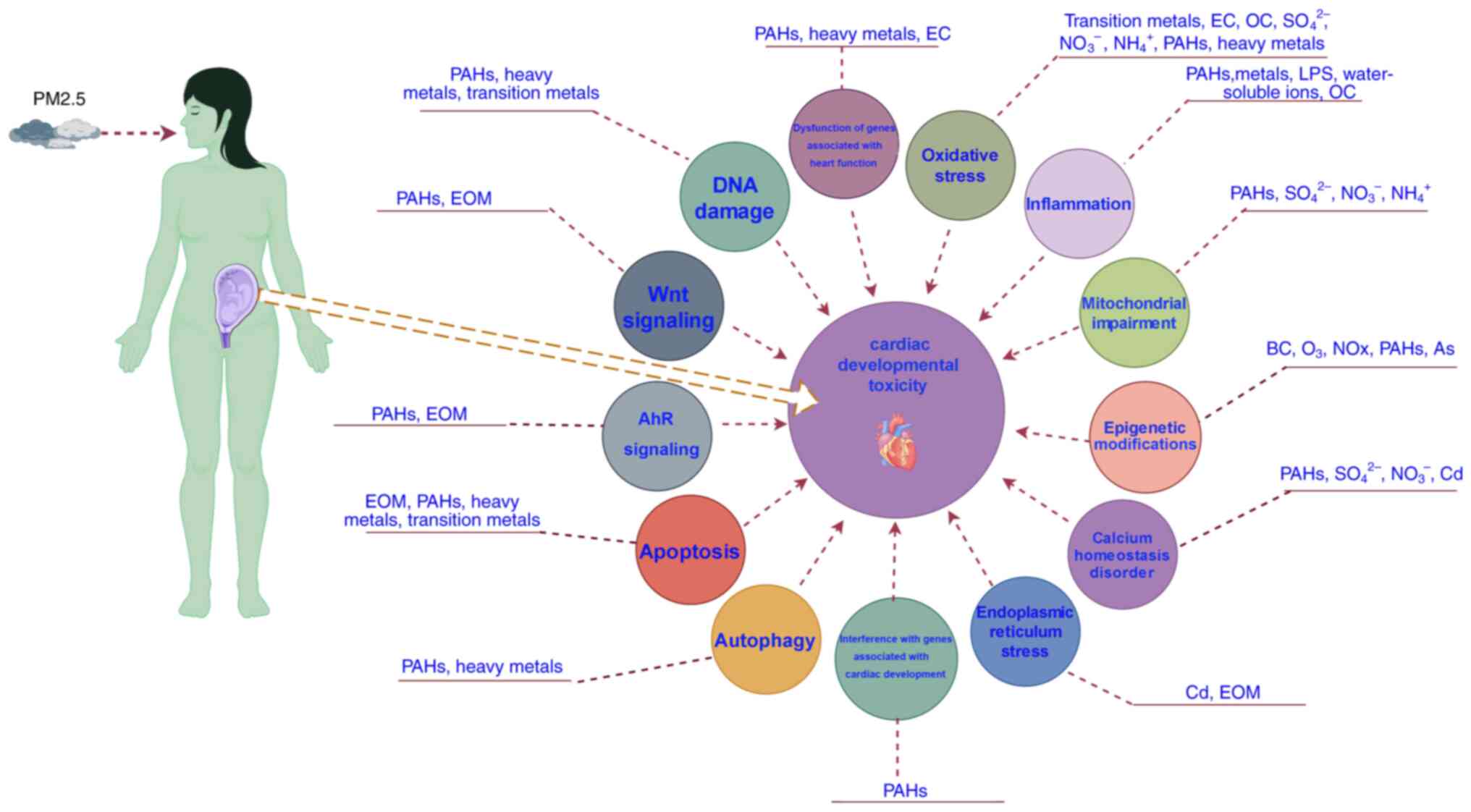
As transcription factors, GATA4 and NKX2.5 perform
crucial roles in fetal cardiac development. Gestational
PM2.5 exposure may increase the risk of GATA4 and NKX2.5
mutations, directly causing fetal cardiac abnormalities. Wu et
al (30) found that
gestational PM2.5 exposure leads to cardiac hypertrophy
with elevated mRNA levels of GATA4 in offspring mice. Moreover, the
important regulatory role of GATA4 in signaling pathways involved
in cardiac development has been confirmed (31,32).
It has been revealed to regulate the expression of key downstream
genes involved in cardiac cell proliferation, development and
hypertrophy, including ANP, CARP, a-MHC and β-MHC (32).
As a key factor in myocardial formation, the
downregulation of GATA4 leads to an increase in the risk of cardiac
structural abnormalities and cardiovascular malformations in the
fetus. Inhibition of GATA4 in the early stage of cardiac
development has been revealed to be associated with myocardial
hypoplasia and CHD, whereas its inactivation in the late stage of
cardiac development leads to decreased cardiac function (32). GATA4 is involved in normal cardiac
development, functional gene expression, and the pathological
processes of cardiac hypertrophy. It has been recognized as a key
effector mediating cardiac gene transcription in response to
hypertrophic stimuli. In addition, during myocardial hypertrophy,
GATA4 serves as a molecular ‘bridge’ connecting multiple nuclear
factors, including myocyte enhancer factor 2C (Mef2c), Nkx2.5 and
AP1 (33,34).
OS, caused by free radicals, serves as a key factor
leading to cellular and tissue oxidative damage, as well as being a
major driver of aging and various diseases. ROS are the most
important free radical that cause oxidative damage to the body. OS
is the most common mechanism underlying PM2.5-induced
damage (45,46). Transition metals [Fe, Cu and Mn]
and organic compounds from PM2.5 are able to induce the
production of ROS and reactive nitrogen species (RNS), and the
ability of PM2.5 to induce ROS has been significantly
correlated with the concentrations of PAHs and specific transition
metals therein (47).
PM2.5 may induce OS in target cells through a variety of
pathways. First, PM2.5 contains persistent free radicals
that are found in the environment, especially combustion-derived
particles (48). Secondly,
numerous organic compounds from PM2.5 can be metabolized
into reactive electrophilic metabolites, which thereby induce the
further generation of ROS (49).
Thirdly, transition metals can induce ROS through the Fenton
reaction (50). Finally, OS may
also be caused by the PM2.5-mediated activation of
inflammatory cells, which are able to produce both ROS and RNS
(51). On the other hand,
PM2.5 may also decrease the cellular antioxidant
capacity through downregulating the expression of antioxidant
enzymes, such as superoxide dismutase and glutathione metabolizing
enzymes (52). ROS react with
biomolecules such as proteins and DNA, resulting in various adverse
effects on cells, including the disruption of their structure and
function, which ultimately leads to damage to target cells and
tissues. Two pathways are mainly involved in the pathogenic
mechanisms underlying: One is gene damage resulting from genetic
mutations, and the other is damage that is caused to the cell
membrane, which results in changes in its permeability through
lipid peroxidation, leading to physiological changes such as
inflammation.
The embryonic development of both humans and
zebrafish is abnormally sensitive to OS induced by ROS, and
excessive ROS production is considered one of the factors
contributing to CHD (53,54). Ren et al (55) demonstrated that extractable organic
matter (EOM) from PM2.5 is able to induce ROS
production, thereby increasing the levels of nuclear factor
erythroid 2-related factor 2 (Nrf2) signaling pathway-associated
genes [namely, SOD2, glutathione S-transferase (GST)P1/2, catalase
(CAT) and heme oxygenase-1 (HO-1)], with the Nrf2 signaling pathway
being the major pathway that is activated by OS.
2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD), an AhR agonist, was
revealed to cause an upregulation of the protein levels and
activity of Nrf2 in mice (56).
The presence of multiple AhR-binding elements located in the
promoter and first intron of Nrf2a and Nrf2b suggests that AhR
exerts a regulatory role with respect to their transcription
(57). According to other research
results, OS, in turn, may inhibit the activity of AhR (58). Elbekai and El-Kadi (59) reported that the ROS scavenger
N-acetylcysteine (NAC) could ameliorate the inhibitory effects of
chromium on AhR activity in human liver cell line (59). NAC treatment led to an increase in
the activity of cytochrome P450, family 1, subfamily A, polypeptide
1 (Cyp1a1), whereas the inhibitory effects of AhR inhibitor,
CH223191, were alleviated (60).
However, Ren et al (55)
found that NAC did not reduce EOM-induced AhR activity, suggesting
that the effects of OS on the AhR signaling pathway may be species-
or cell type-specific (55).
Zebrafish possess two Nrf2 genes (Nrf2a and Nrf2b), whose
downstream genes (SOD2, GSTP1/2, CAT and HO-1) exert a range of
antioxidant effects, and this may represent a negative feedback
mechanism to circumvent EOM-induced excessive ROS (57).
Inflammation is an adaptive response for the body
that both enables the clearance of harmful stimuli and heals
damaged tissues. However, persistent or chronic inflammation may be
detrimental to the body (66). As
an important mechanism that is associated with PM2.5
toxicity, the inflammatory response may impose the negative effects
of PM2.5 on the cardiovascular, pulmonary and nervous
systems (67). The PAHs, metals,
water-soluble ions as well as various bioactive substances (such as
endotoxins) that are contained in PM2.5 may cause
inflammation, a process that is associated with the polarization of
pro-inflammatory macrophages (68). PM2.5 has been revealed
to cause an increase in the levels of ROS in macrophages, and is
recognized by the Toll-like receptors TLR4 and TLR2, leading to the
induction or exacerbation of acute inflammation and thereby
promoting M1 polarization of macrophages (69). This process may also involve the
activation of Notch signaling due to a decreased level of the
microRNA, miR-34a-5p (70). In
addition, exposure to PAHs has been revealed to upregulate the
levels of inducible nitric oxide synthase (iNOS), NLR family pyrin
domain containing 3 (NLRP3) and tissue protease B in macrophages,
demonstrating that pyroptosis provides the basis for the
pro-inflammatory polarization of macrophages induced by exposure to
PAHs (71). Myocardial macrophages
are able to eliminate the defective mitochondria that are released
by cardiomyocytes, thereby maintaining cardiac mitochondrial
homeostasis. However, in the absence of membrane-bound bone marrow
epithelial reproductive receptor tyrosine kinase (MerTK),
myocardial macrophages lose the ability to capture and eliminate
defective mitochondria, leading to dysfunctional cardiac metabolism
and left ventricular dysfunction, suggesting that MerTK fulfills a
crucial role in supporting cardiac homeostasis (72). Pro-inflammatory polarization of
myocardial macrophages promotes the lysis of MerTK, which affects
the ability of myocardial macrophages to participate in cardiac
repair, consequently leading to cardiac homeostasis imbalance,
myocardial injury and decreased cardiac function.
The important pro-inflammatory cytokines tumor
necrosis factor (TNF)-α and interleukin (IL)-6 are involved in the
pathogenesis of heart failure, cardiac hypertrophy and fibrosis
(73,74). It has been revealed that exposure
to PM2.5 in the uterus induces the expression of
pro-inflammatory cytokines in the hearts of the offspring mice,
leading to cardiac inflammation (30,42).
Li et al (75) through
studying the cardiac inflammatory response, demonstrated that the
levels of TNF-α and IL-1β were significantly increased in offspring
mice subjected to uterine PM2.5 exposure. Long-term
exposure to PM2.5 was revealed to cause a marked
upregulation of the levels of intercellular adhesion molecule-1
(ICAM-1) and C-reactive protein in rat myocardial tissues, leading
to ultrastructural changes in myocardial cells and inflammatory
cell influx (76). Among the
signaling molecules that regulate the inflammatory response,
nuclear factor-κB (NF-κB) is the major signaling molecule that is
involved in the production of cytokines, chemokines and growth
factors, regulating the expression of immune and inflammatory
response-associated genes (77).
The NF-κB signaling pathway, involved in tissue damage, has been
reported to have a role in systemic inflammation induced by
PM2.5 (78).
Inflammation fulfills crucial roles in systemic myocardial
hypertrophy and cardiotoxicity induced by particulate matter
(79). Jiao et al (80) found that the
PM2.5-mediated induction of inflammation is dependent on
the activation of the key transcription factor NF-κB, which
enhances the expression of the downstream factors, TNF-α and IL-1β.
Exposure to PM2.5 in vivo has been revealed to
activate the NF-κB signaling pathway, leading to inflammatory
responses in target tissues and organs (81). Interesting, the activation of
NF-κB, with the subsequent inflammatory response that is caused by
exposure to PM2.5, may be suppressed by antioxidants,
suggesting the involvement of ROS and/or RNS in
PM2.5-mediated NF-κB activation (52). Considering that NF-κB also triggers
the generation of ROS and nitric oxide (NO), this may form a
positive feedback loop that amplifies downstream responses upon
PM2.5 exposure (82).
An increased level of OS resulting from exposure to
PM2.5, in turn, mediates the activation of downstream
inflammatory signaling pathways, including the mitogen-activated
protein kinase (MAPK), c-Jun N-terminal kinase (JNK)/p53,
Nrf2/NLRP3, TLR/MyD88 and extracellular signal-regulated kinase
(ERK)/AKT pathways (83).
PM2.5 has been revealed to increase the protein level of
cleaved IL-1β, a key downstream factor for NLRP3 inflammasome
activation, further confirming that PM2.5 can activate
the NLRP3 inflammasome in myocardial tissue; NLRP3 inflammasome
activation, in itself, has a potential role in mediating the
pathological damage resulting from PM2.5 exposure in the
mouse heart (84). The augmented
levels of ROS triggered by exposure to PM2.5 may
activate the MAPK and NF-κB pathways, thereby increasing the
synthesis of inflammatory proteins and leading to changes in
membrane permeability and mitochondrial dysfunction (85). It is worth noting that
mitochondrial DNA (mtDNA) lacks the ability to repair DNA, making
it more susceptible to oxidative damage compared with nuclear DNA.
Mitochondrial dysfunction and subsequent cell death can trigger
inflammation in various types of tissues (86). Mitochondrial dysfunction makes a
key contribution to the PM2.5-mediated inflammatory
response (87). mtDNA and n-formyl
peptides that are released from dysfunctional mitochondria both
trigger inflammation. PM2.5 exposure has also been
revealed to increase the expression and release of adhesion
molecules, including E-selectin, P-selectin and ICAM-1, leading to
monocyte/macrophage adhesion (88), whereas, on the other hand,
diminishing the levels of circulating endothelial progenitor cells
that are involved in postnatal endothelial repair and regeneration
(89), thereby exacerbating the
inflammatory response. Inflammatory factors such as
cyclooxygenase-2 (COX-2) are able to inhibit the activity of
Ca2+ pumps in the endoplasmic reticulum, thereby
inducing ERS through upregulating iNOS expression (90), suggesting that inflammation induced
by PM2.5 can trigger ERS. Furthermore, PM2.5
has been revealed to activate the unfolded protein response (UPR),
which provides an additional mechanism for triggering ERS (91). UPR signaling both stimulates the
expression of inflammatory cytokines and induces the activation of
NF-κB (92), suggesting that UPR
signaling makes an important contribution towards
PM2.5-induced ERS in the inflammatory process, and that
this serves as an inflammatory factor both as a cause and as a
consequence of ERS (93).
Ca2+ leakage from the endoplasmic reticulum directly
drives the production of mitochondrial ROS (mtROS), affecting
downstream signaling pathways and rendering cells more susceptible
to autophagy (94). It is now well
documented that inflammation, ERS and autophagy are closely
interlinked, and that these processes can interact with each other.
Taken together, these aforementioned findings suggest that the
cardiac developmental toxicity that is caused by PM2.5
is associated with inflammation, ERS and autophagy.
The biogenesis and functional improvement of
mitochondria are crucial processes for enabling the differentiation
and maturation of the heart (95).
Previously, investigations of the molecular mechanisms associated
with mitochondria underlying the toxic effects of environmental
pollution have been mainly focused on the mitochondrial
permeability transition pore (mPTP), mitochondrial dynamics, mtDNA
function and the mitochondrial respiratory chain system, along with
mitochondrial damage-associated signaling pathways.
PM2.5 was found to induce mitochondrial impairment in
exposed individuals (96), and
mitochondrial dysfunction has been revealed to mediate the
cardiovascular damage caused by PM2.5 to a certain
extent (97). Enhancing the
production of cardiac energy may be achieved through growing the
mitochondria count (98), and
swelling, disrupted crista and mitochondrial vacuolization
represent the primary manifestations for cardiac mitochondrial
pathological changes (99). Acute
exposure to PM can lead to significant mitochondrial dysfunction,
accompanied by decreased cardiac oxygen consumption, succinate
dehydrogenase activity and mitochondrial membrane potential, as
well as impaired oxidative phosphorylation (100). These findings suggested that
mitochondrial damage caused by PM2.5 exposure may have a
bearing on mitochondrial dysfunction.
Epigenetic modifications, including DNA methylation
(DNAm), histone modifications and RNA-mediated processes, are
sensitive to environmental stress and are considered to serve as a
‘bridge’ between environmental and genetic factors by certain
researchers (117,118). Epigenetic modifications fulfill
an important role in cardiac development and the occurrence of
various diseases, with DNAm as the primary form, which can be
inherited and reversed. To date, however, little is known regarding
the underlying molecular mechanisms through which PM2.5
triggers the epigenetic changes that lead to cardiac developmental
toxicity.
M6A RNA methylation, as the most common form of RNA
modification, accounts for ~60% of the total number of RNA
modifications. M6A RNA methylation, a dynamic and reversible
process, occurs under the regulation of methyltransferases
(including METTL3 and METTL14), demethylases (such as FTO and
ALKBH5,), and binding proteins (including YTHDF1/2/3 and ythdc2/2)
(119). M6A RNA methylation
regulates gene expression through affecting mRNA stability,
selective splicing, nuclear output and protein translation
(120,121). M6A RNA methylation has been
reported to be involved in excessive cellular ROS production and
apoptosis (122-124).
A crucial role of m6A modification in heart development has been
demonstrated (124,125), and PM2.5 has been
revealed to induce changes in m6A RNA methylation in rats and mice
(126,127). Ji et al (128) found that EOM from
PM2.5 caused a significant inhibition of m6A RNA
methylation levels in zebrafish juvenile hearts mediated via the
AhR, although this inhibitory effect was restored by
supplementation with betaine (the predominant methyl donor in the
carbon metabolism cycle). Betaine can also mitigate EOM-induced ROS
generation, cell apoptosis and cardiac defects, suggesting that EOM
inhibits m6A RNA methylation by interfering with mettl14/mettl3
expression, leading to cardiac defects (128). These findings validated the
hypothesis that m6A modification fulfills an important role in
cardiac developmental toxicity induced by PM2.5
exposure, although the antioxidant activity of betaine should not
be overlooked. On the other hand, other studies have revealed that
exposure to PM2.5 leads to an upregulation of the levels
of Mettl3 and total m6A methylation in mice lung tissues (127,129). The differences noted in the
expression levels of m6A methyltransferase may be due to the
differential responses of these genes to PM2.5 exposure
in embryonic/larval and adult tissues (130); another possibility is that
changes in the level of m6A RNA methylation induced by
PM2.5 exposure may be due to species specificity.
Supplementing the diet with AhR inhibitor,
CH223191, has been reported to successfully circumvent the
occurrence of EOM-induced cardiac defects in zebrafish juveniles
(17,55). Either adding betaine or
overexpressing mett13/14 was revealed to ameliorate the effects of
EOM-induced intracellular and mtROS, as well as reducing the level
of apoptosis in zebrafish juvenile cardiomyocytes. Therefore,
changes that occur in the level of m6A RNA methylation may be an
important underlying cause of EOM-induced cardiac abnormalities.
M6A modification has been revealed to regulate OS and cell
apoptosis via regulating the expression of m6A-modified genes
(131,132). Cao et al (133) reported that exposure to
PM2.5 increases ROS generation and apoptosis in rat
cardiomyocytes, leading to cardiac injury. In addition, EOM from
PM2.5 led to OS-mediated cell apoptosis in zebrafish
juvenile hearts (55).
Collectively, these studies have demonstrated that gestational
exposure to PM2.5 may cause OS and cell apoptosis
through altering m6A modification, thereby causing cardiac
developmental toxicity.
Exposure to air pollutants may alter epigenetic
modifications such as DNAm, which, in turn, may affect
inflammation, disease development and the risk of deterioration.
Exposure to several air pollutants associated with transportation,
including PM2.5, black carbon, ozone, nitrogen oxides
and PAHs, leads to a decrease in DNAm. This may be due to both the
reduced expression of methionine adenosyl-transferase 1A and single
carbon metabolism efficiency mediated by oxidative species,
resulting in a scarcity of the methyl donor of S-adenosylmethionine
that is required for establishing and maintaining DNAm (146). Goodson et al (147) found that in utero exposure
to diesel exhaust (DE) induced a decreased level of DNAm in the
first exon of GM6307, suggesting that DE can affect the developing
heart by altering epigenetic patterns. The mechanism(s) through
which PM2.5 exposure leads to DNAm changes, however,
have yet to be fully elucidated. It has been revealed that exposure
to DE increases the production of ROS (148), which, in turn, interact with DNA,
thereby oxidizing methyl-cytosine to hydroxymethyl-cytosine (HMC).
HMC has been revealed to prevent the binding of methyl binding
proteins (MBPs) to methylated cytosine (149), which prevents normal chromatin
silencing from occurring at these sites. In addition, 8-oxoguanine
produced by guanine oxidative damage was also found to inhibit the
binding of MBPs, thereby hindering the silencing of chromatin
regions (149).
Among the histone acetyltransferases (HATs), p300
is closely associated with the transcriptional regulation of
cardiac development (157).
SIRT3, the third type of histone deacetylase, is able to inhibit
the OS response and promote the tricarboxylic acid cycle, which has
the effect of enhancing myocardial ATP energy supply and
contraction, as well as regulating the energy metabolism balance
(158). Knockout of SIRT3 was
revealed to lead to myocardial mitochondrial dysfunction and
cardiac dysfunction (159).
Furthermore, the abnormal expression of HATs and HDAC led to
imbalanced histone acetylation modifications, giving rise to
cardiac developmental disorders (160). Exposure to PM2.5 in
the uterus is known to lead to cardiac hypertrophy in adulthood.
P300/CREB binding protein mediated histone acetylation modification
may exert an important role in the upregulation of thickening
transcription factors, such as GATA binding protein 4 (GATA4) and
Mef2c. To date, the mechanism(s) of PM2.5-induced
histone modification are poorly understood. Environmental chemicals
may directly alter histone methyltransferases or demethylases. For
example, Ni exposure has been revealed to inhibit the activity of
lysine-specific demethylase 3A by binding and substituting
Fe2+ ions, thereby increasing H3K9me2 modification
(161).
The endoplasmic reticulum performs a crucial role
in terms of protein synthesis and folding, and post-translational
modifications. The disruption of endoplasmic reticulum function may
lead to accumulated unfolded or misfolded proteins in the lumen,
which activates the UPR, a complex intracellular signaling pathway
aimed at restoring protein balance. The endoplasmic reticulum is
closely associated with normal development and homeostasis of the
internal environment, and it has a crucial role in cardiac
development and function (162).
Zhu et al (163) found
that Cd exposure increased ERS in myocardial tissue and primary
cardiomyocytes, which was manifested in elevated levels of
stress-associated genes. Impaired cardiac contractility and
prolonged diastolic duration have been revealed to be common
pathological features of the ERS-stimulated heart (164). Previous studies have also
suggested that PM2.5 is capable to induce ERS (165,166); however, the mechanism(s)
underlying PM2.5-induced ERS, and its role in cardiac
development, has yet to be elucidated. EOM from PM2.5
was revealed to induce AhR-mediated ROS production in zebrafish
embryonic hearts (17,55,167). In addition, OS induces ERS
through disrupting the normal processes of protein
folding/transport and altering Ca2+ homeostasis
(168-170).
On the other hand, ERS was also demonstrated to increase the
content of ROS, and to induce OS (171). Early-stage embryos are highly
susceptible to oxidative damage, and excessive ROS is considered
one of the causative agents for CHD (53,54);
therefore, PM2.5 may induce ERS through AhR-mediated ROS
overproduction, thereby inducing cardiac developmental toxicity via
oxidative damage.
Cardiac development is a coordinated process
depending on the subtle balance among cell proliferation, apoptosis
and differentiation. It is well established that long-term or
severe ERS can lead to cell apoptosis, with C/EBP homologous
protein (CHOP) being recognized as one of the most important
mediators. The expression of CHOP may be upregulated through
activating all three ERS sensors, namely: Activating transcription
factor 6, protein kinase RNA like endoplasmic reticulum kinase and
inositol requiring enzyme 1α. As a transcription factor, CHOP
induces cell apoptosis through downregulating members of the
antiapoptotic Bcl protein family and increasing the level of
endoplasmic reticulum oxidoreductin 1α. EOM was reported to induce
apoptosis of zebrafish embryonic cardiomyocytes, although the
increased level of apoptosis was attenuated via inhibiting AhR
activity or ROS production (55,172); furthermore, ERS was found to have
a key role in this process.
Autophagy is crucial for heart development.
Numerous autophagic defects are known to be associated with
cardiovascular diseases, including atherosclerosis and
cardiomyopathy (172). Autophagy
has an important role in the process of cardiac remodeling,
including the morphogenesis of cardiac tissues and their eventual
differentiation into cardiomyocytes. Atg5-deficient mice were
demonstrated to have abnormal heart valves and separated
ventricular (173). In a
zebrafish cardiac development model, knocking down the core
autophagy genes resulted in various defects, including cardiac
blood-flow defects and atrial enlargement, among other defects
(174). In addition, knocking
down these autophagy genes resulted in profound changes in the
levels of developmental genes, including certain key transcription
factors that are necessary for cardiac development. The knockdown
of these genes also led to the accumulation of dead cells in the
developing heart, demonstrating the necessity of autophagic
clearance of dead cells for normal cardiac remodeling.
Cardiac-specific Atg5 deficiency in adult mice was revealed to lead
to mitochondrial aggregation and ventricular dilation,
demonstrating the vital role that autophagy has in cardiac cell
development and homeostasis (175).
As another cellular protective mechanism for UPR,
autophagy contributes to the degradation of the accumulated
unfolded or misfolded proteins in the endoplasmic reticulum,
thereby restoring endoplasmic reticulum homeostasis and further
improving the overall cell survival rate (182). However, autophagy is also a
‘double-edged sword’ since excessive autophagy may promote cell
death through excessive self-digestion and the degradation of
essential cellular components (183), thereby bringing about embryonic
developmental toxicity (184).
Autophagy exerts important roles in the processes of cellular and
tissue balance, specialization, tissue differentiation and
organogenesis (185); in
addition, inflammation, ERS and autophagy are closely associated,
and these processes have been revealed to interact with each other
(186).
As the principal means of cell death, apoptosis has
an important role in maintaining cellular homeostasis. Abnormal
apoptosis can give rise to various diseases, including
cardiovascular diseases. Apoptosis in mammalian cells is mainly
triggered through two pathways: The endogenous (intrinsic) pathway
initiated by mitochondria, and the exogenous (extrinsic) pathway
initiated by death receptors. The former is controlled by the Bcl-2
family of proteins, whereas the latter involves members of the TNF
family of proteins, with the resultant signaling cascade (187). Apoptosis induced by exposure to
PM2.5 has a participatory role in a series of signaling
pathways, including the MAPK (133) and PI3K/Akt (188) pathways, with caspase-3 serving as
a vital biomarker in this process (189). Yang et al (190) demonstrated that the
mitochondria-mediated apoptosis pathway has a key role in the
PM2.5-induced toxicity of AC16 cardiomyocytes, leading
to cardiac dysfunction. The mitochondrial pathway mainly activates
caspase-9 by releasing cytochrome c into the cytoplasm,
triggering downstream cascade reactions, and ultimately activating
downstream caspase-3(191).
However, in zebrafish, 2,3-bromofluoranthene derived from PAHs is
able to induce apoptosis of vascular endothelial cells and cardiac
toxicity through both pathways simultaneously (192). In addition, dysregulation of
cellular Ca2+ homeostasis may also lead to cardiomyocyte
apoptosis. Ca2+ is one of the most important
signal-transduction systems in cells, and a low intracellular
Ca2+ concentration is a prerequisite for normal cellular
function. After PM2.5 has entered the circulatory
system, it leads to an increase in the intracellular
Ca2+ concentration, and an overload of Ca2+
will lead to DNA degradation, free radical production and protein
kinase activation, ultimately leading to cell apoptosis (193).
Apoptosis can be induced by DNA damage and is
crucial for normal cardiac development (194,195). Previous studies have suggested
that excessive production of ROS during early embryonic development
in zebrafish may lead to DNA damage and cell apoptosis (196-198).
Ren et al (55) found that
the levels of 8-hydroxydeoxyguanosine (8-OHdG) and cH2AX were
raised in the embryonic hearts of zebrafish exposed to EOM,
although these increases were significantly reduced in the presence
of the ROS scavenger, NAC. This further demonstrated that NAC is
able to attenuate EOM-induced apoptosis in zebrafish embryonic
cardiomyocyte. Bcl2 binding component 3 (BBC3), a member of the
Bcl-2 family, is an important participant in apoptosis (199). BBC3 is localized at the
mitochondria under apoptotic stimulations, leading to
mitochondria-mediated intrinsic cell apoptosis (200). Traf4a, a zebrafish homolog for
human TNF receptor-associated factor 4, is also involved in the
regulation of apoptosis (201,202). TRAF4 is also essential for
development and can regulate ROS generation by stabilizing NADPH
oxidase complexes (203).
Knocking down BBC3 or TRAF4 leads to the termination of EOM-induced
excessive ROS production and apoptosis in zebrafish embryonic
hearts, suggesting that both genes are required for this process.
Therefore, overexpression of these two genes may exacerbate cardiac
abnormalities in zebrafish juveniles induced by EOM derived from
PM2.5.
However, it is necessary to further investigate
whether other forms of cell death besides apoptosis and autophagic
death, such as ferroptosis, may be associated with the cardiac
developmental toxicity induced by PM2.5 exposure, since
PM2.5 exposure results in excessive amount of ROS, and
severely damaged mitochondria release large amount of Fe, thereby
inducing ferroptosis.
AhR, an essential ligand-activated transcription
factor for the cytochrome P450 pathway, controls the expression of
genes such as CYP1A1, CYP1B1 and CYP1A2 in the
cytochrome P450 family (204).
AhR can be activated by numerous environmental pollutants,
including PM2.5 (55).
Following activation, AhR is dissociated from binding its ligands
and enters the nucleus, forming a dimer with AhR nuclear transport
protein, subsequently binding with enhancers to form heterologous
reaction elements that are involved in the regulation of the
expression of cytochrome P450 family genes. Employing the
P19 cell line as an in vitro model, Chen et al
(167) found that exposure to EOM
derived from PM2.5 for 2 days led to an inhibition of
cardiac differentiation for the next 14 days, demonstrating the
persistent adverse effects of PM2.5 on cardiac
development. Mechanistically, AhR mediates the inhibitory effects
of EOM on P19 cell cardiac differentiation, probably through
dysregulation of cell proliferation, altering the normal processes
of Wnt signaling, and inducing breaks of DNA double strands.
As a typical type of PAHs, exposure to TCDD impairs
the cardiac differentiation of ESCs, and this impairment is mainly
mediated by AhR. The generation of cardiomyocytes was most
significantly inhibited in the case of human ESCs (and not mouse
ESCs) exposed to TCDD during the ESC stage. By contrast, in the
absence of TCDD, AhR is significantly inhibited in mouse ESCs,
which decreases the expression of numerous pluripotent genes
(212). In addition, ESC cardiac
differentiation was found to be suppressed by TCDD exposure during
embryonic formation via disrupting activin, bone morphogenetic
protein and the Wnt signaling pathway, and through altering the
expression of homologous cassette transcription factors (213-216).
These differences suggest that human and mouse ESCs exhibit
different susceptibility to TCDD toxicity, possibly due to
species-specific differential expression patterns of AhR and its
cofactors (217). Furthermore,
AHR may regulate differential target genes in different species or
cells (218,219). Therefore, similarly to TCDD,
PM2.5 can inhibit the activation of mesodermal genes
through AhR binding, interfering with the differentiation and
development of normal cells, and thereby inhibiting mesodermal
differentiation. Jiang et al (220) also found that PM2.5
can activate the PI3K/akt2/mammalian target of rapamycin complex 1
signaling pathway through AhR/ROS induced PTEN inhibition, leading
to activation of the mitochondria-mediated intrinsic apoptotic
pathway and Wnt signaling inhibition, resulting in heart defects in
zebrafish juveniles. It has been documented that folate
supplementation during pregnancy helps to resist
PM2.5-induced cardiac developmental toxicity via
targeting the AhR and Wnt/β catenin signaling pathways (205). This provides theoretical support
for alleviating PM2.5-induced cardiac developmental
toxicity.
The Wnt/β-catenin signaling pathway has an
essential role in the cardiac development of vertebrates (221), and its activation may induce
cardiac specification in the early developmental stages, although
this may be suppressed later (221). As a core transcription factor of
the typical Wnt signaling pathway, β-catenin is able to regulate
the expression of key genes in cardiac development (222). Chen et al (167) found that the mRNA and protein
levels of β-catenin were downregulated in cells exposed to EOM
derived from PM2.5, suggesting a role of Wnt signaling
in EOM-exerted cardiotoxic effects. It is well established that
crosstalk exists between the AhR and Wnt/β-catenin signaling
pathways (223,224), since activated AhR can antagonize
β-catenin in colon cancer cells and zebrafish embryos (224). The Wnt/b-catenin signaling
pathway has also been revealed to be crucial for embryonic cardiac
development. In the absence of Wnt, cytoplasmic β-catenin is
phosphorylated and degraded by a disruption complex composed of
adenomatosis polysaccharide coli, Axin and glycogen synthase
kinase-3β. Upon Wnt stimulation, cytoplasmic β-catenin is
translocated into the nucleus, where it activates the transcription
of genes essential for cardiac specification, such as Nkx2.5 and
Sox9 (224-226).
CH223191 and the Wnt/β-catenin activator, CHIR, were found to
rescue the most of the EOM-induced cardiac defects, suggesting the
involvement of the AhR and Wnt/β-catenin signaling pathways in
cardiac developmental toxicity resulting from PM2.5
exposure, and therefore the feasibility of employing AhR or
Wnt/β-catenin antagonists to prevent the cardiac developmental
toxicity from occurring (17). The
typical Wnt/β-catenin signaling pathway regulates multiple steps in
cardiac differentiation (167).
The activation of Wnt signaling is crucial for both the formation
of the mesoderm in the early stage of development as well as the
morphogenesis of cardiac valve formation in later ones (227). Previous studies have demonstrated
the inhibition of Wnt signaling by EOM in the embryonic heart of
zebrafish (17,205), and both treatment with the ERS
inhibitor 4-phenylbutyric acid and CHOP knockdown significantly
attenuated these inhibitory effects, probably by means of either
affecting β-catenin expression or inhibiting T cell factor
(228).
OS, ionizing radiation and chemical reagents are
all capable of causing DNA damage (233). Components of water-soluble
PM2.5 extracts are more likely to induce DNA oxidative
damage compared with organic compounds (234). OS-induced DNA damage has been
revealed to be a key mechanism of action in urban PM2.5
pollution (235). Excessive ROS
generated by pollutants induces OS, thereby mediating DNA damage in
the mouse heart (236). The
organic components and transition metals (including Fe, Cu, Ni and
Zn) in PM2.5 can directly generate ROS (237), which either directly causes DNA
deamination and base oxidation, or indirectly induces base
alkylation through lipid peroxidation (238). Therefore,
PM2.5-induced OS can lead to DNA damage. DNA damage is
usually associated with cell apoptosis (239). Both ROS- or RNS-mediated DNA
damage and redox-mediated inhibition of DNA-damage response
proteins may lead to changes in DNA structure, thereby activating
DNA-repair signaling. The latter can regulate the activities of
certain apoptosis factors, further demonstrating the close
association between DNA damage and cell apoptosis (240). In human or animal cells, heavy
metals derived from PM2.5 can cause various types of DNA
damage, including chain breakage, diminishing the activity of
endonuclease III, and damaging guanine glycosidase-sensitive sites
(49). Interestingly, antioxidants
and ROS scavengers can significantly block the DNA damage resulting
from PM2.5 exposure.
DNA damage can also cause cell-cycle arrest and
induce apoptosis, and this may extensively disrupt the potential of
progenitor cells, thereby impairing cardiac development (241). There is a large body of evidence
suggesting that environmental pollutants, including
PM2.5, may attack DNA by means of OS (45). Excessive ROS production during
zebrafish embryonic development has been revealed to lead to DNA
damage and apoptosis (196).
Elevated levels of 8-OHdG and γ-h2ax were observed in the zebrafish
embryonic heart, although these were significantly circumvented by
treatment with the ROS scavenge, NAC. However, NAC could not
completely reverse the DNA-damage signaling processes induced by
PM2.5, suggesting that OS is only partly responsible for
causing the DNA damage (242).
The cardiac DNA damage caused by PM2.5 is most likely to
be involved in multiple signaling pathways and synergistic effects
of multiple molecules, and further in-depth exploration in this
regard is required.
As a key factor in cardiac development and
homeostasis, AhR signaling fulfills a key role in cardiac
development. The interruption of AhR function during development
can lead to potential cardiac developmental toxicity, and this
comprises a number of dysregulated signaling pathways that
participate in cardiac development, function and metabolism. At the
same time, AhR is also a target for environmental factors that may
disrupt the homeostasis of AhR, laying the foundation for CHD. Due
to the widespread presence of AhR antagonists and methyl donors
such as flavonoids, curcumin and betaine, that are found in daily
foods, these may serve as favorable candidates for addressing the
abnormal activation of AhR and changes in m6A RNA methylation
derived from PM2.5 exposure. Moreover, phytochemicals
are able to alleviate the adverse effects of PM2.5
exposure on human health, predominantly via inhibiting OS, ERS and
Fe deposition, which subsequently alleviates inflammatory
reactions, along with regulating autophagy. Taken together, these
findings provide a potential approach for therapeutic intervention
with regard to the cardiac developmental toxicity caused by
PM2.5 exposure.
Although mitochondria are the most important source
of ROS, increasing evidence has suggested that other organelles as
the potential sources of ROS, such as the endoplasmic reticulum,
peroxisomes and cell membranes, are also important. Therefore,
understanding the sources of ROS is of great significance for
targeted therapy, dosage selection and pollution control.
Considering the high energy consumption of the heart and the role
of mitochondria as the body's energy production factory,
mitochondria may be a promising therapeutic target for the
prevention and treatment of CHD. Given the deepening understanding
of mitochondrial biology, the widespread application of large-scale
experimental animal models, and the rapid development of new
scientific and technological advancements, mitochondrial medicine
may become a realistic therapeutic option in the near future.
In summary, the present review elaborates the
potential molecular mechanisms underlying the cardiac developmental
toxicity induced by gestational PM2.5 (including some of
its specific components) exposure, and the complexity presents
numerous challenges and opportunities for future investigations.
Understanding the interplay of various signaling pathways during
this process, alongside the concentration and duration of
PM2.5 exposure, will be crucial for advancing our
knowledge in this field. In addition, exploring the individual and
synergistical cardiac developmental toxicity effects induced by
differential PM2.5 components will be vital for
developing effective intervention measures and regulatory
strategies. In order to deepen our understanding of the cardiac
developmental toxicity induced by PM2.5, future
researches should focus on longitudinal studies evaluating the
long-term effects of early exposure on the cardiac outcomes. This
will provide insights into potential interventions to mitigate
these effects. Addressing these multifaceted challenges will
provide supports for public health policies to reduce exposure to
PM2.5 and improve population health outcomes.
Ultimately, a comprehensive understanding of PM2.5's
toxicological effects will contribute to the scientific community
and empower policymakers to implement effective strategies
safeguarding public health, particularly among vulnerable
populations.
Not applicable.
Funding: No funding was received.
The data generated in the present study are
included in the figures and/or tables of this article.
XM, WD and ZS conceived and designed the study. XM
and WD collected the literature. XM, WD and ZS analyzed the
literature, and drafted and reviewed the manuscript. Data
authentication is not applicable. All authors read and approved the
final version of the manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Dominici F, Greenstone M and Sunstein CR:
Science and regulation. Particulate matter matters. Science.
344:257–259. 2014.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Thurston GD, Ahn J, Cromar KR, Shao Y,
Reynolds HR, Jerrett M, Lim CC, Shanley R, Park Y and Hayes RB:
Ambient particulate matter air pollution exposure and mortality in
the NIH-AARP diet and health cohort. Environ Health Perspect.
124:484–490. 2016.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Huang Q, Chi Y, Deng J, Liu Y, Lu Y, Chen
J and Dong S: Fine particulate matter 2.5 exerted its toxicological
effect by regulating a new layer, long non-coding RNA. Sci Rep.
7(9392)2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Long JF, Waldman WJ, Kristovich R,
Williams M, Knight D and Dutta PK: Comparison of ultrastructural
cytotoxic effects of carbon and carbon/iron particulates on human
monocyte-derived macrophages. Environ Health Perspect. 113:170–174.
2005.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Samek L, Furman L, Mikrut M, Regiel-Futyra
A, Macyk W, Stochel G and van Eldik R: Chemical composition of
submicron and fine particulate matter collected in Krakow, Poland.
Consequences for the APARIC project. Chemosphere. 187:430–439.
2017.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Mesquita SR, van Drooge BL, Reche C,
Guimarães L, Grimalt JO, Barata C and Piña B: Toxic assessment of
urban atmospheric particle-bound PAHs: Relevance of composition and
particle size in Barcelona (Spain). Environ Pollut. 184:555–562.
2014.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Wang L, Luo D, Liu X, Zhu J, Wang F, Li B
and Li L: Effects of PM2.5 exposure on reproductive
system and its mechanisms. Chemosphere. 264(128436)2021.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Hoffman JIE, Kaplan S and Liberthson RR:
Prevalence of congenital heart disease. Am Heart J. 147:425–439.
2004.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Olson EN: Gene regulatory networks in the
evolution and development of the heart. Science. 313:1922–1927.
2006.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Li M, Li J, Wei C, Lu Q, Tang X, Erickson
SW, MacLeod SL and Hobbs CA: A three-way interaction among maternal
and fetal variants contributing to congenital heart defects. Ann
Hum Genet. 80:20–31. 2016.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Hu CY, Huang K, Fang Y, Yang XJ, Ding K,
Jiang W, Hua XG, Huang DY, Jiang ZX and Zhang XJ: Maternal air
pollution exposure and congenital heart defects in offspring: A
systematic review and meta-analysis. Chemosphere.
253(126668)2020.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Huang CC, Chen BY, Pan SC, Ho YL and Guo
YL: Prenatal exposure to PM2.5 and congenital heart
diseases in Taiwan. Sci Total Environ. 655:880–886. 2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Li D, Xu W, Qiu Y, Pan F, Lou H, Li J, Jin
Y, Wu T, Pan L, An J, et al: Maternal air pollution exposure and
neonatal congenital heart disease: A multi-city cross-sectional
study in eastern China. Int J Hyg Environ Health.
240(113898)2022.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Zhang Q, Sun S, Sui X, Ding L, Yang M, Li
C, Zhang C, Zhang X, Hao J, Xu Y, et al: Associations between
weekly air pollution exposure and congenital heart disease. Sci
Total Environ. 757(143821)2021.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Jiang Q, Zhang C, Chen S, Shi L, Li DC, Lv
N, Cui L, Chen Y and Zheng Y: Particulate matter 2.5 induced
developmental cardiotoxicity in chicken embryo and hatchling. Front
Pharmacol. 11(841)2020.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Wang H, Peng X, Cao F, Wang Y, Shi H, Lin
S, Zhong W and Sun J: Cardiotoxicity and mechanism of particulate
matter 2.5 (PM2.5) exposure in offspring rats during pregnancy. Med
Sci Monit. 23:3890–3896. 2017.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Zhang H, Yao Y, Chen Y, Yue C, Chen J,
Tong J, Jiang Y and Chen T: Crosstalk between AhR and wnt/β-catenin
signal pathways in the cardiac developmental toxicity of PM2.5 in
zebrafish embryos. Toxicology. 355-356:31–38. 2016.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Lage K, Greenway SC, Rosenfeld JA,
Wakimoto H, Gorham JM, Segrè AV, Roberts AE, Smoot LB, Pu WT,
Pereira AC, et al: Genetic and environmental risk factors in
congenital heart disease functionally converge in protein networks
driving heart development. Proc Natl Acad Sci USA. 109:14035–14040.
2012.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Yan R, Ma D, Liu Y, Wang R, Fan L, Yan Q,
Chen C, Wang W, Ren Z, Ku T, et al: Developmental toxicity of fine
particulate matter: Multifaceted exploration from epidemiological
and laboratory perspectives. Toxics. 12(274)2024.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Feng S, Huang F, Zhang Y, Feng Y, Zhang Y,
Cao Y and Wang X: The pathophysiological and molecular mechanisms
of atmospheric PM2.5 affecting cardiovascular health: A
review. Ecotoxicol Environ Saf. 249(114444)2023.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Liang C, Ding R, Sun Q, Liu S, Sun Z and
Duan J: An overview of adverse outcome pathway links between
PM2.5 exposure and cardiac developmental toxicity.
Environ Health. 2:105–113. 2024.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Agay-Shay K, Friger M, Linn S, Peled A,
Amitai Y and Peretz C: Air pollution and congenital heart defects.
Environ Res. 124:28–34. 2013.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Girguis MS, Strickland MJ, Hu X, Liu Y,
Bartell SM and Vieira VM: Maternal exposure to traffic-related air
pollution and birth defects in Massachusetts. Environ Res. 146:1–9.
2016.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Lavigne E, Lima I, Hatzopoulou M, Van
Ryswyk K, Decou ML, Luo W, van Donkelaar A, Martin RV, Chen H,
Stieb DM, et al: Spatial variations in ambient ultrafine particle
concentrations and risk of congenital heart defects. Environ Int.
130(104953)2019.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Schembari A, Nieuwenhuijsen MJ, Salvador
J, de Nazelle A, Cirach M, Dadvand P, Beelen R, Hoek G, Basagaña X
and Vrijheid M: Traffic-related air pollution and congenital
anomalies in Barcelona. Environ Health Perspect. 122:317–323.
2014.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Zhang B, Liang S, Zhao J, Qian Z, Bassig
BA, Yang R, Zhang Y, Hu K, Xu S, Zheng T and Yang S: Maternal
exposure to air pollutant PM2.5 and PM10 during pregnancy and risk
of congenital heart defects. J Expo Sci Environ Epidemiol.
26:422–427. 2016.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Jenkins KJ, Correa A, Feinstein JA, Botto
L, Britt AE, Daniels SR, Elixson M, Warnes CA and Webb CL: American
Heart Association Council on Cardiovascular Disease in the Young.
Noninherited risk factors and congenital cardiovascular defects:
Current knowledge: A scientific statement from the American heart
association council on cardiovascular disease in the Young:
Endorsed by the American academy of pediatrics. Circulation.
115:2995–3014. 2007.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Lassi ZS, Imam AM, Dean SV and Bhutta ZA:
Preconception care: Caffeine, smoking, alcohol, drugs and other
environmental chemical/radiation exposure. Reprod Health. 11 (Suppl
3)(S6)2014.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Chang YC, Lin YT, Jung CR, Chen KW and
Hwang BF: Maternal exposure to fine particulate matter and
congenital heart defects during preconception and pregnancy period:
A cohort-based case-control study in the Taiwan maternal and child
health database. Environ Res. 231(116154)2023.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Wu X, Pan B, Liu L, Zhao W, Zhu J, Huang X
and Tian J: In utero exposure to PM2.5 during gestation caused
adult cardiac hypertrophy through histone acetylation modification.
J Cell Biochem. 120:4375–4384. 2019.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Watt AJ, Battle MA, Li J and Duncan SA:
GATA4 is essential for formation of the proepicardium and regulates
cardiogenesis. Proc Natl Acad Sci USA. 101:12573–12578.
2004.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Jiang SY, Xu M and Zhang YY: Role of
GATA-4 in cardiac development and remodeling. Sheng Li Ke Xue Jin
Zhan. 39:302–306. 2008.PubMed/NCBI(In Chinese).
|
|
33
|
Akazawa H and Komuro I: Roles of cardiac
transcription factors in cardiac hypertrophy. Circ Res.
92:1079–1088. 2003.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Morimoto T, Hasegawa K, Wada H, Kakita T,
Kaburagi S, Yanazume T and Sasayama S: Calcineurin-GATA4 pathway is
involved in beta-adrenergic agonist-responsive endothelin-1
transcription in cardiac myocytes. J Biol Chem. 276:34983–34989.
2001.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Yang Y, Ruan Z, Wang X, Yang Y, Mason TG,
Lin H and Tian L: Short-term and long-term exposures to fine
particulate matter constituents and health: A systematic review and
meta-analysis. Environ Pollut. 247:874–882. 2019.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Sancini G, Farina F, Battaglia C, Cifola
I, Mangano E, Mantecca P, Camatini M and Palestini P: Health risk
assessment for air pollutants: alterations in lung and cardiac gene
expression in mice exposed to Milano winter fine particulate matter
(PM2.5). PLoS One. 9(e109685)2014.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Qin G, Xia J, Zhang Y, Guo L, Chen R and
Sang N: Ambient fine particulate matter exposure induces reversible
cardiac dysfunction and fibrosis in juvenile and older female mice.
Part Fibre Toxicol. 15(27)2018.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Qi Z, Song Y, Ding Q, Liao X, Li R, Liu G,
Tsang S and Cai Z: Water soluble and insoluble components of
PM2.5 and their functional cardiotoxicities on neonatal
rat cardiomyocytes in vitro. Ecotoxicol Environ Saf. 168:378–387.
2019.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Shaffer F and Ginsberg JP: An overview of
heart rate variability metrics and norms. Front Public Health.
5(258)2017.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Wagner JG, Kamal AS, Morishita M, Dvonch
JT, Harkema JR and Rohr AC: PM2.5-induced cardiovascular
dysregulation in rats is associated with elemental carbon and
temperature-resolved carbon subfractions. Part Fibre Toxicol.
11(25)2014.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Chen R, Qiao L, Li H, Zhao Y, Zhang Y, Xu
W, Wang C, Wang H, Zhao Z, Xu X, et al: Fine particulate matter
constituents, nitric oxide synthase DNA methylation and exhaled
nitric oxide. Environ Sci Technol. 49:11859–11865. 2015.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Tanwar V, Adelstein JM, Grimmer JA, Youtz
DJ, Sugar BP and Wold LE: PM2.5 exposure in utero
contributes to neonatal cardiac dysfunction in mice. Environ
Pollut. 230:116–124. 2017.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Grant AO: Cardiac ion channels. Circ
Arrhythm Electrophysiol. 2:185–194. 2009.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Park KH, Choi YJ, Min WK, Lee SH, Kim J,
Jeong SH, Lee JH, Choi BM and Kim S: Particulate matter induces
arrhythmia-like cardiotoxicity in zebrafish embryos by altering the
expression levels of cardiac development- and ion channel-related
genes. Ecotoxicol Environ Saf. 263(115201)2023.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Gualtieri M, Longhin E, Mattioli M,
Mantecca P, Tinaglia V, Mangano E, Proverbio MC, Bestetti G,
Camatini M and Battaglia C: Gene expression profiling of A549 cells
exposed to Milan PM2.5. Toxicol Lett. 209:136–145. 2012.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Kouassi KS, Billet S, Garçon G, Verdin A,
Diouf A, Cazier F, Djaman J, Courcot D and Shirali P: Oxidative
damage induced in A549 cells by physically and chemically
characterized air particulate matter (PM2.5) collected in Abidjan,
Côte d'Ivoire. J Appl Toxicol. 30:310–320. 2010.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Briedé JJ, De Kok TMCM, Hogervorst JGF,
Moonen EJC, Op Den Camp CLB and Kleinjanst JCS: Development and
application of an electron spin resonance spectrometry method for
the determination of oxygen free radical formation by particulate
matter. Environ Sci Technol. 39:8420–8426. 2005.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Gehling W, Khachatryan L and Dellinger B:
Hydroxyl radical generation from environmentally persistent free
radicals (EPFRs) in PM2.5. Environ Sci Technol. 48:4266–4272.
2014.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Longhin E, Holme JA, Gutzkow KB, Arlt VM,
Kucab JE, Camatini M and Gualtieri M: Cell cycle alterations
induced by urban PM2.5 in bronchial epithelial cells:
characterization of the process and possible mechanisms involved.
Part Fibre Toxicol. 10(63)2013.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Huang Q, Zhang J, Peng S, Tian M, Chen J
and Shen H: Effects of water soluble PM2.5 extracts exposure on
human lung epithelial cells (A549): A proteomic study. J Appl
Toxicol. 34:675–687. 2014.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Kannan S, Misra DP, Dvonch JT and
Krishnakumar A: Exposures to airborne particulate matter and
adverse perinatal outcomes: A biologically plausible mechanistic
framework for exploring potential effect modification by nutrition.
Environ Health Perspect. 114:1636–1642. 2006.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Feng S, Gao D, Liao F, Zhou F and Wang X:
The health effects of ambient PM2.5 and potential mechanisms.
Ecotoxicol Environ Saf. 128:67–74. 2016.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Li SY, Sigmon VK, Babcock SA and Ren J:
Advanced glycation endproduct induces ROS accumulation, apoptosis,
MAP kinase activation and nuclear O-GlcNAcylation in human cardiac
myocytes. Life Sci. 80:1051–1056. 2007.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Moazzen H, Lu X, Ma NL, Velenosi TJ,
Urquhart BL, Wisse LJ, Gittenberger-de Groot AC and Feng Q:
N-Acetylcysteine prevents congenital heart defects induced by
pregestational diabetes. Cardiovasc Diabetol. 13(46)2014.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Ren F, Ji C, Huang Y, Aniagu S, Jiang Y
and Chen T: AHR-mediated ROS production contributes to the cardiac
developmental toxicity of PM2.5 in zebrafish embryos. Sci Total
Environ. 719(135097)2020.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Wang L, He X, Szklarz GD, Bi Y,
Rojanasakul Y and Ma Q: The aryl hydrocarbon receptor interacts
with nuclear factor erythroid 2-related factor 2 to mediate
induction of NAD(P)H:quinoneoxidoreductase 1 by
2,3,7,8-tetrachlorodibenzo-p-dioxin. Arch Biochem Biophys.
537:31–38. 2013.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Rousseau ME, Sant KE, Borden LR, Franks
DG, Hahn ME and Timme-Laragy AR: Regulation of Ahr signaling by
Nrf2 during development: Effects of Nrf2a deficiency on PCB126
embryotoxicity in zebrafish (Danio rerio). Aquat Toxicol.
167:157–171. 2015.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Dalton TP, Puga A and Shertzer HG:
Induction of cellular oxidative stress by aryl hydrocarbon receptor
activation. Chem Biol Interact. 141:77–95. 2002.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Elbekai RH and El-Kadi AOS: The role of
oxidative stress in the modulation of aryl hydrocarbon
receptor-regulated genes by As3+, Cd2+, and Cr6+. Free Radic Biol
Med. 39:1499–1511. 2005.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Mohammadi-Bardbori A, Omidi M and
Arabnezhad MR: Impact of CH223191-induced mitochondrial dysfunction
on its Aryl hydrocarbon receptor agonistic and antagonistic
activities. Chem Res Toxicol. 32:691–697. 2019.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Kopf PG and Walker MK:
2,3,7,8-Tetrachlorodibenzo-p-dioxin increases reactive oxygen
species production in human endothelial cells via induction of
cytochrome P4501A1. Toxicol Appl Pharmacol. 245:91–99.
2010.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Zangar RC, Davydov DR and Verma S:
Mechanisms that regulate production of reactive oxygen species by
cytochrome P450. Toxicol Appl Pharmacol. 199:316–331.
2004.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Zhou B, Wang X, Li F, Wang Y, Yang L, Zhen
X and Tan W: Mitochondrial activity and oxidative stress functions
are influenced by the activation of AhR-induced CYP1A1
overexpression in cardiomyocytes. Mol Med Rep. 16:174–180.
2017.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Pei Y, Jiang R, Zou Y, Wang Y, Zhang S,
Wang G, Zhao J and Song W: Effects of fine particulate matter
(PM2.5) on systemic oxidative stress and cardiac function in
ApoE(-/-) mice. Int J Environ Res Public Health.
13(484)2016.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Yang JL, Lu JY, Zhang MS, Qin G and Li CP:
Involvement of heme oxygenase in PM2.5-toxicity in human umbilical
vein endothelial cells. Zhonghua Xin Xue Guan Bing Za Zhi.
41:955–961. 2013.PubMed/NCBI(In Chinese).
|
|
66
|
Medzhitov R: Origin and physiological
roles of inflammation. Nature. 454:428–435. 2008.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Zhao J, Gao Z, Tian Z, Xie Y, Xin F, Jiang
R, Kan H and Song W: The biological effects of individual-level
PM(2.5) exposure on systemic immunity and inflammatory response in
traffic policemen. Occup Environ Med. 70:426–431. 2013.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Bekki K, Ito T, Yoshida Y, He C,
Arashidani K, He M, Sun G, Zeng Y, Sone H, Kunugita N and Ichinose
T: PM2.5 collected in China causes inflammatory and oxidative
stress responses in macrophages through the multiple pathways.
Environ Toxicol Pharmacol. 45:362–369. 2016.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Shi Q, Zhao L, Xu C, Zhang L and Zhao H:
High molecular weight hyaluronan suppresses macrophage M1
polarization and enhances IL-10 production in
PM2.5-induced lung inflammation. Molecules.
24(1766)2019.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Chen W, Liu Y, Chen J, Song Y, You M and
Yang G: Long-term co-exposure DBP and BaP causes imbalance in liver
macrophages polarization via activation of notch signaling
regulated by miR-34a-5p in rats. Chem Biol Interact.
359(109919)2022.PubMed/NCBI View Article : Google Scholar
|
|
71
|
You M, Song Y, Chen J, Liu Y, Chen W, Cen
Y, Zhao X, Tao Z and Yang G: Combined exposure to benzo(a)pyrene
and dibutyl phthalate aggravates pro-inflammatory macrophage
polarization in spleen via pyroptosis involving cathepsin B. Sci
Total Environ. 881(163460)2023.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Nicolás-Ávila JA, Lechuga-Vieco AV,
Esteban-Martinez L, Sánchez-Díaz M, Díaz-García E, Santiago DJ,
Rubio-Ponce A, Li JL, Balachander A, Quintana JA, et al: A network
of macrophages supports mitochondrial homeostasis in the heart.
Cell. 183:94–109.e23. 2020.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Ueland T, Gullestad L, Nymo SH, Yndestad
A, Aukrust P and Askevold ET: Inflammatory cytokines as biomarkers
in heart failure. Clin Chim Acta. 443:71–77. 2015.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Frati G, Schirone L, Chimenti I, Yee D,
Biondi-Zoccai G, Volpe M and Sciarretta S: An overview of the
inflammatory signalling mechanisms in the myocardium underlying the
development of diabetic cardiomyopathy. Cardiovasc Res.
113:378–388. 2017.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Li R, Zhao Y, Shi J, Zhao C, Xie P, Huang
W, Yong T and Cai Z: Effects of PM2.5 exposure in utero
on heart injury, histone acetylation and GATA4 expression in
offspring mice. Chemosphere. 256(127133)2020.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Ma XN, Li RQ, Xie JL, Li SH, Li JW and Yan
XX: PM2.5-induced inflammation and myocardial cell injury in rats.
Eur Rev Med Pharmacol Sci. 25:6670–6677. 2021.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Fröde-Saleh TS and Calixto JB: Synergistic
antiinflammatory effect of NF-kappaB inhibitors and steroidal or
non steroidal antiinflammatory drugs in the pleural inflammation
induced by carrageenan in mice. Inflamm Res. 49:330–337.
2000.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Ryu YS, Kang KA, Piao MJ, Ahn MJ, Yi JM,
Hyun YM, Kim SH, Ko MK, Park CO and Hyun JW: Particulate matter
induces inflammatory cytokine production via activation of NFκB by
TLR5-NOX4-ROS signaling in human skin keratinocyte and mouse skin.
Redox Biol. 21(101080)2019.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Li H, Shi Y, Wang X, Li P, Zhang S, Wu T,
Yan Y, Zhan Y, Ren Y, Rong X, et al: Piceatannol alleviates
inflammation and oxidative stress via modulation of the Nrf2/HO-1
and NF-κB pathways in diabetic cardiomyopathy. Chem Biol Interact.
310(108754)2019.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Jiao Y, Wang S, Jiang L, Sun X, Li J, Liu
X, Yao X, Zhang C, Wang N, Deng H and Yang G: 2-Undecanone protects
against fine particles-induced heart inflammation via modulating
Nrf2/HO-1 and NF-κB pathways. Environ Toxicol. 37:1642–1652.
2022.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Zhang Y, Ji X, Ku T and Sang N:
Inflammatory response and endothelial dysfunction in the hearts of
mice co-exposed to SO2, NO2, and
PM2.5. Environ Toxicol. 31:1996–2005. 2016.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Chen M, Qin X, Qiu L, Chen S, Zhou H, Xu
Y, Hu Z, Zhang Y, Cao Q and Ying Z: Concentrated ambient
PM2.5-induced inflammation and endothelial dysfunction
in a murine model of neural IKK2 deficiency. Environ Health
Perspect. 126(027003)2018.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Hu B, Tong B, Xiang Y, Li SR, Tan ZX,
Xiang HX, Fu L, Wang H, Zhao H and Xu DX: Acute 1-NP exposure
induces inflammatory responses through activating various
inflammatory signaling pathways in mouse lungs and human A549
cells. Ecotoxicol Environ Saf. 189(109977)2020.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Duan S, Wang N, Huang L, Zhao Y, Shao H,
Jin Y, Zhang R, Li C, Wu W, Wang J and Feng F: NLRP3 inflammasome
activation is associated with PM2.5-induced cardiac
functional and pathological injury in mice. Environ Toxicol.
34:1246–1254. 2019.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Bevan GH, Al-Kindi SG, Brook RD, Münzel T
and Rajagopalan S: Ambient air pollution and atherosclerosis:
Insights into dose, time, and mechanisms. Arterioscler Thromb Vasc
Biol. 41:628–637. 2021.PubMed/NCBI View Article : Google Scholar
|
|
86
|
West AP: Mitochondrial dysfunction as a
trigger of innate immune responses and inflammation. Toxicology.
391:54–63. 2017.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Breda CNS, Davanzo GG, Basso PJ, Saraiva
Câmara NO and Moraes-Vieira PMM: Mitochondria as central hub of the
immune system. Redox Biol. 26(101255)2019.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Wang G, Zhao J, Jiang R and Song W: Rat
lung response to ozone and fine particulate matter (PM2.5)
exposures. Environ Toxicol. 30:343–356. 2015.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Niu J, Liberda EN, Qu S, Guo X, Li X,
Zhang J, Meng J, Yan B, Li N, Zhong M, et al: The role of metal
components in the cardiovascular effects of PM2.5. PLoS One.
8(e83782)2013.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Cardozo AK, Ortis F, Storling J, Feng YM,
Rasschaert J, Tonnesen M, Van Eylen F, Mandrup-Poulsen T, Herchuelz
A and Eizirik DL: Cytokines downregulate the sarcoendoplasmic
reticulum pump Ca2+ ATPase 2b and deplete endoplasmic reticulum
Ca2+, leading to induction of endoplasmic reticulum stress in
pancreatic beta-cells. Diabetes. 54:452–461. 2005.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Baccarelli A, Wright R, Bollati V,
Litonjua A, Zanobetti A, Tarantini L, Sparrow D, Vokonas P and
Schwartz J: Ischemic heart disease and stroke in relation to blood
DNA methylation. Epidemiology. 21:819–828. 2010.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Hotamisligil GS: Endoplasmic reticulum
stress and the inflammatory basis of metabolic disease. Cell.
140:900–917. 2010.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Bettigole SE and Glimcher LH: Endoplasmic
reticulum stress in immunity. Annu Rev Immunol. 33:107–138.
2015.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Song S, Tan J, Miao Y, Li M and Zhang Q:
Crosstalk of autophagy and apoptosis: Involvement of the dual role
of autophagy under ER stress. J Cell Physiol. 232:2977–2984.
2017.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Ding Q, Qi Y and Tsang SY: Mitochondrial
biogenesis, mitochondrial dynamics, and mitophagy in the maturation
of cardiomyocytes. Cells. 10(2463)2021.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Hou L, Zhu ZZ, Zhang X, Nordio F, Bonzini
M, Schwartz J, Hoxha M, Dioni L, Marinelli B, Pegoraro V, et al:
Airborne particulate matter and mitochondrial damage: A
cross-sectional study. Environ Health. 9(48)2010.PubMed/NCBI View Article : Google Scholar
|
|
97
|
Xia T, Kovochich M and Nel AE: Impairment
of mitochondrial function by particulate matter (PM) and their
toxic components: Implications for PM-induced cardiovascular and
lung disease. Front Biosci. 12:1238–1246. 2007.PubMed/NCBI View Article : Google Scholar
|
|
98
|
Lehman JJ, Barger PM, Kovacs A, Saffitz
JE, Medeiros DM and Kelly DP: Peroxisome proliferator-activated
receptor gamma coactivator-1 promotes cardiac mitochondrial
biogenesis. J Clin Invest. 106:847–856. 2000.PubMed/NCBI View Article : Google Scholar
|
|
99
|
Meng Z and Liu Y: Cell morphological
ultrastructural changes in various organs from mice exposed by
inhalation to sulfur dioxide. Inhal Toxicol. 19:543–551.
2007.PubMed/NCBI View Article : Google Scholar
|
|
100
|
Marchini T, Magnani N, D'Annunzio V, Tasat
D, Gelpi RJ, Alvarez S and Evelson P: Impaired cardiac
mitochondrial function and contractile reserve following an acute
exposure to environmental particulate matter. Biochim Biophys Acta.
1830:2545–2552. 2013.PubMed/NCBI View Article : Google Scholar
|
|
101
|
Wang G, Zhen L, Lü P, Jiang R and Song W:
Effects of ozone and fine particulate matter (PM2.5) on rat cardiac
autonomic nervous system and systemic inflammation. Wei Sheng Yan
Jiu. 42:554–560. 2013.PubMed/NCBI(In Chinese).
|
|
102
|
Wang Q and Zhang L, Yuan X, Ou Y, Zhu X,
Cheng Z, Zhang P, Wu X, Meng Y and Zhang L: The relationship
between the Bcl-2/Bax proteins and the mitochondria-mediated
apoptosis pathway in the differentiation of adipose-derived stromal
cells into neurons. PLoS One. 11(e0163327)2016.PubMed/NCBI View Article : Google Scholar
|
|
103
|
Zorzano A, Liesa M, Sebastian D, Segales J
and Palacin M: Mitochondrial fusion proteins: Dual regulators of
morphology and metabolism. Semin Cell Dev Biol. 21:566–574.
2010.PubMed/NCBI View Article : Google Scholar
|
|
104
|
Yang XD, Shi Q, Sun J, Lv Y, Ma Y, Chen C,
Xiao K, Zhou W and Dong XP: Aberrant alterations of mitochondrial
factors Drp1 and Opa1 in the brains of scrapie experiment rodents.
J Mol Neurosci. 61:368–378. 2017.PubMed/NCBI View Article : Google Scholar
|
|
105
|
Westermann B: Mitochondrial fusion and
fission in cell life and death. Nat Rev Mol Cell Biol. 11:872–884.
2010.PubMed/NCBI View Article : Google Scholar
|
|
106
|
Ikeda Y, Sciarretta S, Nagarajan N,
Rubattu S, Volpe M, Frati G and Sadoshima J: New insights into the
role of mitochondrial dynamics and autophagy during oxidative
stress and aging in the heart. Oxid Med Cell Longev.
2014(210934)2014.PubMed/NCBI View Article : Google Scholar
|
|
107
|
Soberanes S, Urich D, Baker CM, Burgess Z,
Chiarella SE, Bell EL, Ghio AJ, De Vizcaya-Ruiz A, Liu J, Ridge KM,
et al: Mitochondrial complex III-generated oxidants activate ASK1
and JNK to induce alveolar epithelial cell death following exposure
to particulate matter air pollution. J Biol Chem. 284:2176–2186.
2009.PubMed/NCBI View Article : Google Scholar
|
|
108
|
Castilho RF, Meinicke AR, Almeida AM,
Hermes-Lima M and Vercesi AE: Oxidative damage of mitochondria
induced by Fe(II)citrate is potentiated by Ca2+ and includes lipid
peroxidation and alterations in membrane proteins. Arch Biochem
Biophys. 308:158–163. 1994.PubMed/NCBI View Article : Google Scholar
|
|
109
|
Packer MA, Porteous CM and Murphy MP:
Superoxide production by mitochondria in the presence of nitric
oxide forms peroxynitrite. Biochem Mol Biol Int. 40:527–534.
1996.PubMed/NCBI View Article : Google Scholar
|
|
110
|
Meyer JN, Leung MCK, Rooney JP, Sendoel A,
Hengartner MO, Kisby GE and Bess AS: Mitochondria as a target of
environmental toxicants. Toxicol Sci. 134:1–17. 2013.PubMed/NCBI View Article : Google Scholar
|
|
111
|
Rodriguez-Enriquez S, He L and Lemasters
JJ: Role of mitochondrial permeability transition pores in
mitochondrial autophagy. Int J Biochem Cell Biol. 36:2463–2472.
2004.PubMed/NCBI View Article : Google Scholar
|
|
112
|
Urrutia PJ, Mena NP and Núñez MT: The
interplay between iron accumulation, mitochondrial dysfunction, and
inflammation during the execution step of neurodegenerative
disorders. Front Pharmacol. 5(38)2014.PubMed/NCBI View Article : Google Scholar
|
|
113
|
Liang H and Ward WF: PGC-1alpha: A key
regulator of energy metabolism. Adv Physiol Educ. 30:145–151.
2006.PubMed/NCBI View Article : Google Scholar
|
|
114
|
Prakash C and Kumar V: Arsenic-induced
mitochondrial oxidative damage is mediated by decreased PGC-1α
expression and its downstream targets in rat brain. Chem Biol
Interact. 256:228–235. 2016.PubMed/NCBI View Article : Google Scholar
|
|
115
|
Chen J, Zhang M, Aniagu S, Jiang Y and
Chen T: PM2.5 induces cardiac defects via
AHR-SIRT1-PGC-1α mediated mitochondrial damage. Environ Toxicol
Pharmacol. 106(104393)2024.PubMed/NCBI View Article : Google Scholar
|
|
116
|
Chen J, Zhang M, Zou H, Aniagu S, Jiang Y
and Chen T: PM2.5 induces mitochondrial dysfunction via
AHR-mediated cyp1a1 overexpression during zebrafish heart
development. Toxicology. 487(153466)2023.PubMed/NCBI View Article : Google Scholar
|
|
117
|
Cavalli G and Heard E: Advances in
epigenetics link genetics to the environment and disease. Nature.
571:489–499. 2019.PubMed/NCBI View Article : Google Scholar
|
|
118
|
Marcho C, Oluwayiose OA and Pilsner JR:
The preconception environment and sperm epigenetics. Andrology.
8:924–942. 2020.PubMed/NCBI View Article : Google Scholar
|
|
119
|
Sun T, Wu R and Ming L: The role of m6A
RNA methylation in cancer. Biomed Pharmacother.
112(108613)2019.PubMed/NCBI View Article : Google Scholar
|
|
120
|
Liu Q and Gregory RI: RNAmod: An
integrated system for the annotation of mRNA modifications. Nucleic
Acids Res. 47:W548–W555. 2019.PubMed/NCBI View Article : Google Scholar
|
|
121
|
Wang Y, Li Y, Toth JI, Petroski MD, Zhang
Z and Zhao JC: N6-methyladenosine modification destabilizes
developmental regulators in embryonic stem cells. Nat Cell Biol.
16:191–198. 2014.PubMed/NCBI View Article : Google Scholar
|
|
122
|
Liu L, Li H, Hu D, Wang Y, Shao W, Zhong
J, Yang S, Liu J and Zhang J: Insights into N6-methyladenosine and
programmed cell death in cancer. Mol Cancer. 21(32)2022.PubMed/NCBI View Article : Google Scholar
|
|
123
|
Tang F, Chen L, Gao H, Xiao D and Li X:
m6A: An emerging role in programmed cell death. Front
Cell Dev Biol. 10(817112)2022.PubMed/NCBI View Article : Google Scholar
|
|
124
|
Yang Y, Shen S, Cai Y, Zeng K, Liu K, Li
S, Zeng L, Chen L, Tang J, Hu Z, et al: Dynamic patterns of
N6-methyladenosine profiles of messenger RNA correlated with the
cardiomyocyte regenerability during the early heart development in
mice. Oxid Med Cell Longev. 2021(5537804)2021.PubMed/NCBI View Article : Google Scholar
|
|
125
|
Shen S, Liu K, Li S, Rampes S, Yang Y,
Huang Y, Tang J, Xia Z, Ma D and Zhang L: N6-methyladenosine
modulates long non-coding RNA in the developing mouse heart. Cell
Death Discov. 8(329)2022.PubMed/NCBI View Article : Google Scholar
|
|
126
|
Guo X, Lin Y, Lin Y, Zhong Y, Yu H, Huang
Y, Yang J, Cai Y, Liu F, Li Y, et al: PM2.5 induces pulmonary
microvascular injury in COPD via METTL16-mediated m6A modification.
Environ Pollut. 303(119115)2022.PubMed/NCBI View Article : Google Scholar
|
|
127
|
He X, Zhang L, Liu S, Wang J, Liu Y, Xiong
A, Jiang M, Luo L, Ying X and Li G: Methyltransferase-like 3 leads
to lung injury by up-regulation of interleukin 24 through
N6-methyladenosine-dependent mRNA stability and translation
efficiency in mice exposed to fine particulate matter 2.5. Environ
Pollut. 308(119607)2022.PubMed/NCBI View Article : Google Scholar
|
|
128
|
Ji C, Tao Y, Li X, Wang J, Chen J, Aniagu
S, Jiang Y and Chen T: AHR-mediated m6A RNA methylation
contributes to PM2.5-induced cardiac malformations in
zebrafish larvae. J Hazard Mater. 457(131749)2023.PubMed/NCBI View Article : Google Scholar
|
|
129
|
Ning J, Du H, Zhang Y, Liu Q, Jiang T,
Pang Y, Tian X, Yan L, Niu Y and Zhang R: N6-Methyladenosine
modification of CDH1 mRNA promotes PM2.5-induced pulmonary fibrosis
via mediating epithelial mesenchymal transition. Toxicol Sci.
185:143–157. 2022.PubMed/NCBI View Article : Google Scholar
|
|
130
|
Batista PJ, Molinie B, Wang J, Qu K, Zhang
J, Li L, Bouley DM, Lujan E, Haddad B, Daneshvar K, et al: m(6)A
RNA modification controls cell fate transition in mammalian
embryonic stem cells. Cell Stem Cell. 15:707–719. 2014.PubMed/NCBI View Article : Google Scholar
|
|
131
|
Zhao T, Sun D, Zhao M, Lai Y, Liu Y and
Zhang Z: N6-methyladenosine mediates arsenite-induced
human keratinocyte transformation by suppressing p53 activation.
Environ Pollut. 259(113908)2020.PubMed/NCBI View Article : Google Scholar
|
|
132
|
Zhuang C, Zhuang C, Luo X, Huang X, Yao L,
Li J, Li Y, Xiong T, Ye J, Zhang F and Gui Y: N6-methyladenosine
demethylase FTO suppresses clear cell renal cell carcinoma through
a novel FTO-PGC-1α signalling axis. J Cell Mol Med. 23:2163–2173.
2019.PubMed/NCBI View Article : Google Scholar
|
|
133
|
Cao J, Qin G, Shi R, Bai F, Yang G, Zhang
M and Lv J: Overproduction of reactive oxygen species and
activation of MAPKs are involved in apoptosis induced by PM2.5 in
rat cardiac H9c2 cells. J Appl Toxicol. 36:609–617. 2016.PubMed/NCBI View Article : Google Scholar
|
|
134
|
Dong W, Matsumura F and Kullman SW: TCDD
induced pericardial edema and relative COX-2 expression in medaka
(Oryzias Latipes) embryos. Toxicol Sci. 118:213–223.
2010.PubMed/NCBI View Article : Google Scholar
|
|
135
|
Shi Y, Zhao T, Yang X, Sun B, Li Y, Duan J
and Sun Z: PM2.5-induced alteration of DNA methylation
and RNA-transcription are associated with inflammatory response and
lung injury. Sci Total Environ. 650:908–921. 2019.PubMed/NCBI View Article : Google Scholar
|
|
136
|
Avilla MN, Malecki KMC, Hahn ME, Wilson RH
and Bradfield CA: The Ah receptor: Adaptive metabolism, ligand
diversity, and the xenokine model. Chem Res Toxicol. 33:860–879.
2020.PubMed/NCBI View Article : Google Scholar
|
|
137
|
Hahn ME, Karchner SI and Merson RR:
Diversity as opportunity: Insights from 600 million years of AHR
evolution. Curr Opin Toxicol. 2:58–71. 2017.PubMed/NCBI View Article : Google Scholar
|
|
138
|
Jeuken A, Keser BJG, Khan E, Brouwer A,
Koeman J and Denison MS: Activation of the Ah receptor by extracts
of dietary herbal supplements, vegetables, and fruits. J Agric Food
Chem. 51:5478–5487. 2003.PubMed/NCBI View Article : Google Scholar
|
|
139
|
Aluru N, Kuo E, Helfrich LW, Karchner SI,
Linney EA, Pais JE and Franks DG: Developmental exposure to
2,3,7,8-tetrachlorodibenzo-p-dioxin alters DNA methyltransferase
(dnmt) expression in zebrafish (Danio rerio). Toxicol Appl
Pharmacol. 284:142–151. 2015.PubMed/NCBI View Article : Google Scholar
|
|
140
|
Jiang Y, Li J, Ren F, Ji C, Aniagu S and
Chen T: PM2.5-induced extensive DNA methylation changes in the
heart of zebrafish embryos and the protective effect of folic acid.
Environ Pollut. 255(113331)2019.PubMed/NCBI View Article : Google Scholar
|
|
141
|
Soberanes S, Gonzalez A, Urich D,
Chiarella SE, Radigan KA, Osornio-Vargas A, Joseph J, Kalyanaraman
B, Ridge KM, Chandel NS, et al: Particulate matter air pollution
induces hypermethylation of the p16 promoter via a mitochondrial
ROS-JNK-DNMT1 pathway. Sci Rep. 2(275)2012.PubMed/NCBI View Article : Google Scholar
|
|
142
|
Al-Saleh I, Shinwari N, Mashhour A,
Mohamed Gel D and Rabah A: Heavy metals (lead, cadmium and mercury)
in maternal, cord blood and placenta of healthy women. Int J Hyg
Environ Health. 214:79–101. 2011.PubMed/NCBI View Article : Google Scholar
|
|
143
|
Maccani JZJ, Koestler DC, Lester B,
Houseman EA, Armstrong DA, Kelsey KT and Marsit CJ: Placental DNA
methylation related to both infant toenail mercury and adverse
neurobehavioral outcomes. Environ Health Perspect. 123:723–729.
2015.PubMed/NCBI View Article : Google Scholar
|
|
144
|
Mohanty AF, Farin FM, Bammler TK,
MacDonald JW, Afsharinejad Z, Burbacher TM, Siscovick DS, Williams
MA and Enquobahrie DA: Infant sex-specific placental cadmium and
DNA methylation associations. Environ Res. 138:74–81.
2015.PubMed/NCBI View Article : Google Scholar
|
|
145
|
Li W, Liu H, Yu M, Zhang X, Zhang Y, Liu
H, Wilson JX and Huang G: Folic acid alters methylation profile of
JAK-STAT and long-term depression signaling pathways in Alzheimer's
disease models. Mol Neurobiol. 53:6548–6556. 2016.PubMed/NCBI View Article : Google Scholar
|
|
146
|
Maghbooli Z, Hossein-Nezhad A, Adabi E,
Asadollah-Pour E, Sadeghi M, Mohammad-Nabi S, Zakeri Rad L, Malek
Hosseini AA, Radmehr M, Faghihi F, et al: Air pollution during
pregnancy and placental adaptation in the levels of global DNA
methylation. PLoS One. 13(e0199772)2018.PubMed/NCBI View Article : Google Scholar
|
|
147
|
Goodson JM, Weldy CS, MacDonald JW, Liu Y,
Bammler TK, Chien WM and Chin MT: In utero exposure to diesel
exhaust particulates is associated with an altered cardiac
transcriptional response to transverse aortic constriction and
altered DNA methylation. FASEB J. 31:4935–4945. 2017.PubMed/NCBI View Article : Google Scholar
|
|
148
|
Wauters A, Dreyfuss C, Pochet S, Hendrick
P, Berkenboom G, van de Borne P and Argacha JF: Acute exposure to
diesel exhaust impairs nitric oxide-mediated endothelial vasomotor
function by increasing endothelial oxidative stress. Hypertension.
62:352–358. 2013.PubMed/NCBI View Article : Google Scholar
|
|
149
|
Valinluck V, Tsai HH, Rogstad DK, Burdzy
A, Bird A and Sowers LC: Oxidative damage to methyl-CpG sequences
inhibits the binding of the methyl-CpG binding domain (MBD) of
methyl-CpG binding protein 2 (MeCP2). Nucleic Acids Res.
32:4100–4108. 2004.PubMed/NCBI View Article : Google Scholar
|
|
150
|
Panni T, Mehta AJ, Schwartz JD, Baccarelli
AA, Just AC, Wolf K, Wahl S, Cyrys J, Kunze S, Strauch K, et al:
Genome-wide analysis of DNA methylation and fine particulate matter
air pollution in three study populations: KORA F3, KORA F4, and the
normative aging study. Environ Health Perspect. 124:983–990.
2016.PubMed/NCBI View Article : Google Scholar
|
|
151
|
Wei S, Segura S, Vendrell J, Aviles FX,
Lanoue E, Day R, Feng Y and Fricker LD: Identification and
characterization of three members of the human
metallocarboxypeptidase gene family. J Biol Chem. 277:14954–14964.
2002.PubMed/NCBI View Article : Google Scholar
|
|
152
|
Bellavia A, Urch B, Speck M, Brook RD,
Scott JA, Albetti B, Behbod B, North M, Valeri L, Bertazzi PA, et
al: DNA hypomethylation, ambient particulate matter, and increased
blood pressure: Findings from controlled human exposure
experiments. J Am Heart Assoc. 2(e000212)2013.PubMed/NCBI View Article : Google Scholar
|
|
153
|
Shahbazian MD and Grunstein M: Functions
of site-specific histone acetylation and deacetylation. Annu Rev
Biochem. 76:75–100. 2007.PubMed/NCBI View Article : Google Scholar
|
|
154
|
Chervona Y, Hall MN, Arita A, Wu F, Sun H,
Tseng HC, Ali E, Uddin MN, Liu X, Zoroddu MA, et al: Associations
between arsenic exposure and global posttranslational histone
modifications among adults in Bangladesh. Cancer Epidemiol
Biomarkers Prev. 21:2252–2260. 2012.PubMed/NCBI View Article : Google Scholar
|
|
155
|
Zhang Z, Chen L, Xing X, Li D, Gao C, He
Z, Li J, Zhu X, Xiao X, Wang S, et al: Specific histone
modifications were associated with the PAH-induced DNA damage
response in coke oven workers. Toxicol Res (Camb). 5:1193–1201.
2016.PubMed/NCBI View Article : Google Scholar
|
|
156
|
Wang Z, Zhao YT and Zhao TC: Histone
deacetylases in modulating cardiac disease and their clinical
translational and therapeutic implications. Exp Biol Med (Maywood).
246:213–225. 2021.PubMed/NCBI View Article : Google Scholar
|
|
157
|
Sun H, Yang X, Zhu J, Lv T, Chen Y, Chen
G, Zhong L, Li Y, Huang X, Huang G and Tian J: Inhibition of
p300-HAT results in a reduced histone acetylation and
down-regulation of gene expression in cardiac myocytes. Life Sci.
87:707–714. 2010.PubMed/NCBI View Article : Google Scholar
|
|
158
|
Hu DX, Liu XB, Song WC and Wang JA: Roles
of SIRT3 in heart failure: From bench to bedside. J Zhejiang Univ
Sci B. 17:821–830. 2016.PubMed/NCBI View Article : Google Scholar
|
|
159
|
Li Y, Ma Y, Song L, Yu L, Zhang L, Zhang
Y, Xing Y, Yin Y and Ma H: SIRT3 deficiency exacerbates
p53/Parkin-mediated mitophagy inhibition and promotes mitochondrial
dysfunction: Implication for aged hearts. Int J Mol Med.
41:3517–3526. 2018.PubMed/NCBI View Article : Google Scholar
|
|
160
|
Wang Y, Miao X, Liu Y, Li F, Liu Q, Sun J
and Cai L: Dysregulation of histone acetyltransferases and
deacetylases in cardiovascular diseases. Oxid Med Cell Longev.
2014(641979)2014.PubMed/NCBI View Article : Google Scholar
|
|
161
|
Chen H, Giri NC, Zhang R, Yamane K, Zhang
Y, Maroney M and Costa M: Nickel ions inhibit histone demethylase
JMJD1A and DNA repair enzyme ABH2 by replacing the ferrous iron in
the catalytic centers. J Biol Chem. 285:7374–7383. 2010.PubMed/NCBI View Article : Google Scholar
|
|
162
|
Prins D and Michalak M: Endoplasmic
reticulum proteins in cardiac development and dysfunction. Can J
Physiol Pharmacol. 87:419–425. 2009.PubMed/NCBI View Article : Google Scholar
|
|
163
|
Zhu Y, Guan H, Zhu X, Cai J, Jiao X, Shan
J, Li Y, Wu Q and Zhang Z: Astilbin antagonizes developmental
cardiotoxicity after cadmium exposure in chicken embryos by
inhibiting endoplasmic reticulum stress and maintaining calcium
homeostasis. Ecotoxicol Environ Saf. 270(115847)2024.PubMed/NCBI View Article : Google Scholar
|
|
164
|
Minamino T and Kitakaze M: ER stress in
cardiovascular disease. J Mol Cell Cardiol. 48:1105–1110.
2010.PubMed/NCBI View Article : Google Scholar
|
|
165
|
Li R, Zhang M, Wang Y, Yung KKL, Su R, Li
Z, Zhao L, Dong C and Cai Z: Effects of sub-chronic exposure to
atmospheric PM2.5 on fibrosis, inflammation, endoplasmic
reticulum stress and apoptosis in the livers of rats. Toxicol Res
(Camb). 7:271–282. 2018.PubMed/NCBI View Article : Google Scholar
|
|
166
|
Wang Y and Tang M: PM2.5 induces autophagy
and apoptosis through endoplasmic reticulum stress in human
endothelial cells. Sci Total Environ. 710(136397)2020.PubMed/NCBI View Article : Google Scholar
|
|
167
|
Chen T, Jin H, Wang H, Yao Y, Aniagu S,
Tong J and Jiang Y: Aryl hydrocarbon receptor mediates the cardiac
developmental toxicity of EOM from PM2.5 in P19
embryonic carcinoma cells. Chemosphere. 216:372–378.
2019.PubMed/NCBI View Article : Google Scholar
|
|
168
|
de la Harpe A, Beukes N and Frost CL: CBD
activation of TRPV1 induces oxidative signaling and subsequent ER
stress in breast cancer cell lines. Biotechnol Appl Biochem.
69:420–430. 2022.PubMed/NCBI View Article : Google Scholar
|
|
169
|
Malhotra JD and Kaufman RJ: Endoplasmic
reticulum stress and oxidative stress: A vicious cycle or a
double-edged sword? Antioxid Redox Signal. 9:2277–2293.
2007.PubMed/NCBI View Article : Google Scholar
|
|
170
|
Ozgur R, Uzilday B, Sekmen AH and Turkan
I: The effects of induced production of reactive oxygen species in
organelles on endoplasmic reticulum stress and on the unfolded
protein response in arabidopsis. Ann Bot. 116:541–553.
2015.PubMed/NCBI View Article : Google Scholar
|
|
171
|
Burgos-Morón E, Abad-Jiménez Z, Marañón
AM, Iannantuoni F, Escribano-López I, López-Domènech S, Salom C,
Jover A, Mora V, Roldan I, et al: Relationship between oxidative
stress, ER stress, and inflammation in type 2 diabetes: The battle
continues. J Clin Med. 8(1385)2019.PubMed/NCBI View Article : Google Scholar
|
|
172
|
Jiang B, Zhou X, Yang T, Wang L, Feng L,
Wang Z, Xu J, Jing W, Wang T, Su H, et al: The role of autophagy in
cardiovascular disease: Cross-interference of signaling pathways
and underlying therapeutic targets. Front Cardiovasc Med.
10(1088575)2023.PubMed/NCBI View Article : Google Scholar
|
|
173
|
Lavandero S, Chiong M, Rothermel BA and
Hill JA: Autophagy in cardiovascular biology. J Clin Invest.
125:55–64. 2015.PubMed/NCBI View Article : Google Scholar
|
|
174
|
Lee E, Koo Y, Ng A, Wei Y, Luby-Phelps K,
Juraszek A, Xavier RJ, Cleaver O, Levine B and Amatruda JF:
Autophagy is essential for cardiac morphogenesis during vertebrate
development. Autophagy. 10:572–587. 2014.PubMed/NCBI View Article : Google Scholar
|
|
175
|
Nakai A, Yamaguchi O, Takeda T, Higuchi Y,
Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, et
al: The role of autophagy in cardiomyocytes in the basal state and
in response to hemodynamic stress. Nat Med. 13:619–624.
2007.PubMed/NCBI View
Article : Google Scholar
|
|
176
|
Wang Y, Fang J, Leonard SS and Rao KM:
Cadmium inhibits the electron transfer chain and induces reactive
oxygen species. Free Radic Biol Med. 36:1434–1443. 2004.PubMed/NCBI View Article : Google Scholar
|
|
177
|
Høyer-Hansen M and Jäättelä M: Connecting
endoplasmic reticulum stress to autophagy by unfolded protein
response and calcium. Cell Death Differ. 14:1576–1582.
2007.PubMed/NCBI View Article : Google Scholar
|
|
178
|
Biagioli M, Pifferi S, Ragghianti M, Bucci
S, Rizzuto R and Pinton P: Endoplasmic reticulum stress and
alteration in calcium homeostasis are involved in cadmium-induced
apoptosis. Cell Calcium. 43:184–195. 2008.PubMed/NCBI View Article : Google Scholar
|
|
179
|
Zheng Q, Chen Y, Chen D, Zhao H, Feng Y,
Meng Q, Zhao Y and Zhang H: Calcium transients on the ER surface
trigger liquid-liquid phase separation of FIP200 to specify
autophagosome initiation sites. Cell. 185:4082–4098.e22.
2022.PubMed/NCBI View Article : Google Scholar
|
|
180
|
Sun M, Jiang Z, Gu P, Guo B, Li J, Cheng
S, Ba Q and Wang H: Cadmium promotes colorectal cancer metastasis
through EGFR/Akt/mTOR signaling cascade and dynamics. Sci Total
Environ. 899(165699)2023.PubMed/NCBI View Article : Google Scholar
|
|
181
|
Lian J, Wu X, He F, Karnak D, Tang W, Meng
Y, Xiang D, Ji M, Lawrence TS and Xu L: A natural BH3 mimetic
induces autophagy in apoptosis-resistant prostate cancer via
modulating Bcl-2-Beclin1 interaction at endoplasmic reticulum. Cell
Death Differ. 18:60–71. 2011.PubMed/NCBI View Article : Google Scholar
|
|
182
|
Plácido AI, Pereira CM, Duarte AI,
Candeias E, Correia SC, Santos RX, Carvalho C, Cardoso S, Oliveira
CR and Moreira PI: The role of endoplasmic reticulum in amyloid
precursor protein processing and trafficking: Implications for
Alzheimer's disease. Biochim Biophys Acta. 1842:1444–1453.
2014.PubMed/NCBI View Article : Google Scholar
|
|
183
|
Su R, Jin X, Lyu L, Tian J, Amin S and Li
Z: The potential immunotoxicity of fine particulate matter based on
SD rat spleen. Environ Sci Pollut Res Int. 26:23958–23966.
2019.PubMed/NCBI View Article : Google Scholar
|
|
184
|
Rubiolo JA, López-Alonso H, Martinez P,
Millán A, Cagide E, Vieytes MR, Vega FV and Botana LM: Yessotoxin
induces ER-stress followed by autophagic cell death in glioma cells
mediated by mTOR and BNIP3. Cell Signal. 26:419–432.
2014.PubMed/NCBI
|
|
185
|
Carloni S, Favrais G, Saliba E, Albertini
MC, Chalon S, Longini M, Gressens P, Buonocore G and Balduini W:
Melatonin modulates neonatal brain inflammation through endoplasmic
reticulum stress, autophagy, and miR-34a/silent information
regulator 1 pathway. J Pineal Res. 61:370–380. 2016.PubMed/NCBI View Article : Google Scholar
|
|
186
|
Zhang S, Jiang C, Liu H, Guan Z, Zeng Q,
Zhang C, Lei R, Xia T, Gao H, Yang L, et al: Fluoride-elicited
developmental testicular toxicity in rats: Roles of endoplasmic
reticulum stress and inflammatory response. Toxicol Appl Pharmacol.
271:206–215. 2013.PubMed/NCBI View Article : Google Scholar
|
|
187
|
Dagher Z, Garçon G, Billet S, Gosset P,
Ledoux F, Courcot D, Aboukais A and Shirali P: Activation of
different pathways of apoptosis by air pollution particulate matter
(PM2.5) in human epithelial lung cells (L132) in culture.
Toxicology. 225:12–24. 2006.PubMed/NCBI View Article : Google Scholar
|
|
188
|
Yang X, Zhao T, Feng L, Shi Y, Jiang J,
Liang S, Sun B, Xu Q, Duan J and Sun Z: PM2.5-induced
ADRB2 hypermethylation contributed to cardiac dysfunction through
cardiomyocytes apoptosis via PI3K/Akt pathway. Environ Int.
127:601–614. 2019.PubMed/NCBI View Article : Google Scholar
|
|
189
|
Elmore S: Apoptosis: A review of
programmed cell death. Toxicol Pathol. 35:495–516. 2007.PubMed/NCBI View Article : Google Scholar
|
|
190
|
Yang X, Feng L, Zhang Y, Hu H, Shi Y,
Liang S, Zhao T, Fu Y, Duan J and Sun Z: Cytotoxicity induced by
fine particulate matter (PM2.5) via
mitochondria-mediated apoptosis pathway in human cardiomyocytes.
Ecotoxicol Environ Saf. 161:198–207. 2018.PubMed/NCBI View Article : Google Scholar
|
|
191
|
Bock FJ and Tait SWG: Mitochondria as
multifaceted regulators of cell death. Nat Rev Mol Cell Biol.
21:85–100. 2020.PubMed/NCBI View Article : Google Scholar
|
|
192
|
Su CH, Chen SP, Chen LY, Yang JJ, Lee YC,
Lee SS, Chen HH, Ng YY and Kuan YH: 3-Bromofluoranthene-induced
cardiotoxicity of zebrafish and apoptosis in the vascular
endothelial cells via intrinsic and extrinsic caspase-dependent
pathways. Ecotoxicol Environ Saf. 228(112962)2021.PubMed/NCBI View Article : Google Scholar : (Epub ahead of
print).
|
|
193
|
Gombedza FC, Shin S, Kanaras YL and
Bandyopadhyay BC: Abrogation of store-operated Ca2+
entry protects against crystal-induced ER stress in human proximal
tubular cells. Cell Death Discov. 5(124)2019.PubMed/NCBI View Article : Google Scholar
|
|
194
|
Dlamini Z, Tshidino SC and Hull R:
Abnormalities in alternative splicing of apoptotic genes and
cardiovascular diseases. Int J Mol Sci. 16:27171–27190.
2015.PubMed/NCBI View Article : Google Scholar
|
|
195
|
Yan L, Zhou Y, Yu S, Ji G, Wang L, Liu W
and Gu A: 8-Oxoguanine DNA glycosylase 1 (ogg1) maintains the
function of cardiac progenitor cells during heart formation in
zebrafish. Exp Cell Res. 319:2954–2963. 2013.PubMed/NCBI View Article : Google Scholar
|
|
196
|
Zhao X, Ren X, Zhu R, Luo Z and Ren B:
Zinc oxide nanoparticles induce oxidative DNA damage and
ROS-triggered mitochondria-mediated apoptosis in zebrafish embryos.
Aquat Toxicol. 180:56–70. 2016.PubMed/NCBI View Article : Google Scholar
|
|
197
|
Zhao X, Wang S, Wu Y, You H and Lv L:
Acute ZnO nanoparticles exposure induces developmental toxicity,
oxidative stress and DNA damage in embryo-larval zebrafish. Aquat
Toxicol. 136-137:49–59. 2013.PubMed/NCBI View Article : Google Scholar
|
|
198
|
Zhu L, Dong X, Xie H, Wang J, Wang J, Su J
and Yu C: DNA damage and effects on glutathione-S-transferase
activity induced by atrazine exposure in zebrafish (Danio rerio).
Environ Toxicol. 26:480–488. 2011.PubMed/NCBI View Article : Google Scholar
|
|
199
|
Liu H, Cheng Y, Yang J, Wang W, Fang S,
Zhang W, Han B, Zhou Z, Yao H, Chao J and Liao H: BBC3 in
macrophages promoted pulmonary fibrosis development through
inducing autophagy during silicosis. Cell Death Dis.
8(e2657)2017.PubMed/NCBI View Article : Google Scholar
|
|
200
|
Huang DC and Strasser A: BH3-Only
proteins-essential initiators of apoptotic cell death. Cell.
103:839–842. 2000.PubMed/NCBI View Article : Google Scholar
|
|
201
|
Kedinger V, Alpy F, Tomasetto C, Thisse C,
Thisse B and Rio MC: Spatial and temporal distribution of the traf4
genes during zebrafish development. Gene Expr Patterns. 5:545–552.
2005.PubMed/NCBI View Article : Google Scholar
|
|
202
|
Sax JK and El-Deiry WS: Identification and
characterization of the cytoplasmic protein TRAF4 as a
p53-regulated proapoptotic gene. J Biol Chem. 278:36435–36444.
2003.PubMed/NCBI View Article : Google Scholar
|
|
203
|
Ruan X, Zhang R, Li R, Zhu H, Wang Z, Wang
C, Cheng Z and Peng H: The research progress in physiological and
pathological functions of TRAF4. Front Oncol.
12(842072)2022.PubMed/NCBI View Article : Google Scholar
|
|
204
|
Zhang J, Cui S, Shen L, Gao Y, Liu W,
Zhang C and Zhuang S: Promotion of bladder cancer cell metastasis
by 2-mercaptobenzothiazole via its activation of Aryl hydrocarbon
receptor transcription: Molecular dynamics simulations, cell-based
assays, and machine learning-driven prediction. Environ Sci
Technol. 56:13254–13263. 2022.PubMed/NCBI View Article : Google Scholar
|
|
205
|
Yue C, Ji C, Zhang H, Zhang LW, Tong J,
Jiang Y and Chen T: Protective effects of folic acid on
PM2.5-induced cardiac developmental toxicity in zebrafish embryos
by targeting AhR and Wnt/β-catenin signal pathways. Environ
Toxicol. 32:2316–2322. 2017.PubMed/NCBI View Article : Google Scholar
|
|
206
|
Bello SM, Heideman W and Peterson RE:
2,3,7,8-Tetrachlorodibenzo-p-dioxin inhibits regression of the
common cardinal vein in developing zebrafish. Toxicol Sci.
78:258–266. 2004.PubMed/NCBI View Article : Google Scholar
|
|
207
|
Fu H, Wang L, Wang J, Bennett BD, Li JL,
Zhao B and Hu G: Dioxin and AHR impairs mesoderm gene expression
and cardiac differentiation in human embryonic stem cells. Sci
Total Environ. 651:1038–1046. 2019.PubMed/NCBI View Article : Google Scholar
|
|
208
|
Lund AK, Goens MB, Nuñez BA and Walker MK:
Characterizing the role of endothelin-1 in the progression of
cardiac hypertrophy in aryl hydrocarbon receptor (AhR) null mice.
Toxicol Appl Pharmacol. 212:127–135. 2006.PubMed/NCBI View Article : Google Scholar
|
|
209
|
Evans BR, Karchner SI, Franks DG and Hahn
ME: Duplicate aryl hydrocarbon receptor repressor genes (ahrr1 and
ahrr2) in the zebrafish Danio rerio: Structure, function,
evolution, and AHR-dependent regulation in vivo. Arch Biochem
Biophys. 441:151–167. 2005.PubMed/NCBI View Article : Google Scholar
|
|
210
|
Jenny MJ, Karchner SI, Franks DG, Woodin
BR, Stegeman JJ and Hahn ME: Distinct roles of two zebrafish AHR
repressors (AHRRa and AHRRb) in embryonic development and
regulating the response to 2,3,7,8-tetrachlorodibenzo-p-dioxin.
Toxicol Sci. 110:426–441. 2009.PubMed/NCBI View Article : Google Scholar
|
|
211
|
Jayasundara N, Van Tiem Garner L, Meyer
JN, Erwin KN and Di Giulio RT: AHR2-mediated transcriptomic
responses underlying the synergistic cardiac developmental toxicity
of PAHs. Toxicol Sci. 143:469–481. 2015.PubMed/NCBI View Article : Google Scholar
|
|
212
|
Ko CI, Fan Y, de Gannes M, Wang Q, Xia Y
and Puga A: Repression of the Aryl hydrocarbon receptor is required
to maintain mitotic progression and prevent loss of pluripotency of
embryonic stem cells. Stem Cells. 34:2825–2839. 2016.PubMed/NCBI View Article : Google Scholar
|
|
213
|
Jiang Y, Wang D, Zhang G, Wang G, Tong J
and Chen T: Disruption of cardiogenesis in human embryonic stem
cells exposed to trichloroethylene. Environ Toxicol. 31:1372–1380.
2016.PubMed/NCBI View Article : Google Scholar
|
|
214
|
Wang Q, Chen J, Ko CI, Fan Y, Carreira V,
Chen Y, Xia Y, Medvedovic M and Puga A: Disruption of aryl
hydrocarbon receptor homeostatic levels during embryonic stem cell
differentiation alters expression of homeobox transcription factors
that control cardiomyogenesis. Environ Health Perspect.
121:1334–1343. 2013.PubMed/NCBI View Article : Google Scholar
|
|
215
|
Carreira VS, Fan Y, Kurita H, Wang Q, Ko
CI, Naticchioni M, Jiang M, Koch S, Zhang X, Biesiada J, et al:
Disruption of Ah receptor signaling during mouse development leads
to abnormal cardiac structure and function in the adult. PLoS One.
10(e0142440)2015.PubMed/NCBI View Article : Google Scholar
|
|
216
|
Wang Q, Kurita H, Carreira V, Ko CI, Fan
Y, Zhang X, Biesiada J, Medvedovic M and Puga A: Ah receptor
activation by dioxin disrupts activin, BMP, and WNT signals during
the early differentiation of mouse embryonic stem cells and
inhibits cardiomyocyte functions. Toxicol Sci. 149:346–357.
2016.PubMed/NCBI View Article : Google Scholar
|
|
217
|
Ginis I, Luo Y, Miura T, Thies S,
Brandenberger R, Gerecht-Nir S, Amit M, Hoke A, Carpenter MK,
Itskovitz-Eldor J and Rao MS: Differences between human and mouse
embryonic stem cells. Dev Biol. 269:360–380. 2004.PubMed/NCBI View Article : Google Scholar
|
|
218
|
Dere E, Lee AW, Burgoon LD and Zacharewski
TR: Differences in TCDD-elicited gene expression profiles in human
HepG2, mouse Hepa1c1c7 and rat H4IIE hepatoma cells. BMC Genomics.
12(193)2011.PubMed/NCBI View Article : Google Scholar
|
|
219
|
Suzuki T and Nohara K: Regulatory factors
involved in species-specific modulation of arylhydrocarbon receptor
(AhR)-dependent gene expression in humans and mice. J Biochem.
142:443–452. 2007.PubMed/NCBI View Article : Google Scholar
|
|
220
|
Jiang Y, Zhao X, Chen J, Aniagu S and Chen
T: PM2.5 induces cardiac malformations via PI3K/akt2/mTORC1
signaling pathway in zebrafish larvae. Environ Pollut.
323(121306)2023.PubMed/NCBI View Article : Google Scholar
|
|
221
|
Ozhan G and Weidinger G: Wnt/β-catenin
signaling in heart regeneration. Cell Regen. 4(3)2015.PubMed/NCBI View Article : Google Scholar
|
|
222
|
Ueno S, Weidinger G, Osugi T, Kohn AD,
Golob JL, Pabon L, Reinecke H, Moon RT and Murry CE: Biphasic role
for Wnt/beta-catenin signaling in cardiac specification in
zebrafish and embryonic stem cells. Proc Natl Acad Sci USA.
104:9685–9690. 2007.PubMed/NCBI View Article : Google Scholar
|
|
223
|
Schneider AJ, Branam AM and Peterson RE:
Intersection of AHR and Wnt signaling in development, health, and
disease. Int J Mol Sci. 15:17852–17885. 2014.PubMed/NCBI View Article : Google Scholar
|
|
224
|
Wincent E, Stegeman JJ and Jönsson ME:
Combination effects of AHR agonists and Wnt/β-catenin modulators in
zebrafish embryos: Implications for physiological and toxicological
AHR functions. Toxicol Appl Pharmacol. 284:163–179. 2015.PubMed/NCBI View Article : Google Scholar
|
|
225
|
Blache P, van de Wetering M, Duluc I,
Domon C, Berta P, Freund JN, Clevers H and Jay P: SOX9 is an
intestine crypt transcription factor, is regulated by the Wnt
pathway, and represses the CDX2 and MUC2 genes. J Cell Biol.
166:37–47. 2004.PubMed/NCBI View Article : Google Scholar
|
|
226
|
Liu Z, Li T, Liu Y, Jia Z, Li Y, Zhang C,
Chen P, Ma K, Affara N and Zhou C: WNT signaling promotes Nkx2.5
expression and early cardiomyogenesis via downregulation of Hdac1.
Biochim Biophys Acta. 1793:300–311. 2009.PubMed/NCBI View Article : Google Scholar
|
|
227
|
Lin X and Xu X: Distinct functions of
Wnt/beta-catenin signaling in KV development and cardiac asymmetry.
Development. 136:207–217. 2009.PubMed/NCBI View Article : Google Scholar
|
|
228
|
Chiu CS, Tsai CH, Hsieh MS, Tsai SC, Jan
YJ, Lin WY, Lai DW, Wu SM, Hsing HY, Arbiser JL and Sheu ML:
Exploiting Honokiol-induced ER stress CHOP activation inhibits the
growth and metastasis of melanoma by suppressing the MITF and
β-catenin pathways. Cancer Lett. 442:113–125. 2019.PubMed/NCBI View Article : Google Scholar
|
|
229
|
Valavanidis A, Fiotakis K and Vlachogianni
T: Airborne particulate matter and human health: Toxicological
assessment and importance of size and composition of particles for
oxidative damage and carcinogenic mechanisms. J Environ Sci Health
C Environ Carcinog Ecotoxicol Rev. 26:339–362. 2008.PubMed/NCBI View Article : Google Scholar
|
|
230
|
Fu Y, Niu Y, Pan B, Liu Y, Zhang B, Li X,
Yang A, Nie J, Wang R and Yang J: OGG1 methylation mediated the
effects of cell cycle and oxidative DNA damage related to PAHs
exposure in Chinese coke oven workers. Chemosphere. 224:48–57.
2019.PubMed/NCBI View Article : Google Scholar
|
|
231
|
Zhang Z, Xing X, Jiang S, Qiu C, Mo Z,
Chen S, Chen L, Wang Q, Xiao Y, Dong G, et al: Global H3K79
di-methylation mediates DNA damage response to PAH exposure in
Chinese coke oven workers. Environ Pollut.
268(115956)2021.PubMed/NCBI View Article : Google Scholar
|
|
232
|
Zhao L, Zhang L, Chen M, Dong C, Li R and
Cai Z: Effects of ambient atmospheric PM2.5,
1-nitropyrene and 9-nitroanthracene on DNA damage and oxidative
stress in hearts of rats. Cardiovasc Toxicol. 19:178–190.
2019.PubMed/NCBI View Article : Google Scholar
|
|
233
|
Wang W, Li Y, Liu X, Jin M, Du H, Liu Y,
Huang P, Zhou X, Yuan L and Sun Z: Multinucleation and cell
dysfunction induced by amorphous silica nanoparticles in an L-02
human hepatic cell line. Int J Nanomedicine. 8:3533–3541.
2013.PubMed/NCBI View Article : Google Scholar
|
|
234
|
Gutiérrez-Castillo ME, Roubicek DA,
Cebrián-García ME, De Vizcaya-Ruíz A, Sordo-Cedeño M and
Ostrosky-Wegman P: Effect of chemical composition on the induction
of DNA damage by urban airborne particulate matter. Environ Mol
Mutagen. 47:199–211. 2006.PubMed/NCBI View Article : Google Scholar
|
|
235
|
Risom L, Møller P and Loft S: Oxidative
stress-induced DNA damage by particulate air pollution. Mutat Res.
592:119–137. 2005.PubMed/NCBI View Article : Google Scholar
|
|
236
|
Zhang P, Yi LH, Meng GY, Zhang HY, Sun HH
and Cui LQ: Apelin-13 attenuates cisplatin-induced cardiotoxicity
through inhibition of ROS-mediated DNA damage and regulation of
MAPKs and AKT pathways. Free Radic Res. 51:449–459. 2017.PubMed/NCBI View Article : Google Scholar
|
|
237
|
Ayres JG, Borm P, Cassee FR, Castranova V,
Donaldson K, Ghio A, Harrison RM, Hider R, Kelly F, Kooter IM, et
al: Evaluating the toxicity of airborne particulate matter and
nanoparticles by measuring oxidative stress potential-a workshop
report and consensus statement. Inhal Toxicol. 20:75–99.
2008.PubMed/NCBI View Article : Google Scholar
|
|
238
|
Meira LB, Bugni JM, Green SL, Lee CW, Pang
B, Borenshtein D, Rickman BH, Rogers AB, Moroski-Erkul CA, McFaline
JL, et al: DNA damage induced by chronic inflammation contributes
to colon carcinogenesis in mice. J Clin Invest. 118:2516–2525.
2008.PubMed/NCBI View Article : Google Scholar
|
|
239
|
Zhang X, Jiang Y and Yang J:
p53-independent signaling pathway in DNA damage-induced cell
apoptosis. Zhejiang Da Xue Xue Bao Yi Xue Ban. 42:217–223.
2013.PubMed/NCBI(In Chinese).
|
|
240
|
De Zio D, Cianfanelli V and Cecconi F: New
insights into the link between DNA damage and apoptosis. Antioxid
Redox Signal. 19:559–571. 2013.PubMed/NCBI View Article : Google Scholar
|
|
241
|
Lorda-Diez CI, Solis-Mancilla ME,
Sanchez-Fernandez C, Garcia-Porrero JA, Hurle JM and Montero JA:
Cell senescence, apoptosis and DNA damage cooperate in the
remodeling processes accounting for heart morphogenesis. J Anat.
234:815–829. 2019.PubMed/NCBI View Article : Google Scholar
|
|
242
|
Huang Y, Tao Y, Cai C, Chen J, Ji C,
Aniagu S, Jiang Y and Chen T: Using immunofluorescence to Detect
PM2.5-induced DNA damage in zebrafish embryo hearts. J Vis Exp,
2021.
|
|
243
|
Cartwright EJ, Oceandy D, Austin C and
Neyses L: Ca2+ signalling in cardiovascular disease: The role of
the plasma membrane calcium pumps. Sci China Life Sci. 54:691–698.
2011.PubMed/NCBI View Article : Google Scholar
|
|
244
|
Cai C, Huang J, Lin Y, Miao W, Chen P,
Chen X, Wang J and Chen M: Particulate matter 2.5 induced
arrhythmogenesis mediated by TRPC3 in human induced pluripotent
stem cell-derived cardiomyocytes. Arch Toxicol. 93:1009–1020.
2019.PubMed/NCBI View Article : Google Scholar
|
|
245
|
Wang Y, Wu T and Tang M: Ambient
particulate matter triggers dysfunction of subcellular structures
and endothelial cell apoptosis through disruption of redox
equilibrium and calcium homeostasis. J Hazard Mater.
394(122439)2020.PubMed/NCBI View Article : Google Scholar
|
|
246
|
Dong L, Sun W, Li F, Shi M, Meng X, Wang
C, Meng M, Tang W, Liu H, Wang L and Song L: The harmful effects of
acute PM2.5 exposure to the heart and a novel preventive
and therapeutic function of CEOs. Sci Rep. 9(3495)2019.PubMed/NCBI View Article : Google Scholar
|
|
247
|
Xu R, Cao JW, Xu TC, Liu TJ, Zhu MR and
Guo MY: Selenium deficiency induced inflammation and apoptosis via
NF-κB and MAPKs pathways in muscle of common carp (Cyprinus carpio
L.). Fish Shellfish Immunol. 138(108847)2023.PubMed/NCBI View Article : Google Scholar
|
|
248
|
Nowak WN, Deng J, Ruan XZ and Xu Q:
Reactive oxygen species generation and atherosclerosis.
Arterioscler Thromb Vasc Biol. 37:e41–e52. 2017.PubMed/NCBI View Article : Google Scholar
|
|
249
|
Rajakumar S, Bhanupriya N, Ravi C and
Nachiappan V: Endoplasmic reticulum stress and calcium imbalance
are involved in cadmium-induced lipid aberrancy in Saccharomyces
cerevisiae. Cell Stress Chaperones. 21:895–906. 2016.PubMed/NCBI View Article : Google Scholar
|
|
250
|
Vohra K, Vodonos A, Schwartz J, Marais EA,
Sulprizio MP and Mickley LJ: Global mortality from outdoor fine
particle pollution generated by fossil fuel combustion: Results
from GEOS-chem. Environ Res. 195(110754)2021.PubMed/NCBI View Article : Google Scholar
|
|
251
|
Maciejczyk P, Chen LC and Thurston G: The
role of fossil fuel combustion metals in PM air pollution health
associations. Atmosphere. 12(1086)2021.
|
|
252
|
Fuller R, Landrigan PJ, Balakrishnan K,
Bathan G, Bose-O'Reilly S, Brauer M, Caravanos J, Chiles T, Cohen
A, Corra L, et al: Pollution and health: A progress update. Lancet
Planet Health. 6:e535–e547. 2022.PubMed/NCBI View Article : Google Scholar
|
|
253
|
Landrigan PJ, Britt M, Fisher S, Holmes A,
Kumar M, Mu J, Rizzo I, Sather A, Yousuf A and Kumar P: Assessing
the human health benefits of climate mitigation, pollution
prevention, and biodiversity preservation. Ann Glob Health.
90(1)2024.PubMed/NCBI View Article : Google Scholar
|
|
254
|
Landrigan PJ, Fuller R, Acosta NJR, Adeyi
O, Arnold R, Basu NN, Baldé AB, Bertollini R, Bose-O'Reilly S,
Boufford JI, et al: The lancet commission on pollution and health.
Lancet. 391:462–512. 2018.PubMed/NCBI View Article : Google Scholar
|
|
255
|
Yang BY, Qu Y, Guo Y, Markevych I,
Heinrich J, Bloom MS, Bai Z, Knibbs LC, Li S, Chen G, et al:
Maternal exposure to ambient air pollution and congenital heart
defects in China. Environ Int. 153(106548)2021.PubMed/NCBI View Article : Google Scholar
|