Vascular calcification (VC), which is associated
with increased cardiovascular morbidity and mortality, is
characterized by abnormal calcium phosphate deposition on blood
vessel walls and osteogenic transdifferentiation of vascular smooth
muscle cells (VSMCs) (1) VC can
occur in elderly individuals and patients with chronic kidney
disease (CKD), atherosclerosis, diabetes, or systemic lupus
erythematosus (2). VC is a major
factor associated with cardiovascular disease in patients with CKD.
The Kidney Disease: Improving Global Outcomes 2017 Clinical
Practice Guideline suggests that patients with CKD and with known
VC have the highest risk among patients with CKD for cardiovascular
events (3). VC increases in
patients with either type 1 diabetes mellitus (DM) or type 2 DM
(4) and the prevalence of VC is
far greater in DM patients than in individuals without DM (5-7).
Calcification is a hallmark of atherosclerosis (8). Coronary arterial calcification is
indeed a process involved in of atherosclerosis development and
occurs almost exclusively in atherosclerotic arteries (9,10).
In addition, calcification can occur in the abdominal aorta and
heart valves (11,12). VC induces atherosclerosis,
increases vascular stiffness and decreases vascular compliance
(13).
Similar to osteogenesis, VC involves diverse factors
and mechanisms. Exposure to various stimuli leads to an imbalance
between the promotion and inhibition of osteogenesis, ultimately
leading to VC (14). VSMCs undergo
a phenotypic switch during VC (15). This switch is accompanied by the
upregulation of bone-related proteins, including runt-related
transcription factor 2 (Runx2), bone morphogenetic protein 2
(BMP-2) (16) and osteocalcin and
the downregulation of Sirtuins (SIRTs) (17,18)
and contractile proteins, including smooth muscle actin α and
smooth muscle 22α (14). In
addition, long noncoding RNAs are involved in the occurrence of VC
(19).
VC is very common and leads to increases in the
incidence and mortality of cardiovascular diseases (20). Traditional pathogenic factors
cannot fully explain the high prevalence of VC (21,22).
Therefore, identifying new key regulators and new therapeutic
targets involved in the pathogenesis of VC is a crucial
requirement.
Post-translational modifications (PTMs) are chemical
modifications of specific amino acid residues after protein
synthesis and markedly influence the biological function of
proteins (23). Different types of
PTMs, as well as the same modification at different sites, often
have different effects on the biological behavior and mediate
functions of the modified protein (24). In addition, PTMs may interact with
each other and co-operatively regulate the function of the protein,
suggesting that the effects of PTMs are quite complex. PTMs
regulate the activity, stability and function of numerous proteins,
including transcription factors, and have been reported to be
involved in the progression of diseases such as cancer, autoimmune
diseases and neurological diseases (25-27).
New insights into protein modifications and its related key roles
could lead to new strategies to improve management of multiple
human diseases. Recent advances in proteomics and bioinformatics
approaches have led to increased recognition that aberrant PTMs
play important roles in VC. This review presents the latest
progress in clarifying the roles of PTMs in VC.
VC is an actively regulated process involving
multiple calcification-related factors and signaling pathways,
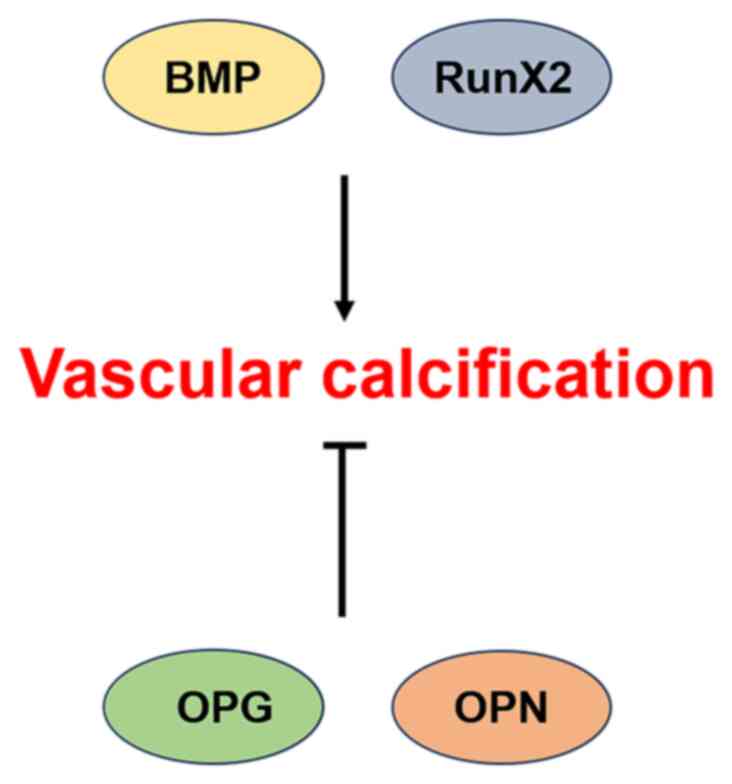
which remain incompletely elucidated. BMP-2, Runx2 and other
calcification factors can promote VC, whereas osteoprotegerin
(OPG), osteopontin (OPN) and other anticalcification factors can
inhibit VC. Pathways such as the BMP signaling pathway,
Wnt/β-catenin pathway and AKT pathway regulate the development of
VC (28). In addition, apoptosis,
mitochondrial metabolism, inflammation, oxidative stress and
autophagy are involved in the occurrence and development of VC
(29-31).
BMPs are members of the transforming growth factor
beta (TGF-β) superfamily and have been reported to play a causal
role in osteogenesis and VC (32).
BMP-2 has been widely studied in vivo and in vitro
for its procalcification properties. The main BMP-2 signaling axis
involved in osteogenic differentiation and VC is the
BMP-2/Smad1/Runx2 axis, in which BMP-2 binds to the corresponding
receptor to activate the intracellular BMP effector proteins
Smad1/5/8. The activated Smad complex then enters the nucleus to
positively regulate Runx2, the transcription factor controlling
bone formation (33). In addition,
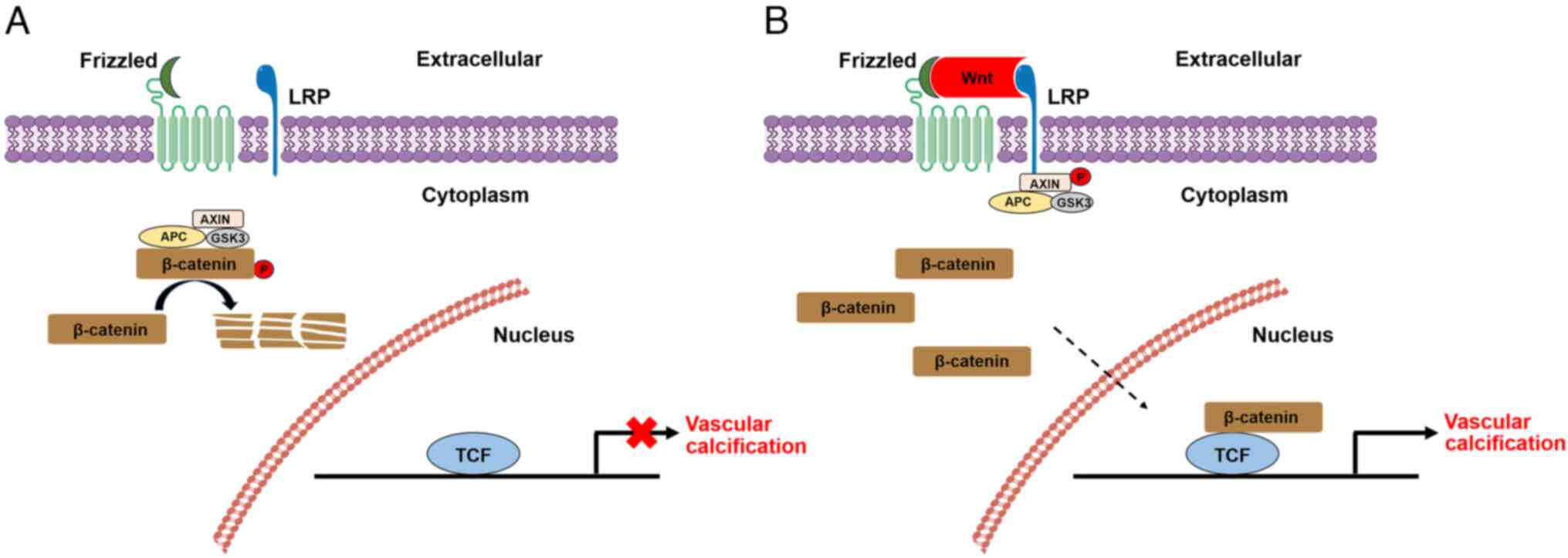
BMP-2 can activate the Wnt/β-catenin pathway, thereby inducing
VC-associated processes in VSMCs (34). Multiple experiments have shown that
the expression of BMP2 is upregulated in the setting of VC. When
BMP2 was used to stimulate human coronary artery smooth muscle
cells, the expression of Runx2 was upregulated within 24 h and
intracellular calcium deposition was significantly increased
(35). Treatment with a BMP
antagonist (LDN-193189 or ALK3-Fc20) to inhibit BMP signaling
attenuated VC in low density lipoprotein receptor-deficient
(LDLR-/-) mice (36). A
report indicating that atherosclerotic intimal calcification is
significantly accelerated in BMP-2-transgenic mice with high
expression of BMP-2 provided further support for a role of BMP in
VC (37). Runx2 is a transcription
factor involved in osteoblast differentiation and bone formation.
The expression level of Runx2 is low in normal blood vessels but
high in calcified blood vessels (38). Runx2 can induce VSMC-mediated
calcification in vitro and plays a key role in oxidative
stress-induced VSMC-mediated calcification (39). Decreased expression of Runx2 was
found to strongly inhibit intimal VC in atherosclerosis-susceptible
mice and medial VC in mice with CKD, further supporting the
critical role of Smooth muscle cell-specific Runx2 in the
development of VC (Fig. 1)
(40,41).
OPG can directly inhibit the differentiation and
maturation of osteoclasts and induce the differentiation of
osteoblasts, which are key inhibitors of calcification (42). OPG has been shown to be highly
expressed in blood vessels, but its expression is low in calcified
blood vessels in mice. OPG knockout mice were found to develop
early and severe VC (43) and
treatment of atherogenic mice with OPG resulted in a reduced
calcified lesion area (44).
However, the results of numerous clinical studies are opposite
those reported in animal studies. For example, a meta-analysis
involving 26,442 patients in the general population revealed that
an elevated OPG level is associated with an increased risk of
cardiovascular disease (45). In
addition, Morena et al (46) reported that high expression of OPG
is closely associated with coronary artery calcification.
OPN is a phosphoglycoprotein adhesion molecule found
primarily in mineral-rich teeth and bone and is found to alleviate
VC by inhibiting the formation and growth of mineral crystals
(47). OPN expression is increased
in calcified plaques, but OPN is not expressed in normal arteries
(48). OPN deficiency does not
cause calcification in wild-type mice but increases VC in mice
deficient in the matrix protein Gla, suggesting that OPN plays an
inducible inhibitory role in VC (Fig.
1) (49).
Apoptosis is another common factor that affects VC.
Reports have indicated that initial calcium phosphate precipitation
could be associated with apoptosis via the release of phosphorous
via intracellular metabolism and calcification-promoting membrane
phospholipid-rich microparticles (53,54).
Further studies revealed that after cell death, cells can release
free DNA to precipitate calcium phosphate crystals on blood vessel
walls, leading to VC (55,56). In addition, in vitro
experiments have suggested that calcification can be blocked by
inhibiting apoptosis and that apoptosis induction can increase the
incidence of calcification by 10-fold (53).
Mitochondria are important bioenergetic powerhouses
and biosynthetic centers in the cell and various stimuli, such as
hyperphosphataemia, hyperglycaemia and increased mitochondrial
outer membrane permeability (57),
can induce mitochondrial dysfunction. Mitochondrial dysfunction
results in reduced production of adenosine triphosphate (ATP),
abnormal production of active oxides, abnormal regulation of
apoptosis and changes in the autophagic ability of cells, all of
which are involved in the occurrence of VC (58). Previous studies have shown that
inhibiting mitochondrial fission with melatonin can abrogate
β-GP-induced VSMC-mediated calcification (59,60).
Various PTMs, such as ubiquitination, acetylation,
carbamylation and glycosylation, play different roles in VC. In
addition, different PTMs may interact with each other to
co-operatively control the occurrence and development of VC
(Table I).
Ubiquitination is a process by which target proteins
are degraded through the ubiquitin-proteasome pathway via a process
regulated by a cascade comprising an E1 ubiquitin-activating
enzyme, an E2 ubiquitin-conjugating enzyme and an E3 ubiquitin
ligase that binds to specific target proteins (61,62).
Ubiquitination is widely involved in physiological processes such
as DNA damage repair, transcriptional regulation, cell cycle
progression, apoptosis and vesicle transport by regulating the
stability, localization, activity and interactions of proteins
(63,64).
The osteogenic transcription factor Runx2 plays a
key role in regulating VC. Studies in osteocytes have shown that
the degradation and stability of the Runx2 protein are regulated
via the ubiquitin-proteasome pathway. The molecular mechanisms that
promote Runx2 protein degradation involve Smad ubiquitination
regulatory factor 1 (Smurf1) and cyclin D1. Smurf1 binds to Runx2
and mediates the conjugation of ubiquitin to Runx2, leading to its
degradation (65,66). Cyclin D1 induces the C-terminal
phosphorylation of Runx2 through cyclin D1/Cdk4 and promotes its
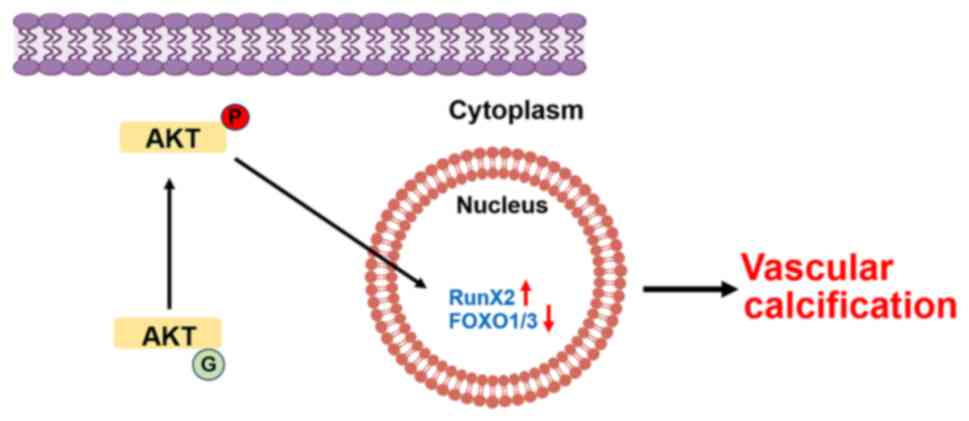
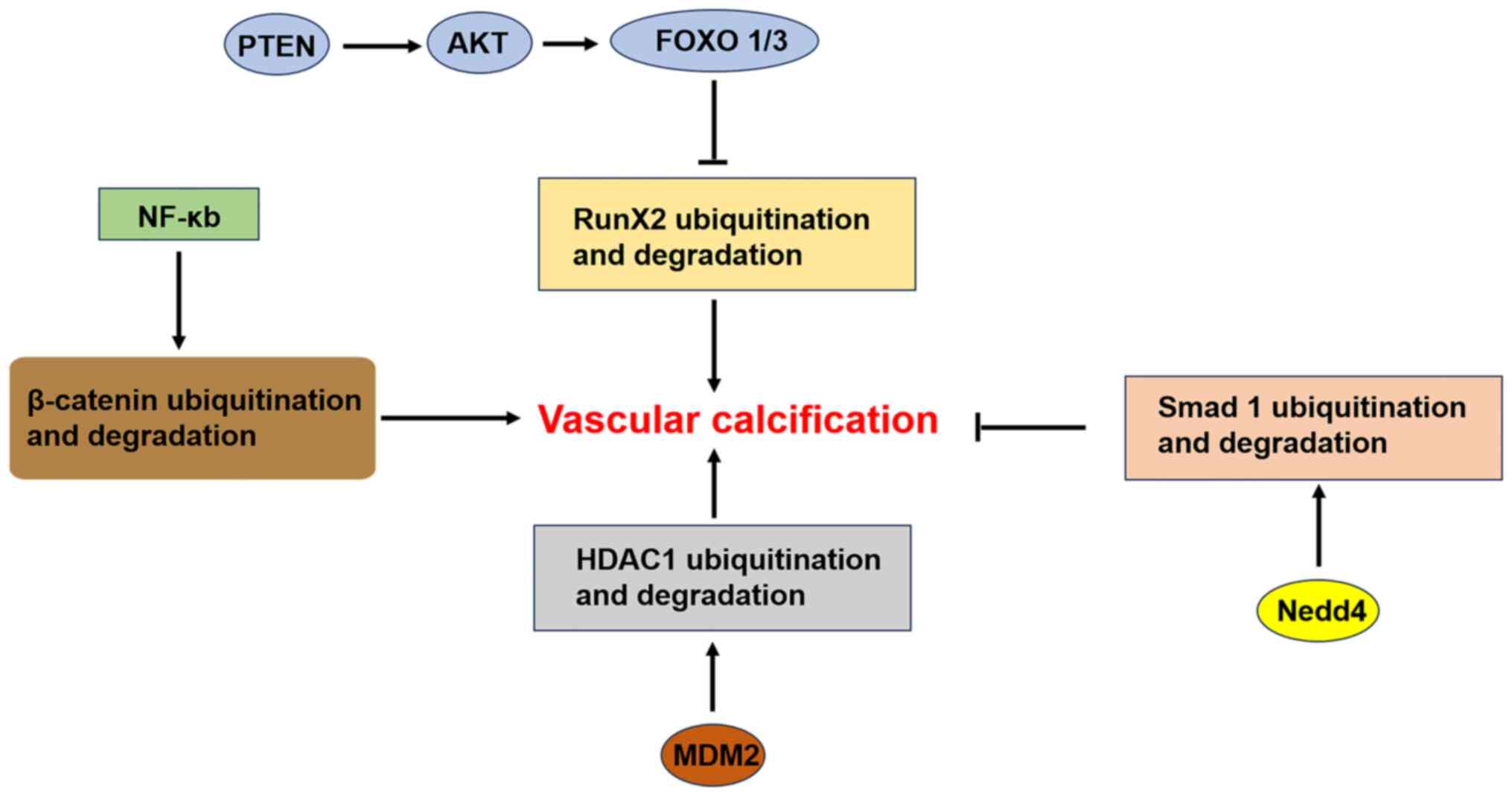
proteasome-dependent degradation (67). Deng et al (52) demonstrated in vitro and
in vivo that the phosphatase and tensin homologue
(PTEN)/AKT/forkhead box O1/3 (FOXO1/3) signaling axes regulate VC
by regulating Runx2 ubiquitination. Specifically, PTEN deficiency
activates AKT, phosphorylates FOXO1/3, inhibits Runx2
ubiquitination, upregulates Runx2 and promotes VC without affecting
the expression of common regulators of Runx2 ubiquitination, such
as Smurf1 and cyclin-D1. Knockdown of FOXO1/3 in VSMCs mimics the
aforementioned effects of PTEN deficiency, including the inhibition
of Runx2 ubiquitination, the upregulation of Runx2 and the
promotion of VC. In addition, a recent study revealed that
kynurenine, a major product of indoleamine 2,3-dioxygenase 1
(IDO1)-mediated tryptophan metabolism, promoted Runx2
ubiquitination and delayed the progression of intimal calcification
in transgenic apolipoprotein E-/- mice. Moreover,
ubiquitination-mediated proteasomal degradation of Runx2 was found
to be associated with the kynurenine-mediated aryl hydrocarbon
receptor-dependent nongenomic pathway (68). Therefore, the molecular mechanisms
regulating Runx2 ubiquitination in VSMCs may be novel and worthy of
further exploration.
In cell models, the neuronal precursor
cell-expressed developmentally downregulated 4 (Nedd4) E3 ubiquitin
ligase was found to negatively regulate VC induced by a
high-phosphorus environment through the ubiquitination of Smad1 and
the development of VC is induced by the specific inhibition of
Nedd4(69). Paradoxically, another
study showed that Murine double minute 2 (MDM2) can promote VC
through the ubiquitination of histone deacetylase 1 (HDAC1) via its
E3 ligase activity. In both in vitro and in vivo
models of VC, the HDAC1 protein level is significantly reduced, a
phenomenon related to the ubiquitination of HDAC1 at lysine (K) 73.
Coimmunoprecipitation assays revealed that the E3 ubiquitin ligase
MDM2 induces the polyubiquitination of HDAC1. The overexpression of
MDM2 significantly increases VC in a dose-dependent manner, whereas
the knockdown of MDM2 inhibits VC (70). In addition, that study confirms
that blocking proteasomal degradation by treatment with various
ubiquitination inhibitors ameliorates VC in both in vitro
and in vivo models (70).
The nuclear factor-κB (NF-κB) family plays important
roles in inflammation and atherosclerosis. In unstimulated cells,
NF-kB is present in the cytoplasm and binds to inhibitor of κB
(IκB). In response to various stimuli, including inflammation,
specific kinases, such as IκB-kinase β (IκKβ), mediate the
phosphorylation of IκB. This leads to its ubiquitination and
degradation, which are followed by the translocation of NF-κB to
the nucleus and the activation of transcription (71). Knockdown of IκKβ in VSMCs reduces
the ubiquitination of β-catenin, upregulates β-catenin and Runx2
signaling and accelerates calcification. By contrast, persistent
activation of IκKβ inhibits calcification by increasing the
ubiquitination of β-catenin (72).
Dysregulation of the autophagy-lysosome pathway in VSMCs mediates
VC induced by a high-phosphorus environment and this pathway has
become a new target for VC therapy. Transcription factor EB (TFEB)
is considered the master regulator of lysosomal biogenesis. A
high-phosphorus environment can result in the degradation of TFEB
through the ubiquitin-proteasome system and subsequently promote VC
in vitro and in vivo (73).
The results of studies on the effects of
ubiquitination on VC are inconsistent. Moreover, the related
mechanisms are complex and need further study. Identifying the
ubiquitination sites in calcification-related factors and targeting
these sites is expected to lead to approaches for suppressing VC
(Fig. 4).
Protein acetylation is a reversible modification
that is catalyzed by acetyltransferases (N-terminal
acetyltransferases/lysine acetyltransferases, NATs/KATs), which
transfer acetyl groups from acetyl-CoA to amino acid residues in
proteins such as histones and transcription factors (74,75).
The reverse process, deacetylation, is catalyzed by lysine
deacetylases. Histone acetylation is the most common type of
acetylation. The main histone acetyltransferases are the members of
the P300/CBP, Gcn5-related N-acetyltransferase, Steroid receptor
coactivator and MYST (MOZ-YBF2/SAS3-SAS2-TIP60) families. There are
four subfamilies of histone deacetylases (HDACs): Classes I, II and
IV, which are Zn2+ dependent and class III (for example,
SIRTs), which are NAD+ dependent. In addition, acetyltransferases
catalyze the acetylation of nonhistone proteins, such as
transcription factors and regulate transcription factor stability
and DNA binding (31). In some
cases, ubiquitination competes with acetylation for the same lysine
residues (75).
The levels of histone 3 and 4 (H3 and H4)
acetylation are elevated in the aortic valve calcification model.
However, in this model, treatment with an inhibitor of the histone
acetyltransferase P300, C646, was found to attenuate calcification
in vivo and in vitro by significantly decreasing the
acetylation levels of H3 and H4 (76,77).
Studies have shown that β-catenin and Runx2 are also targets of
P300-mediated acetylation and that acetylation of β-catenin by P300
can enhance the activation of β-catenin signaling (78,79).
The acetylation of Runx2 can increase its stability and its
osteogenic transcriptional activity, as well as osteoblast
differentiation (80,81). By contrast, HDACs can directly bind
to Runx2 and act as core inhibitors of its transcriptional
activity. HDACs catalyze the deacetylation of Runx2, mediate its
ubiquitination and degradation and inhibit osteoblast
differentiation (80,82). HDAC inhibitors have been shown to
promote Runx2 acetylation, thereby promoting osteoblast
differentiation in vitro and osteogenesis in vivo
(80,83). However, no evidence indicates that
the aforementioned acetylation events are directly related to
VC.
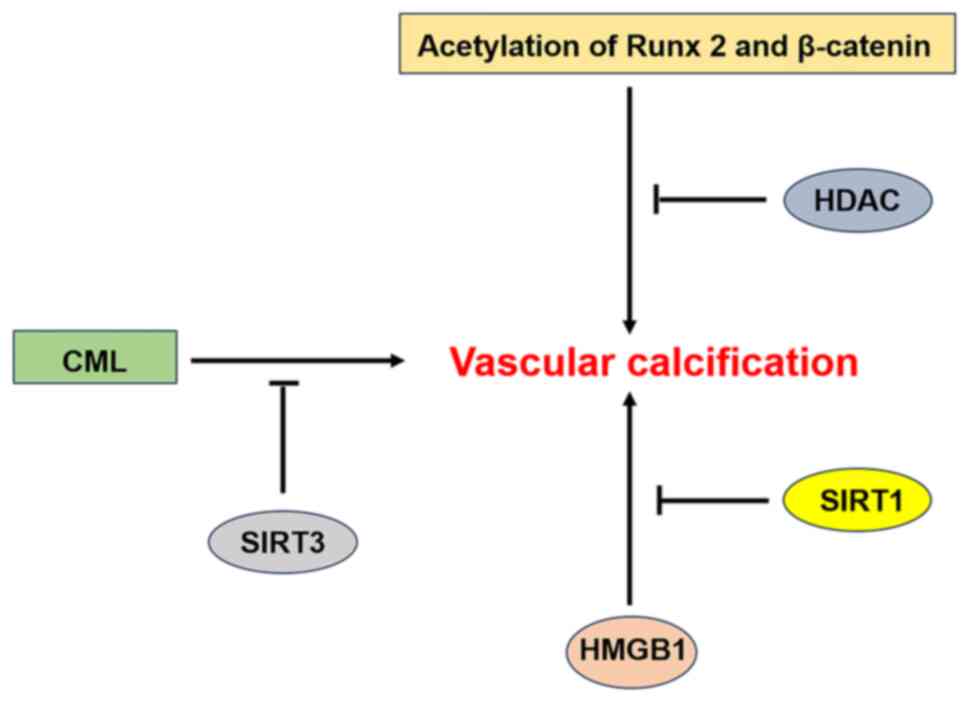
The class III HDAC sirtuin 1 (SIRT1) is an important
factor in the regulation of VC, with three main related functions.
i) SIRT1 can regulate Wnt/β-catenin signaling. Under physiological
conditions, SIRT1 binds to p300 and the acetylation and nuclear
translocation of β-catenin are reduced, thereby inhibiting
osteogenic activity and VC. However, in a high-glucose environment,
the low abundance of SIRT1 cannot prevent P300-mediated acetylation
of β-catenin, thus promoting VC (84). Similarly, hyperphosphataemia can
downregulate SIRT1 expression (85), allow constitutive acetylation of
downstream proteins and accelerate calcification by promoting
hyperacetylation of β-catenin and Runx2 via P300 (80,86).
ii) In a high-glucose environment, the decreased expression of
SIRT1 results in an increase in Runx2 expression and the promotion
of VC via a direct increase in the acetylation level of the Runx2
promoter (87). iii) Other studies
have shown that high mobility group box 1 (HMGB1), an activator of
BMP-2 and protein kinase RNA-like ER kinase (PERK) are targets of
SIRT1 for deacetylation. SIRT1 can inhibit the inflammatory
response through the deacetylation of HMGB1, thereby inhibiting VC
(88,89). Moreover, in both in vitro
and in vivo models, SIRT1 was found to ameliorate
endoplasmic reticulum (ER) stress-induced VC by deacetylating K889
in PERK (90). In addition to
SIRT1, SIRT3 also plays an important role in VC. Sun et al
(91) report that SIRT3 inhibits
VC by regulating Nε-carboxymethyl-lysine (CML).
SIRT1 and SIRT3 play important roles in VC.
Identifying their targets will help us to understand the mechanism
of VC and develop protective agents against VC that will benefit
the majority of patients (Fig.
5).
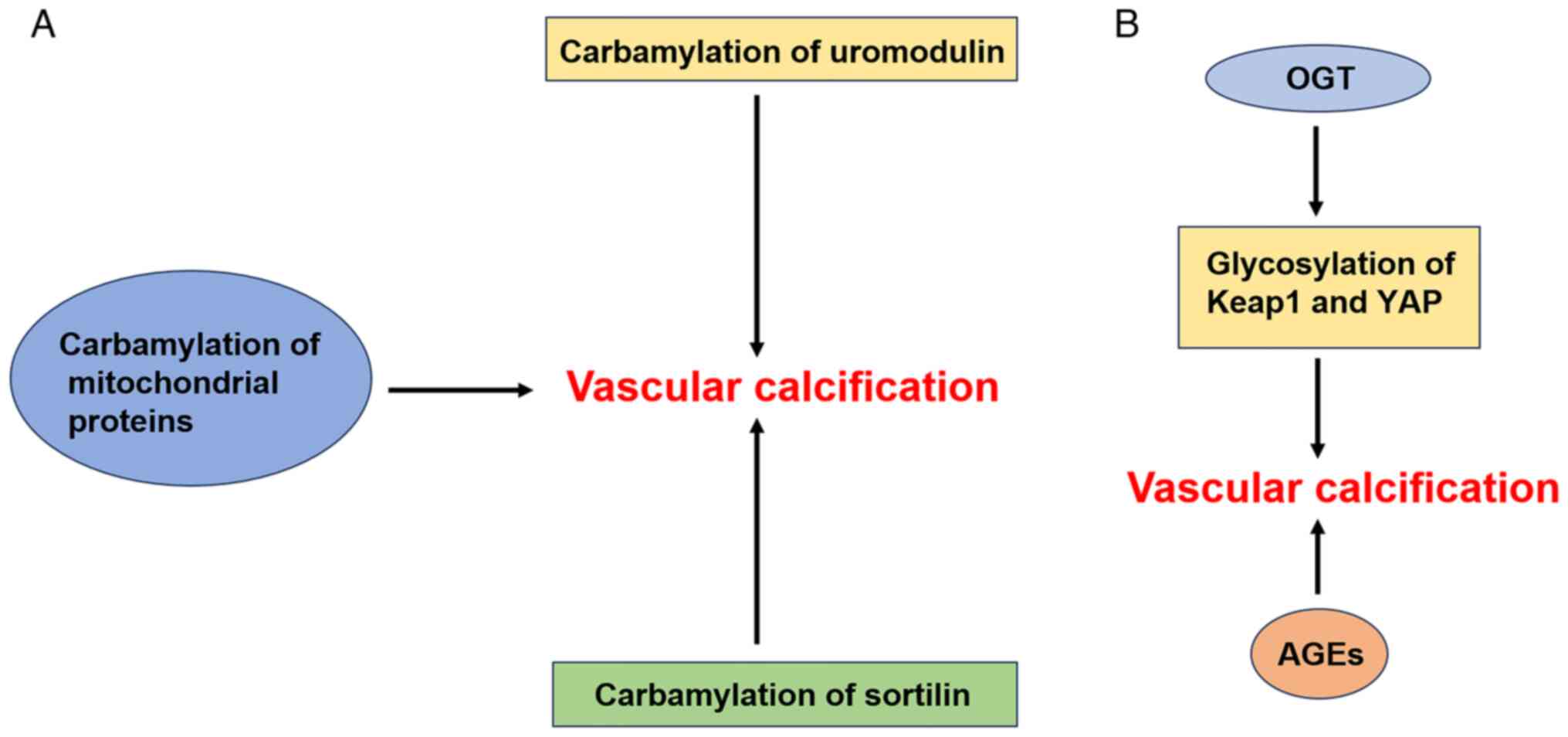
Sortilin is an intracellular sorting receptor that
has been identified as a cardiovascular risk factor in humans
(96). Compared with control
individuals with normal renal function, patients with CKD have
increased serum levels of sortilin and harbor carbamylated sortilin
in the circulation (93),
localized to calcified areas. By generating hVSMCs and establishing
an isolated rat aortic ring model of calcification, Jankowski et
al (97) found that
carbamylated sortilin can promote VC. Further studies revealed that
sortilin carbamylation promotes VC mediated by hVSMCs, possibly due
to an increase in their binding affinity for leukocytes. Similarly,
sortilin carbamylation was found to be associated with the volume
and progression of coronary artery calcification in patients with
CKD (97,98).
O-GlcNAcylation, in which a single GlcNAc
monosaccharide is transferred to serine and threonine residues in
the target intracellular protein, is the most commonly studied type
of O-glycosylation. This modification is catalyzed by O-GlcNAc
transferase (OGT) and studies have reported that abnormal OGT
activity can lead to cardiovascular complications (100). For instance, Xu et al
(101) reported that OGT
expression is significantly increased in both high-phosphorus
diet-induced 5/6 nephrectomized rats and VSMC calcification models
and that OGT knockdown inhibits VC induced by a high-phosphorus
environment. Further study of the mechanism by which OGT promotes
VC in CKD revealed that OGT overexpression increases the
glycosylation of Kelch-like ECH-associated protein 1 (Keap1), which
leads to the degradation of nuclear factor erythroid 2-related
factor 2 and inhibits VSMC autophagy, in turn promoting VC in
vitro and in vivo. Xu et al (102) also report that OGT promotes the
glycosylation of Yes-associated protein (YAP) to increase its
stability and upregulates the expression of YAP to inhibit
autophagy, thus accelerating VC induced by a high-phosphorus
environment. Therefore, OGT knockdown is expected to inhibit VC in
CKD by reducing the glycosylation of target proteins and activating
autophagy. In addition, Heath et al (51) report that activating the
O-GlcNAcylation of AKT increases Runx2 activity and promotes the
development of VC in diabetic mice.
The relationship between N-glycosylation and VC is
another research hotspot. Insulin-like growth factor-I (IGF-I) has
been identified as a major inhibitor of VC. Siddals et al
(103) used statins to deplete
the substrates required for N-glycosylation and used tunicamycin to
inhibit N-glycosylation. They found that statins and tunicamycin
disrupt the inhibitory effect of IGF-I on
β-glycerophosphate-induced VC by altering the glycosylation of the
IGF receptor (IGFR). TGF-β is a multifunctional cytokine that has
been shown to regulate VC and the differentiation of VSMCs in
vivo (104,105). Studies have shown that TGF-β1 is
a target of N-glycosylation. Sha et al (106) reported that tunicamycin inhibits
TGF-β1 secretion and leads to an increase in the level of the
cell-associated non-glycosylated form of TGF-β1. In addition, the
TGF-β receptor TGF-βR is regulated by N-glycosylation. The removal
of glycosylation from TGF-βR affects its interaction with its
ligand, resulting in failure to activate downstream signaling
pathways (107). Although a few
reports have addressed TGF-β or TGF-β receptor N-glycosylation, no
relevant research has been conducted on the direct relationship
between TGF-β or TGF-β receptor N-glycosylation and VC. Notably,
blocking core fucosylation, a specific form of N-glycosylation,
suppresses VC-associated processes in VSMCs. Inhibition of TGFβR
fucosylation significantly dysregulates downstream TGFβ/Smad2/3
signaling (108).
Accumulated studies have shown that advanced
glycation end products (AGEs), which are predictors of
cardiovascular disease mortality, may play an important role in VC.
Previous studies have shown that the local and circulating levels
of AGEs in patients with diabetic nephropathy and nondialysis CKD
are significantly elevated (109,110). Moreover, recent clinical studies
show that the measured levels of AGEs in the skin of 122 patients
with type 2 diabetes and in the radial arteries of 54 patients with
CKD are positively correlated with the degree of arterial
calcification (111,112). In addition, several animal
studies show that the knockdown of the receptor for AGEs (RAGE) has
therapeutic benefits in inhibiting the development of VC (113-115).
B4GALNT3-mediated LacdiNAc (LDN) glycosylation of sclerostin may be
a bone-specific osteoporosis target (116), suggesting that glycosylated
sclerostin may have potential research value in VC.
In general, glycosylation contributes to the
development of VC. Predicting glycosylation events and sites
through machine learning models (117) and identifying inhibitors of
glycosylation may lead to the development of a therapeutic strategy
to target VC in patients with CKD and diabetes (Fig. 6B).
Several auxiliary tests are currently used for the
diagnosis of VC. The ankle-brachial index test is the preferred
first-line screening method for VC in patients with peripheral
artery disease (118). Recently,
a simple VC score on the basis of a planar X-ray of the foot in two
projections was proposed. Via evaluation of five vascular sites and
the length of the ‘pipe-steam’, patients can be divided into three
VC categories: Absent, moderate and severe (119). Computed tomography (CT)
exquisitely visualizes peripheral and coronary artery
calcifications as high-density signals and provides a whole-body
estimate of the VC burden (120).
Recently, several novel biological markers, such as Fibroblast
growth factor-23 and Klotho, have also been used to predict VC
(120).
Studies show that mutation or deletion of certain
genes is closely related to VC. Mutations in the ABCC6 and ENPP1
genes are responsible for generalized arterial calcification in 14
of 28 children and patients with idiopathic infantile arterial
calcification, respectively (121,122). Loss of the RAGE gene attenuated
atherosclerosis in apolipoprotein-deficient mice (123). Moreover, Klotho gene deficiency
is widely recognized to be responsible for VC in CKD (124). Therefore, genetic testing is
valuable for predicting VC.
Numerous preclinical and clinical studies have been
conducted to find further approaches for the management of VC.
Potentially effective therapeutic drugs include calcium channel
blockers, renin-angiotensin system inhibitors, statins,
bisphosphonates, denosumab and myoinositol hexaphosphate (125). However, the therapeutic effects
of these drugs are still unsatisfactory. Therefore, more effective
drugs to delay or treat VC are needed.
In the present review, the important role of PTMs in
the occurrence and development of VC were reviewed. In general,
acetylation, carbamylation and glycosylation can promote VC,
whereas deacetylation suppresses VC. Therefore, future inhibitors
of acetylation, carbamylation and glycosylation and agonists of
SIRT family deacetylases are expected to ameliorate VC. However,
the results of studies reporting the effect of ubiquitination on VC
are inconsistent. More clearly, increasing the
ubiquitination-mediated degradation of the procalcification factors
Runx2 and β-catenin is expected to delay the development of VC.
VC is a common complication of atherosclerotic
cardiovascular disease, DM and CKD and there is still no effective
treatment. PTMs have recently become a popular topic across medical
research. Studies have shown that PTMs play important roles in the
development of VC by participating in the regulation of
calcification-related pathways, such as the Wnt/β-catenin and
PTEN/AKT pathways; calcification-related factors, such as Runx2 and
BMP2; mitochondrial function; inflammation; oxidative stress; and
autophagy (29-31,34,52,68).
In addition, various PTMs interact with each other and
co-operatively control the occurrence and development of VC.
However, few studies have been conducted on PTMs in VC and the
results of the existing studies are inconsistent. In addition, the
role of PTMs in the transdifferentiation of VSMCs to osteoblasts
remains to be elucidated.
It is well known that PTMs involve the chemical
modification of certain amino acids in target proteins, which can
alter the biological function of the modified proteins. Therefore,
the modification sites on target proteins play a crucial role in
PTMs. Currently, a large number of protein databases have been
established, such as the UniProt database, which contains 189
million protein sequence records (126). Given the vast amount of data,
using traditional molecular biology experimental techniques to
identify specific PTMs and their modification sites requires
significant human resources and time. Recently, machine learning
has been increasingly applied in the fields of medicine and life
sciences. Computational methods based on machine learning offer an
alternative strategy for predicting PTMs and their modification
sites in a cost-effective and efficient manner. Moreover,
identifying the specific types of PTMs and the amino acid
modification sites on target proteins can provide novel insights
into disease treatment strategies (127). Further studies on PTMs will help
us understand the mechanism underlying the development and
progression of VC to support the development of new strategies and
targets for VC prevention and treatment.
Not applicable.
Funding: No funding was received.
Not applicable.
DW and QL designed the review and conducted the
literature search. CX wrote and revised the manuscript. All authors
have read and approved the final manuscript. Data authentication is
not applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Li J, Li C, Huang Z, Huang C, Liu J, Wu T,
Xu S, Mai P, Geng D, Zhou S, et al: Empagliflozin alleviates
atherosclerotic calcification by inhibiting osteogenic
differentiation of vascular smooth muscle cells. Front Pharmacol.
14(1295463)2023.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Urbain F, Ponnaiah M, Ichou F, Lhomme M,
Materne C, Galier S, Haroche J, Frisdal E, Mathian A, Durand H, et
al: Impaired metabolism predicts coronary artery calcification in
women with systemic lupus erythematosus. EBioMedicine.
96(104802)2023.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Zhang H, Li G, Yu X, Yang J, Jiang A,
Cheng H, Fu J, Liang X, Liu J, Lou J, et al: Progression of
vascular calcification and clinical outcomes in patients receiving
maintenance dialysis. JAMA Netw Open. 6(e2310909)2023.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Snell-Bergeon JK, Budoff MJ and Hokanson
JE: Vascular calcification in diabetes: Mechanisms and
implications. Curr Diab Rep. 13:391–402. 2013.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Alman AC, Maahs DM, Rewers MJ and
Snell-Bergeon JK: Ideal cardiovascular health and the prevalence
and progression of coronary artery calcification in adults with and
without type 1 diabetes. Diabetes Care. 37:521–528. 2014.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Berry C, Tardif JC and Bourassa MG:
Coronary heart disease in patients with diabetes: Part I: Recent
advances in prevention and noninvasive management. J Am Coll
Cardiol. 49:631–642. 2007.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Yahagi K, Kolodgie FD, Lutter C, Mori H,
Romero ME, Finn AV and Virmani R: Pathology of human coronary and
carotid artery atherosclerosis and vascular calcification in
diabetes mellitus. Arterioscler Thromb Vasc Biol. 37:191–204.
2017.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Doherty TM, Fitzpatrick LA, Inoue D, Qiao
JH, Fishbein MC, Detrano RC, Shah PK and Rajavashisth TB:
Molecular, endocrine, and genetic mechanisms of arterial
calcification. Endocr Rev. 25:629–672. 2004.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Stary HC, Chandler AB, Dinsmore RE, Fuster
V, Glagov S, Insull W Jr, Rosenfeld ME, Schwartz CJ, Wagner WD and
Wissler RW: A definition of advanced types of atherosclerotic
lesions and a histological classification of atherosclerosis. A
report from the committee on vascular lesions of the council on
arteriosclerosis, American heart association. Circulation.
92:1355–1374. 1995.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Ross R: The pathogenesis of
atherosclerosis: A perspective for the 1990s. Nature. 362:801–809.
1993.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Sharif N, Gilani SZ, Suter D, Reid S,
Szulc P, Kimelman D, Monchka BA, Jozani MJ, Hodgson JM, Sim M, et
al: Machine learning for abdominal aortic calcification assessment
from bone density machine-derived lateral spine images.
EBioMedicine. 94(104676)2023.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Wang M, Niu G, Chen Y, Zhou Z, Feng D,
Zhang Y and Wu Y: China-DVD2 Study Group (Standard Evaluation and
Optimal Treatment for Elderly Patients with Valvular Heart Disease,
National Key R&D Program of China, NCT05044338):. Development
and validation of a deep learning-based fully automated algorithm
for pre-TAVR CT assessment of the aortic valvular complex and
detection of anatomical risk factors: A retrospective, multicentre
study. EBioMedicine. 96(104794)2023.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Park S, Choi ES, Jung HW, Lee JY, Park JW,
Bang JS and Jeon YT: Preoperative serum alkaline phosphatase and
neurological outcome of cerebrovascular surgery. J Clin Med.
11(2981)2022.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Bardeesi ASA, Gao J, Zhang K, Yu S, Wei M,
Liu P and Huang H: A novel role of cellular interactions in
vascular calcification. J Transl Med. 15(95)2017.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Durham AL, Speer MY, Scatena M, Giachelli
CM and Shanahan CM: Role of smooth muscle cells in vascular
calcification: Implications in atherosclerosis and arterial
stiffness. Cardiovasc Res. 114:590–600. 2018.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Niu Z, Su G, Li T, Yu H, Shen Y, Zhang D
and Liu X: Vascular calcification: New insights into BMP type I
receptor A. Front Pharmacol. 13(887253)2022.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Pan X, Pi C, Ruan X, Zheng H, Zhang D and
Liu X: Mammalian sirtuins and their relevance in vascular
calcification. Front Pharmacol. 13(907835)2022.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Feng S, Qi Y, Xiao Z, Chen H, Liu S, Luo
H, Wu H and Zhang W: CircHIPK3 relieves vascular calcification via
mediating SIRT1/PGC-1α/MFN2 pathway by interacting with FUS. BMC
Cardiovasc Disord. 23(583)2023.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Song GY, Guo XN, Yao J, Lu ZN, Xie JH, Wu
F, He J, Fu ZL and Han J: Differential expression profiles and
functional analysis of long non-coding RNAs in calcific aortic
valve disease. BMC Cardiovasc Disord. 23(326)2023.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Wu M, Rementer C and Giachelli CM:
Vascular calcification: An update on mechanisms and challenges in
treatment. Calcif Tissue Int. 93:365–373. 2013.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Demer LL and Tintut Y: Vascular
calcification: Pathobiology of a multifaceted disease. Circulation.
117:2938–2948. 2008.PubMed/NCBI View Article : Google Scholar
|
|
22
|
McCullough PA, Chinnaiyan KM, Agrawal V,
Danielewicz E and Abela GS: Amplification of atherosclerotic
calcification and Mönckeberg's sclerosis: A spectrum of the same
disease process. Adv Chronic Kidney Dis. 15:396–412.
2008.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Beltrao P, Bork P, Krogan NJ and van Noort
V: Evolution and functional cross-talk of protein
post-translational modifications. Mol Syst Biol.
9(714)2013.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Wang R and Wang G: Protein modification
and autophagy activation. Adv Exp Med Biol. 1206:237–259.
2019.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Liu Y and Peng FX: Research progress on
O-GlcNAcylation in the occurrence, development, and treatment of
colorectal cancer. World J Gastrointest Surg. 13:96–115.
2021.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Mir AR and Moinuddin : Glycoxidation
of histone proteins in autoimmune disorders. Clin Chim Acta.
450:25–30. 2015.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Tweedie-Cullen RY, Reck JM and Mansuy IM:
Comprehensive mapping of post-translational modifications on
synaptic, nuclear, and histone proteins in the adult mouse brain. J
Proteome Res. 8:4966–4982. 2009.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Voelkl J, Lang F, Eckardt KU, Amann K,
Kuro-O M, Pasch A, Pieske B and Alesutan I: Signaling pathways
involved in vascular smooth muscle cell calcification during
hyperphosphatemia. Cell Mol Life Sci. 76:2077–2091. 2019.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Lee HY, Lim S and Park S: Role of
inflammation in arterial calcification. Korean Circ J. 51:114–125.
2021.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Neutel CHG, Hendrickx JO, Martinet W, De
Meyer GRY and Guns PJ: The protective effects of the autophagic and
lysosomal machinery in vascular and valvular calcification: A
systematic review. Int J Mol Sci. 21(8933)2020.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Kwon DH, Ryu J, Kim YK and Kook H: Roles
of histone acetylation modifiers and other epigenetic regulators in
vascular calcification. Int J Mol Sci. 21(3246)2020.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Wozney JM, Rosen V, Celeste AJ, Mitsock
LM, Whitters MJ, Kriz RW, Hewick RM and Wang EA: Novel regulators
of bone formation: Molecular clones and activities. Science.
242:1528–1534. 1988.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Cai J, Pardali E, Sánchez-Duffhues G and
ten Dijke P: BMP signaling in vascular diseases. FEBS Lett.
586:1993–2002. 2012.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Rong S, Zhao X, Jin X, Zhang Z, Chen L,
Zhu Y and Yuan W: Vascular calcification in chronic kidney disease
is induced by bone morphogenetic protein-2 via a mechanism
involving the Wnt/β-catenin pathway. Cell Physiol Biochem.
34:2049–2060. 2014.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Balderman JA, Lee HY, Mahoney CE, Handy
DE, White K, Annis S, Lebeche D, Hajjar RJ, Loscalzo J and Leopold
JA: Bone morphogenetic protein-2 decreases microRNA-30b and
microRNA-30c to promote vascular smooth muscle cell calcification.
J Am Heart Assoc. 1(e003905)2012.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Derwall M, Malhotra R, Lai CS, Beppu Y,
Aikawa E, Seehra JS, Zapol WM, Bloch KD and Yu PB: Inhibition of
bone morphogenetic protein signaling reduces vascular calcification
and atherosclerosis. Arterioscler Thromb Vasc Biol. 32:613–622.
2012.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Nakagawa Y, Ikeda K, Akakabe Y, Koide M,
Uraoka M, Yutaka KT, Kurimoto-Nakano R, Takahashi T, Matoba S,
Yamada H, et al: Paracrine osteogenic signals via bone
morphogenetic protein-2 accelerate the atherosclerotic intimal
calcification in vivo. Arterioscler Thromb Vasc Biol. 30:1908–1915.
2010.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Graciolli FG, Neves KR, dos Reis LM,
Graciolli RG, Noronha IL, Moysés RM and Jorgetti V: Phosphorus
overload and PTH induce aortic expression of Runx2 in experimental
uraemia. Nephrol Dial Transplant. 24:1416–1421. 2009.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Byon CH, Javed A, Dai Q, Kappes JC,
Clemens TL, Darley-Usmar VM, McDonald JM and Chen Y: Oxidative
stress induces vascular calcification through modulation of the
osteogenic transcription factor Runx2 by AKT signaling. J Biol
Chem. 283:15319–15327. 2008.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Lin ME, Chen T, Leaf EM, Speer MY and
Giachelli CM: Runx2 expression in smooth muscle cells is required
for arterial medial calcification in mice. Am J Pathol.
185:1958–1969. 2015.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Sun Y, Byon CH, Yuan K, Chen J, Mao X,
Heath JM, Javed A, Zhang K, Anderson PG and Chen Y: Smooth muscle
cell-specific runx2 deficiency inhibits vascular calcification.
Circ Res. 111:543–552. 2012.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Simonet WS, Lacey DL, Dunstan CR, Kelley
M, Chang MS, Lüthy R, Nguyen HQ, Wooden S, Bennett L, Boone T, et
al: Osteoprotegerin: A novel secreted protein involved in the
regulation of bone density. Cell. 89:309–319. 1997.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Bucay N, Sarosi I, Dunstan CR, Morony S,
Tarpley J, Capparelli C, Scully S, Tan HL, Xu W, Lacey DL, et al:
Osteoprotegerin-deficient mice develop early onset osteoporosis and
arterial calcification. Genes Dev. 12:1260–1268. 1998.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Morony S, Tintut Y, Zhang Z, Cattley RC,
Van G, Dwyer D, Stolina M, Kostenuik PJ and Demer LL:
Osteoprotegerin inhibits vascular calcification without affecting
atherosclerosis in ldlr(-/-) mice. Circulation. 117:411–420.
2008.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Tschiderer L, Willeit J, Schett G, Kiechl
S and Willeit P: Osteoprotegerin concentration and risk of
cardiovascular outcomes in nine general population studies:
Literature-based meta-analysis involving 26, 442 participants. PLoS
One. 12(e0183910)2017.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Morena M, Jaussent I, Halkovich A, Dupuy
AM, Bargnoux AS, Chenine L, Leray-Moragues H, Klouche K, Vernhet H,
Canaud B and Cristol JP: Bone biomarkers help grading severity of
coronary calcifications in non dialysis chronic kidney disease
patients. PLoS One. 7(e36175)2012.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Valdivielso JM: Vascular calcification:
Types and mechanisms. Nefrologia. 31:142–147. 2011.PubMed/NCBI View Article : Google Scholar : (In Spanish).
|
|
48
|
Fitzpatrick LA, Severson A, Edwards WD and
Ingram RT: Diffuse calcification in human coronary arteries.
Association of osteopontin with atherosclerosis. J Clin Invest.
94:1597–1604. 1994.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Speer MY, McKee MD, Guldberg RE, Liaw L,
Yang HY, Tung E, Karsenty G and Giachelli CM: Inactivation of the
osteopontin gene enhances vascular calcification of matrix Gla
protein-deficient mice: Evidence for osteopontin as an inducible
inhibitor of vascular calcification in vivo. J Exp Med.
196:1047–1055. 2002.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Johnson ML and Rajamannan N: Diseases of
Wnt signaling. Rev Endocr Metab Disord. 7:41–49. 2006.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Heath JM, Sun Y, Yuan K, Bradley WE,
Litovsky S, Dell'Italia LJ, Chatham JC, Wu H and Chen Y: Activation
of AKT by O-linked N-acetylglucosamine induces vascular
calcification in diabetes mellitus. Circ Res. 114:1094–1102.
2014.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Deng L, Huang L, Sun Y, Heath JM, Wu H and
Chen Y: Inhibition of FOXO1/3 promotes vascular calcification.
Arterioscler Thromb Vasc Biol. 35:175–183. 2015.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Proudfoot D, Skepper JN, Hegyi L, Bennett
MR, Shanahan CM and Weissberg PL: Apoptosis regulates human
vascular calcification in vitro: Evidence for initiation of
vascular calcification by apoptotic bodies. Circ Res. 87:1055–1062.
2000.PubMed/NCBI View Article : Google Scholar
|
|
54
|
New SEP and Aikawa E: Role of
extracellular vesicles in de novo mineralization: An additional
novel mechanism of cardiovascular calcification. Arterioscler
Thromb Vasc Biol. 33:1753–1758. 2013.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Coscas R, Bensussan M, Jacob MP, Louedec
L, Massy Z, Sadoine J, Daudon M, Chaussain C, Bazin D and Michel
JB: Free DNA precipitates calcium phosphate apatite crystals in the
arterial wall in vivo. Atherosclerosis. 259:60–67. 2017.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Li M, Wang ZW, Fang LJ, Cheng SQ, Wang X
and Liu NF: Programmed cell death in atherosclerosis and vascular
calcification. Cell Death Dis. 13(467)2022.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Chen Y, Wei L, Zhang X, Liu X, Chen Y,
Zhang S, Zhou L, Li Q, Pan Q, Zhao S and Liu H: 3-Bromopyruvate
sensitizes human breast cancer cells to TRAIL-induced apoptosis via
the phosphorylated AMPK-mediated upregulation of DR5. Oncol Rep.
40:2435–2444. 2018.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Chiong M, Cartes-Saavedra B,
Norambuena-Soto I, Mondaca-Ruff D, Morales PE, García-Miguel M and
Mellado R: Mitochondrial metabolism and the control of vascular
smooth muscle cell proliferation. Front Cell Dev Biol.
2(72)2014.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Chen WR, Zhou YJ, Yang JQ, Liu F, Wu XP
and Sha Y: Melatonin attenuates calcium deposition from vascular
smooth muscle cells by activating mitochondrial fusion and
mitophagy via an AMPK/OPA1 signaling pathway. Oxid Med Cell Longev.
2020(5298483)2020.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Chen WR, Zhou YJ, Sha Y, Wu XP, Yang JQ
and Liu F: Melatonin attenuates vascular calcification by
inhibiting mitochondria fission via an AMPK/Drp1 signalling
pathway. J Cell Mol Med. 24:6043–6054. 2020.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Scheffner M, Nuber U and Huibregtse JM:
Protein ubiquitination involving an E1-E2-E3 enzyme ubiquitin
thioester cascade. Nature. 373:81–83. 1995.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Kaoutari AE, Fraunhoffer NA, Audebert S,
Camoin L, Berthois Y, Gayet O, Roques J, Bigonnet M, Bongrain C,
Ciccolini J, et al: Pancreatic ductal adenocarcinoma ubiquitination
profiling reveals specific prognostic and theranostic markers.
EBioMedicine. 92(104634)2023.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Frezza M, Schmitt S and Dou QP: Targeting
the ubiquitin-proteasome pathway: An emerging concept in cancer
therapy. Curr Top Med Chem. 11:2888–2905. 2011.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Xu Q, Pan G, Wang Z, Wang L, Tang Y, Dong
J and Qin JJ: Platycodin-D exerts its anti-cancer effect by
promoting c-Myc protein ubiquitination and degradation in gastric
cancer. Front Pharmacol. 14(1138658)2023.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Zhao M, Qiao M, Oyajobi BO, Mundy GR and
Chen D: E3 ubiquitin ligase Smurf1 mediates core-binding factor
alpha1/Runx2 degradation and plays a specific role in osteoblast
differentiation. J Biol Chem. 278:27939–27944. 2003.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Jiang Y, Zhang J, Li Z and Jia G: Bone
marrow mesenchymal stem cell-derived exosomal miR-25 regulates the
ubiquitination and degradation of Runx2 by SMURF1 to promote
fracture healing in mice. Front Med (Lausanne).
7(577578)2020.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Choi YH, Kim YJ, Jeong HM, Jin YH, Yeo CY
and Lee KY: Akt enhances Runx2 protein stability by regulating
Smurf2 function during osteoblast differentiation. FEBS J.
281:3656–3666. 2014.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Ouyang L, Yu C, Xie Z, Su X, Xu Z, Song P,
Li J, Huang H, Ding Y and Zou MH: Indoleamine 2,3-dioxygenase 1
deletion-mediated kynurenine insufficiency in vascular smooth
muscle cells exacerbates arterial calcification. Circulation.
145:1784–1798. 2022.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Kim BG, Lee JH, Yasuda J, Ryoo HM and Cho
JY: Phospho-Smad1 modulation by nedd4 E3 ligase in BMP/TGF-β
signaling. J Bone Miner Res. 26:1411–1424. 2011.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Kwon DH, Eom GH, Ko JH, Shin S, Joung H,
Choe N, Nam YS, Min HK, Kook T, Yoon S, et al: MDM2 E3
ligase-mediated ubiquitination and degradation of HDAC1 in vascular
calcification. Nat Commun. 7(10492)2016.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Weng X, Luo X, Dai X, Lv Y, Zhang S, Bai
X, Bao X, Wang Y, Zhao C, Zeng M, et al: Apigenin inhibits
macrophage pyroptosis through regulation of oxidative stress and
the NF-κB pathway and ameliorates atherosclerosis. Phytother Res.
37:5300–5314. 2023.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Al-Huseini I, Ashida N and Kimura T:
Deletion of IκB-kinase β in smooth muscle cells induces vascular
calcification through β-catenin-runt-related transcription factor 2
signaling. J Am Heart Assoc. 7(e007405)2018.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Ishiwata R and Morimoto Y:
Hyperphosphatemia-induced degradation of transcription factor EB
exacerbates vascular calcification. Biochim Biophys Acta Mol Basis
Dis. 1868(166323)2022.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Weinert BT, Iesmantavicius V, Moustafa T,
Schölz C, Wagner SA, Magnes C, Zechner R and Choudhary C:
Acetylation dynamics and stoichiometry in Saccharomyces cerevisiae.
Mol Syst Biol. 10(716)2014.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Lin R, Tao R, Gao X, Li T, Zhou X, Guan
KL, Xiong Y and Lei QY: Acetylation stabilizes ATP-citrate lyase to
promote lipid biosynthesis and tumor growth. Mol Cell. 51:506–518.
2013.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Gu J, Lu Y, Deng M, Qiu M, Tian Y, Ji Y,
Zong P, Shao Y, Zheng R, Zhou B, et al: Inhibition of acetylation
of histones 3 and 4 attenuates aortic valve calcification. Exp Mol
Med. 51:1–14. 2019.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Li SJ, Kao YH, Chung CC, Chen WY, Cheng WL
and Chen YJ: Activated p300 acetyltransferase activity modulates
aortic valvular calcification with osteogenic transdifferentiation
and downregulation of Klotho. Int J Cardiol. 232:271–279.
2017.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Bouras T, Fu M, Sauve AA, Wang F, Quong
AA, Perkins ND, Hay RT, Gu W and Pestell RG: SIRT1 deacetylation
and repression of p300 involves lysine residues 1020/1024 within
the cell cycle regulatory domain 1. J Biol Chem. 280:10264–10276.
2005.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Hecht A, Vleminckx K, Stemmler MP, van Roy
F and Kemler R: The p300/CBP acetyltransferases function as
transcriptional coactivators of beta-catenin in vertebrates. EMBO
J. 19:1839–1850. 2000.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Jeon EJ, Lee KY, Choi NS, Lee MH, Kim HN,
Jin YH, Ryoo HM, Choi JY, Yoshida M, Nishino N, et al: Bone
morphogenetic protein-2 stimulates Runx2 acetylation. J Biol Chem.
281:16502–16511. 2006.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Jun JH, Yoon WJ, Seo SB, Woo KM, Kim GS,
Ryoo HM and Baek JH: BMP2-activated Erk/MAP kinase stabilizes Runx2
by increasing p300 levels and histone acetyltransferase activity. J
Biol Chem. 285:36410–36419. 2010.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Zhang Z, Deepak V, Meng L, Wang L, Li Y,
Jiang Q, Zeng X and Liu W: Analysis of HDAC1-mediated regulation of
Runx2-induced osteopontin gene expression in C3h10t1/2 cells.
Biotechnol Lett. 34:197–203. 2012.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Bae HS, Yoon WJ, Cho YD, Islam R, Shin HR,
Kim BS, Lim JM, Seo MS, Cho SA, Choi KY, et al: An HDAC inhibitor,
entinostat/MS-275, partially prevents delayed cranial suture
closure in heterozygous Runx2 null mice. J Bone Miner Res.
32:951–961. 2017.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Bartoli-Leonard F, Wilkinson FL,
Langford-Smith AWW, Alexander MY and Weston R: The interplay of
SIRT1 and Wnt signaling in vascular calcification. Front Cardiovasc
Med. 5(183)2018.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Takemura A, Iijima K, Ota H, Son BK, Ito
Y, Ogawa S, Eto M, Akishita M and Ouchi Y: Sirtuin 1 retards
hyperphosphatemia-induced calcification of vascular smooth muscle
cells. Arterioscler Thromb Vasc Biol. 31:2054–2062. 2011.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Lévy L, Wei Y, Labalette C, Wu Y, Renard
CA, Buendia MA and Neuveut C: Acetylation of beta-catenin by p300
regulates beta-catenin-Tcf4 interaction. Mol Cell Biol.
24:3404–3414. 2004.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Bartoli-Leonard F, Wilkinson FL, Schiro A,
Inglott FS, Alexander MY and Weston R: Suppression of SIRT1 in
diabetic conditions induces osteogenic differentiation of human
vascular smooth muscle cells via RUNX2 signalling. Sci Rep.
9(878)2019.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Rabadi MM, Xavier S, Vasko R, Kaur K,
Goligorksy MS and Ratliff BB: High-mobility group box 1 is a novel
deacetylation target of Sirtuin1. Kidney Int. 87:95–108.
2015.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Hwang JS, Choi HS, Ham SA, Yoo T, Lee WJ,
Paek KS and Seo HG: Deacetylation-mediated interaction of
SIRT1-HMGB1 improves survival in a mouse model of endotoxemia. Sci
Rep. 5(15971)2015.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Zhang Y, He L, Tu M, Huang M, Chen Y, Pan
D, Peng J and Shen X: The ameliorative effect of terpinen-4-ol on
ER stress-induced vascular calcification depends on SIRT1-mediated
regulation of PERK acetylation. Pharmacol Res.
170(105629)2021.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Sun Z, Zhang L, Yin K, Zang G, Qian Y, Mao
X, Li L, Jing Q and Wang Z: SIRT3-and FAK-mediated
acetylation-phosphorylation crosstalk of NFATc1 regulates
Nε-carboxymethyl-lysine-induced vascular calcification
in diabetes mellitus. Atherosclerosis. 377:43–59. 2023.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Jaisson S, Pietrement C and Gillery P:
Carbamylation-derived products: Bioactive compounds and potential
biomarkers in chronic renal failure and atherosclerosis. Clin Chem.
57:1499–1505. 2011.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Berg AH, Drechsler C, Wenger J, Buccafusca
R, Hod T, Kalim S, Ramma W, Parikh SM, Steen H, Friedman DJ, et al:
Carbamylation of serum albumin as a risk factor for mortality in
patients with kidney failure. Sci Transl Med.
5(175ra29)2013.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Mori D, Matsui I, Shimomura A, Hashimoto
N, Matsumoto A, Shimada K, Yamaguchi S, Oka T, Kubota K, Yonemoto
S, et al: Protein carbamylation exacerbates vascular calcification.
Kidney Int. 94:72–90. 2018.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Alesutan I, Luong TTD, Schelski N, Masyout
J, Hille S, Schneider MP, Graham D, Zickler D, Verheyen N, Estepa
M, et al: Circulating uromodulin inhibits vascular calcification by
interfering with pro-inflammatory cytokine signalling. Cardiovasc
Res. 117:930–941. 2021.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Goettsch C, Kjolby M and Aikawa E:
Sortilin and its multiple roles in cardiovascular and metabolic
diseases. Arterioscler Thromb Vasc Biol. 38:19–25. 2018.PubMed/NCBI View Article : Google Scholar
|
|
97
|
Jankowski V, Saritas T, Kjolby M, Hermann
J, Speer T, Himmelsbach A, Mahr K, Marina Heuschkel A, Schunk SJ,
Thirup S, et al: Carbamylated sortilin associates with
cardiovascular calcification in patients with chronic kidney
disease. Kidney Int. 101:574–584. 2022.PubMed/NCBI View Article : Google Scholar
|
|
98
|
Massy ZA and Liabeuf S: Sortilin,
carbamylation, and cardiovascular calcification in chronic kidney
disease. Kidney Int. 101:456–459. 2022.PubMed/NCBI View Article : Google Scholar
|
|
99
|
Lumibao JC, Tremblay JR, Hsu J and Engle
DD: Altered glycosylation in pancreatic cancer and beyond. J Exp
Med. 219(e20211505)2022.PubMed/NCBI View Article : Google Scholar
|
|
100
|
Karunakaran U and Jeoung NH: O-GlcNAc
modification: Friend or foe in diabetic cardiovascular disease.
Korean Diabetes J. 34:211–219. 2010.PubMed/NCBI View Article : Google Scholar
|
|
101
|
Xu TH, Du Y, Sheng Z, Li Y, Qiu X, Tian B
and Yao L: OGT-mediated KEAP1 glycosylation accelerates NRF2
degradation leading to high phosphate-induced vascular
calcification in chronic kidney disease. Front Physiol.
11(1092)2020.PubMed/NCBI View Article : Google Scholar
|
|
102
|
Xu TH, Sheng Z, Li Y, Qiu X, Tian B and
Yao L: OGT knockdown counteracts high phosphate-induced vascular
calcification in chronic kidney disease through autophagy
activation by downregulating YAP. Life Sci.
261(118121)2020.PubMed/NCBI View Article : Google Scholar
|
|
103
|
Siddals KW, Allen J, Sinha S, Canfield AE,
Kalra PA and Gibson JM: Apposite insulin-like growth factor (IGF)
receptor glycosylation is critical to the maintenance of vascular
smooth muscle phenotype in the presence of factors promoting
osteogenic differentiation and mineralization. J Biol Chem.
286:16623–16630. 2011.PubMed/NCBI View Article : Google Scholar
|
|
104
|
Watson KE, Boström K, Ravindranath R, Lam
T, Norton B and Demer LL: TGF-beta 1 and 25-hydroxycholesterol
stimulate osteoblast-like vascular cells to calcify. J Clin Invest.
93:2106–2113. 1994.PubMed/NCBI View Article : Google Scholar
|
|
105
|
Kanno Y, Into T, Lowenstein CJ and
Matsushita K: Nitric oxide regulates vascular calcification by
interfering with TGF-signalling. Cardiovasc Res. 77:221–230.
2008.PubMed/NCBI View Article : Google Scholar
|
|
106
|
Sha X, Brunner AM, Purchio AF and Gentry
LE: Transforming growth factor beta 1: Importance of glycosylation
and acidic proteases for processing and secretion. Mol Endocrinol.
3:1090–1098. 1989.PubMed/NCBI View Article : Google Scholar
|
|
107
|
Watanabe S, Misawa M, Matsuzaki T, Sakurai
T, Muramatsu T and Sato M: A novel glycosylation signal regulates
transforming growth factor beta receptors as evidenced by
endo-beta-galactosidase C expression in rodent cells. Glycobiology.
21:482–492. 2011.PubMed/NCBI View Article : Google Scholar
|
|
108
|
Wen X, Liu A, Yu C, Wang L, Zhou M, Wang
N, Fang M, Wang W and Lin H: Inhibiting post-translational core
fucosylation prevents vascular calcification in the model of
uremia. Int J Biochem Cell Biol. 79:69–79. 2016.PubMed/NCBI View Article : Google Scholar
|
|
109
|
Miyata T, van Ypersele de Strihou C,
Kurokawa K and Baynes JW: Alterations in nonenzymatic biochemistry
in uremia: Origin and significance of ‘carbonyl stress’ in
long-term uremic complications. Kidney Int. 55:389–399.
1999.PubMed/NCBI View Article : Google Scholar
|
|
110
|
Rabbani N and Thornalley PJ: Advanced
glycation end products in the pathogenesis of chronic kidney
disease. Kidney Int. 93:803–813. 2018.PubMed/NCBI View Article : Google Scholar
|
|
111
|
Hangai M, Takebe N, Honma H, Sasaki A,
Chida A, Nakano R, Togashi H, Nakagawa R, Oda T, Matsui M, et al:
Association of advanced glycation end products with coronary artery
calcification in japanese subjects with type 2 diabetes as assessed
by skin autofluorescence. J Atheroscler Thromb. 23:1178–1187.
2016.PubMed/NCBI View Article : Google Scholar
|
|
112
|
Janda K, Krzanowski M, Gajda M, Dumnicka
P, Jasek E, Fedak D, Pietrzycka A, Kuźniewski M, Litwin JA and
Sułowicz W: Vascular effects of advanced glycation end-products:
Content of immunohistochemically detected AGEs in radial artery
samples as a predictor for arterial calcification and
cardiovascular risk in asymptomatic patients with chronic kidney
disease. Dis Markers. 2015(153978)2015.PubMed/NCBI View Article : Google Scholar
|
|
113
|
Koike S, Yano S, Tanaka S, Sheikh AM,
Nagai A and Sugimoto T: Advanced glycation end-products induce
apoptosis of vascular smooth muscle cells: A mechanism for vascular
calcification. Int J Mol Sci. 17(1567)2016.PubMed/NCBI View Article : Google Scholar
|
|
114
|
Wei Q, Ren X, Jiang Y, Jin H, Liu N and Li
J: Advanced glycation end products accelerate rat vascular
calcification through RAGE/oxidative stress. BMC Cardiovasc Disord.
13(13)2013.PubMed/NCBI View Article : Google Scholar
|
|
115
|
Bro S, Flyvbjerg A, Binder CJ, Bang CA,
Denner L, Olgaard K and Nielsen LB: A neutralizing antibody against
receptor for advanced glycation end products (RAGE) reduces
atherosclerosis in uremic mice. Atherosclerosis. 201:274–280.
2008.PubMed/NCBI View Article : Google Scholar
|
|
116
|
Movérare-Skrtic S, Voelkl J, Nilsson KH,
Nethander M, Luong TTD, Alesutan I, Li L, Wu J, Horkeby K,
Lagerquist MK, et al: B4GALNT3 regulates glycosylation of
sclerostin and bone mass. EBioMedicine. 91(104546)2023.PubMed/NCBI View Article : Google Scholar
|
|
117
|
Tong YT, Gao GJ, Chang H, Wu XW and Li MT:
Development and economic assessment of machine learning models to
predict glycosylated hemoglobin in type 2 diabetes. Front
Pharmacol. 14(1216182)2023.PubMed/NCBI View Article : Google Scholar
|
|
118
|
Hoek AG, Zwakenberg SR, Elders PJM, de
Jong PA, Spiering W, Bartstra JW, Doesburg T, van der Heijden AA,
van der Schouw YT and Beulens JWJ: SMART Study Group. An elevated
ankle-brachial index is not a valid proxy for peripheral medial
arterial calcification. Atherosclerosis. 323:13–19. 2021.PubMed/NCBI View Article : Google Scholar
|
|
119
|
Ferraresi R, Ucci A, Pizzuto A, Losurdo F,
Caminiti M, Minnella D, Casini A, Clerici G, Montero-Baker M and
Mills J: A novel scoring system for small artery disease and medial
arterial calcification is strongly associated with major adverse
limb events in patients with chronic limb-threatening ischemia. J
Endovasc Ther. 28:194–207. 2021.PubMed/NCBI View Article : Google Scholar
|
|
120
|
Lanzer P, Hannan FM, Lanzer JD, Janzen J,
Raggi P, Furniss D, Schuchardt M, Thakker R, Fok PW, Saez-Rodriguez
J, et al: Medial arterial calcification: JACC state-of-the-art
review. J Am Coll Cardiol. 78:1145–1165. 2021.PubMed/NCBI View Article : Google Scholar
|
|
121
|
Ruf N, Uhlenberg B, Terkeltaub R, Nürnberg
P and Rutsch F: The mutational spectrum of ENPP1 as arising after
the analysis of 23 unrelated patients with generalized arterial
calcification of infancy (GACI). Hum Mutat. 25(98)2005.PubMed/NCBI View Article : Google Scholar
|
|
122
|
Nitschke Y, Baujat G, Botschen U,
Wittkampf T, du Moulin M, Stella J, Le Merrer M, Guest G, Lambot K,
Tazarourte-Pinturier MF, et al: Generalized arterial calcification
of infancy and pseudoxanthoma elasticum can be caused by mutations
in either ENPP1 or ABCC6. Am J Hum Genet. 90:25–39. 2012.PubMed/NCBI View Article : Google Scholar
|
|
123
|
Harja E, Bu DX, Hudson BI, Chang JS, Shen
X, Hallam K, Kalea AZ, Lu Y, Rosario RH, Oruganti S, et al:
Vascular and inflammatory stresses mediate atherosclerosis via RAGE
and its ligands in apoE-/- mice. J Clin Invest. 118:183–194.
2008.PubMed/NCBI View Article : Google Scholar
|
|
124
|
Lin Y and Sun Z: Klotho deficiency-induced
arterial calcification involves osteoblastic transition of VSMCs
and activation of BMP signaling. J Cell Physiol. 237:720–729.
2022.PubMed/NCBI View Article : Google Scholar
|
|
125
|
Pan W, Jie W and Huang H: Vascular
calcification: Molecular mechanisms and therapeutic interventions.
MedComm (2020). 4(e200)2023.PubMed/NCBI View Article : Google Scholar
|
|
126
|
UniProt Consortium: UniProt: The universal
protein knowledgebase in 2021. Nucleic Acids Res. 49
(D1):D480–D489. 2021.PubMed/NCBI View Article : Google Scholar
|
|
127
|
Luo S, Kong C, Zhao S, Tang X, Wang Y,
Zhou X, Li R, Liu X, Tang X, Sun S, et al: Endothelial
HDAC1-ZEB2-NuRD complex drives aortic aneurysm and dissection
through regulation of protein S-sulfhydration. Circulation.
147:1382–1403. 2023.PubMed/NCBI View Article : Google Scholar
|