocardial I/R
injury by regulating energy synthesis and ROS generation.
Mitochondria and the ER play a fundamental role in
controlling cellular physiology and regulating diverse signal
transduction pathways. The mitochondria-associated membranes (MAMs)
represent the first discovered connection between these two
organelles, forming specialized lipid raft-like structures
(30). MAMs possess distinct
structures and serve as platforms for various signal transduction
functions, including lipid synthesis, transport and calcium
transfer from the ER to the mitochondria (31). Consequently, they regulate crucial
signaling pathways and maintain cellular homeostasis. During
myocardial I/R, an extensive UPR occurs (32). ER stress, triggered by the
accumulation of unfolded or misfolded proteins in the ER, activates
various cellular processes, including oxidative stress leading to
mitochondrial dysfunction (33).
Evidence suggests that myocardial cells experience increased ER
stress following I/R, with ATF6 knockdown exacerbating heart damage
and functional decline post-I/R (34). The present study showed that
targeting TSPO triggered ER stress in cardiomyocytes following A/R
stimulation, and Co-IP analysis revealed an interaction between
TSPO and ATF6. Notably, ER stress-induced autophagy is a
compensatory response to cellular stress; however, prolonged or
severe ER stress can precipitate cell death (35). The activation of the UPR can lead
to apoptosis, but it can also trigger protective mechanisms, such
as autophagy (36). According to
Vanhoutte et al (37), the
Thbs1-mediated PERK-eIF2α-ATF4 signaling pathway plays a crucial
role in inducing autophagy and regulating cardiomyocyte size in the
stressed heart. Margariti et al (38) identified the IRE1α-XBP1-S axis,
which promotes the conversion of LC3 I to LC3 II in endothelial
cells, thereby promoting autophagy. Dang et al (39) found that under ER stress, ATF6
enhances autophagy and connects UPR-associated pathways to maintain
ER homeostasis. Overexpression of activated ATF6 also rescues
defects in autophagy regulation (39). These studies substantiate the role
of ER stress in modulating autophagy, in agreement with the
findings of the present study. Autophagy is suppressed in the
myocardium during I/R (40). This
process maintains cardiomyocyte function by removing damaged
organelles and proteins. Appropriate levels of autophagy are
essential for myocardial recovery (41); however, excessive autophagy can
lead to cardiomyocyte death and reduced myocardial function. In the
present study, following A/R, autophagy in H9c2 cardiomyocytes was
significantly decreased, and autophagy further decreased following
TSPO knockdown. Concurrently, the PI3K-Akt-mTOR pathway was notably
activated following A/R stimulation and further enhanced following
TSPO knockdown, suggesting that TSPO may modulate autophagy via
this pathway.
In prior studies, the role of DRP1 promotes the
production of ROS (42). During
I/R injury, both Fis1 and DRP1 are upregulated in neonatal and
adult cardiomyocytes (43,44). In this scenario, upstream signaling
events promote the activation of DRP1, which then interacts with
Fis1 at the OMM (45). This
interaction leads to an excessive accumulation of fragmented and
dysfunctional mitochondria, resulting in a redox imbalance
(46). Fis1 also binds to
mitochondrial fusion proteins (MFN1, MFN2 and OPA1) and inhibits
their GTPase activity, consequently impeding mitochondrial fusion
in mammalian cells (47). The
expression of MFN2 and DRP1 shows a negative correlation and
jointly contributes to the regulation of mitochondrial size
(48). In the present study, the
inconsistency in the expression of ROS and DRP1 may be attributed
to the function of TSPO. The interaction between TSPO and VDAC1 can
influence ROS production (15,16).
Previous studies have shown that TSPO knockdown significantly
reduces ROS expression (15,16).
Hence, it is plausible to suggest that TSPO knockdown could
potentially inhibit the DRP1-mediated increase in ROS.
Notably, in addition to autophagy, mitophagy, which
is crucial for removing damaged mitochondria and maintaining
cellular homeostasis (49), is
inversely associated with TSPO levels (17). In the present study, it was
observed that A/R treatment significantly reduced mitophagy.
Following TSPO knockdown, the extent of mitophagy
significantly increased. In general, impaired mitochondrial
function hampers ATP synthesis (50). However, the present study revealed
that cardiomyocytes with reduced TSPO expression produced more ATP
following A/R stimulation. This could be attributed to enhanced
clearance of damaged mitochondria and restoration of mitochondrial
function (51). Additionally, the
activation of ER stress may assist in the elimination of
accumulated unfolded proteins, requiring a significant amount of
ATP (52). These findings suggest
that mitochondrial function is partially restored in cardiomyocytes
following A/R stimulation with TSPO expression knocked down.
Based on these observations, it is postulated that TSPO
knockdown would mitigate damage in cardiomyocytes following A/R
stimulation, potentially by triggering persistent ER stress and
concomitant induction of mitophagy. However, the precise mechanisms
of TSPO warrant further investigation.
It is necessary to acknowledge the limitations of
the present work. This study only used an in vitro model.
Although this provides mechanistic insights, further validation of
the reliability and general applicability of the research results
using animal models and clinical samples is required. Furthermore,
other unexplored signaling pathways and mechanisms related to TSPO
in myocardial I/R injury may require further investigation for
clarification in future studies.
In conclusion, the role of TSPO was investigated in
an in vitro model of A/R injury using H9c2 cardiomyocytes.
The results establish a potential mechanistic association between
ER stress and TSPO-induced mitochondrial dysfunction in the process
of myocardial I/R injury. Taken together, these results suggest
that targeting TSPO may be a potential strategy for alleviating
myocardial I/R injury in patients with IHD.
Supplementary Material
Knockdown of TSPO. (A) Western
blotting and densitometry analysis of TSPO protein expression
levels in control, si-NC, and si-TSPO H9c2 cells. (B) TSPO gene
expression level determined by reverse transcription-quantiative
PCR. *P<0.05, **P<0.01,
***P<0.001, ****P<0.0001. TSPO,
translocator protein; si, small interfering; NC, negative control;
A/R, anoxia/reoxygenation.
ATP5B protein expression levels
following TSPO knockdown. Western blotting and densitometry
analysis showing ATP5B protein expression levels in si-NC and
si-TSPO H9c2 cells with or without A/R stimulation.
*P<0.05. TSPO, translocator protein; si, small
interfering; NC, negative control; A/R, anoxia/reoxygenation;
ATP5B, ATP synthase F1 β subunit.
Acknowledgements
The authors would like to thank Dr Xu Duo,
Department of Oncology, The First Affiliated Hospital of Nanjing
Medical University, Nanjing, China, for her assistance in
performing experiments.
Funding
Funding: This study was supported by The Nanjing Scientific
Research Project for Outstanding Overseas Students (grant no.
#2021) and The Key Medical Research Project from the Jiangsu
Provincial Health Commission (grant no. K2023039).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
CW, ZQ and SL conceived and designed the
experiments. CW, YJ, SL, ZQ, WC, YX, GC, QZ, HJ, YL, YY and XC
carried out the experiments. CW and YJ cultured the cells. ZQ and
SL treated the cells. GC and YL designed the primers and performed
the reverse transcription-quantitative PCR. YJ, WC and YX performed
the flow cytometric analysis. CW, SL and ZQ performed Co-IP
experiment. QZ and HJ performed the mitochondrial membrane
potential, ROS and mtROS assay. CW and YJ performed LHD and CCK8
assay. XC and YY performed MDA and SOD assays. CW, YJ, SL, WC, YX,
QZ, YY and XC performed the western blot analysis. QL and RZ
quantified and analyzed the raw data. ZQ and SL confirmed the
authenticity of all raw data. CW drafted the original manuscript.
All authors have read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Roth GA, Mensah GA, Johnson CO, Addolorato
G, Ammirati E, Baddour LM, Barengo NC, Beaton AZ, Benjamin EJ,
Benziger CP, et al: Global burden of cardiovascular diseases and
risk factors, 1990-2019: Update from the GBD 2019 study. J Am Coll
Cardiol. 76:2982–3021. 2020.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Doenst T, Thiele H, Haasenritter J,
Wahlers T, Massberg S and Haverich A: The treatment of coronary
artery disease. Dtsch Arztebl Int. 119:716–723. 2022.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Bhatt DL, Lopes RD and Harrington RA:
Diagnosis and treatment of acute coronary syndromes: A review.
JAMA. 327:662–675. 2022.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Martí-Pàmies Í, Thoonen R, Morley M,
Graves L, Tamez J, Caplan A, McDaid K, Yao V, Hindle A, Gerszten
RE, et al: Brown adipose tissue and BMP3b decrease injury in
cardiac ischemia-reperfusion. Circ Res. 133:353–365.
2023.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Heusch G: Myocardial ischaemia-reperfusion
injury and cardioprotection in perspective. Nat Rev Cardiol.
17:773–789. 2020.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Jiang L, Yin X, Chen YH, Chen Y, Jiang W,
Zheng H, Huang FQ, Liu B, Zhou W, Qi LW and Li J: Proteomic
analysis reveals ginsenoside Rb1 attenuates myocardial
ischemia/reperfusion injury through inhibiting ROS production from
mitochondrial complex I. Theranostics. 11:1703–1720.
2021.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Xiang Q, Yi X, Zhu XH, Wei X and Jiang DS:
Regulated cell death in myocardial ischemia-reperfusion injury.
Trends Endocrinol Metab. 35:219–234. 2024.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Chen CL, Zhang L, Jin Z, Kasumov T and
Chen YR: Mitochondrial redox regulation and myocardial
ischemia-reperfusion injury. Am J Physiol Cell Physiol.
322:C12–C23. 2022.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Papadopoulos V, Baraldi M, Guilarte TR,
Knudsen TB, Lacapère JJ, Lindemann P, Norenberg MD, Nutt D, Weizman
A, Zhang MR and Gavish M: Translocator protein (18kDa): New
nomenclature for the peripheral-type benzodiazepine receptor based
on its structure and molecular function. Trends Pharmacol Sci.
27:402–409. 2006.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Banati RB, Middleton RJ, Chan R, Hatty CR,
Kam WWY, Quin C, Graeber MB, Parmar A, Zahra D, Callaghan P, et al:
Positron emission tomography and functional characterization of a
complete PBR/TSPO knockout. Nat Commun. 5(5452)2014.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Selvaraj V and Stocco DM: The changing
landscape in translocator protein (TSPO) function. Trends
Endocrinol Metab. 26:341–348. 2015.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Braestrup C, Albrechtsen R and Squires RF:
High densities of benzodiazepine receptors in human cortical areas.
Nature. 269:702–704. 1977.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Farhan F, Almarhoun M, Wong A, Findlay AS,
Bartholomew C, Williams MTS, Hurd TW and Shu X: Deletion of TSPO
causes dysregulation of cholesterol metabolism in mouse retina.
Cells. 10(3066)2021.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Liu GJ, Middleton RJ, Kam WWY, Chin DY,
Hatty CR, Chan RHC and Banati RB: Functional gains in energy and
cell metabolism after TSPO gene insertion. Cell Cycle. 16:436–447.
2017.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Gatliff J, East D, Crosby J, Abeti R,
Harvey R, Craigen W, Parker P and Campanella M: TSPO interacts with
VDAC1 and triggers a ROS-mediated inhibition of mitochondrial
quality control. Autophagy. 10:2279–2296. 2014.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Meng Y, Tian M, Yin S, Lai S, Zhou Y, Chen
J, He M and Liao Z: Downregulation of TSPO expression inhibits
oxidative stress and maintains mitochondrial homeostasis in
cardiomyocytes subjected to anoxia/reoxygenation injury. Biomed
Pharmacother. 121(109588)2020.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Scaini G, Barichello T, Fries GR, Kennon
EA, Andrews T, Nix BR, Zunta-Soares G, Valvassori SS, Soares JC and
Quevedo J: TSPO upregulation in bipolar disorder and concomitant
downregulation of mitophagic proteins and NLRP3 inflammasome
activation. Neuropsychopharmacology. 44:1291–1299. 2019.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Ma XH, Liu JH, Liu CY, Sun WY, Duan WJ,
Wang G, Kurihara H, He RR, Li YF, Chen Y and Shang H:
ALOX15-launched PUFA-phospholipids peroxidation increases the
susceptibility of ferroptosis in ischemia-induced myocardial
damage. Signal Transduct Target Ther. 7(288)2022.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Pooja S, Pushpanathan M, Gunasekaran P and
Rajendhran J: Endocytosis-mediated invasion and pathogenicity of
streptococcus agalactiae in rat cardiomyocyte (H9C2). PLoS One.
10(e0139733)2015.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Tong Z, Xie Y, He M, Ma W, Zhou Y, Lai S,
Meng Y and Liao Z: VDAC1 deacetylation is involved in the
protective effects of resveratrol against mitochondria-mediated
apoptosis in cardiomyocytes subjected to anoxia/reoxygenation
injury. Biomed Pharmacother. 95:77–83. 2017.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Martucciello S, Masullo M, Cerulli A and
Piacente S: Natural products targeting ER stress, and the
functional link to mitochondria. Int J Mol Sci.
21(1905)2020.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Salvagno C, Mandula JK, Rodriguez PC and
Cubillos-Ruiz JR: Decoding endoplasmic reticulum stress signals in
cancer cells and antitumor immunity. Trends Cancer. 8:930–943.
2022.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Wang CC, Li Y, Qian XQ, Zhao H, Wang D,
Zuo GX and Wang K: Empagliflozin alleviates myocardial I/R injury
and cardiomyocyte apoptosis via inhibiting ER stress-induced
autophagy and the PERK/ATF4/Beclin1 pathway. J Drug Target.
30:858–872. 2022.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Ba L, Gao J, Chen Y, Qi H, Dong C, Pan H,
Zhang Q, Shi P, Song C, Guan X, et al: Allicin attenuates
pathological cardiac hypertrophy by inhibiting autophagy via
activation of PI3K/Akt/mTOR and MAPK/ERK/mTOR signaling pathways.
Phytomedicine. 58(152765)2019.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Rizwan H, Pal S, Sabnam S and Pal A: High
glucose augments ROS generation regulates mitochondrial dysfunction
and apoptosis via stress signalling cascades in keratinocytes. Life
Sci. 241(117148)2020.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Wang J and Zhou H: Mitochondrial quality
control mechanisms as molecular targets in cardiac
ischemia-reperfusion injury. Acta Pharm Sin B. 10:1866–1879.
2020.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Paradis S, Leoni V, Caccia C, Berdeaux A
and Morin D: Cardioprotection by the TSPO ligand 4'-chlorodiazepam
is associated with inhibition of mitochondrial accumulation of
cholesterol at reperfusion. Cardiovasc Res. 98:420–427.
2013.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Livingston MJ, Wang J, Zhou J, Wu G,
Ganley IG, Hill JA, Yin XM and Dong Z: Clearance of damaged
mitochondria via mitophagy is important to the protective effect of
ischemic preconditioning in kidneys. Autophagy. 15:2142–2162.
2019.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Wang N, Wang C, Zhao H, He Y, Lan B, Sun L
and Gao Y: The MAMs structure and its role in cell death. Cells.
10(657)2021.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Pinton P: Mitochondria-associated
membranes (MAMs) and pathologies. Cell Death Dis.
9(413)2018.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Glembotski CC, Rosarda JD and Wiseman RL:
Proteostasis and beyond: ATF6 in ischemic disease. Trends Mol Med.
25:538–550. 2019.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Senft D and Ronai ZA: UPR, autophagy, and
mitochondria crosstalk underlies the ER stress response. Trends
Biochem Sci. 40:141–148. 2015.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Jin JK, Blackwood EA, Azizi K, Thuerauf
DJ, Fahem AG, Hofmann C, Kaufman RJ, Doroudgar S and Glembotski CC:
ATF6 decreases myocardial ischemia/reperfusion damage and links ER
stress and oxidative stress signaling pathways in the heart. Circ
Res. 120:862–875. 2017.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Zhang J, Guo J, Yang N, Huang Y, Hu T and
Rao C: Endoplasmic reticulum stress-mediated cell death in liver
injury. Cell Death Dis. 13(1051)2022.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Bhardwaj M, Leli NM, Koumenis C and
Amaravadi RK: Regulation of autophagy by canonical and
non-canonical ER stress responses. Semin Cancer Biol. 66:116–128.
2020.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Vanhoutte D, Schips TG, Vo A, Grimes KM,
Baldwin TA, Brody MJ, Accornero F, Sargent MA and Molkentin JD:
Thbs1 induces lethal cardiac atrophy through PERK-ATF4 regulated
autophagy. Nat Commun. 12(3928)2021.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Margariti A, Li H, Chen T, Martin D,
Vizcay-Barrena G, Alam S, Karamariti E, Xiao Q, Zampetaki A, Zhang
Z, et al: XBP1 mRNA splicing triggers an autophagic response in
endothelial cells through BECLIN-1 transcriptional activation. J
Biol Chem. 288:859–872. 2013.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Dang TT, Kim MJ, Lee YY, Le HT, Kim KH,
Nam S, Hyun SH, Kim HL, Chung SW, Chung HT, et al: Phosphorylation
of EIF2S1 (eukaryotic translation initiation factor 2 subunit
alpha) is indispensable for nuclear translocation of TFEB and TFE3
during ER stress. Autophagy. 19:2111–2142. 2023.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Gu S, Tan J, Li Q, Liu S, Ma J, Zheng Y,
Liu J, Bi W, Sha P, Li X, et al: Downregulation of LAPTM4B
contributes to the impairment of the autophagic flux via unopposed
activation of mTORC1 signaling during myocardial
ischemia/reperfusion injury. Circ Res. 127:e148–e165.
2020.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Del Re DP, Amgalan D, Linkermann A, Liu Q
and Kitsis RN: Fundamental mechanisms of regulated cell death and
implications for heart disease. Physiol Rev. 99:1765–1817.
2019.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Zeng X, Zhang YD, Ma RY, Chen YJ, Xiang
XM, Hou DY, Li XH, Huang H, Li T and Duan CY: Activated Drp1
regulates p62-mediated autophagic flux and aggravates inflammation
in cerebral ischemia-reperfusion via the ROS-RIP1/RIP3-exosome
axis. Mil Med Res. 9(25)2022.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Hom J, Yu T, Yoon Y, Porter G and Sheu SS:
Regulation of mitochondrial fission by intracellular Ca2+ in rat
ventricular myocytes. Biochim Biophys Acta. 1797:913–921.
2010.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Zhou H, Zhu P, Wang J, Zhu H, Ren J and
Chen Y: Pathogenesis of cardiac ischemia reperfusion injury is
associated with CK2α-disturbed mitochondrial homeostasis via
suppression of FUNDC1-related mitophagy. Cell Death Differ.
25:1080–1093. 2018.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Yu Y, Peng XD, Qian XJ, Zhang KM, Huang X,
Chen YH, Li YT, Feng GK, Zhang HL, Xu XL, et al: Fis1
phosphorylation by Met promotes mitochondrial fission and
hepatocellular carcinoma metastasis. Signal Transduct Target Ther.
6(401)2021.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Disatnik MH, Ferreira JCB, Campos JC,
Gomes KS, Dourado PMM, Qi X and Mochly-Rosen D: Acute inhibition of
excessive mitochondrial fission after myocardial infarction
prevents long-term cardiac dysfunction. J Am Heart Assoc.
2(e000461)2013.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Yu R, Jin SB, Lendahl U, Nistér M and Zhao
J: Human Fis1 regulates mitochondrial dynamics through inhibition
of the fusion machinery. EMBO J. 38(e99748)2019.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Ziviani E, Tao RN and Whitworth AJ:
Drosophila parkin requires PINK1 for mitochondrial translocation
and ubiquitinates mitofusin. Proc Natl Acad Sci USA. 107:5018–5023.
2010.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Lu Y, Li Z, Zhang S, Zhang T, Liu Y and
Zhang L: Cellular mitophagy: Mechanism, roles in diseases and small
molecule pharmacological regulation. Theranostics. 13:736–766.
2023.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Tanno S, Yamamoto K, Kurata Y, Adachi M,
Inoue Y, Otani N, Mishima M, Yamamoto Y, Kuwabara M, Ogino K, et
al: Protective effects of topiroxostat on an ischemia-reperfusion
model of rat hearts. Circ J. 82:1101–1111. 2018.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Wu D, Wang Y, Hu J, Xu Y, Gong D, Wu P,
Dong J, He B, Qian H and Wang G: Rab26 promotes macrophage
phagocytosis through regulation of MFN2 trafficking to
mitochondria. FEBS J. 290:4023–4039. 2023.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Yong J, Bischof H, Burgstaller S, Siirin
M, Murphy A, Malli R and Kaufman RJ: Mitochondria supply ATP to the
ER through a mechanism antagonized by cytosolic Ca2.
Elife. 8(e49682)2019.PubMed/NCBI View Article : Google Scholar
|














