Introduction
Sepsis, a severe medical condition typically
triggered by a bacterial infection, presents with symptoms such as
elevated fever, hypotension and tachycardia (1). Despite advances in medical
treatments, sepsis continues to pose a significant public health
challenge, with a morbidity rate of 535/100,000 individuals/year
and mortality rates ranging from 25-30% (2). Therefore, it is essential to
thoroughly investigate the molecular and cellular mechanisms
underlying sepsis (3). Such
studies are crucial for enhancing the current understanding of its
pathogenesis and developing more effective treatment
strategies.
Amino acid metabolism (AAM) serves a crucial role in
protein synthesis and various metabolic pathways, thus being
essential for understanding disease mechanisms and developing new
therapies. For instance, it has been shown that mutations in
glutamate metabolism-related genes can lead to metabolic disorders
that severely impact liver function in certain liver diseases
(4). Additionally, AAM is
associated with tumor growth and invasion in certain cancer types
(5). Similarly, AAM is involved in
the progression of sepsis. For example, circulating N-lactoyl amino
acids and N-formylmethionine can predict mortality in patients with
septic shock, and glutamine helps to maintain energy metabolism and
alleviate liver injury in burn-related sepsis (6,7).
Consequently, a number of studies have explored the role of
AAM-related genes (AAMGs) in various diseases. For instance, one
study analyzed five AAMGs and found them to be potential biomarkers
for predicting the prognosis and efficacy of immunotherapy in
colorectal cancer (8). Similar
research has been conducted in certain inflammatory diseases, such
as identifying glutamine metabolism-related genes as diagnostic
markers for diabetic foot ulcers (9). However, such studies are less common
for sepsis. It is noteworthy that traditional bioinformatics
methods often utilize approaches such as differential expression
analysis and protein-protein interaction networks to identify hub
genes (10,11). In contrast to these traditional
bioinformatics methods, machine learning algorithms, despite their
lower interpretability, offer improved performance, the ability to
handle large-scale data and the automatic extraction of complex
patterns, making them widely applicable in clinical research
(12). In machine learning, the
least absolute shrinkage and selection operator (LASSO) algorithm
is used for feature selection, employing L1 regularization to
reduce non-significant features and to enhance model
interpretability and predictive performance (13). Support vector machine-recursive
feature elimination (SVM-RFE) recursively selects important
features to optimize feature selection for classification tasks,
thereby improving classification accuracy (14). Random forests (RFs) are used for
feature importance evaluation and classification, providing feature
importance rankings and achieving efficient classification
(15). The use of these machine
learning algorithms, either individually or in combination, has
yielded promising results in studies on various diseases,
identifying biomarkers with high diagnostic and prognostic value,
such as in sepsis (16,17). However, to the best of our
knowledge, the application of these three machine learning methods
in combination to investigate the role of AAMGs in sepsis remains
to be investigated.
The present study, by using machine learning
algorithms, identified methionine synthase (MTR) and
methionine-R-isomerase 1 (MRI1) as hub AAMGs with
significant diagnostic and prognostic potential in sepsis. The
expression levels of these genes in patients with sepsis was
validated using clinical samples from Xianning Central Hospital
(Hubei, China). Additionally, the preliminary roles of MTR
were investigated through an in vitro sepsis model, which
demonstrated the crucial function of MTR in the disease's
pathophysiology. The present study highlighted the importance of
AAMGs in understanding the pathophysiology of sepsis and could
potentially be used in the future for the development of novel
targeted therapeutic interventions.
Materials and methods
Data acquisition and clinical
samples
The present study utilized three datasets from the
Gene Expression Omnibus (GEO) database (www.ncbi.nlm.nih.gov/geo) containing mRNA
transcriptome sequencing of peripheral blood from patients with
sepsis. The datasets used were GSE65682, which included samples
from 760 sepsis patients and 42 healthy controls; GSE154918,
comprising 40 healthy and 20 sepsis samples; and GSE185263, which
consisted of 44 healthy and 26 sepsis samples. For the GSE65682
dataset, patients lacking survival data were excluded, which
resulted in 479 sepsis samples with complete survival information
for survival analysis. The preprocessing of the three datasets
included averaging the expression values of the same gene and
removing values ≤0. Principal component analysis (PCA) was
conducted to confirm the differences between the sepsis and healthy
groups and to demonstrate the heterogeneity among the three
datasets. PCA was performed using the plot PCA function in R
(http://www.bioconductor.org/) and
visualized using the ggplot2 package (version 3.42) (https://cran.r-project.org/web/packages/ggplot2/index.html).
Furthermore, 448 AAMGs were identified based on previous literature
(8).
In the present study, whole blood samples were
collected from five patients with sepsis who were admitted to the
Department of Emergency and Intensive Care Unit of Xianning Central
Hospital (Hubei, China) from March to April 2024 were selected
(Table I). Inclusion criteria were
as follows: i) Age, ≥18 years; ii) a definitive infection source or
positive bacterial culture; and iii) compliance with sepsis
criteria (18). Exclusion criteria
were as follows: i) Hospitalization for <24 h; and ii) presence
of malignant tumors, coronary heart disease, acute coronary
syndrome, myocardial infarction, cardiogenic shock, major surgery
or significant trauma. The healthy control group consisted of five
individuals undergoing routine health examinations at the same
hospital during aforementioned time period. The entire workflow was
illustrated in Fig. 1.
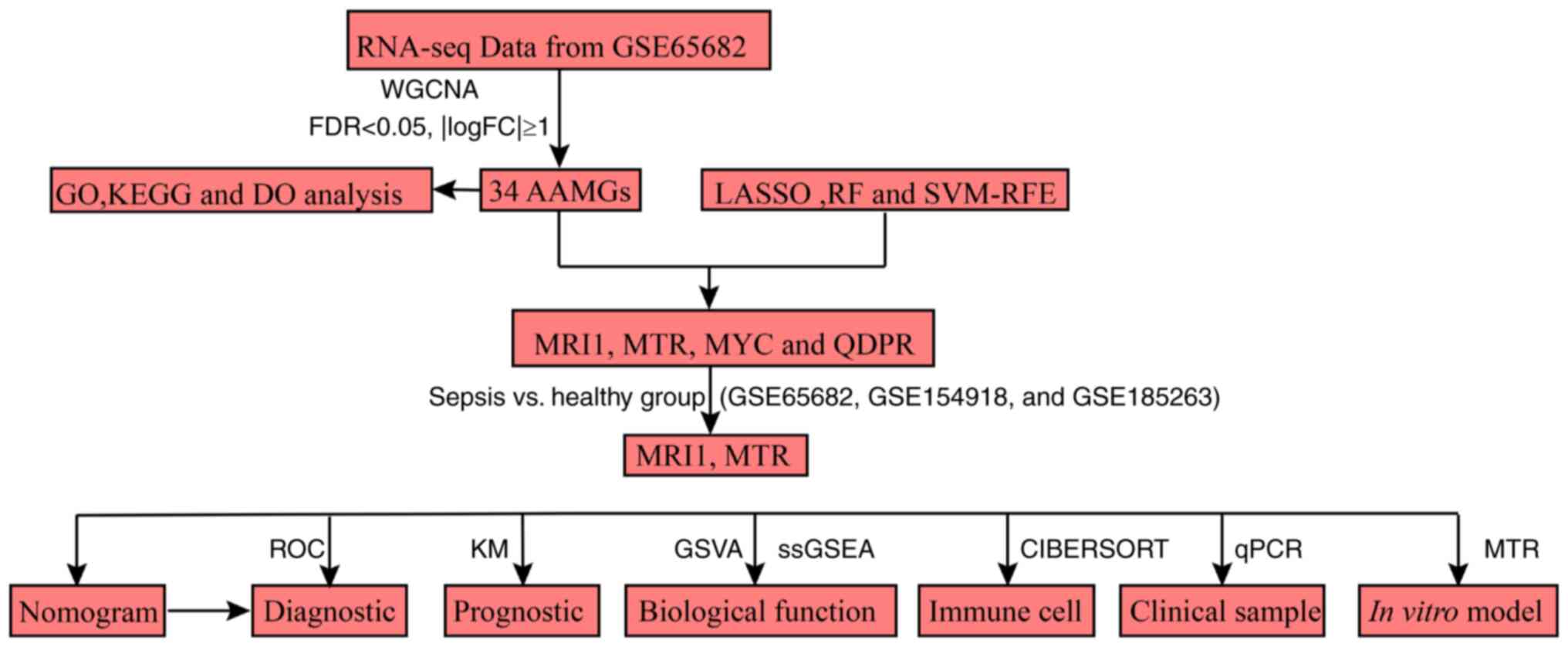 | Figure 1Flow chart of the present study.
RNA-seq, RNA sequencing; WGCNA, weighted gene co-expression network
analysis; FDR, false discovery rate; FC, fold-change; GO, Gene
Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; DO,
Disease Ontology; AAMG, amino acid metabolism-related genes; LASSO,
least absolute shrinkage and selection operator; RF, random forest;
SVM-RFE, support vector machine-recursive feature elimination;
MRI1, methionine-R-isomerase 1; MTR, methionine synthase; MYC, MYC
proto-oncogene; QDPR, quinoid dihydropteridine reductase; ROC,
receiver operating characteristic; KM, Kaplan-Meier; GSVA, gene set
variation analysis; ssGSEA, single-sample gene set enrichment
analysis; RT-qPCR, reverse transcription-quantitative PCR. |
 | Table IGeneral clinical information of
sepsis and healthy control groups. |
Table I
General clinical information of
sepsis and healthy control groups.
| Patient
characteristic | Patients with
sepsis | Healthy
controls | P-value |
|---|
| Age, years (mean ±
SD) | 61.2±5.4 | 62.8±5.9 | 0.67 |
| Male, n | 4 | 2 | 0.52 |
| Female, n | 1 | 3 | 0.52 |
Weighted gene co-expression network
analysis (WGCNA) and differentially expressed genes analysis
The GSE65682 dataset was employed to identify
critical AAMGs in sepsis. Initially, a co-expression network was
constructed using the ‘WGCNA’ package (version 1.69) (19) to perform WGCNA. A total of 11,205
genes were categorized into 12 modules, with the module exhibiting
the highest correlation with sepsis being selected for further
analysis. Subsequently, the ‘limma’ package (version 3.52.1)
(20) was used to obtain the
differentially expressed AAMGs between sepsis and healthy samples,
applying the criteria of log2 |fold change| (logFC) >1 and
adjusted P<0.05. Finally, the AAMGs were acquired from the
intersections of the module most highly correlated with sepsis and
the genes that were significantly differentially expressed between
sepsis and normal samples, and they were selected through machine
learning techniques, specifically using the LASSO, SVM-RFE and RF
algorithm, for detailed investigation.
Biological analysis
Gene Ontology (GO), Kyoto Encyclopedia of Genes and
Genomes (KEGG) and Disease Ontology (DO) analyses were performed to
elucidate the biological functions of the key AAMG. These analyses
were conducted using the ‘clusterProfiler’ (version 4.4.1)
(21) and ‘DOSE’ (version 3.22.0)
(22) packages. Additionally,
single-sample Gene Set Enrichment Analysis (ssGSEA) analysis was
performed on the GSE65682 dataset to identify different pathway
activations between sepsis and healthy samples using the ‘GSVA’
package (version 4.4.1) (23).
Machine learning algorithm to identify
hub AAMGs
To identify AAMGs most closely associated with
sepsis, a robust feature selection procedure was employed: First,
L1 regularization via LASSO analysis was performed using the
‘glmnet’ package (version 4.1-4) (24). The optimal penalty parameter (λ)
was determined based on 10-fold cross-validation with the lowest
deviance probability. Genes with non-zero coefficients were
retained to reduce data dimensionality, ultimately obtaining a
model with fewer variables (25).
The present study calculated LASSO-Cox coefficients using a Lasso
regression model to select key AAMGs in sepsis. Next, the SVM-RFE
algorithm was used, which is a backward elimination method designed
to optimize classifier performance by selecting the best subset of
features. SVM-RFE analysis was conducted using the ‘e1071’ package
(version 1.7-4) (26) to determine
the most relevant AAMGs associated with sepsis. Subsequently, the
RF algorithm was used for feature importance evaluation and
classification. The ‘randomForest’ package (version 4.7-1.1)
(27) was used to identify AAMGs
with a relative importance >0.5, and these AAMGs were
subsequently ranked by importance from highest to lowest for
further analysis. The RF algorithm was suitable for both
classification and regression tasks, which was ideal for the
present analysis (28). By
integrating results from LASSO, SVM-RFE and RF analyses, key AAMGs
in sepsis were identified. These candidate AAMGs were validated in
the GSE65682, GSE154918 and GSE185263 cohorts, assessing their
expression levels between sepsis and healthy samples. Genes with
inconsistent expression levels across the GSE65682, GSE154918 and
GSE185263 datasets were excluded. Ultimately, through the methods
described as aforementioned, two critical AAMGs were identified:
MTR and MRI1.
Assessment of the diagnostic and
prognostic value of AAMGs in sepsis
The diagnostic value of MTR and MRI1
in sepsis was assessed in the GSE65682, GSE154918 and GSE185263
cohorts. Furthermore, based on the expression levels of MTR
and MRI1, a nomogram was also built using the ‘rms’ package
(version 6.3-0) (https://CRAN.R-project.org/package=rms) for these
cohorts. The effectiveness of the nomogram was evaluated using the
area under the curve (AUC) derived from the receiver operating
characteristic curve. The prognostic value of MTR and
MRI1 for sepsis was demonstrated in the GSE65682 dataset,
where the samples were divided into two datasets using random
sampling (3:7) for bootstrap-based internal validation, a method
used to assess the reliability and stability of the results within
the same dataset. Moreover, the combined prognostic value of
MTR and MRI1 was evaluated using the same validation
approaches. Kaplan-Meier survival curves were generated to
illustrate prognosis using the ‘survival’ (version 3.5-5)
(https://CRAN.R-project.org/package=survival) and
‘survminer’ packages (version 0.4-9) (https://CRAN.R-project.org/package=survminer).
Cell culture and in vitro sepsis model
establishment
Macrophages act as the first line of host immune
defense during sepsis, responding rapidly to injury (29). The balance between pro-inflammatory
and anti-inflammatory cytokines that macrophages produce influences
the inflammatory response and patient outcomes in sepsis (30). Therefore, macrophages are widely
used in in vitro studies of sepsis. Commonly used cell lines
include RAW 264.7 and THP-1. RAW 264.7 cells can be used directly
without differentiation, providing simplicity and stability for
experimental use, and are widely adopted in many studies (31-33).
Although THP-1 cells are more relevant in simulating the human
response to sepsis, they require differentiation into macrophages
using inducers such as PMA, which adds complexity and time to the
experiments (34). Hence, the
present study selected RAW 264.7 cells for in vitro sepsis
model studies. IRAW 264.7 cells, a murine macrophage cell line
derived from the ascites of male BALB/c mice with an Abelson murine
leukemia virus-induced tumor, were obtained from the National
Collection of Authenticated Cell Cultures (https://www.cellbank.org.cn/index.php). Cells were
cultured in sterile conditions using DMEM supplemented with 10% FBS
and 1% penicillin/streptomycin. Cells were then seeded into either
96- or 6-well plates and incubated at 37˚C until they reached
80-90% confluency. Following previously reported methodologies
(35), an in vitro sepsis
model was established by treating RAW 264.7 cells with 500 ng/ml of
lipopolysaccharide (LPS) for 4 h to simulate bacterial infection,
followed by 30 min treatment with 5 mM ATP, which mimicked the
cellular damage and inflammation response observed in sepsis. DMEM,
FBS, 1% penicillin/streptomycin, LPS and ATP were procured from
MedChemExpress.
Cell transfection
Cell transfection was performed using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) at 37˚C for 48 h. A total of 5.0 µg of the
pcDNA3.1 MTR overexpression (OE) vector was used, with the
sequence containing both the coding region of the MTR gene (in
uppercase) and necessary flanking sequences (in lowercase) as
follows: forward (F)
5'-TACCGAGCTCGGATCCGCCACCATGAAGAAAACCCTGCAGGATG-3' and reverse (R)
5'-GATATCTGCAGAATTCTCAGTCTGTGTCATAGCCCAG-3'. The negative control
(NC), referred to as OE-NC, consisted of an empty pcDNA3.1 vector
without any insert and was used as a baseline control to ensure the
observed effects were due to MTR overexpression (Data S1). All vectors were purchased from
Sangon Biotech Co., Ltd. After 48 h of transfection at 37˚C, cells
were collected by centrifugation at 300 x g for 5 min at 4˚C, and
the transfection efficiency was assessed using reverse
transcription-quantitative PCR (RT-qPCR) and western blotting
analysis.
RT-qPCR
For whole blood samples from sepsis patients and
healthy controls, as well as cell lines treated with LPS and APT,
including those in the control, NC-OE and OE-MTR groups, total RNA
was extracted using TRIzol® reagent (Invitrogen; Thermo
Fisher Scientific, Inc.) according to the manufacturer's
instructions. The quality and concentration of RNA were assessed
before proceeding. RNA was reverse transcribed into cDNA using the
RevertAid RT Reverse Transcription Kit (Thermo Fisher Scientific,
Inc.). RT-qPCR was performed using the SYBR GREEN kit (Takara Bio,
Inc.). The 15 µl qPCR reaction mixture contained 1 µg RNA, 10 µl
reaction solution, 1 µl RT primers (Tables II and III) and 1 µl deoxyribonucleotide
triphosphates. The following thermocycling conditions were used for
PCR: 95˚C for 3 min; 40 cycles of 95˚C for 30 sec, 55˚C for 30 sec
and 72˚C for 30 sec. Primers were synthesized by Sangon Biotech
Co., Ltd., and GAPDH was used as the internal reference
gene. The relative expression levels of the target genes were
calculated using the 2-ΔΔCq method (36).
| Table IIPrimer sequences for detection of
human genes. |
Table II
Primer sequences for detection of
human genes.
| Gene | Sequence
(5'-3') |
|---|
| MTR | F:
GAACGCCTTGAGTATGCCCTTG |
| | R:
CCGGGCTGACTTTATAACCTGAG |
| MRI1 | F:
GCCCCGCTCCCAAGTGCGCGCGGAC |
| | R:
GTCCGCGCGCACTTGGGAGCGGGGC |
| GAPDH | F:
GCACCGTCAAGGCTGAGAAC |
| | R:
TGGTGAAGACGCCAGTGGA |
| Table IIIPrimer sequences for detection of
mouse genes. |
Table III
Primer sequences for detection of
mouse genes.
| Gene | Sequence
(5'-3') |
|---|
| MTR | F:
TTCCTTTAGTCTGTCGCTGCGGCCT |
| | R:
AGGCCGCAGCGACAGACTAAAGGAA |
| GAPDH | F:
CTCTGAGCCTCCTCCAATTCAACCC |
| | R:
GGGTTGAATTGGAGGAGGCTCAGAG |
Cell Counting Kit-8 (CCK-8) assay
RAW 264.7 cells were seeded into 96-well plates at a
concentration of 8,000-10,000 cells per well, and cultured for 6,
12, 24 and 48 h at 37˚C with 5% CO2. Following each
incubation period, CCK-8 solution (Beyotime Institute of
Biotechnology) was added to each well and the plates were further
incubated for 2 h. Absorbance was subsequently measured at 450 nm
using a microplate reader to assess cell viability.
Western blotting
Total proteins were extracted from cells using RIPA
lysis buffer (Beyotime Institute of Biotechnology) and the total
protein concentration was measured using a BCA assay kit (Beyotime
Institute of Biotechnology). A total of 40 µg of protein was loaded
per lane on a 10% gel, separated by SDS-PAGE and transferred onto
PVDF membranes. Membranes were blocked with 5% fat-free milk for 1
h at room temperature and then incubated with primary antibodies
against MTR (1:1,000; cat. no. ab66039; Abcam) and GAPDH
(1:1,000; cat. no. GB12002-100; Wuhan Servicebio Technology Co.,
Ltd.) overnight at 4˚C. Next, membranes were treated with
HRP-conjugated secondary antibodies (1:20,000; cat. no. SA00001-2;
Wuhan Sanying Biotechnology Co., Ltd.) for 2 h at room temperature.
Protein bands were visualized using an ECL detection kit (Wuhan
Sanying Biotechnology Co., Ltd.) and protein expression levels were
quantified using ImageJ software (version 1.8.0; National
Institutes of Health) using GAPDH as the loading control.
Colony formation and Transwell
assays
In the logarithmic growth phase, cells were detached
using 0.25% trypsin, and adjusted to a concentration of
2.5x102 cells/ml. Next, cells were seeded into a 6-well
plate and incubated 37˚C with 5% CO2 for 2-3 weeks. The
culture medium was changed every 3 days. After the incubation
period, cells were fixed using methanol at room temperature (~25˚C)
for 15 min. Following fixation, cells were stained with 1 ml of
Giemsa solution at room temperature for 30 min. After staining, the
cells were gently rinsed with water, and excess water was removed
using filter paper. Colonies were defined as groups of at least 50
cells that were clearly distinguishable from one another. For the
migration assay, a Transwell assay was conducted using 8 µm pore
size Transwell inserts (Beijing Solarbio Science & Technology
Co., Ltd.) without Matrigel. Briefly, 4x104 cells in 200
µl FBS-free medium were added to the upper chamber, while the lower
chamber was filled with 700 µl 10% FBS medium. The cells were
incubated at 37˚C with 5% CO2 for 24 h. Following
incubation, cells on the lower side of the membrane were stained
with Wright-Giemsa dye (Shanghai Canspec Scientific Instruments
Co., Ltd.) at room temperature for 15 min. After staining, the
cells were observed using a Nikon light microscope (Nikon Corp).
Both colony formation and migration assays were quantified using
ImageJ software (version 1.45s/Java1.6.0_20; National Institutes of
Health).
Statistical analysis
All cell experiments were repeated three times to
ensure reliability. The final results were presented as mean ±
standard deviation. Statistical analyses and graphical
presentations were performed using R (version 4.2.1) and GraphPad
Prism (version 9.0; Dotmatics). Comparison analysis between groups
for clinical RT-qPCR data were performed using an unpaired
Student's t-test. For data involving multiple group comparisons, a
one-way ANOVA followed by Tukey's post hoc test was used. The
Wilcoxon rank-sum test was applied for bioinformatics data,
including the comparison of MRI1 and MTR levels between the healthy
group and individuals with sepsis across three datasets, as well as
the comparison of different pathways between the sepsis and healthy
groups. Differences in survival rates were assessed using
Kaplan-Meier method and analyzed with the log-rank test.
Correlations between gene expressions and pathways were analyzed
using Pearson's correlation analysis. P<0.05 was considered to
indicate a statistically significant difference.
Results
Biological analysis of the
differentially expressed AAMGs
To illustrate sample differences and dataset
heterogeneity, PCA was performed on each dataset (Fig. S1A-C). The clear separation
indicated significant differences between patients with sepsis and
healthy controls. Additionally, the datasets demonstrated distinct
separations, which indicated that there was significant
heterogeneity among the datasets (Fig. S1D). WGCNA was used to screen the
differentially expressed genes between patients with sepsis and
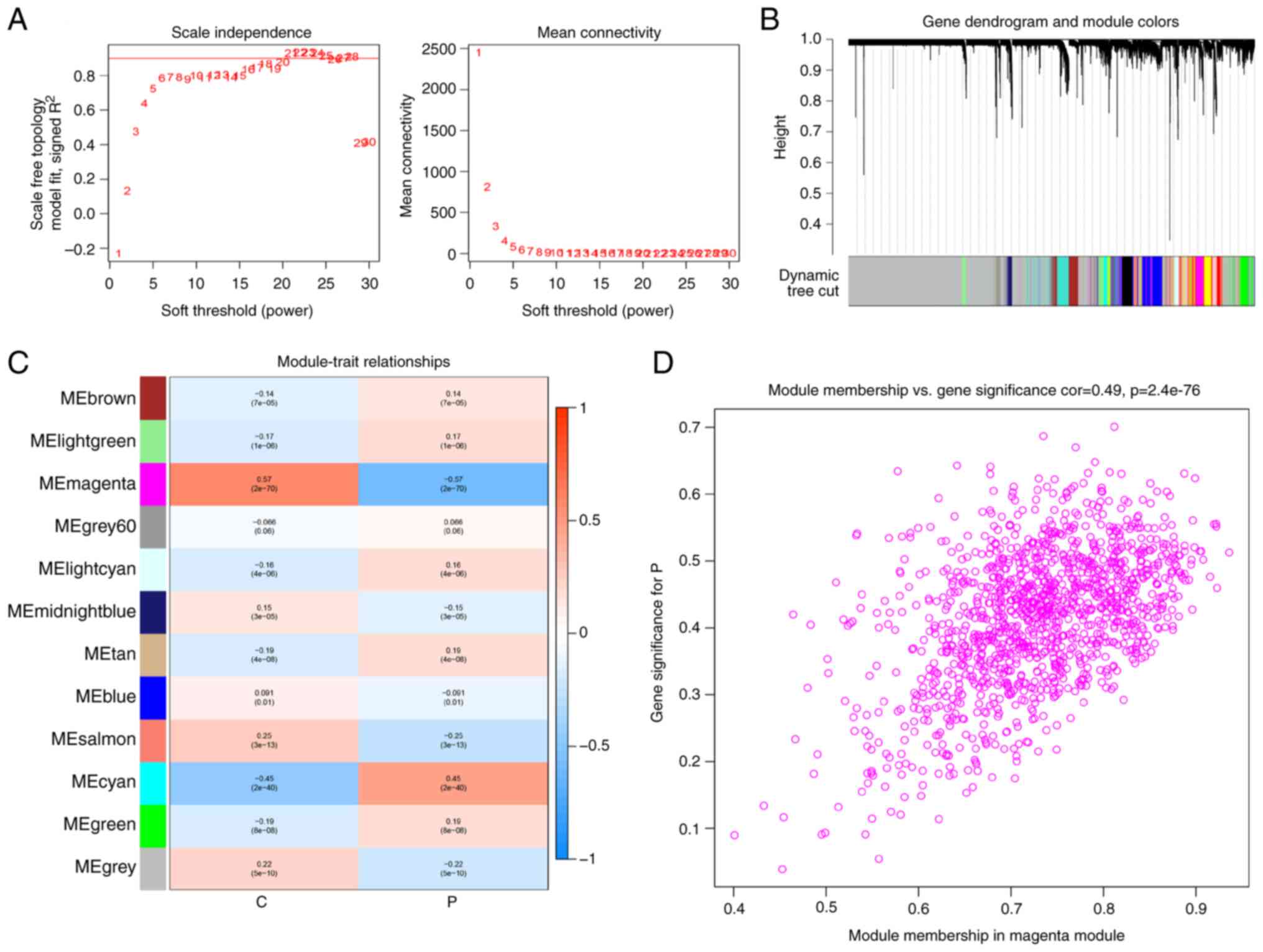
healthy subjects. A soft threshold of β=21 was selected to
construct a scale-free network (Fig.
2A). Through this analysis, 11 gene modules were acquired
(Fig. 2B), with the magenta module
showing the highest association with sepsis (r=-5.77; P<0.05;
Fig. 2C). The genes within this
module exhibited a significant correlation with the gene expression
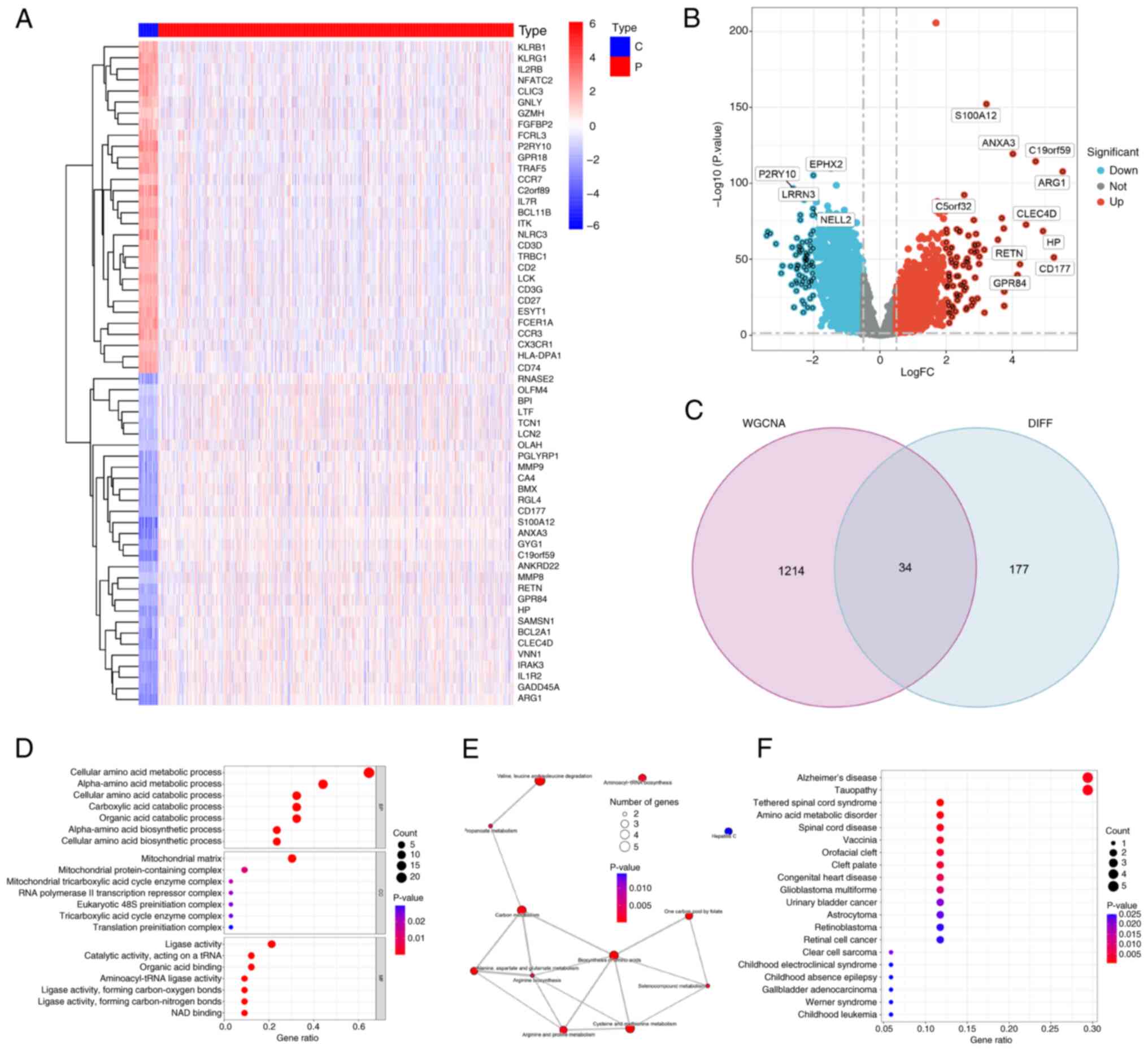
profiles of patients with sepsis (r=0.49; P<0.05; Fig. 2D). In total, 211 AAMGs that were
differentially expressed between sepsis and healthy subjects were
identified and visualized using a heatmap (Fig. 3A) and volcano plot (Fig. 3B). From these, 34 AAMGs were
obtained at the intersections of genes in the magenta module and
differentially expressed AAMGs (Fig.
3C). GO analysis demonstrated enrichment of these genes in AAM
(Fig. 3D). Additionally, KEGG
analysis presented pathways, including aminoacyl-transfer RNA
(tRNA) biosynthesis and cysteine and methionine metabolism,
associated with these genes (Fig.
3E). DO analysis further confirmed that these 34 AAMGs were
enriched in amino acid metabolic disorder (Fig. 3F). These results collectively
demonstrated the close association between 34 AAMGs and AAM in
sepsis.
Identification of hub AAMGs
Machine learning algorithms are increasingly
prevalent for identifying robust diagnosis biomarkers in
bioinformatics analysis (37). In
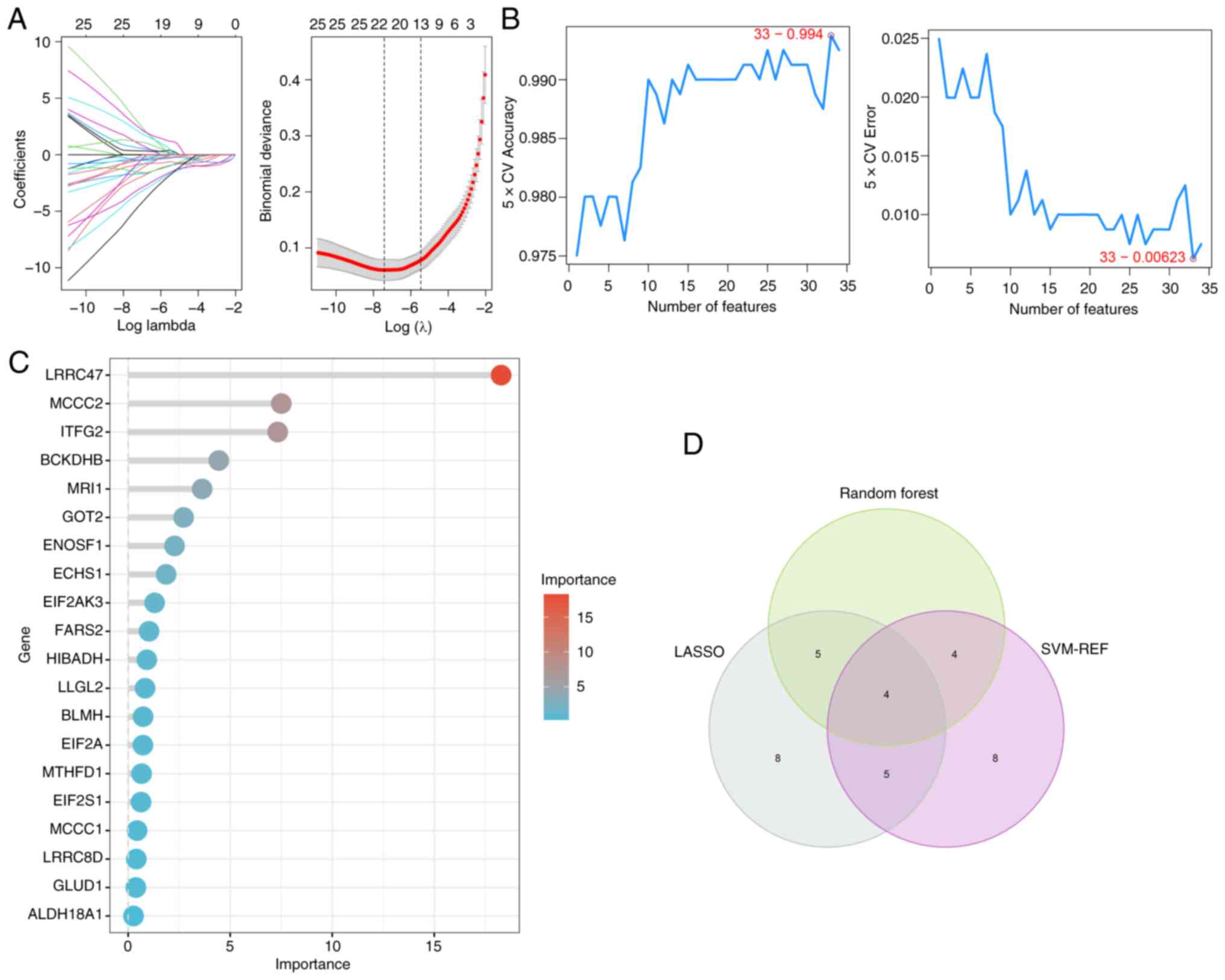
the present study, these methods were performed to screen for hub
AAMGs in sepsis. Using LASSO, SVM-RFE and RF algorithms, 22, 21 and
13 candidate AAMGs were identified, respectively (Fig. 4A-C). Through a Venn diagram
analysis, an overlap among results of these machine learning
algorithms showed four potential AAMGs as diagnostic candidates,
namely MRI1, MTR, MYC and quinoid
dihydropteridine reductase (QDPR) (Fig. 4D). To verify the robustness of
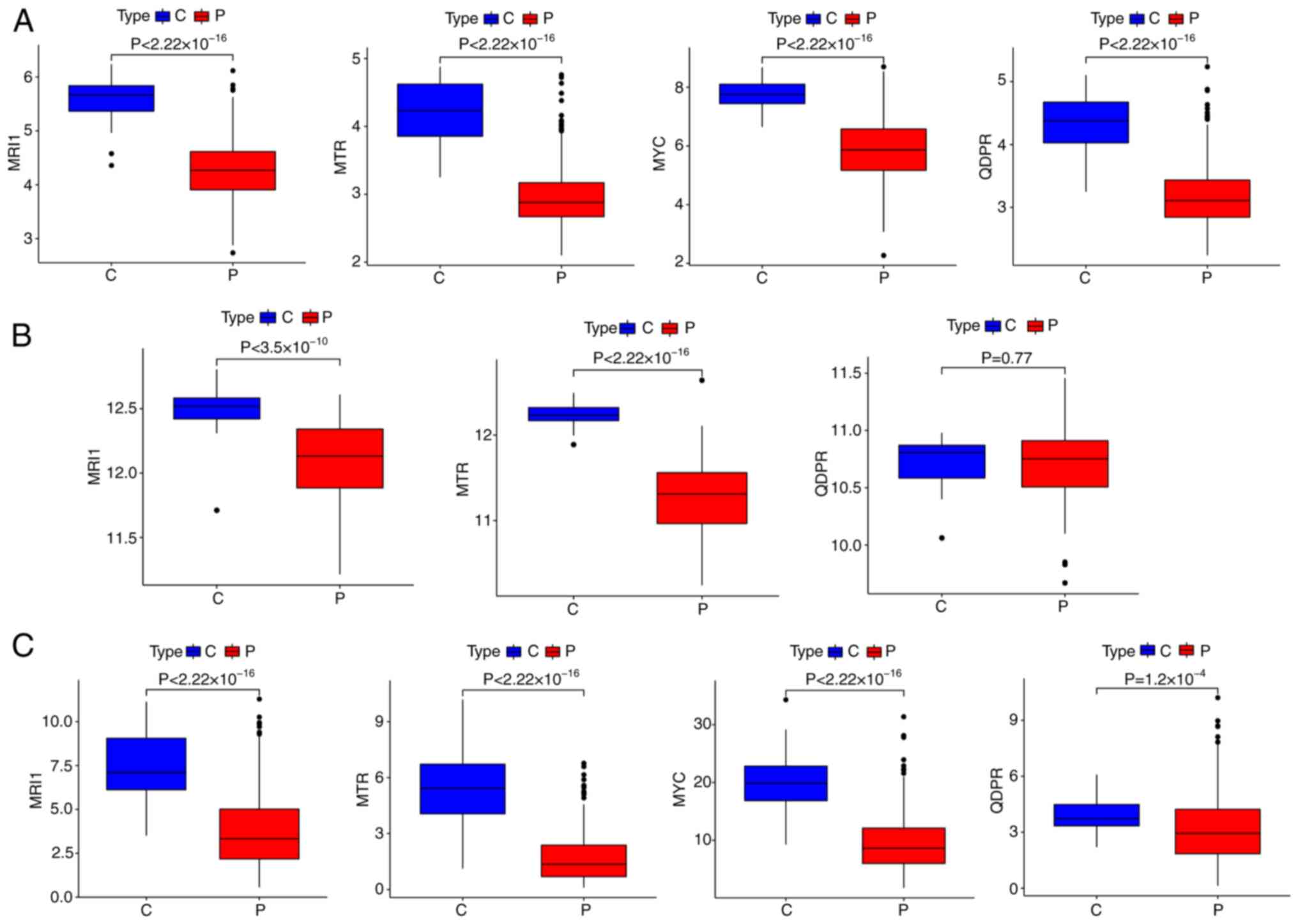
theses AAMGs in sepsis, their expressions levels across three
cohorts of healthy subjects and patients with sepsis were assessed.
While the expression levels of MRI1 and MTR remained
consistent across all three cohorts, MYC and QDPR
showed decreased expression in sepsis compared with healthy
controls in the GSE65682 and GSE185263 cohorts. However, MYC
was undetected and QDPR expression levels demonstrated no
significant differences between healthy subjects and patients with
sepsis in the GSE154918 cohort (Fig.
5). Although elevated methylation levels in MRI1 have
been observed in patients with severe asthma (38) and genetic polymorphisms in
MTR have been strongly linked to various diseases (39,40),
their roles in sepsis remain understudied. Thus, MRI1 and
MTR were selected as the hub AAMGs for further analysis.
Diagnostic and prognostic value of hub
AAMGs in sepsis
Similar to a previous study (41) on the diagnostic value of certain
biomarkers for sepsis, the present study also demonstrated the
potential diagnostic value of hub AAMGS (MRI1 and
MTR) for sepsis. A nomogram was constructed based on the
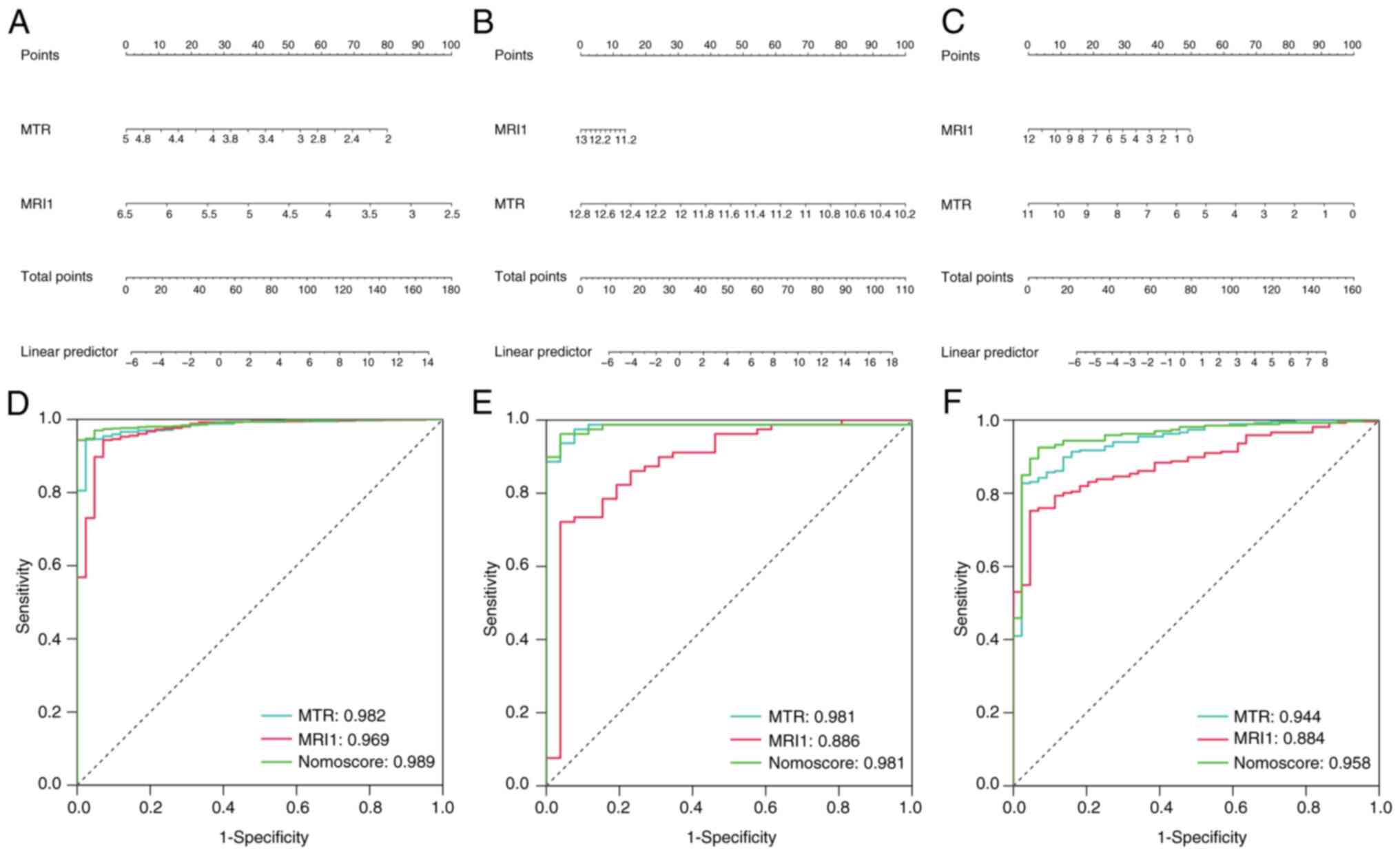
expression levels of MRI1 and MTR (Fig. 6A-C) and the AUC values of each gene
and the nomogram were evaluated across the three cohorts. MRI1,
MTR and the nomogram all exhibited consistently high diagnostic
values for sepsis in the GSE65682 (AUC=0.969, 0.982 and 0.989,
respectively), GSE154918 (AUC=0.886, 0.981 and 0.981, respectively)
and GSE185263 datasets (AUC=0.884, 0.944 and 0.958, respectively)
(Fig. 6D-F). Given the diagnostic
value of the two AAMGs, their prognostic predictive value in sepsis
was further explored. Patients with sepsis were divided into a
high-expression group and a low-expression group according to the
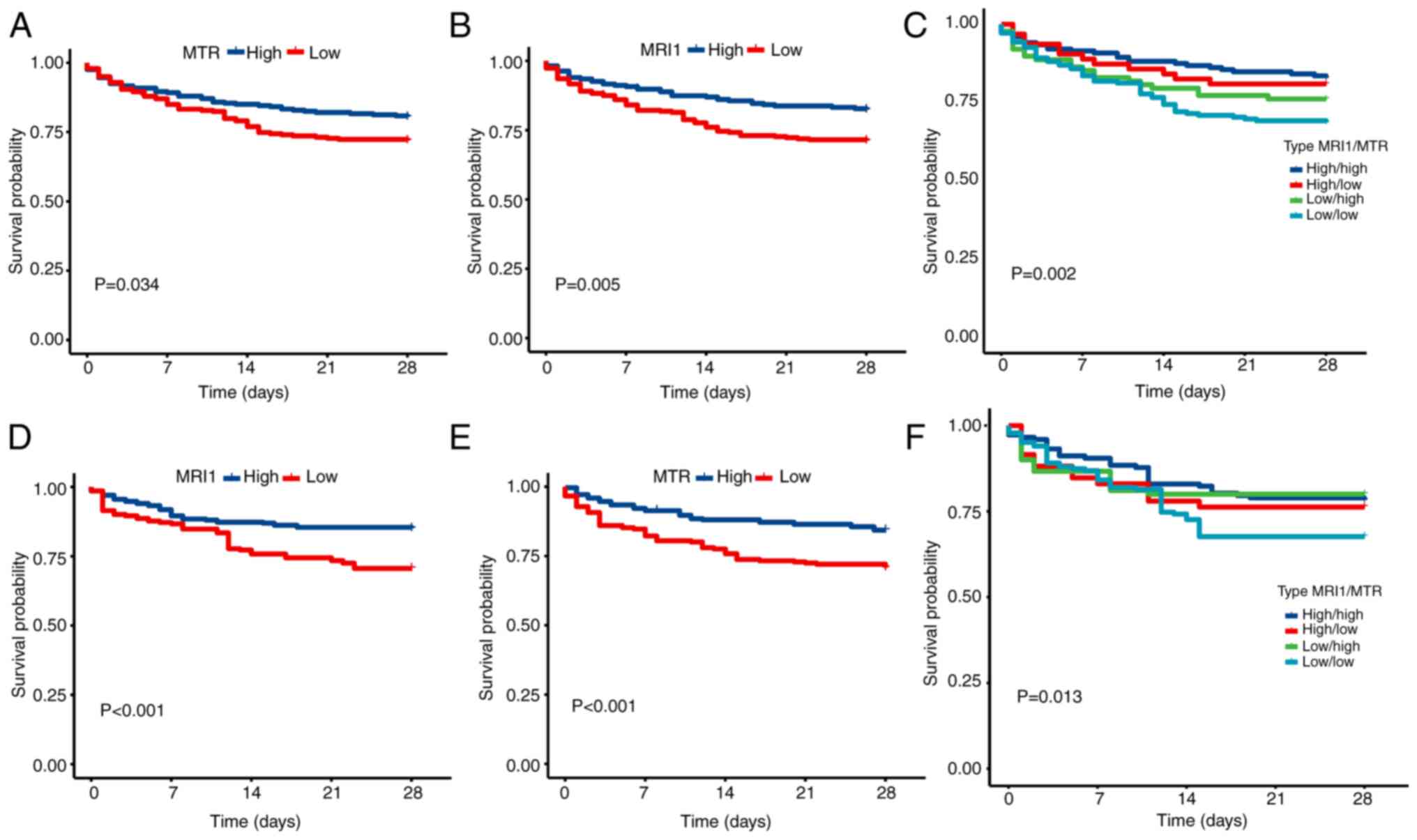
best cutoff point. The high-expression MTR group had a
better prognosis than the low-expression MTR group (Fig. 7A), and this result was confirmed in
the internal validation assay (Fig.
7D). Similar results were observed for MRI1 (Fig. 7B and E). Furthermore, among the four groups,
those with high expression levels of both MTR and
MRI1 exhibited the most favorable prognosis, whereas the
opposite was observed for those with low expression levels of both
MTR and MRI1 (Fig.
7C and F). To further validate
the diagnostic and prognostic models constructed in the present
study, external patient cohorts were used, specifically GSE95233
(22 healthy samples and 102 sepsis samples) and GSE4607 (15 healthy
samples and 108 sepsis samples). The diagnostic model results
demonstrated that MRI1, MTR and the nomogram
maintained high diagnostic value for sepsis. In GSE95233, the AUC
values for MRI1, MTR and the nomogram were 0.963,
0.972 and 0.987, respectively, and in GSE4607, the AUC values were
0.845, 0.864 and 0.879, respectively (data not shown). However, the
prognostic model did not perform well, demonstrating no significant
difference in survival time between the high and low expression
groups for MTR and MRI1 in both datasets (data not
shown). Overall, MRI1 and MTR demonstrated robust
diagnostic and prognosis values for patients with sepsis, and they
could potentially be used as predictive prognostic factors for
patients with sepsis.
Biological functions, potential
pathways and immune cell infiltration analysis of MRI1 and MTR in
sepsis
Given the demonstrated diagnostic and prognosis
values of MRI1 and MTR in sepsis, ssGSEA was utilized
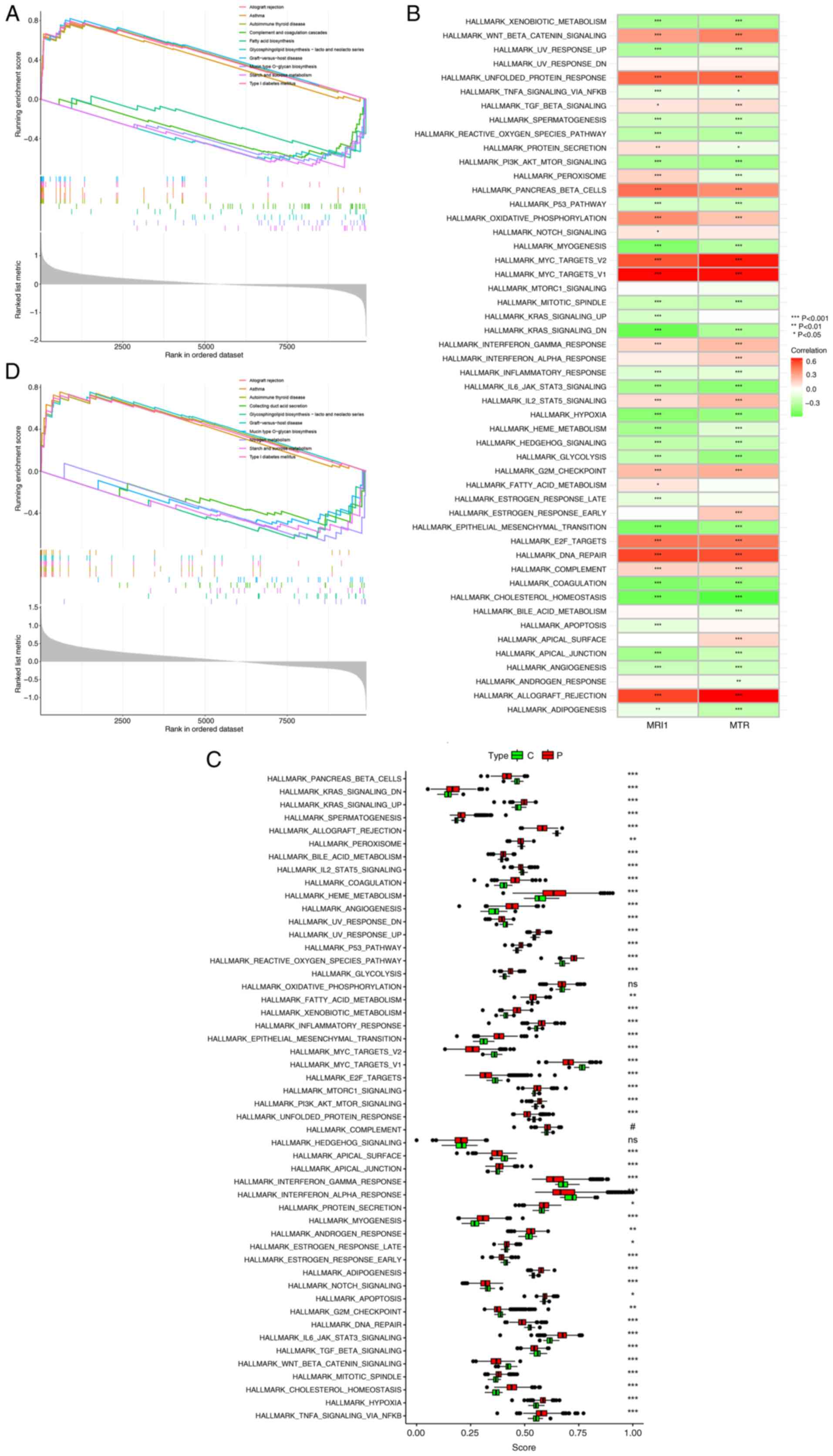
to clarify their potential roles. Activation of immune-related
diseases, including allograft rejection, asthma and autoimmune
thyroid disease, were observed with both MRI1 and MTR
(Fig. 8A and D). Conversely, metabolic pathways such as
fatty acid biosynthesis and glycosphingolipid biosynthesis were
inhibited with MRI1 and MTR expression. Additionally,
significant associations were found between MRI1 and
MTR expression and various inflammation-related pathways,
such as PI3K/AKT/mTOR signaling,
IL-6/JAK/STAT3 signaling and hypoxia, which
highlights their potential role in sepsis (Fig. 8B). Given the high mortality rate of
sepsis, identifying effective treatment methods is imperative.
Thus, potential pathways associated with sepsis were explored to
identify novel targets. Apart from oxidative phosphorylation,
complement and hedgehog signaling, all other pathways showed
significant differences between healthy subjects and patients with
sepsis (Fig. 8C). Some known
pathways, such as PI3K/AKT/mTOR (42), p53 (43), Notch (44) and hypoxia (45), have been previously reported to
have a strong association with sepsis. Additionally, novel pathways
such as KRAS and IL2/STAT5 were first reported in the
current study. This expansion of our understanding of
sepsis-related pathways underscores the complexity of its
pathophysiology and highlights the need to explore further how
these pathways influence immune responses. To this end, considering
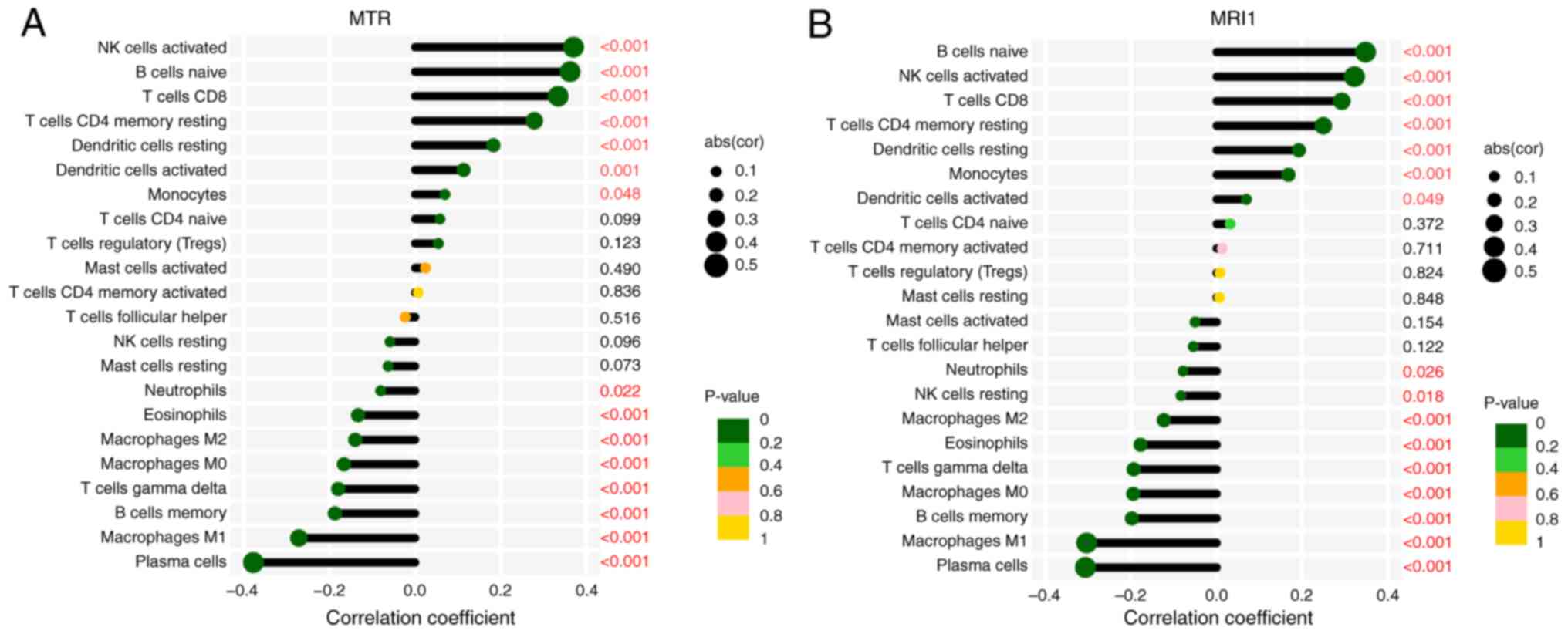
the crucial role of immune cells in sepsis, the CIBERSORT algorithm
was further applied to analyze the associations between
MRI1, MTR and immune cell infiltration. The present
findings demonstrated that these genes were negatively correlated
with the infiltration of pro-inflammatory cells, such as M1
macrophages, neutrophils and eosinophils, while exhibiting positive
correlations with anti-inflammatory cells, such as activated
natural killer, CD8+ T and dendritic cells (Fig. 9A and B). Collectively, the current study
identified MRI1 and MTR as potential promising
therapeutic targets for sepsis treatment, providing a foundation
for further exploration into their roles in modulating immune
responses in this complex condition.
Role of AAMGs in sepsis
Further validation was conducted using samples from
five patients with sepsis and five healthy individuals from
Xianning Central Hospital (Hubei, China) to verify the expression
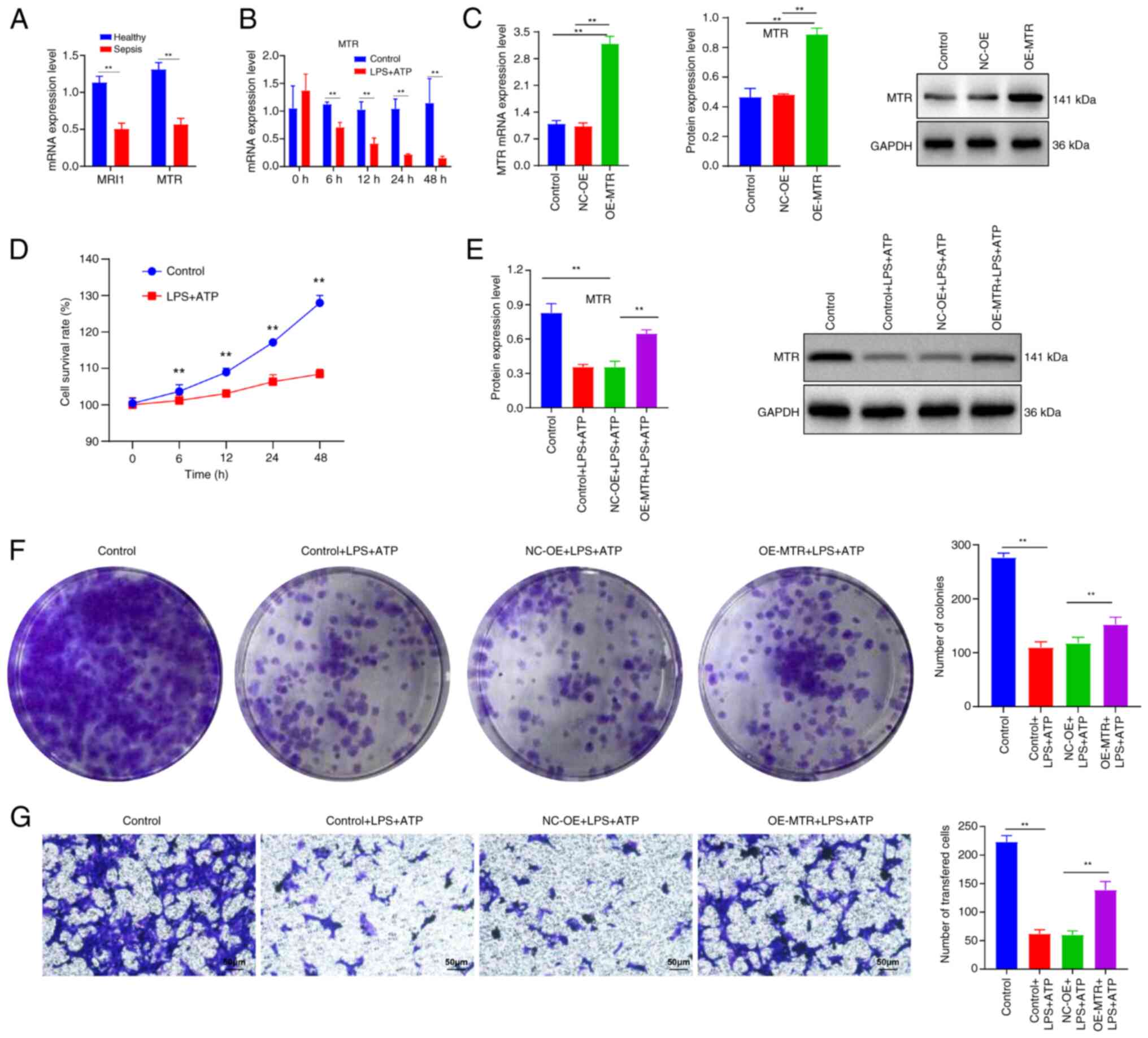
trends of MRI1 and MTR. The results indicated that
both genes were downregulated in the peripheral blood of patients
with sepsis compared with healthy individuals (Fig. 10A). As MTR exhibited
consistently high diagnostic values for sepsis in the
aforementioned three datasets (all AUC>0.94), the present study
focused on the mechanism of MTR in sepsis. In an in
vitro sepsis model induced by LPS and ATP, the mRNA expression
levels of MTR and cell viability gradually decreased over
time at 6, 12, 24 and 48 h compared with the control group (all
P<0.05) (Fig. 10B and D). Subsequently, RAW 264.7 cells were
transfected with MTR-OE plasmid. The RT-qPCR and western blot
results demonstrated that the MTR mRNA and protein expression
levels in the transfected cell line were higher than those compared
with the control and NC-OE groups (Fig. 10C), which confirmed successful
construction of the MTR-OE plasmid. Further experiments using LPS
and ATP to simulate sepsis in vitro showed that, compared
with the control group, MTR protein expression decreased after 4 h
of LPS stimulation followed by 30 min of ATP treatment (Fig. 10E). By contrast, the cell line
transfected with the MTR-OE plasmid exhibited increased MTR protein
levels compared with the NC-OE group. Additionally, LPS and ATP
gradually inhibited cell clonogenic and proliferative abilities,
whereas MTR-OE treatment reversed these trends (all P<0.05)
(Fig. 10F and G). In summary, these findings suggested
that MTR may serve a protective role in the LPS and ATP-induced
sepsis model.
Discussion
Sepsis is a critical medical condition characterized
by high incidence and mortality rates (2). The need for biomarkers with strong
diagnostic and prognostic predictive value has emerged as a central
focus in contemporary sepsis research. Previously, widely-used
machine learning algorithms, such as WGCNA, LASSO, SVM-RFE and RF
analysis, have facilitated the identification of key genes for
sepsis biomarkers (46,47). For instance, Dai et al
(48) employed WGCNA to identify
LPIN1 as a potential biomarker for sepsis, while another
study combined LASSO, SVM-RFE and RF algorithms to screen for four
sensitive diagnostic genes for sepsis (17). In the present study, WGCNA, LASSO,
SVM-RFE and RF analyses were integrated. WGCNA focuses on specific
phenotypes and co-expression modules, where genes within the same
module are functionally related and biologically significant
(49). LASSO enhances model
performance by effectively selecting features and addressing
multicollinearity issues (50).
SVM-RFE optimizes feature selection by recursively eliminating
unimportant features (51), while
RF evaluates the importance of variables in classification
(52). Traditional bioinformatics
analyses rely on manually designed rules and processes, which can
introduce subjectivity and limitations, potentially hindering the
discovery of patterns and information within the data and making it
difficult to handle complex data structures and relationships
(53). The present study combined
WGCNA, LASSO, SVM-RFE and RF algorithms to enable the
identification of more efficient biomarkers for sepsis, which
resulted in the identification of two AAMGs, MTR and
MIR1. These genes exhibited downregulated expression in
patients with sepsis across various public datasets and clinical
samples and were demonstrated to be valuable for both diagnostic
and prognostic purposes. Additionally, the current study
demonstrated that MTR and MIR1 were closely
associated with certain pathways, such as KRAS and
IL2/STAT5, as well as immune cell infiltration,
including M1 macrophages and neutrophils. Collectively, the present
findings potentially offer a novel approach to sepsis biomarker
identification with high sensitivity and reliability, thus
providing a promising avenue for sepsis diagnosis and
prognosis.
A number of previous in vitro and in
vivo studies have elucidated the mechanisms underlying AAM in
sepsis. These studies have reported significant reductions in
muscle amino acid uptake, which is regulated by various circulating
factors such as growth hormone, cytokines like TNF-α and
IL-6, stress hormones including cortisol, and metabolic
regulators such as insulin (54),
as well as impaired leucine-induced mTOR activation in
muscles during sepsis (55). To
the best of our knowledge, bioinformatics analysis was applied for
the first time to evaluate the diagnostic and prognostic value of
AAMGs in patients with sepsis. The present study identified two
AAMGs, MTR and MIR1, with high diagnostic value
across all three publicly available datasets. Moreover, additional
analyses were conducted to determine their prognostic significance.
Although MTR and MIR1 did not show prognostic
predictive value for sepsis in the GSE95233 and GSE4607 datasets,
this lack of performance could potentially be attributed to the
short follow-up duration in these datasets, which was only 3 days.
Furthermore, considering that previous studies lacked internal
validation or randomized splitting (56), the present study employed a more
reliable bootstrap approach, which confirmed the robust prognostic
predictive ability of MTR and MIR1 in patients with
sepsis. Clinical samples from a local hospital also supported the
trend of reduced expression of MTR and MIR1 in the
peripheral blood of patients with sepsis compared with healthy
individuals, which may potentially provide a solid foundation for
the development of personalized treatment plans in future clinical
practice. Furthermore, in vitro experiments involving the
overexpression of MTR demonstrated its potential to mitigate
LPS-induced damage, which further supported the consistency and
validity of the aforementioned findings.
Currently, research on MTR and MIR1
in the context of sepsis is limited. Considering that MTR is
a critical enzyme responsible for converting homocysteine to
methionine in methylation reactions and MIR1 acts as
methionyl-tRNA synthetase 1, their potential mechanisms of action
in sepsis may involve the regulation of methionine metabolism
(57). A previous study reported
that levels of methionine and its derivatives, particularly
N-formyl-L-methionine, were significantly elevated in the plasma of
patients with septic shock compared to those with cardiogenic shock
or non-septic bacteremia and healthy controls (6). Furthermore, the methionine cycle and
its associated antioxidant and anti-inflammatory processes may
present new potential targets for the treatment of sepsis (58). Methionine sulfoxide, an oxidized
form of methionine, has been recognized as a biomarker in the
progression of sepsis and subsequent acute kidney injury (59). Furthermore, methionine and certain
immune cells serve a role in impacting the development of sepsis.
For instance, in in vivo models of sepsis, the oxidation of
methionine exacerbates the release of pro-inflammatory mediators
and the inflammatory response by altering macrophage metabolism
towards glycolysis and increasing reactive oxygen species levels
(60). The methionine pathway is
also inhibited by the long non-coding RNA MALAT1, which in
turn increases the levels of pro-inflammatory cytokines and the
antioxidant capacity of macrophages, thereby exacerbating the
inflammatory response in sepsis (58). These findings underscore the
research potential of methionine in sepsis and provide direction
for further investigation into the specific roles of MTR and
MIR1 in AAM within the context of sepsis.
Furthermore, the present study demonstrated that
MTR and MIR1 were negatively correlated with
signaling pathways such as PI3K/AKT/mTOR,
whose activation has been reported by a number of previous studies
to mitigate the severity of sepsis (61,62).
The PI3K/AKT/mTOR pathway regulates cellular
metabolism, proliferation and survival, and its activation leads to
the suppression of pro-inflammatory cytokine production and the
enhancement of anti-inflammatory cytokines, thereby reducing the
severity of sepsis (63,64). In the present study, two pathways
previously unreported in sepsis research were also identified,
namely the KRAS and IL2/STAT5 pathways. KRAS
activation leads to the phosphorylation of MAPK, which in
turn activates transcription factors such as NF-κB (65). NF-κB then translocates to
the nucleus and promotes the transcription of pro-inflammatory
cytokines, including TNF-α and IL-6, which serve
crucial roles in the inflammatory response in sepsis by promoting
systemic inflammation, fever and tissue damage (66-70).
Furthermore, the IL2/STAT5 pathway serves a critical
role in regulating the immune response, particularly the T cell
response in sepsis. Binding of IL-2 to its receptor leads to
the activation of JAK3, which phosphorylates STAT5.
Activated STAT5 dimerizes and translocates to the nucleus,
where it promotes the expression of genes involved in T cell
proliferation, differentiation and survival (71). This enhances the immune system's
response to infection by increasing the number and activity of T
cells, which are essential for clearing pathogens and modulating
the immune response in sepsis (72). Additionally, the interaction
between immune cells and AAM serves a crucial role in sepsis. For
instance, it has been reported that, when macrophages are
stimulated by α-aminobutyric acid, oxidative phosphorylation and
the metabolism of glutamine and arginine are activated, thereby
inhibiting the polarization of M1 macrophages and ultimately
prolonging the survival time of mice with sepsis (73). Another previous study reported that
L-lysine could regulate the neutrophil and lymphocyte counts in the
peripheral blood of animal models with sepsis-induced acute lung
injury (74). Glutamine has been
shown to balance the polarization of T helper cells in the blood of
patients with sepsis, maintain T cell populations, prevent splenic
CD4+ T cell apoptosis and reduce late-stage kidney
injury in polymicrobial sepsis (75).
Given the importance of immune cells and AAM in
sepsis, the association between MTR and MIR1 and
immune cells was further analyzed in the present study. These
results demonstrated that MTR and MIR1 were
negatively correlated with the infiltration of pro-inflammatory
cells, such as neutrophils and M1 macrophages, and were positively
correlated with anti-inflammatory cells, such as CD8+ T
and dendritic cells. Specific immune cells, neutrophils, M1
macrophages, CD8+ T cells and dendritic cells, are
closely associated with sepsis, laying the foundation for research
targeting MTR and MIR1 to modulate immune cell
responses in sepsis (76-80).
Overall, these findings support the research potential of
MTR and MIR1 in sepsis and provide direction for
further investigation into their specific mechanisms of action.
Although the present study has filled a gap in
research regarding AAMGs in sepsis, it is not without limitations.
Firstly, despite using multiple datasets for validation and
applying standardized preprocessing procedures to minimize batch
effects and heterogeneity, inherent variations between datasets,
including differences in sample collection, processing methods and
demographic disparities, could still impact the results. The
preprocessing steps included averaging the expression values of the
same gene and removing values ≤0. PCA was conducted on each dataset
individually to compare the distribution differences between the
sepsis and healthy groups, which ensured significant distribution
differences before further analysis. Since datasets for analysis
were not merged, batch effect correction methods such as Combat
were not applied. The PCA results indicated significant
heterogeneity between datasets, potentially confounding the present
findings. Specifically, different sample collection methods may
lead to variations in gene expression levels, with potential
contamination or degradation affecting measurements. Processing
protocol differences among laboratories, such as storage
conditions, extraction methods and sequencing technologies, could
reduce data consistency. Different sequencing platforms may have
varying sequencing depths and accuracies, which may impact data
comparability. Patient demographics such as age, sex and disease
severity could differ between datasets, influencing gene expression
and confounding the identification of sepsis-related genes. Future
studies should integrate data from multiple centers, standardize
sample processing and sequencing methods, and account for clinical
factors such as age and infection-related variables to reduce batch
effects and heterogeneity, enhancing research credibility.
Furthermore, while machine learning algorithms exhibit excellent
performance in feature selection and classification, they also have
certain limitations. The interpretability of machine learning
models is often lower compared with traditional statistical models,
which typically have simpler, more transparent mechanisms that are
easier to directly understand and explain (81). In the present study, various
machine learning algorithms such as LASSO, SVM-RFE and RF were
used, which, although improving the efficiency of feature
selection, also increased model complexity. Therefore, future
research needs larger datasets and more extensive validation to
ensure the generalizability and robustness of the models. The
current validation using local data was limited by a small sample
size and single-center origin, which may affect the robustness of
the present findings; thus, expanding sample sizes and including
multicenter studies are essential. Additionally, while the present
in vitro experiments provided preliminary evidence, they did
not explore downstream mechanisms in depth. It was identified that
MTR and MIR1 may regulate methionine metabolism in
sepsis and may be associated with certain immune cells, but these
mechanisms were not validated through further experiments. Future
research should investigate the specific mechanisms of action of
these genes to better understand their roles and applications in
sepsis.
In conclusion, the present study identified
MTR and MIR1 as potential promising diagnostic and
prognostic biomarkers for sepsis. These genes were linked with
critical signaling pathways, including KRAS and
IL2/STAT5, and correlated with the infiltration of
immune cells, such as M1 macrophages and neutrophils. By modulating
methionine metabolism, MTR and MIR1 may serve a
pivotal role in sepsis pathophysiology.
Supplementary Material
Supplementary Data
Heterogeneity between sepsis and
normal samples across different datasets. PCA results demonstrated
differences between sepsis and healthy samples in (A) GSE65682, (B)
GSE154918 and (C) GSE185263 datasets. (D) PCA showed heterogeneity
among the GSE65682, GSE154918 and GSE185263 datasets. PCA,
principal component analysis; C, control; P, patient with sepsis;
Dim, Dimension.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by the Scientific
Research Fund of The First Affiliated Hospital of Hubei University
of Science and Technology (Xianning Central Hospital; grant no.
2021XYBO11).
Availability of data and materials
The data supporting the findings of the present
study are openly available in the GEO repository (www.ncbi.nlm.nih.gov/gds/). This includes three
microarray datasets, specifically GSE65682 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?),
GSE154918 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?)
and GSE185263 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?).
The data generated in the present study may be requested from the
corresponding author.
Authors' contributions
YW and QL designed the study, QL wrote the
manuscript and acquired the data, YW and WX produced the figures,
YW and WX performed the experiments and analyzed the data, YW and
QL confirm the authenticity of all the raw data. All authors read
and approved the final version of the manuscript.
Ethics approval and consent to
participate
Adhering to the ethical guidelines of the
Declaration of Helsinki, the present study was approved by the
Ethics Committee of Xianning Central Hospital (Xianning, China)
[approval no. K (2024)005]. Written informed consent was obtained
from all the participants.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Becker JU, Theodosis C, Jacob ST, Wira CR
and Groce NE: Surviving sepsis in low-income and middle-income
countries: New directions for care and research. Lancet Infect Dis.
9:577–582. 2009.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Fleischmann C, Scherag A, Adhikari NKJ,
Hartog CS, Tsaganos T, Schlattmann P, Angus DC and Reinhart K:
International Forum of Acute Care Trialists. Assessment of global
incidence and mortality of hospital-treated sepsis. Current
estimates and limitations. Am J Respir Crit Care Med. 193:259–272.
2016.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Guliciuc M, Porav-Hodade D, Chibelean BC,
Voidazan ST, Ghirca VM, Maier AC, Marinescu M and Firescu D: The
role of biomarkers and scores in describing urosepsis. Medicina
(Kaunas). 59(597)2023.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Dai W, Shen J, Yan J, Bott AJ, Maimouni S,
Daguplo HQ, Wang Y, Khayati K, Guo JY, Zhang L, et al: Glutamine
synthetase limits β-catenin-mutated liver cancer growth by
maintaining nitrogen homeostasis and suppressing mTORC1. J Clin
Invest. 132(e161408)2022.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Sivanand S and Vander Heiden MG: Emerging
roles for branched-chain amino acid metabolism in cancer. Cancer
Cell. 37:147–156. 2020.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Rogers RS, Sharma R, Shah HB, Skinner OS,
Guo XA, Panda A, Gupta R, Durham TJ, Shaughnessy KB, Mayers JR, et
al: Circulating N-lactoyl-amino acids and N-formyl-methionine
reflect mitochondrial dysfunction and predict mortality in septic
shock. Metabolomics. 20(36)2024.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Yang Y, Chen Q, Fan S, Lu Y, Huang Q, Liu
X and Peng X: Glutamine sustains energy metabolism and alleviates
liver injury in burn sepsis by promoting the assembly of
mitochondrial HSP60-HSP10 complex via SIRT4 dependent protein
deacetylation. Redox Rep. 29(2312320)2024.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Peng X, Zheng T, Guo Y and Zhu Y: Amino
acid metabolism genes associated with immunotherapy responses and
clinical prognosis of colorectal cancer. Front Mol Biosci.
9(955705)2022.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Shi H, Yuan X, Yang X, Huang R, Fan W and
Liu G: A novel diabetic foot ulcer diagnostic model: Identification
and analysis of genes related to glutamine metabolism and immune
infiltration. BMC Genomics. 25(125)2024.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Choi H, Lee JY, Yoo H and Jeon K:
Bioinformatics analysis of gene expression profiles for diagnosing
sepsis and risk prediction in patients with sepsis. Int J Mol Sci.
24(9362)2023.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Xu X, Bu B, Tian H, Wu R and Yang J:
MicroRNAs combined with the TLR4/TDAG8 mRNAs and proinflammatory
cytokines are biomarkers for the rapid diagnosis of sepsis. Mol Med
Rep. 26(334)2022.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Zhou M, Li T, Lv S, Gan W, Zhang F, Che Y,
Yang L, Hou Y, Yan Z, Zeng Z, et al: Identification of
immune-related genes and small-molecule drugs in
hypertension-induced left ventricular hypertrophy based on machine
learning algorithms and molecular docking. Front Immunol.
15(1351945)2024.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Daneshvar A and Mousa G: Regression
shrinkage and selection via least quantile shrinkage and selection
operator. PLoS One. 18(e0266267)2023.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Özer ME, Özbek Sarica P and Arğa KY:
SVM-DO: Identification of tumor-discriminating mRNA signatures via
support vector machines supported by disease ontology. Turk J Biol.
47:349–365. 2023.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Ignatenko V, Surkov A and Koltcov S:
Random forests with parametric entropy-based information gains for
classification and regression problems. PeerJ Comput Sci.
10(e1775)2024.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Long G and Yang C: A six-gene support
vector machine classifier contributes to the diagnosis of pediatric
septic shock. Mol Med Rep. 21:1561–1571. 2020.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Jiang Z, Luo Y, Wei L, Gu R, Zhang X, Zhou
Y and Zhang S: Bioinformatic analysis and machine learning methods
in neonatal sepsis: Identification of biomarkers and immune
infiltration. Biomedicines. 11(1853)2023.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Rangel-Frausto MS, Pittet D, Costigan M,
Hwang T, Davis CS and Wenzel RP: The natural history of the
systemic inflammatory response syndrome (SIRS). A prospective
study. JAMA. 273:117–123. 1995.PubMed/NCBI
|
|
19
|
Langfelder P and Horvath S: WGCNA: An R
package for weighted correlation network analysis. BMC
Bioinformatics. 9(559)2008.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW,
Shi W and Smyth GK: limma powers differential expression analyses
for RNA-sequencing and microarray studies. Nucleic Acids Res.
43(e47)2015.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z,
Feng T, Zhou L, Tang W, Zhan L, et al: clusterProfiler 4.0: A
universal enrichment tool for interpreting omics data. Innovation
(Camb). 2(100141)2021.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Yu G, Wang LG, Yan GR and He QY: DOSE: an
R/bioconductor package for disease ontology semantic and enrichment
analysis. Bioinformatics. 31:608–609. 2015.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Hänzelmann S, Castelo R and Guinney J:
GSVA: Gene set variation analysis for microarray and RNA-seq data.
BMC Bioinformatics. 14(7)2013.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Friedman J, Hastie T and Tibshirani R:
Regularization paths for generalized linear models via coordinate
descent. J Stat Softw. 33:1–22. 2010.PubMed/NCBI
|
|
25
|
Tibshirani R, Bien J, Friedman J, Hastie
T, Simon N, Taylor J and Tibshirani RJ: Strong rules for discarding
predictors in lasso-type problems. J R Stat Soc Series B Stat
Methodol. 74:245–266. 2012.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Wang Q and Liu X: Screening of feature
genes in distinguishing different types of breast cancer using
support vector machine. Onco Targets Ther. 8:2311–2317.
2015.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Alderden J, Pepper GA, Wilson A, Whitney
JD, Richardson S, Butcher R, Jo Y and Cummins MR: Predicting
pressure injury in critical care patients: A machine-learning
model. Am J Crit Care. 27:461–468. 2018.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Wang H and Zhou L: Random survival forest
with space extensions for censored data. Artif Intell Med.
79:52–61. 2017.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Tao X, Wang J, Liu B, Cheng P, Mu D, Du H
and Niu B: Plasticity and crosstalk of mesenchymal stem cells and
macrophages in immunomodulation in sepsis. Front Immunol.
15(1338744)2024.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Wang Z and Wang Z: The role of macrophages
polarization in sepsis-induced acute lung injury. Front Immunol.
14(1209438)2023.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Han Y, Wang J, Zhang J, Zheng X, Jiang Y,
Liu W and Li W: VX-702 ameliorates the severity of
sepsis-associated acute kidney injury by downregulating
inflammatory factors in macrophages. J Inflamm Res. 17:4037–4054.
2024.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Yu Y, Li Z, Liu C, Bu Y, Gong W, Luo J and
Yue Z: Danlou tablet alleviates sepsis-induced acute lung and
kidney injury by inhibiting the PARP1/HMGB1 pathway. Heliyon.
10(e30172)2024.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Chang BT, Wang Y, Tu WL, Zhang ZQ, Pu YF,
Xie L, Yuan F, Gao Y, Xu N and Yao Q: Regulatory effects of
mangiferin on LPS-induced inflammatory responses and intestinal
flora imbalance during sepsis. Food Sci Nutr. 12:2068–2080.
2023.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Lund ME, To J, O'Brien BA and Donnelly S:
The choice of phorbol 12-myristate 13-acetate differentiation
protocol influences the response of THP-1 macrophages to a
pro-inflammatory stimulus. J Immunol Methods. 430:64–70.
2016.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Zhang S, Guan X, Liu W, Zhu Z, Jin H, Zhu
Y, Chen Y, Zhang M, Xu C, Tang X, et al: YTHDF1 alleviates sepsis
by upregulating WWP1 to induce NLRP3 ubiquitination and inhibit
caspase-1-dependent pyroptosis. Cell Death Discov.
8(244)2022.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Yan L, Chen C, Wang L, Hong H, Wu C, Huang
J, Jiang J, Chen J, Xu G and Cui Z: Analysis of gene expression in
microglial apoptotic cell clearance following spinal cord injury
based on machine learning algorithms. Exp Ther Med.
28(292)2024.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Wysocki K, Conley Y and Wenzel S:
Epigenome variation in severe asthma. Biol Res Nurs. 17:263–269.
2015.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Kumari R, Kumar S, Thakur VK, Singh K and
Kumar U: MTHFR C677T and MTR A2756G gene polymorphism in neural
tube defect patients and its association with red blood cell folate
level in Eastern Indian population. J Indian Assoc Pediatr Surg.
27:699–706. 2022.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Jing HW, Yin L, Yu HY, Zuo L and Liu T:
MTR D919G variant is associated with prostate adenocarcinoma risk:
Evidence based on 51106 subjects. Eur Rev Med Pharmacol Sci.
24:8329–8340. 2020.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Nitsch L, Ehrentraut SF, Grobe-Einsler M,
Bode FJ, Banat M, Schneider M, Lehmann F, Zimmermann J and Weller
J: The diagnostic value of cerebrospinal fluid lactate for
detection of sepsis in community-acquired bacterial meningitis.
Diagnostics (Basel). 13(1313)2023.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Gao H, Ren Y and Liu C: Aloe-emodin
suppresses oxidative stress and inflammation via a PI3K-dependent
mechanism in a murine model of sepsis. Evid Based Complement
Alternat Med. 2022(9697887)2022.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Gao N, Tang AL, Liu XY, Chen J and Zhang
GQ: p53-Dependent ferroptosis pathways in sepsis. Int
Immunopharmacol. 118(110083)2023.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Bai X, He T, Liu Y, Zhang J, Li X, Shi J,
Wang K, Han F, Zhang W, Zhang Y, et al: Acetylation-dependent
regulation of notch signaling in macrophages by SIRT1 affects
sepsis development. Front Immunol. 9(762)2018.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Chou KT, Cheng SC, Huang SF, Perng DW,
Chang SC, Chen YM, Hsu HS and Hung SC: Impact of intermittent
hypoxia on sepsis outcomes in a murine model. Sci Rep.
9(12900)2019.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Ding W, Huang L, Wu Y, Su J, He L, Tang Z
and Zhang M: The role of pyroptosis-related genes in the diagnosis
and subclassification of sepsis. PLoS One.
18(e0293537)2023.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Wang Y, Fan Y, Jiang Y, Wang E, Song Y,
Chen H, Xu F, Xie K and Yu Y: APOA2: New target for molecular
hydrogen therapy in sepsis-related lung injury based on proteomic
and genomic analysis. Int J Mol Sci. 24(11325)2023.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Dai W, Zheng P, Luo D, Xie Q, Liu F, Shao
Q, Zhao N and Qian K: LPIN1 is a regulatory factor associated with
immune response and inflammation in sepsis. Front Immunol.
13(820164)2022.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Sánchez-Baizán N, Ribas L and Piferrer F:
Improved biomarker discovery through a plot twist in transcriptomic
data analysis. BMC Biol. 20(208)2022.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Alhamzawi R and Ali HTM: The Bayesian
adaptive lasso regression. Math Biosci. 303:75–82. 2018.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Chen H, Chen E, Lu Y and Xu Y:
Identification of immune-related genes in diagnosing retinopathy of
prematurity with sepsis through bioinformatics analysis and machine
learning. Front Genet. 14(1264873)2023.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Yu J, Wu X, Huang K, Zhu M, Zhang X, Zhang
Y, Chen S, Xu X and Zhang Q: Bioinformatics identification of
lncRNA biomarkers associated with the progression of esophageal
squamous cell carcinoma. Mol Med Rep. 19:5309–5320. 2019.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Chicco D and Jurman G: The advantages of
the Matthews correlation coefficient (MCC) over F1 score and
accuracy in binary classification evaluation. BMC Genomics.
21(6)2020.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Safránek R, Holecek M, Sispera L and
Muthný T: Aspects of protein and amino acid metabolism in a model
of severe glutamine deficiency in sepsis. Ann Nutr Metab.
50:361–367. 2006.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Laufenberg LJ, Pruznak AM, Navaratnarajah
M and Lang CH: Sepsis-induced changes in amino acid transporters
and leucine signaling via mTOR in skeletal muscle. Amino Acids.
46:2787–2798. 2014.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Zhang J, Ding N, He Y, Tao C, Liang Z, Xin
W, Zhang Q and Wang F: Bioinformatic identification of genomic
instability-associated lncRNAs signatures for improving the
clinical outcome of cervical cancer by a prognostic model. Sci Rep.
11(20929)2021.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Pokushalov E, Ponomarenko A, Bayramova S,
Garcia C, Pak I, Shrainer E, Ermolaeva M, Kudlay D, Johnson M and
Miller R: Effect of methylfolate, pyridoxal-5'-phosphate, and
methylcobalamin (SolowaysTM) supplementation on
homocysteine and low-density lipoprotein cholesterol levels in
patients with methylenetetrahydrofolate reductase, methionine
synthase, and methionine synthase reductase polymorphisms: A
randomized controlled trial. Nutrients. 16(1550)2024.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Chen J, Tang S, Ke S, Cai JJ, Osorio D,
Golovko A, Morpurgo B, Guo S, Sun Y, Winkle M, et al: Ablation of
long noncoding RNA MALAT1 activates antioxidant pathway and
alleviates sepsis in mice. Redox Biol. 54(102377)2022.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Ping F, Li Y, Cao Y, Shang J, Zhang Z,
Yuan Z, Wang W and Guo Y: Metabolomics analysis of the development
of sepsis and potential biomarkers of sepsis-induced acute kidney
injury. Oxid Med Cell Longev. 2021(6628847)2021.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Yoo HJ, Choi DW, Roh YJ, Lee YM, Lim JH,
Eo S, Lee HJ, Kim NY, Kim S, Cho S, et al: MsrB1-regulated GAPDH
oxidation plays programmatic roles in shaping metabolic and
inflammatory signatures during macrophage activation. Cell Rep.
41(111598)2022.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Bi CF, Liu J, Hao SW, Xu ZX, Ma X, Kang
XF, Yang LS and Zhang JF: Xuebijing injection protects against
sepsis-induced myocardial injury by regulating apoptosis and
autophagy via mediation of PI3K/AKT/mTOR signaling pathway in rats.
Aging (Albany NY). 15:4374–4390. 2023.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Geng H, Zhang H, Cheng L and Dong S:
Sivelestat ameliorates sepsis-induced myocardial dysfunction by
activating the PI3K/AKT/mTOR signaling pathway. Int
Immunopharmacol. 128(111466)2024.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Manning BD and Cantley LC: AKT/PKB
signaling: Navigating downstream. Cell. 129:1261–1274.
2007.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Jiang L, Yang D, Zhang Z, Xu L, Jiang Q,
Tong Y and Zheng L: Elucidating the role of Rhodiola rosea L. in
sepsis-induced acute lung injury via network pharmacology: Emphasis
on inflammatory response, oxidative stress, and the PI3K-AKT
pathway. Pharm Biol. 62:272–284. 2024.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Wang Y, Wang D, Dai Y, Kong X, Zhu X, Fan
Y, Wang Y, Wu H, Jin J, Yao W, et al: Positive crosstalk between
hedgehog and NF-κB pathways is dependent on KRAS mutation in
pancreatic ductal adenocarcinoma. Front Oncol.
11(652283)2021.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Ibrahim MA, Khalifa AM, Abd El-Fadeal NM,
Abdel-Karim RI, Elsharawy AF, Ellawindy A, Galal HM, Nadwa EH,
Abdel-Shafee MA and Galhom RA: Alleviation of doxorubicin-induced
cardiotoxicity in rat by mesenchymal stem cells and olive leaf
extract via MAPK/TNF-α pathway: Preclinical, experimental and
bioinformatics enrichment study. Tissue Cell.
85(102239)2023.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Tokumaru Y, Oshi M, Katsuta E, Yan L,
Satyananda V, Matsuhashi N, Futamura M, Akao Y, Yoshida K and
Takabe K: KRAS signaling enriched triple negative breast cancer is
associated with favorable tumor immune microenvironment and better
survival. Am J Cancer Res. 10:897–907. 2020.PubMed/NCBI
|
|
68
|
Xu M, Feng Y, Xiang X, Liu L and Tang G:
MZB1 regulates cellular proliferation, mitochondrial dysfunction,
and inflammation and targets the PI3K-Akt signaling pathway in
acute pancreatitis. Cell Signal. 118(111143)2024.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Singh SP, Dosch AR, Mehra S, De Castro
Silva I, Bianchi A, Garrido VT, Zhou Z, Adams A, Amirian H, Box EW,
et al: Tumor cell-intrinsic p38 MAPK signaling promotes
IL1α-mediated stromal inflammation and therapeutic resistance in
pancreatic cancer. Cancer Res. 84:1320–1332. 2024.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Mo JS, Lamichhane S, Yun KJ and Chae SC:
MicroRNA 452 regulates SHC1 expression in human colorectal cancer
and colitis. Genes Genomics. 45:1295–1304. 2023.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Li J, Xu F, Li S, Xie M and Li N:
Gentamicin promoted the production of
CD4+CD25+ Tregs via the STAT5 signaling
pathway in mice sepsis. BMC Immunol. 23(47)2022.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Ge Y, Huang M, Wu Y, Dong N and Yao YM:
Interleukin-38 protects against sepsis by augmenting
immunosuppressive activity of CD4+ CD25+
regulatory T cells. J Cell Mol Med. 24:2027–2039. 2020.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Li F, Xia Y, Yuan S, Xie X, Li L, Luo Y,
Du Q, Yuan Y and He R: α-Aminobutyric acid constrains
macrophage-associated inflammatory diseases through metabolic
reprogramming and epigenetic modification. Int J Mol Sci.
24(10444)2023.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Zhang Y, Yu W, Han D, Meng J, Wang H and
Cao G: L-lysine ameliorates sepsis-induced acute lung injury in a
lipopolysaccharide-induced mouse model. Biomed Pharmacother.
118(109307)2019.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Hou YC, Wu JM, Chen KY, Chen PD, Lei CS,
Yeh SL and Lin MT: Effects of prophylactic administration of
glutamine on CD4+ T cell polarisation and kidney injury
in mice with polymicrobial sepsis. Br J Nutr. 122:657–665.
2019.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Steinacher E, Lenz M, Krychtiuk KA,
Hengstenberg C, Huber K, Wojta J, Heinz G, Niessner A, Speidl WS
and Koller L: Decreased percentages of plasmacytoid dendritic cells
predict survival in critically ill patients. J Leukoc Biol.
115:902–912. 2024.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Bi Q, Liu Y, Yuan T, Wang H, Li B, Jiang
Y, Mo X, Lei Y, Xiao Y, Dong S, et al: Predicted CD4+ T
cell infiltration levels could indicate better overall survival in
sarcoma patients. J Int Med Res. 49(300060520981539)2021.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Zhu W, Ou Y, Wang C, An R, Lai J, Shen Y,
Ye X and Wang H: A neutrophil elastase inhibitor, sivelestat,
attenuates sepsis-induced acute kidney injury by inhibiting
oxidative stress. Heliyon. 10(e29366)2024.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Bacârea A, Coman O, Bacârea VC, Văsieşiu
AM, Săplăcan I, Fodor RŚ and Grigorescu BL: Immune profile of
patients-a new approach in management of sepsis and septic shock?
Exp Ther Med. 27(203)2024.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Zhang S, Liu Y, Zhang XL, Sun Y and Lu ZH:
ANKRD22 aggravates sepsis-induced ARDS and promotes pulmonary M1
macrophage polarization. J Transl Autoimmun.
8(100228)2023.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Weckbecker M, Anžel A, Yang Z and Hattab
G: Interpretable molecular encodings and representations for
machine learning tasks. Comput Struct Biotechnol J. 23:2326–2336.
2024.PubMed/NCBI View Article : Google Scholar
|