Introduction
Fanconi anemia (FA) is a genetic instability
syndrome that increases the risk of cancer, bone marrow failure and
congenital anomalies. It is the most common type of hereditary bone
marrow failure worldwide, with an estimated population incidence of
1 in 5,000,000 (1,2).
This disease is caused by an alteration in one of
the 23 FA complementation group A (FANCA) genes associated
with the FA/BRCA repair pathway, responsible for 60-70% of all
mutations. Inefficient DNA damage repair creates genomic
instability, which promotes the development and spread of cancer;
within these DNA repair mechanisms are the genes involved in the
FA/BRCA pathway (3). This pathway
repairs interstrand crosslinks (ICL), endogenously created by
reactive oxygen species, or exogenously by exposure to ICL-inducing
agents. It is crucial for the ICL to be remediated, since they
covalently join opposite DNA strands to block both replication and
transcription (4). If the damage
cannot be repaired, it leads to massive apoptosis that generates
bone marrow failure and leads to an increased risk of developing
certain types of malignancies, such as acute myeloid leukemia or
squamous cell carcinoma of the head and neck (5,6).
In addition to the previously mentioned cancers, FA
has been associated with a predisposition to other types of
neoplasms. Mutations within the FA/BRCA repair pathway have been
shown to be associated with a predisposition to breast, uterine,
prostate and liver cancer, due to alterations in FANCC and
FANCD1 (7), or as a late
onset after treatment with immunosuppressive chemotherapy for
aplastic anemia (8).
This disease is diagnosed based on clinical
suspicion and a chromosome breakage test, and molecular
confirmation allows variants to be detected. In the case of
FANCA, 40% of variants can be detected using multiplex
ligation-dependent probe amplification. In addition,
next-generation sequencing (NGS) provides a detection capacity of
≤90% of all variants (7).
Nucleotide variations, small insertions/deletions and large
deletions are the most common causes of mutations in FANCA
(9).
The current study aims to present a pathogenic
variant in FANCA, which was detected by NGS and was observed
in three Mexican siblings diagnosed with FA.
Materials and methods
Clinical and hematological report
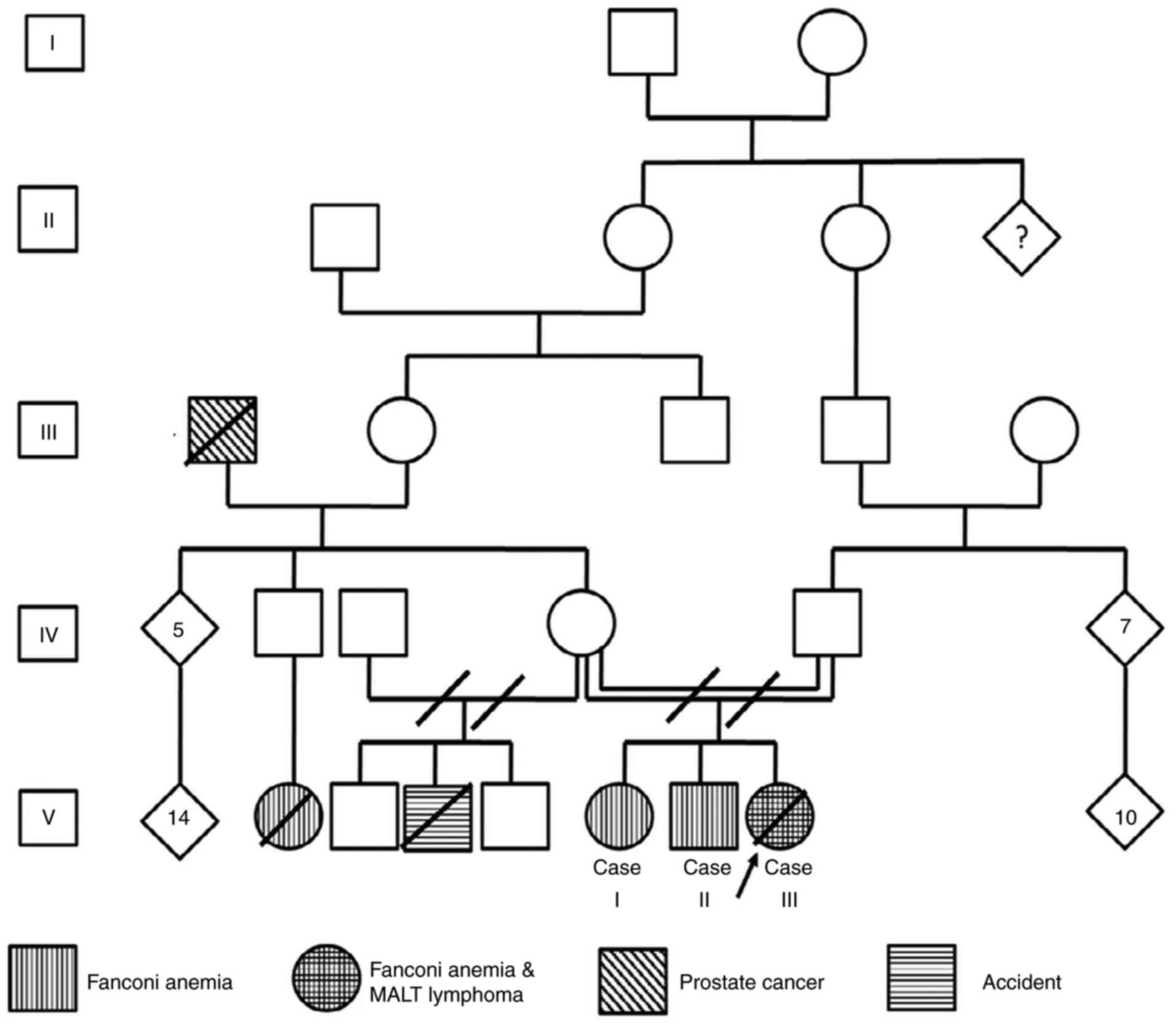
The current study presents the case of a family
diagnosed with FA; the pedigree had an endogamic ancestry (Fig. 1). In total, 3 of 3 children were
diagnosed with FA and 1 of them developed a suggestive
mucosa-assisted lymphoid tissue (MALT) lymphoma during childhood.
Written informed consent was obtained from all 3 children, who are
of legal age, and from the father of the deceased patient.
Case 1: The patient, who was 21 years old at the
time of writing, presented with clinical criteria compatible with
FA. The patient developed pancytopenia at 13 years of age.
Chromosomal breakage tests were performed with diepoxybutane and
the result was positive. The features found in this patient are
listed in Table I.
 | Table IClinical features found in the Fanconi
anemia family. |
Table I
Clinical features found in the Fanconi
anemia family.
| Feature | Case I | Case II | Case III |
|---|
| Small stature | + | + | + |
| Low birth weight | - | - | - |
| Microcephaly | + | + | + |
| Ear anomaly | - | - | - |
| Deafness | - | - | - |
| Strabismus | - | - | - |
| Microphthalmia | - | - | - |
| Congenital heart
defect | - | - | - |
| Cryptorchidism | N/A | - | N/A |
| Kidney
malformation | - | - | - |
| Radial aplasia | - | - | - |
| Thumb
deformity | - | - | - |
| Lymphoma | - | - | + |
| Pigmentary
changes | + | + | + |
|
Hyperpigmentation | + | + | + |
| Cafe-au-lait
spots | + | + | + |
| Mental
retardation | - | - | - |
| Anemia | + | + | + |
| Neutropenia | - | - | - |
|
Thrombocytopenia | + | + | + |
|
Reticulocytopenia | - | - | - |
| Bleeding
pancytopenia | + | + | + |
| Leukemia | - | - | - |
| Multiple
chromosomal breaks | + | + | + |
| CBT by DEB/MMC | + | + | + |
Case 2: The patient, who was 19 years old at the
time of writing, had epistaxis since birth until 5 years of age,
with a clinical suspicion of familiar thrombocytopenic purpura;
however, the patient was diagnosed with bone marrow failure at 11
years of age, alongside positive chromosomal breakage test results.
The patient's features are provided in Table I.
Case 3: The patient, who was a female deceased at 12
years of age, had developed epistaxis and ecchymosis in the lower
limbs from birth until 6 years of age, and was diagnosed with
thrombocytopenic purpura vs. megaloblastic anemia. At 9 years of
age, the patient was subjected to a chromosomal breakage diagnostic
test with diepoxybutane, which revealed bone marrow failure. The
clinical manifestations are shown in Table I. At 11 years of age (March 2018),
the patient presented at the Hospital Civil de Guadalajara ‘Dr Juan
I. Menchaca’ (Guadalajara, Mexico) with abdominal pain, hyporexia
and difficulty swallowing. Gastric endoscopy revealed digestive
tract bleeding associated with chronic gastritis but not with
Helicobacter pylori. Endoscopic colon biopsies were
subjected to hematoxylin and eosin staining performed according to
standard procedures, with the use of a Primo Star 1 microscope
(Carl Zeiss AG).
Sample preparation
Two samples of peripheral blood (PB) (heparin and
EDTA) were obtained from each patient. The first sample underwent a
karyotype and chromosomal breakage test using standard procedures
according to Bobabilla-Morales et al (10), while the second sample was used for
DNA extraction using a QIAamp® DNA Blood Mini kit
(Qiagen GmbH) according to the manufacturer's instructions.
Cytogenetics analysis and chromosome
breakage test
Conventional karyotype analysis was performed from
PB. In brief, the PB were incubated for 72 h in Gibco RPMI 1640
medium (Thermo Fisher Scientific, Inc.), supplemented with
glutamine, fetal bovine serum and antibiotics (Gibco; Thermo Fisher
Scientific, Inc.). Next, Giemsa-Trypsin- Wright (GTW) banding was
performed according to standard procedures (11). The chromosomes were analyzed and
karyotyped following the International System for Human Cytogenetic
Nomenclature (12).
For the chromosome breakage assay, PB samples were
cultured and treated with mitomycin c (Sigma Aldrich; Merck KGaA)
at a concentration of 50 ng/ml, and in certain cases with 0.1 µg/ml
diepoxybutane (Sigma-Aldrich; Merck KGaA), for 72 h at 37˚C. For
each sample, 100 metaphases were analyzed to calculate the mean
number of chromosomal aberrations and the percentage of aberrant
cells. Scoring of chromosomal aberrations was performed according
to Bobabilla-Morales et al (10). The diagnosis of AF was positive if
≥15% of cells possessed aberrant metaphases.
Cultures of blood samples collected from healthy
individuals were used in parallel as negative controls, healthy
patients were recruited from the outpatient clinic and were invited
to participate, the procedure to be performed was explained to them
as indicated in the informed consent form, and once they signed it,
the PB sample was taken.
Nucleic acid extraction
Genomic DNA was isolated from bone marrow using the
QIAamp® DNA Blood Mini kit (Qiagen GmbH) according to
the manufacturer's instructions. The DNA concentration was
determined using ultraviolet light spectrophotometry, with the
absorbance (A) at 260 nm (A260) measured, while its purity was
assessed by reading the A230 and A280, resulting in values of 1.8
and 2.1 for A260/A280 and A260/A230, respectively, according to the
results obtained with the NanoDrop™ One (NanoDrop
Technologies; Thermo Fisher Scientific, Inc.).
NGS
Genetic DNA analysis was performed using a targeted
sequencing panel designed for germline cancer hereditary-associated
mutations [panel size: 403 kb, 113 genes (covering all exons), 125
SNPs (48 ID SNPs and 77 SNPs for polygenic risk score), 10,341
oligo probes (TruSight Hereditary Cancer Panel; Illumina, Inc.)].
The libraries were prepared using Illumina DNA Prep with Enrichment
library prep chemistry, which combines library preparation and
enrichment processes. The analysis enables the detection of
single-nucleotide variants, insertions/deletions and copy-number
variants in a single assay. Assay quality data were evaluated using
the BaseSpace Enrichment App v3.1.0 (Illumina, Inc.). Analysis was
performed in Franklyn on the Genoox platform (https://franklin.genoox.com/clinical-db/home) and
results were reported following the recommendations of the Human
Genome Variation Society (HGVS) Nomenclature 2016 based on the
human assembly GRCh37 (also known as hg19) (https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_000001405.13/).
Bioinformatic analysis
The FASTQ files obtained were evaluated with FastQC
software to analyze the quality of the sequences, which revealed
sufficient quality of the sequences without evident problems.
Next, the sequences were assembled using SPAdeS
(http://bioinf.spbau.ru/spades) with
Burrows-Wheeler Aligner assembly parameters to correct errors in
the sequences before performing the assembly ‘spades.py-1
R1.fastq-2 R2.fastq careful’. The obtained assembly was saved in
the file ‘contigs.fasta’, which was used to perform variant
analysis. The quality of the obtained assembly was sufficient and
met the expected metrics (Table
II).
| Table IIResults regarding the quality of the
obtained genome assembly from Case III. |
Table II
Results regarding the quality of the
obtained genome assembly from Case III.
| Metric | Value |
|---|
| Contigs, bp | 1,945 |
|
0 | 67,302 |
|
1,000 | 282 |
|
5,000 | 3 |
|
10,000 | 1 |
| Total length,
bp | 1,555,717 |
|
0 | 18,706,218 |
|
1,000 | 471,909 |
|
5,000 | 30,440 |
|
10,000 | 16,644 |
| Largest contig | 16,644 |
| GC, % | 47.03 |
| N50 | 742 |
| L50 | 647 |
| Ns per 100 Kbp | 0.00 |
Once the assembly was obtained, alignment analysis
was conducted with BLAST software using the following parameters:
‘blastn-query contigs.fasta-subject fanca.fasta-dbtype nucl-out
blast_results.txt’.
In silico predictive analysis was performed
using MutationTaster software (https://www.genecascade.org/MutationTaster2021/),
which predicts the effect of a variant in the protein encoded by a
gene.
Upon in silico predictive analysis, molecular
modeling of the FANCA variant was performed by using the
Biopython library (13) to
translate the nucleotide sequence into an amino acid sequence.
Subsequently, the amino acid sequence files were stored in ‘.fasta’
format for further use. Next, the AlphaFold Protein Structure
Database (https://alphafold.ebi.ac.uk) was used
to model the structure of the protein encoded by the FANCA
gene with the variant reported against the reference protein.
Immunohistochemical analysis
Tissue had been paraffin-embedded for 12 h at room
temperature and the thickness of sections was 3 µm. For dewaxing,
the slides with the samples were placed in xylene at 60˚C for 16 h
and subsequently, the slides were subjected to two consecutive
washes with xylene for 10 min each. The slides were then washed in
ethanol at 70, 80 and 100% for 2 min each. Subsequently, the
samples were processed in a steamer with citrate buffer for 15 min.
The blocking reagent used was IntelliPATH FLX Peroxidase Blocking
Reagent (cat. no. IPB5000; Biocare Medical), applied for 5 min at
room temperature. The CD20 antibody (cat. no. CM 004; Biocare
Medical) at a dilution of 1:100 at 36˚C for 16 min, the CD3
antibody (cat. no. ACI 3170; Biocare Medical) at a dilution of
1:100 at 36˚C for 15 min and the Ki67 antibody (cat. no. CRM 325;
Biocare Medical) at a dilution of 1:50 at 36˚C for 32 min were
applied; the ultraView Universal DAB Detection Kit (cat. no.
760-500; Roche Tissue Diagnostics; Roche Diagnostics, Ltd.) was
used to detect the primary antibodies according to the
manufacturer's instruction. Visualization and observation were
carried out under a Primo Star 1 light microscope (Carl Zeiss
AG).
Results
Cytogenetics and chromosome breakage
analyses
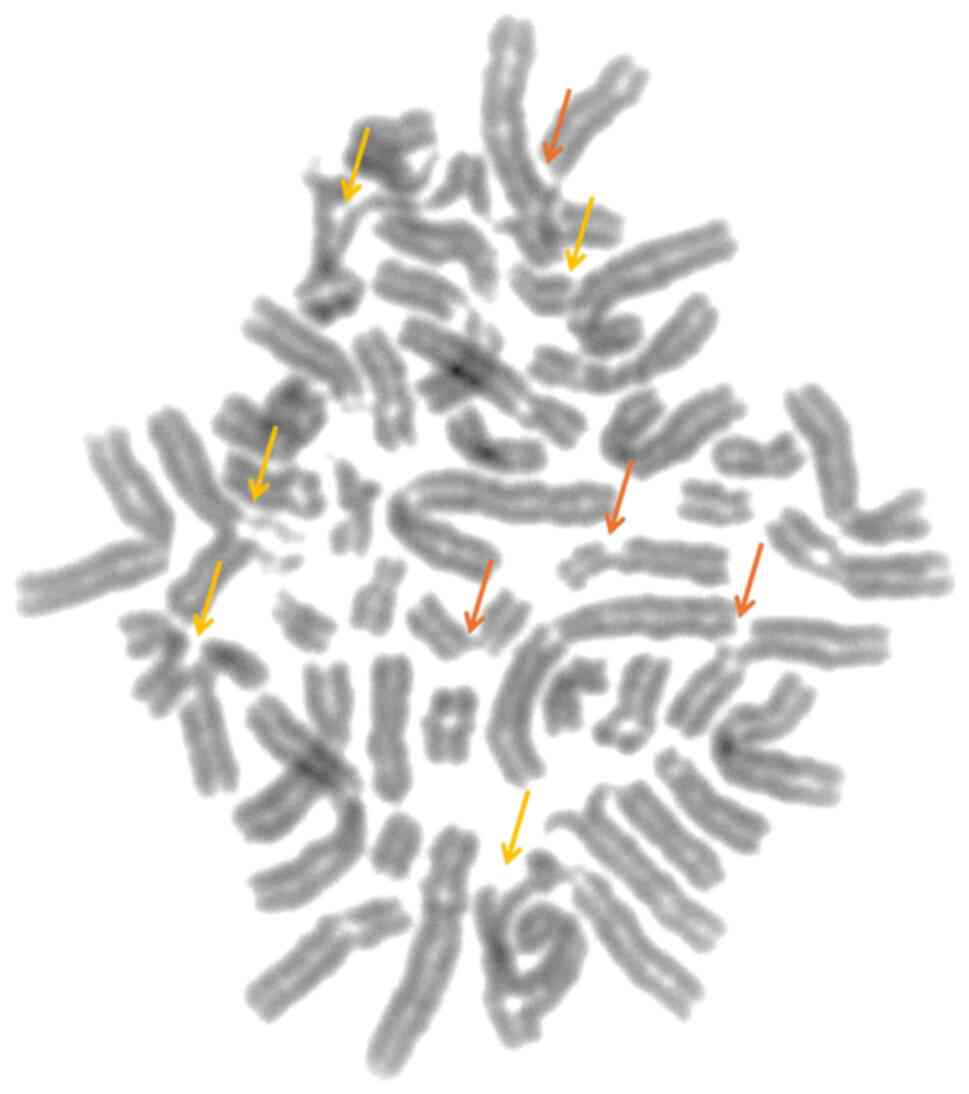
Chromosomal breakage analysis was performed in three
pediatric patients with clinical suspicion of FA. The chromosomal
breakage test was positive, indicating an increased number of
chromosomal rearrangements (mean, 4.2 breaks/cell; Fig. 2), compared with the control
subjects (non-FA, mean, 0.2 breaks/cell). The chromosomal breakage
test confirmed the diagnosis of FA in all three siblings.
Cytogenetic analysis showed a normal karyotype.
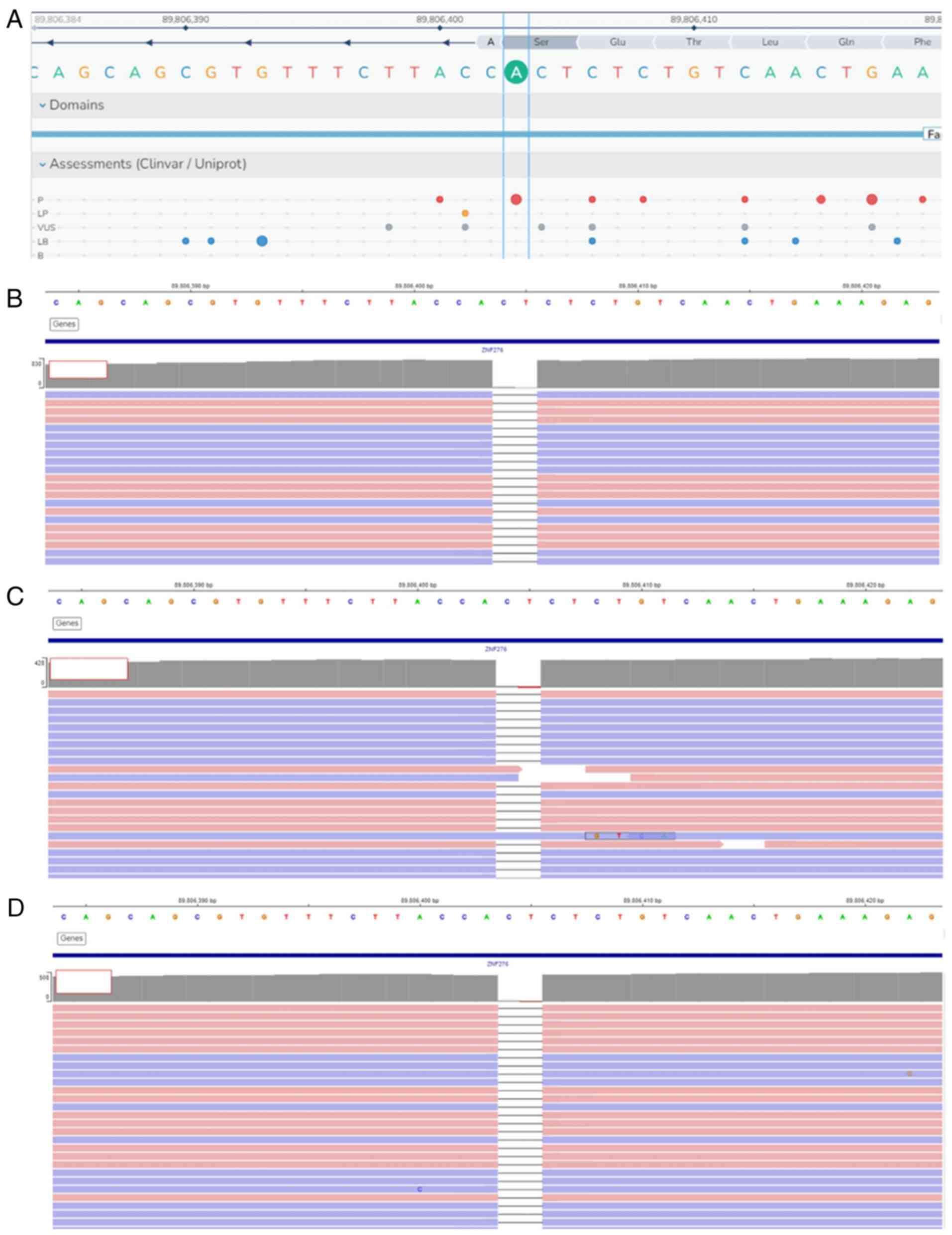
NGS
NGS analysis showed that all three siblings had a
homozygous deletion of two base pairs in FANCA. The
FANCA:c.3931_3932delAG (p.S1311*) variant is
shown in Fig. 3.
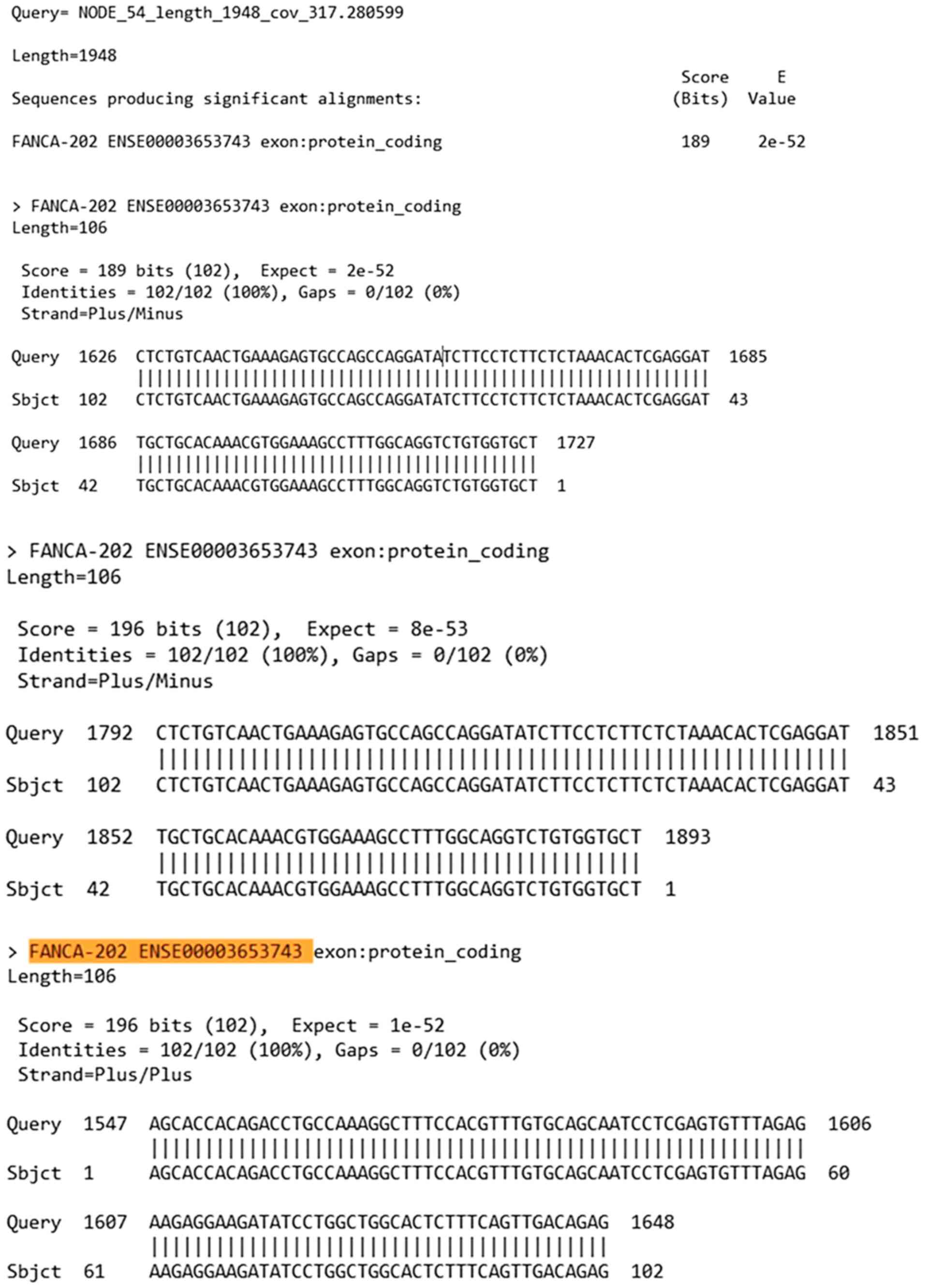
Bioinformatic analysis of the pathogenic variant
c.3931_3932delAG found in FANCA. Once the alignment analysis
was conducted with BLAST software, the results compared based on
the FANCA reference sequence (NM_000135.4) yielded the
outcomes shown in Fig. 4.
The variant present in the patients was of a
deleterious type and was predicted to affect the protein encoded by
the FANCA gene, causing a pronounced change in the protein
sequence. Since the variant caused a frameshift in the protein
sequence, this led to a change in the amino acid sequence, which
may cause a nonsense-mediated mRNA decay in the protein.
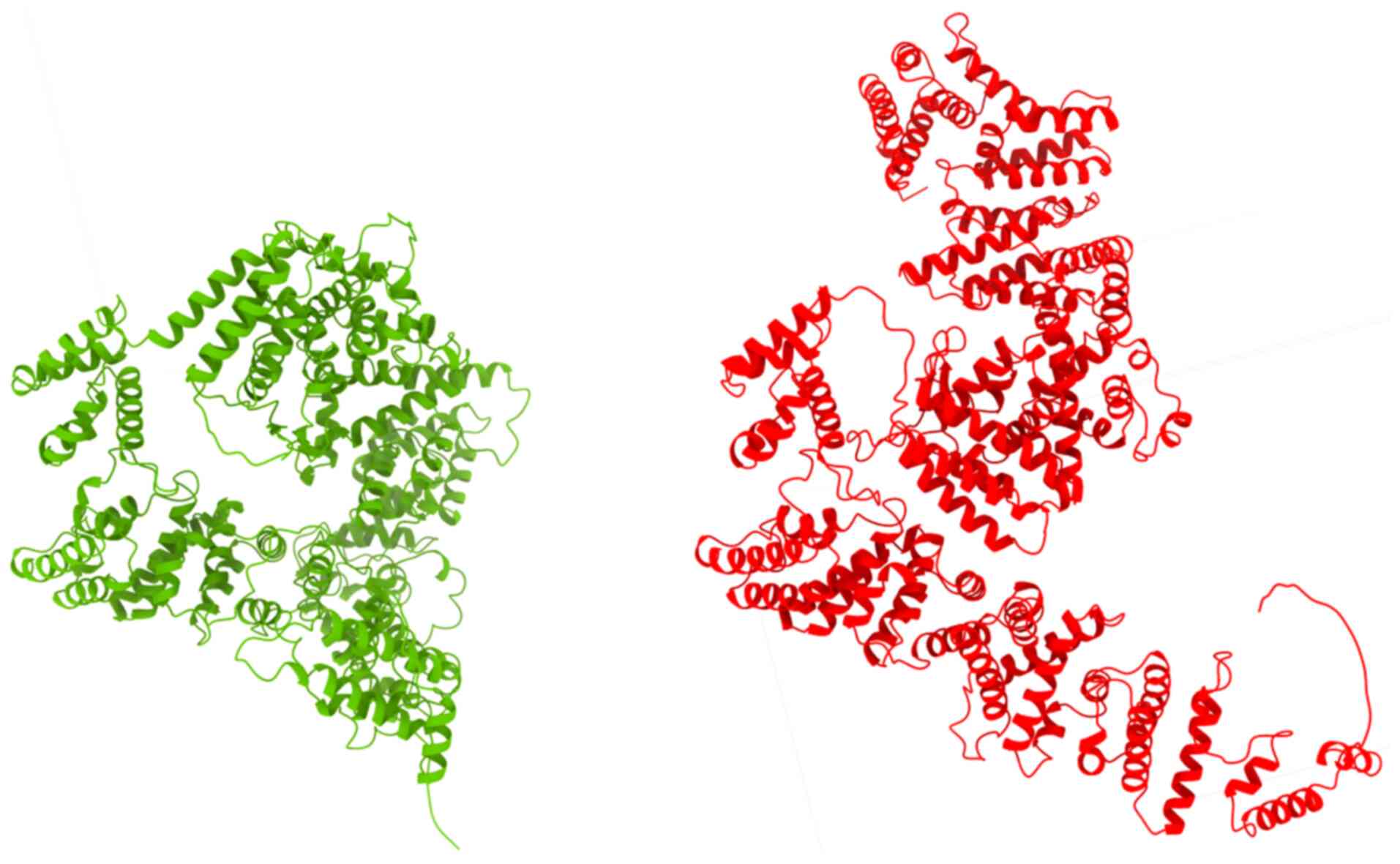
After molecular modeling of the
FANCA:c.3931_393232delAG variant was performed by using the
Biopython library (13), the
AlphaFold Protein Structure Database was used to model the
structure of the protein encoded by the FANCA gene with the
variant reported against the reference protein (Fig. 5). In the AlphaFold- generated
three-dimensional structure, a significant conformational
alteration in the protein was observed. This structural change led
to a profound functional impairment of the protein, resulting in,
among other effects, nonsense-mediated mRNA decay.
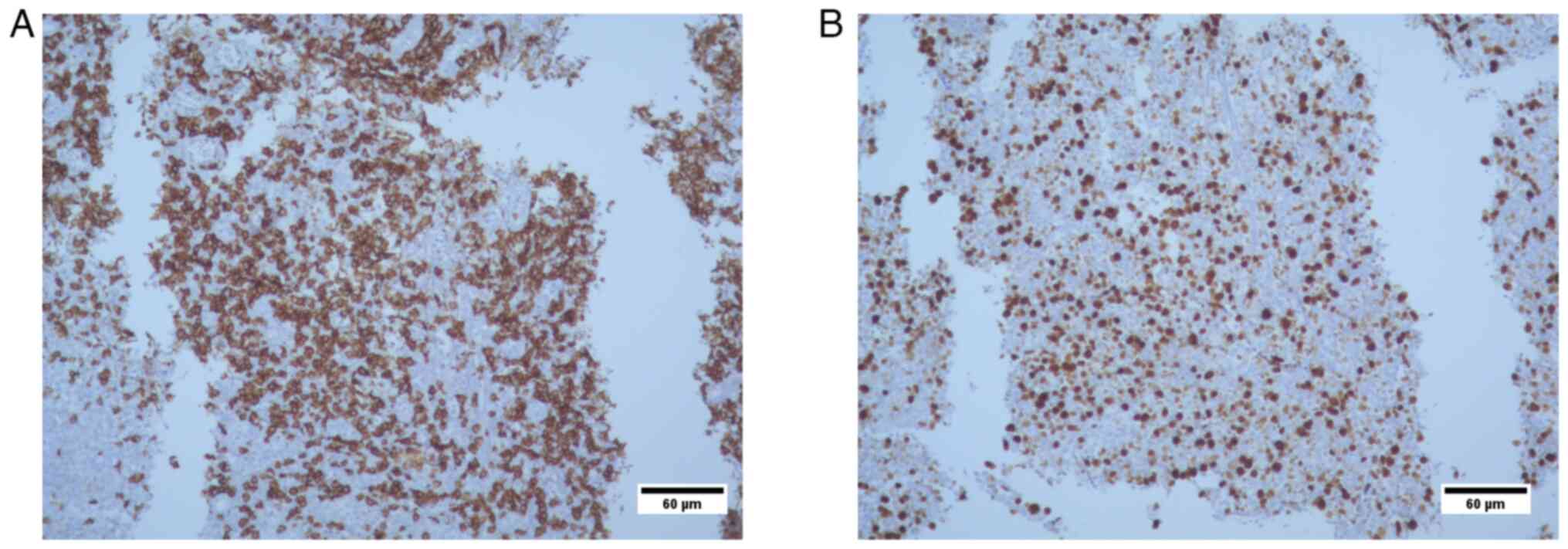
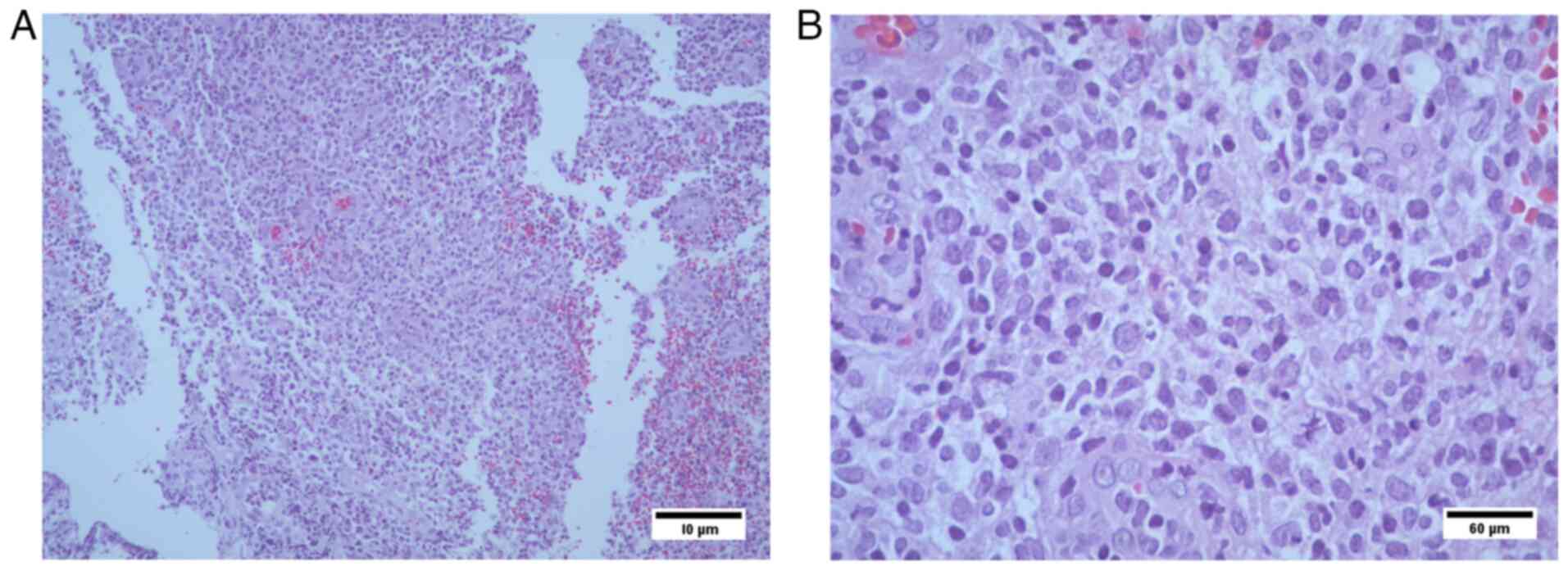
Histopathological analysis
The results revealed i) lymphocyte infiltration due
to lymphoproliferative lesion (transverse colon biopsies); presence
of CD20-positive tumor cells; and CD3 reactivity, which was
suggestive of a MALT type non-Hodgkin lymphoma; and ii) Ki67
positivity in 70% of tumor cell nuclei. At 12 years of age, the
patient succumbed to bone marrow failure, non-Hodgkin lymphoma
progression, severe febrile neutropenia and acute respiratory
failure. Immunohistochemical analysis of the transverse colon
biopsy from Case 3 showed CD20+ tumor cells, CD3
reactivity and 70% KI67+ nuclei in tumor cells (Figs. 6 and 7). The results were suggestive of
non-Hodkin MALT lymphoma.
Discussion
To date, 2,065 variants in the FANCA gene
have been reported, of which 688 have been described only once
according to the Fanconi Anemia Mutation Database (14). It has been demonstrated that
selecting an appropriate method of detection is fundamental to
identify new findings (7).
FANC genes became relevant to the occurrence of
cancer in the general population when bi-allelic mutations in the
breast and ovarian cancer susceptibility genes BRCA1
(FANCS), PALB2 (FANCN) and BRCA2
(FANCD1) were identified in patients with FA, as germline
monoallelic mutations or promoter hypermethylation of FA genes in
patients without FA confer an increased risk of developing multiple
cancer types, since the canonical function of FA proteins is to
collaborate with other DNA repair proteins to eliminate chromosomal
breaks in DNA ICLs. Similarly, in a genomic analysis of nine common
cancer types from The Cancer Genome Atlas, FA genes were altered in
40% of tumors, with the majority belonging to the FA/BRCA pathway
(3). FANCA is a gene that
creates a multi-functional protein that participates in the FA/BRCA
pathway, which repairs ICLs and performs homologous recombination
and nucleotide excision repair in coordination with other
genes.
The present study describes three siblings diagnosed
with FA, as confirmed not only by their clinical manifestations but
also by chromosomal breakage tests. The NGS TruSight Hereditary
Cancer Panel was used to identify changes in the FANC genes
available in this panel. All three patients had the variant
FANCA:c.3931_3932delAG in a homozygous state. This variant
has been reported in a single patient in a study on Japanese
patients with FA (15). Another
study carried out by Zhu et al (16) that had reported the variant was a
study on hereditary breast cancer. It is known that certain
pathogenic variants in breast cancer are located in the FANC genes,
since they belong to the FA/BRCA pathway (16). The current bioinformatic analysis
and the results of Franklin by Genoox indicated that this variant
had a pathogenic classification (17). Since the variant caused a
frameshift in the protein sequence, it was predicted that the
variant present in the aforementioned patients could have a
deleterious effect on the protein encoded by the FANCA gene,
resulting in a pronounced change in the protein sequence. This
change in the amino acid sequence may lead to a nonsense-mediated
mRNA decay in the protein.
Although the histopathological findings were
initially suggestive of MALT lymphoma, the diagnosis remained
inconclusive due to insufficient tissue for further
characterization. Therefore, other differential diagnoses,
including alternative lymphoma subtypes, could not be excluded. The
literature has reported only two cases of FA and lymphoma. The
first one describes a 30-month-old male patient who was diagnosed
with T-cell lymphoblastic lymphoma, presented high sensitivity to
cytotoxic chemotherapy and was diagnosed with FA. In addition to
t(11;14)(p13;q32), cytogenetic analysis showed chromosomal
instability with chromosomal breaks, as well as radial figures
(18). The second report in the
literature describes a male patient who, at 5 years of age, was
diagnosed with FA. At 11 years of age, the patient underwent a bone
marrow transplant, and only 1 year later, the patient's clinical
data were suggestive of lymphoma. The biopsy indicated T-cell
lymphoblastic lymphoma and human leukocyte antigen analysis
revealed tumorigenic cells derived from the same patient (19). To the best of our knowledge, no
reports on chemotherapy for lymphoid tumors in patients with FA
exist in the literature. In total, 30-40% of all extranodal
lymphomas are primary gastric lymphoma (PGL), which is the most
prevalent extranodal location of non-Hodgkin lymphoma. Furthermore,
it accounts for 4-20% of all non-Hodgkin lymphoma and ~5% of
primary gastric neoplasm cases. The two most common types of PGL
based on histology are marginal zone B-cell lymphoma of MALT and
diffuse large B-cell lymphoma. The highest incidence of MALT
lymphoma occurs in individuals aged 50-60 years (20).
FANCA is considered hypermutable and
polymorphic (21). The
FANCA variant is responsible for causing FA; however, it is
not considered to be able to cause MALT lymphoma in Case 3, since
the literature does not refer to any association between this type
of lymphoma and the FANC genes.
In conclusion, the present study reported a variant
in FANCA, which was determined by NGS in three siblings
diagnosed with FA. The detected variant,
FANCA:c.3931_3932delAG, was classified as pathogenic by
bioinformatic analysis.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used are publicly available at the
National Center for Biotechnology Information (NCBI), the
BioProject accession no. is PRJNA1169362 (https://www.ncbi.nlm.nih.gov/sra/PRJNA1169362);
the Sequence Read Archive (SRA) (https://www.ncbi.nlm.nih.gov/sra) accession nos. of
each of the samples are SRX26297315, SRX26297314 and SRX26297313.
All other data may be requested from the corresponding author.
Authors' contributions
Conceptualization, LBM, SABJ, ICQ and ACR;
methodology, SABJ, ICQ and FAFL; clinical management of the cases,
as well as the follow-up and evaluation of the patient's progress,
MMOS and JRCR; bioinformatic analysis, LJZ; histopathological
analysis, FDJBR; writing-original draft preparation, ICQ and FAFL;
writing, review and editing, ACR; supervision, LBM. LBM, SABJ and
ICQ checked and confirm the authenticity of all the raw data. All
authors have read and agreed to the final version of the
manuscript.
Ethics approval and consent to
participate
The study was conducted in accordance with the
Declaration of Helsinki and approved by the Institutional Ethics
Committee of the Hospital Civil de Guadalajara ‘Dr. Juan I.
Menchaca’. Written informed consent and assent were obtained from
the patients and their parents for participation in this study and
genetic testing. Healthy control blood donors provided written
informed consent to their blood being used for scientific
experimentation.
Patient consent for publication
Written informed consent was obtained from the
patients and their parents to publish the present study.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Fiesco-Roa MO, Giri N, McReynolds LJ, Best
AF and Alter BP: Genotype-phenotype associations in Fanconi anemia:
A literature review. Blood Rev. 37(100589)2019.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Molina B, Frías S and Ramos S: Anemia de
Fanconi, Parte 1. Diagnóstico citogenético. Acta Pediatr. Méx.
43:102–128. 2022.
|
|
3
|
Niraj J, Färkkilä A and D'Andrea AD: The
Fanconi anemia pathway in cancer. Annu Rev Cancer Biol. 3:457–478.
2019.PubMed/NCBI View Article : Google Scholar
|
|
4
|
García-de-Teresa B, Rodríguez A and Frias
S: Chromosome instability in Fanconi anemia: From breaks to
phenotypic consequences. Genes (Basel). 11(1528)2020.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Lach FP, Singh S, Rickman KA, Ruiz PD,
Noonan RJ, Hymes KB, DeLacure MD and Kennedy JA: Esophageal cancer
as initial presentation of Fanconi anemia in patients with a
hypomorphic FANCA variant. Cold Spring Harb Mol Case Stud.
6(a005595)2020.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Repczynska A, Pastorczak A, Babol-Pokora
K, Skalska-Sadowska J, Drozniewska M, Mlynarski W and Haus O: Novel
FANCA mutation in the first fully-diagnosed patient with Fanconi
anemia in Polish population-case report. Mol Cytogenet.
13(33)2020.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Joshi G, Arthur NBJ, Geetha TS, Datari
PVR, Modak K, Roy D, Chaudhury AD, Sundaraganesan P, Priyanka S, Na
F, et al: Comprehensive laboratory diagnosis of Fanconi anaemia:
Comparison of cellular and molecular analysis. J Med Genet.
60:801–809. 2023.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Colović M, Todorović M, Colović N, Terzic
T, Karadzic K and Jurišić V: Appearance of estrogen positive
bilateral breast carcinoma with HER2 gene amplification in a
patient with aplastic anemia. Pol J Pathol. 65:66–69.
2014.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Kimble DC, Lach FP, Gregg SQ, Donovan FX,
Flynn EK, Kamat A, Young A, Vemulapalli M and Thomas JW: A
comprehensive approach to identification of pathogenic FANCA
variants in Fanconi anemia patients and their families. Hum Mutat.
39:237–254. 2018.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Bobabilla-Morales L, Corona-Rivera A,
Corona-Rivera JR, Buenrostro C, García-Cobián TA, Corona-Rivera E,
Cantú-Garza JM and García-Cruz D: Chromosome instability induced in
vitro with mitomycin C in five Seckel syndrome patients. Am J Med
Genet A. 123A:148–152. 2003.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Lawce HJ and Brown MG: Cytogenetics: An
overview. In: The AGT Cytogenetics Laboratory Manual. John Wiley
& Sons, Inc., Hoboken NJ, pp25-85, 2017.
|
|
12
|
McGowan-Jordan J, Hastings RJ and Moore S
(eds): ISCN 2020-An International System for Human Cytogenomic
Nomenclature. Basel, Switzerland: Karger Publishers, 2020.
|
|
13
|
Cock PJ, Antao T, Chang JT, Chapman BA,
Cox CJ, Dalke A, Friedberg I, Hamelryck T, Kauff F, Wilczynski B
and de Hoon MJ: Biopython: Freely available Python tools for
computational molecular biology and bioinformatics. Bioinformatics.
25:1422–1423. 2009.PubMed/NCBI View Article : Google Scholar
|
|
14
|
LOVD v.3.0: The next generation in gene
variant databases. Retrieved April 25, Lovd, NL, 2024. Available
from: https://www.lovd.nl/3.0/home.
|
|
15
|
Mori M, Hira A, Yoshida K, Muramatsu H,
Okuno Y, Shiraishi Y, Anmae M, Yasuda J, Tadaka S, Kinoshita K, et
al: Pathogenic mutations identified by a multimodality approach in
117 Japanese Fanconi anemia patients. Haematologica. 104:1962–1973.
2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Zhu QY, Li PC, Zhu YF, Pan JN, Wang R, Li
XL, Ye WW, Ding XW, Wang XJ and Cao WM: A comprehensive analysis of
Fanconi anemia genes in Chinese patients with high-risk hereditary
breast cancer. J Cancer Res Clin Oncol. 149:14303–14313.
2023.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Goldsby RE, Perkins SL, Virshup DM,
Brothman AR and Bruggers CS: Lymphoblastic lymphoma and excessive
toxicity from chemotherapy: An unusual presentation for Fanconi
anemia. J Pediatr Hematol Oncol. 21:240–143. 1999.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Suzuki D, Kobayashi R, Yasuda K, Nakagawa
A, Morimoto T, Yabe M, Yabe H and Kobayashi K: Precursor-T
lymphoblastic lymphoma after unrelated bone marrow transplantation
in a patient with Fanconi anemia. J Pediatr Hematol Oncol.
33:22–24. 2011.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Violeta Filip P, Cuciureanu D, Sorina
Diaconu L, Maria Vladareanu A and Silvia Pop C: MALT lymphoma:
Epidemiology, clinical diagnosis and treatment. J Med Life.
11:187–193. 2018.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Shahid M and Firasat S: FANCA and
contribution of studies from Asian populations to the understanding
of fanca mediated Fanconi anemia. Genetika. 51:1197–1225. 2019.
|