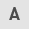Mechanisms of cisplatin sensitivity and resistance in testicular germ cell tumors and potential therapeutic agents (Review)
- Authors:
- Published online on: February 24, 2025 https://doi.org/10.3892/etm.2025.12832
- Article Number: 82
-
Copyright: © Zhan et al. This is an open access article distributed under the terms of Creative Commons Attribution License.
Abstract
1. Introduction
Testicular germ cell tumors (TGCTs) are a heterogeneous group of tumors that occur primarily in children and young men and are the most common tumor types in men aged 20-40 years. Based on histology, TGCTs are classified as seminoma (SE-TGCT) or non-seminoma (NSE-TGCT) tumors (1,2). The latter can be further classified as undifferentiated embryonal carcinoma (EC), choriocarcinoma, yolk sac tumor, differentiated teratoma or mixed tumors (a mixture of two or more components) (3-5). TGCTs develop from precancerous germ cell tumors in the renal tubule, mainly because of the failure of lymphocytes to mature properly during fetal or postnatal development. It progresses to aggressive TGCTs such as seminomas and non-seminomas after puberty, and environmental and genetic risk factors are significant factors in the susceptibility to TGCTs (6). Studies have shown that most testicular germ cell tumors originate from germ cell neoplasia in situ, which is thought to be due to the stagnation and transformation of the original germ cells. Seminomas have the same characteristics as germ cells or primordial germ cell formation in situ, whereas non-seminomas exhibit differential differentiation. Neoplasms and embryonic cell carcinomas are pluripotent and are thought to be responsible for the histological heterogeneity and mixed pathology of testicular germ cell tumors (7).
Platinum-based therapies are often used as first-line treatment for TGCTs in children and adults. TGCTs have a very high cure rate and are highly sensitive to cisplatin chemotherapy (8,9). The sensitivity of TGCTs to cisplatin may correlate with two key reactions: Inadequate repair of cisplatin-induced DNA damage and a hypersensitive apoptotic response (10). Various sources of endogenous and exogenous damage constantly assault the genome, among which DNA double-strand breaks (DSBs) are the most cytotoxic DNA lesions (11-13). To maintain chromatin stability, cells have developed a complex system of biochemical pathways called the DNA damage response (DDR) (14). DNA repair mechanisms have an indispensable role in cisplatin-induced cytotoxicity. DNA repair systems, including nucleotide excision repair (NER), homologous recombination (HR), nonhomologous end joining (NHEJ) and mismatch repair (15).
For metastatic disease, the overall disease-free survival rate of TGCTs is ~80%. Even in patients with advanced metastatic disease, complete remission can be achieved with systemic therapy and secondary removal of residual mass. However, ~10-20% of advanced TGCTs are resistant to platinum-based chemotherapy and have a poor prognosis or recurrence, and the treatments for these patients are poorly understood. Studies have shown that cisplatin resistance is mainly related to the apoptotic pathway, tumor cell cycle regulation-related factors, DNA methylation and DNA damage repair pathways. Among them, the regulated death pathway is mainly related to murine double minute 2 (MDM2)/p53, octamer-binding transcription factor 4 (OCT4)/phorbol-12-myristate-13-acetate-induced protein 1 (NOXA), platelet-derived growth factor receptor (PDGFR)/PI3K/AKT and insulin-like growth factor 1 receptor (IGF1R)/AKT, and certain related microRNA (miRNA) pathways. The main cell cycle regulatory factors were cyclin-dependent kinase (CDK)4/6 inhibitors and CDK2 inhibitor p21. During DNA methylation, hypermethylation can promote cisplatin resistance; therefore, since vir-like N6-methyladenosine (m6A) methyltransferase associated (VIRMA) is positively correlated with m6A in TGCTs, it stimulates the methylation process and thus cisplatin resistance. However, the DNA hypomethylating agent 5-azacytidine (5-aza) and the demethylating agent guadecitabine (SGI-110) promoted cisplatin sensitivity and inhibited cisplatin resistance. In the process of DNA damage repair, cisplatin resistance is positively correlated with NHEJ inhibition and increased HR, and a decrease in DNA damage response protein p53-binding protein 1 (53BP1) and DNA-dependent protein kinases (DNA-PKCs) can inhibit NHEJ and promote HR, thus promoting cisplatin resistance.
This review describes the sensitivity of TGCTs to cisplatin (also referred to as CDDP), the underlying mechanisms related to CDDP and the potential therapeutic agents for cisplatin-resistant TGCTs.
2. Molecular mechanisms involved in the sensitivity of TGCTs to cisplatin
In cisplatin-sensitive TGCT cells, cisplatin interacts with the DNA base to produce different forms of cisplatin-DNA adducts, of which in-chain crosslinking (>90%) is the dominant DNA adduct (15,16). The NER pathway can repair intra-chain crosslinking that can destroy DNA structures induced by cisplatin, and intra-chain crosslinking can be recognized by proteins associated with DNA repair (17). Certain proteins also act as inhibitors. For instance, high-mobility-group (HMG) proteins can inhibit NER by binding to DNA adducts. HMG box protein 4 (HMGB4) enhances the sensitivity of TGCTs to cisplatin by blocking the excision repair of cisplatin-DNA adducts (18). Furthermore, numerous essential proteins involved in NER have low expression levels in TGCTs, including xeroderma pigmentosum group A (XPA), xeroderma pigmentosum group F (XPF) and excision repair cross-complementing 1 (ERCC1) (19). A study showed that high levels of XPF and ERCC could decrease the sensitivity to cisplatin in TGCT cell lines, indicating that XPF and ERCC are rate-limiting factors for the repair of cisplatin-DNA cross-links in TGCTs, which contributes to remarkable cisplatin sensitivity (20). Furthermore, homologous recombination repair defects of DSB also cause high cisplatin sensitivity in TGCT cell lines (18). Of note, a related study showed that compared to normal testes, poly(ADP-ribose) polymerase (PARP) is overexpressed in TGCT and is involved in the repair of DNA single-strand breaks by base excision repair (21). Meanwhile, PARP inhibitors can promote the response of resistant EC cells to cisplatin by reducing their ability to repair damage, providing a potential therapeutic approach for resistant TGCTs (22).
The high sensitivity of TGCTs to cisplatin is due to the various apoptotic pathways induced by cisplatin, in which wild-type p53 plays a major role (23). p53 is frequently mutated in cancer as a tumor suppressor, but is not mutated in TGCTs, retains its wild-type configuration and is activated following exposure to chemotherapeutic agents (8). Cisplatin treatment increases the expression of p53 transcriptional target FAS death receptors and subsequently activates exogenous apoptotic pathways through the interaction of FAS with its ligand (24,25). It has also been shown that cisplatin treatment can upregulate the pro-apoptotic protein p53 upregulated modulator of apoptosis (PUMA) and NOXA (26). As a target protein of p53, NOXA is a Bcl-2 family protein that belongs to a subclass of BH3-only proteins that induce apoptosis via p53-dependent and/or p53-independent mechanisms (27), thus playing a crucial role in cisplatin-induced apoptosis of TGCTs cells (28). In addition, high expression of the pluripotent factor OCT4 (also known as POU5F1) is positively correlated with NOXA, leading to a hypersensitive apoptotic reaction in EC cells (23). OCT4 is also related to the regulation of p21 expression and p21 is associated with cisplatin sensitivity both in vivo and in vitro (28). Furthermore, crosstalk between exogenous and intrinsic apoptotic pathways may further enhance the apoptotic response (10) (Fig. 1).
3. Mechanisms of cisplatin resistance in TGCTs
Cisplatin resistance is a major clinical challenge and a thorough understanding of the mechanisms underlying the development of TGCT resistance will help improve the efficacy of TGCT therapy. Several mechanisms of cisplatin resistance have been identified, mainly related to apoptotic pathways, tumor cell cycle regulation (Fig. 2), methylation and DNA repair systems (Fig. 3). Specific mechanisms are outlined and potential treatments that currently exist are discussed in this chapter.
Apoptotic pathways. MDM2/p53 pathway
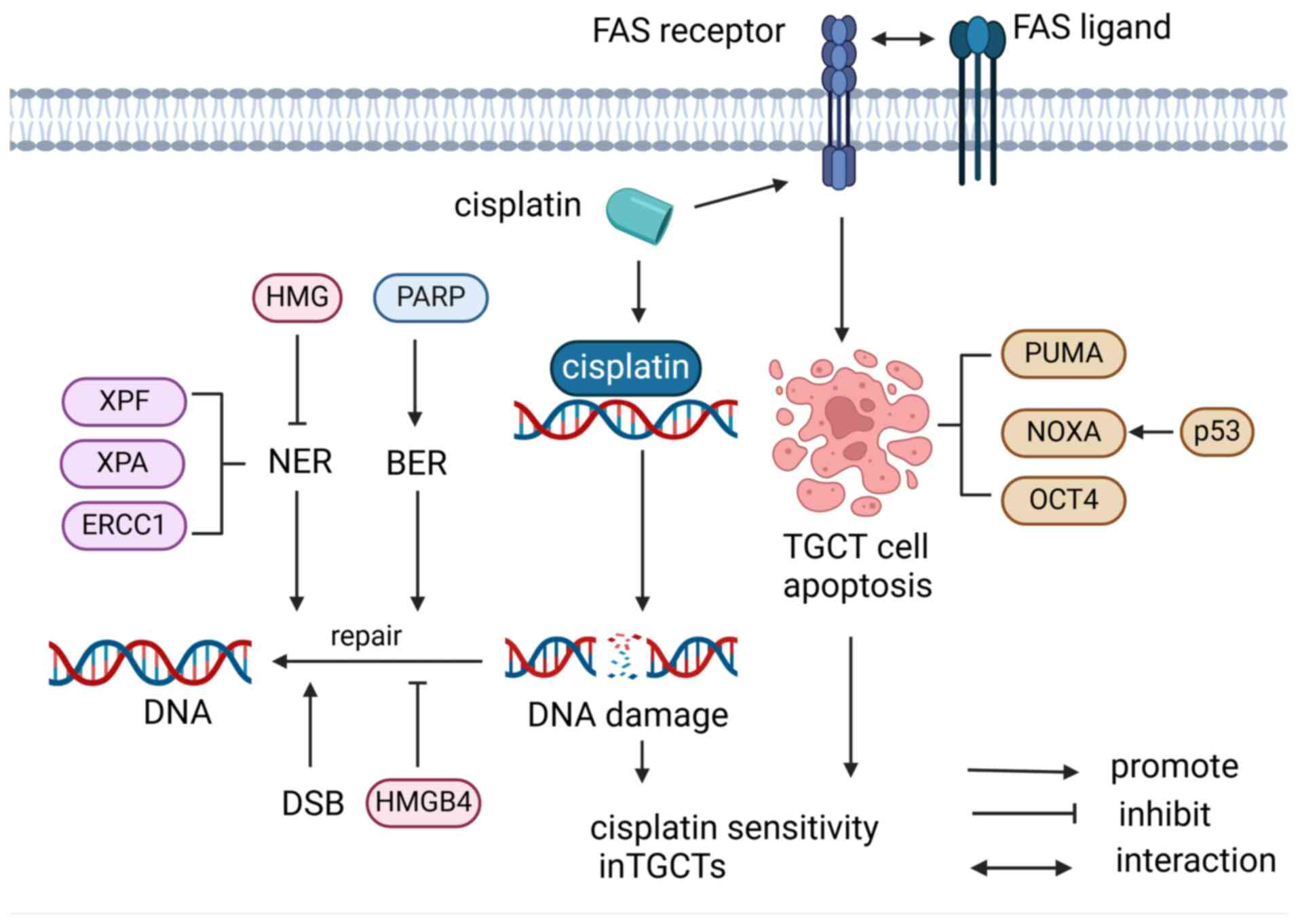
Signal changes in response to apoptotic transduction pathways mediated by DNA damage are important factors in cisplatin resistance in TGCTs. Overexpression of anti-apoptotic factors as well as decreased expression or dysfunction of pro-apoptotic factors can alter the induction of apoptosis. MDM2, a ubiquitin ligase, mediates ubiquitination and degradation of p53, inhibiting apoptosis and cell-cycle arrest for DNA repair (10,24) and recruit transcriptional co-repressors of p53(24). Loss of p53 function results in a lack of cell-cycle regulation and contributes to more aggressive tumors and chemotherapy resistance (29). p53 is a tumor suppressor and inactivation of p53 at the genetic or protein level is ascribed to cisplatin resistance in numerous tumor types, particularly in a subset of refractory TGCTs (28). In addition, p53 regulates MDM2 expression by binding to and positively regulating its promoter. More than 17% of human tumors exhibit MDM2 gene amplification, leading to cisplatin resistance (30). Compared with cisplatin-sensitive tumors, chemotherapy-resistant tumors showed a higher positive tendency for p53 and MDM2. However, inhibition of p53 activity was mediated by MDM2. A study on cisplatin-sensitive and resistant testicular carcinoma (TC) cell lines showed that the interaction between p53 and MDM2 requires higher doses of cisplatin to be disrupted in resistant cell lines (24). Besides, it has been shown that the reduced p53-induced apoptotic response detected in different TGCT cell lines in their relative cisplatin resistance is different, which is associated with the formation of MDM2-p53 complex (28).
OCT4/NOXA pathway. OCT4, a key stem cell transcription factor, is highly expressed in TGCTs and pluripotent stem cells (31,32). However, OCT4 and NOXA are underexpressed in cisplatin-resistant TGCT cells (28). OCT4 is a key regulator of cisplatin resistance (33). Knockdown of OCT4 reduces NOXA transcript levels, leading to the loss of NOXA protein, which decreases cisplatin hypersensitivity (23). In addition, it has been indicated that hypoxia induces resistance to TGCT, which may be related to the hypoxia-induced downregulation of OCT4. Under hypoxic conditions, overexpression of SUMO1 increases the sumoylation of OCT4, thereby reducing OCT4 protein stability (34).
PDGFR/PI3K/AKT pathway. Several studies have demonstrated that the PDGFR/PI3K/AKT signaling pathway has an important role in cisplatin resistance. PDGF has been shown to bind and phosphorylate its receptor PDGFR, subsequently activating the downstream PI3K/AKT pathway (35-38) In the activation of the PDGFR/PI3K/AKT signaling pathway, in which activated PI3K is recruited to the plasma membrane and phosphorylates phosphatidylinositol 4,5-diphosphate (PIP2), thus producing the second messenger phosphatidylinositol 3,4,5-triphosphate (PIP3), which can activate the serine/threonine kinase AKT through 3-phosphoinositide dependent protein kinase-1 (PDPK1) (39-41). Of note, this signal transduction process can be inhibited by phosphatase and tensin homolog deleted on chromosome 10 (PTEN) (42). Di Vizio et al (43) found that the tumor suppressor gene PTEN is widely expressed in germ cells and germ-cell neoplasia in situ but is virtually absent in 56% of seminomas, 86% of embryonal carcinomas and all teratomas. In cisplatin-resistant TGCTs cells, AKT is overactivated due to increased levels of PDGFRb and PDGF-b ligands at both the mRNA and protein levels. Subsequently, the PDGFR/PI3K/AKT pathway is over-activated, leading to the phosphorylation of p21 and its cytoplasmic accumulation, while increasing the phosphorylation of MDM2 which leads to the inhibition of P53-mediated apoptosis (44,45). Furthermore, phosphorylation of AKT directly inhibits forkhead box O3 (FOXO3a), which is responsible for the transcription of pro-apoptotic proteins such as PUMA and FAS ligands; thus, AKT indirectly negatively affects apoptosis (46).
IGF1R/AKT pathway. IGF1R has been implicated in numerous carcinogenic processes and can activate anti-apoptotic proteins and improve cell survival by promoting the PI3K/AKT pathway. Furthermore, the MAPK (Ras/Raf/MEK/ERK) pathway can be activated to promote proteins involved in the cell cycle and drive cell proliferation (47). IGF1R signaling is also associated with numerous cellular processes that promote metastasis. For instance, it is involved in the integrin pathway to induce RhoA-dependent movement through focal adhesion kinase and receptor for activated C kinase 1, and is associated with TGCT survival and migration. A study has shown that IGF1R is highly expressed in TGCT cell lines and IGF1R knockdown can reduce the growth of non-seminoma cells, leading to apoptosis (48). In cisplatin-resistant TGCTs, IGF1R levels are upregulated and the upregulation of IGF1R could promote the overexpression of AKT and phospho-AKT, leading to increased translocation of p21 in the cytoplasm, thereby inhibiting apoptosis and promoting cisplatin resistance (49). However, IGF1R silencing leads to decreased phosphorylated AKT levels, which allows the translocation of p21 to the cytoplasm for transfer to the nucleus, thereby restoring cisplatin sensitivity. Therefore, inhibition of the IGF1R in combination with chemotherapy may promote re-sensitization in the treatment of chemotherapy-resistant diseases (47).
Factors associated with tumor cell cycle regulation. CDK4/CDK6
CDKs are key enzymes that control cell cycle progression, and CDK4/6 are targets of cell cycle checkpoint inhibitors such as palbociclib (50). It has been shown that CDK4/6 inhibitors can induce cell cycle arrest and apoptosis in different types of TGCTs, indicating that CDK4/6 inhibitors have therapeutic potential for TGCTs. Palbociclib has a concentration-dependent inhibitory effect on TGCT cell survival and can induce apoptosis by decreasing the phosphorylation of retinoblastoma protein (RB). However, palbociclib can induce a rapid increase in CDK4/6 protein levels in NT2/D1 cells, a TGCT cell line, thus limiting its cytotoxic effects. It was demonstrated that the NT2/D1 cell line could overcome the effect of palbocinb and develop resistance, but the specific mechanism remains elusive and requires further investigation (51).
p21. A previous study showed that high cytoplasmic expression of the CDK inhibitor p21 is a critical factor in cisplatin resistance in testicular EC. Nucleic p21 controls the cell cycle and DNA replication, whereas cytoplasmic p21 is involved in the inhibition of apoptosis. In most patients with refractory testicular cancer, cytoplasmic p21 is highly expressed and high cytoplasmic p21 can form complexes with CDK2 and apoptosis signal-regulating kinase 1 (ASK1). The formation of complexes inhibits the pro-apoptotic functions of CDK2 and ASK1, thereby protecting EC cells from cisplatin-induced apoptosis, leading to cisplatin resistance. Phosphorylated (P-)AKT-mediated p21 phosphorylation is critical for the localization of p21 in the cytoplasm, whereas p-AKT inhibition (LY294002) promotes p21 translocation to the nucleus, thus reducing p21-CDK2 complex formation and sensitizing EC cells to cisplatin in a CDK2-dependent manner (45).
Different types of TGCT sensitive to cisplatin have different p21 expression levels. For instance, p21 is almost undetectable in cisplatin-sensitive seminomas and EC but is significantly higher in cisplatin-sensitive choriocarcinoma and mature teratoma than in cisplatin-resistant ones (45). In mature teratomas, intrinsic chemotherapy resistance is associated with the upregulation of various factors that affect p21-induced cell cycle arrest (52).
Cisplatin is a highly cytotoxic drug that is effective against EC by inducing apoptosis in numerous cell types. Cisplatin treatment of EC cells increased p53 and MDM2 levels, activated the Fas apoptotic pathway and induced apoptosis; however, the expression level of p21 was almost unaffected. However, in EC cells, gamma irradiation increased the levels of p53 and MDM2 and significantly induced cytoplasmic p21 without inducing cell cycle arrest or apoptosis. These results suggested that cytoplasmic p21 has an important role in preventing DNA damage-induced apoptosis of EC cells (45). Studies have also shown that high cytoplasmic p21 expression and cisplatin resistance of EC/TC are negatively associated with the expression of OCT4 and miR-106b but positively associated with p53 and MDM2 (28,45,53). Therefore, targeting cytoplasmic p21 may provide a new strategy for the treatment of chemotherapy-resistant testicular cancer (45) (Fig. 2).
Role of epigenetic mechanisms
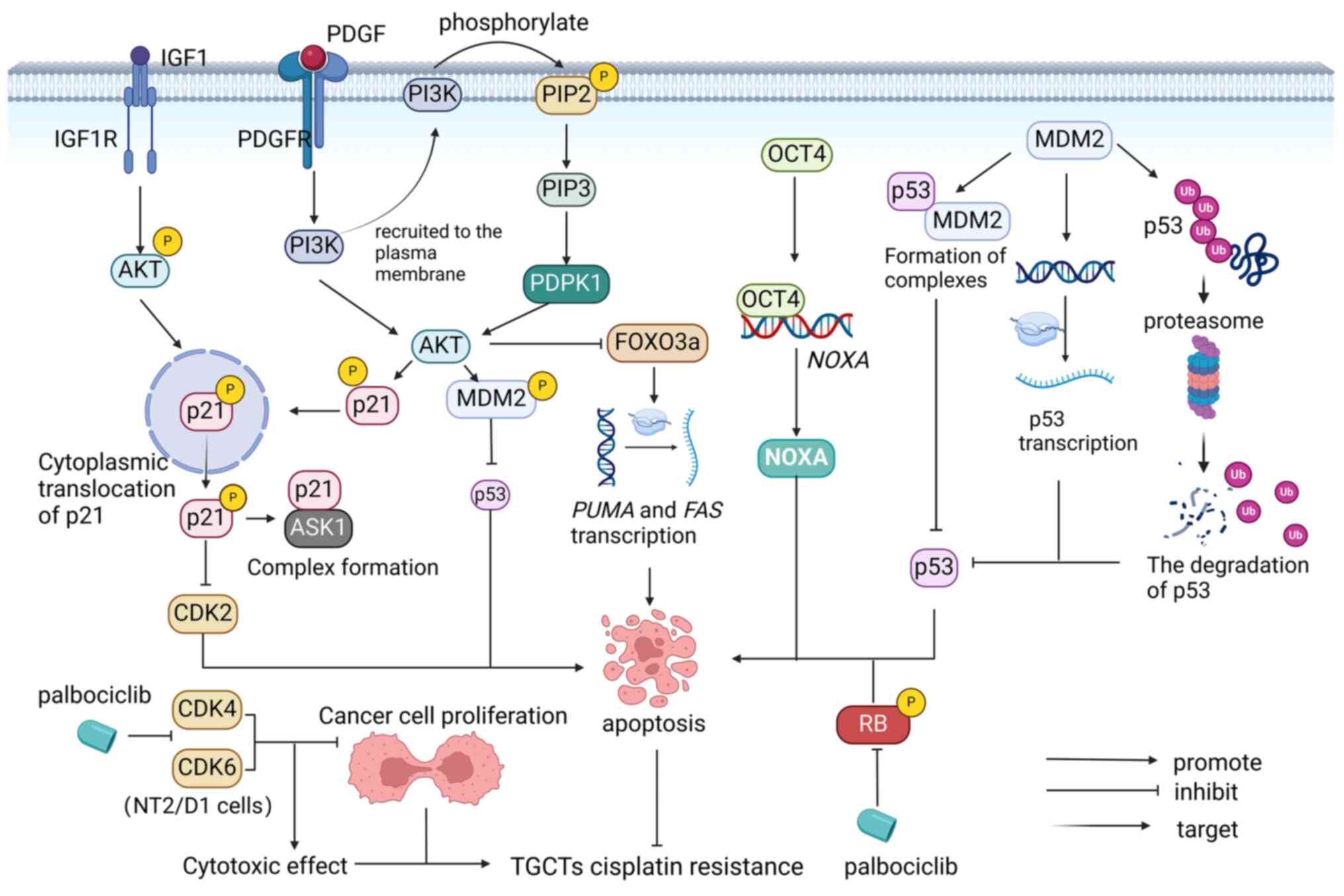
Epigenetics plays an important role in the development and classification of TGCTs (54). DNA methylation is one of the major epigenetic modifications that have a key role in cisplatin resistance in TGCTs (55,56). Seminomas are undifferentiated tumors that are hypomethylated and exhibit low cisplatin resistance (18). More differentiated non-seminomas have higher DNA methylation levels (18). Embryonic cancers have moderate levels of DNA methylation and are sensitive to cisplatin, but may also acquire cisplatin resistance (18). Teratomas, choriocarcinomas and yolk sac tumors have the highest levels of DNA methylation, which is closely related to cisplatin resistance (18). A study has shown that global remodeling of DNA methylation is a critical factor that mediates TGCT resistance and cisplatin hypersensitivity (56).
m6A. m6A is the most abundant of modifications in mRNA, and this process includes ‘writers’, ‘erasers’ and ‘readers’ of m6A methylation, proteins that can add, remove or recognize m6A-modified sites, respectively, and alter biological processes accordingly (57). It can be introduced into the mRNA by different families of enzymes. They are assembled to form methyltransferase complexes that participate in methylation modification, mainly by the catalytically active component Methyltransferase-like protein 3 (METTL3) and other cofactors that recruit the complex, such as VIRMA, Wilms' tumor 1-associating protein (WTAP) and METTL14. Various m6A-related proteins have been shown to be differentially expressed in TGCT subtypes (as biomarkers for the disease) and in cisplatin-resistant and cisplatin-sensitive TGCT cells. VIRMA and METTL3 were found to be more important. In the SE subtype of TGCTs, VIRMA is significantly upregulated at the mRNA and protein levels, promoting the progression of multiple malignancies by regulating cell cycle progression, migration, invasion, apoptotic resistance and tumor growth in an m6A-dependent manner. Furthermore, VIRMA introduced m6A modifications to TGCTs and contributed to tumor aggressiveness and cisplatin resistance in TGCT cells both in vitro and in vivo by modulating the DDR (58).
METTL3 and IGF2 mRNA-binding protein 1 (IGF2BP1) play key roles in enhancing cisplatin resistance in seminoma by enhancing transcription factors to activate m6A methylation of enhancer binding protein 2C (TFAP2C).
METTL3, as the ‘writer’ of m6A, enhanced the m6A methylation level of TFAP2C mRNA and improved the stability of TFAP2C mRNA, thus enhancing the viability of TGCT-resistant cells under cisplatin treatment. Furthermore, IGF2BP1 functions as a TFAP2C m6A ‘reader’, enhances TFAP2C mRNA stability and is involved in cisplatin resistance (59).
Regulatory mechanisms of miRNAs in TGCTs. Of note, in addition to the signaling pathways mentioned above, miRNAs also play a critical role in oncogenesis in TGCTs by interacting with different mechanisms. In general, miRNAs are involved in the p53, epidermal growth factor receptor, Wnt/β-catenin and PTEN/AKT/mTOR pathway [see ref. (60) to review the regulatory mechanisms of miRNAs in TGCTs]. In addition, miRNAs play different roles in CDDP-resistant and -sensitive TGCT cells. On the one hand, miR-302a and miR-106b mainly inhibit the resistance to cisplatin by targeting p21, while miR-371-373 increases cisplatin resistance by targeting p53. By contrast, miR-106b-5p reverses resistance to cisplatin by targeting testis development related 1. miR-383 can improve cisplatin sensitivity by targeting histone H2AX, the adaptor protein of PP1 phosphatase and CDK4. miR-514a-3p decreases cisplatin resistance by targeting paternally expressed gene 3. Furthermore, miR-27b-3p, miR-31-5p, miR-125b-5p, miR-218-5p, miR-199a-5p and miR-324 were upregulated in cisplatin-resistant TCGT cells, while miR-374b-5p, miR-320a, miR-20b-5p, miR-375-5p, miR-17-5p, miR-106a, miR-378a-3p and miR-30e-3p were downregulated in cisplatin-resistant TCGT cells. However, miR-378a-3p was downregulated only in the CDDP-resistant TGCT cell line 1411HP, and miR-30e-3p was only down-regulated in the CDDP-resistant TGCT cell line 1777NRpmet (Table I).
DNA repair systems. The balance of DNA repair pathways
Cisplatin can cause several types of DNA damage, including inter-strand crosslinking (ICLs) (61). ICLs covalently join two strands of a DNA double-strand, blocking basic cellular processes such as DNA replication and leading to cell death. Cisplatin can also induce ICL-associated formation of DSB, which can be repaired by HR during the S/G2 phase of the cell cycle using sister chromatids as a repair template. One mechanism of TGCT resistance to cisplatin is its enhanced ability to repair DSB through HR (62).
In addition to HR, NHEJ is also involved in the restoration process and is the main DSB repair pathway throughout the cell cycle, accounting for almost all DSB repair, except in the S and G2 phases. NHEJ relies on Ku binding to the end of DNA, thereby improving the affinity of the NHEJ enzyme component consisting of polymerase (Pol µ and Pol λ), nuclease (Artemis· DNA-PKCs complex) and ligase (XRCC4-like factor-X-ray repair cross-complementing protein 4-DNA ligase IV complex), promoting DNA repair (63). NHEJ acts throughout the cell cycle, mediating the direct reconnection of DSB ends in an error-prone manner (64). In the process of repair, HR and NHEJ compete with each other to repair DSB, and the choice of repair pathway depends on the balance of pro-NHEJ and pro-HR factors at the DSB site. Key pro-NHEJ factors include 53BP1, the ATP-dependent DNA helicase 2 subunit KU70/KU80 and DNA-PKCs. 53BP1 inhibits extensive excision of DNA ends by assembly on DSB-side chromatin, repairing far away from HR, whereas KU70/KU80 and DNA-PKCs are involved in the later steps of DSB blunt end ligation. Cisplatin resistance was found to be positively associated with NHEJ inhibition and an increase in HR, and both often occur together (65).
Cisplatin-resistant cell lines have an NHEJ-less phenotype, with a decreased basal expression of 53BP1 and DNA-PKC proteins, thus forming fewer 53BP1 lesions after cisplatin treatment. Similarly, reduced expression of DNA-PKCs and 53BP1 inhibited the repair of DSB through the action of NHEJ, but could be effectively repaired by HR. Therefore, inhibition of 53BP1 protein expression and DNA-PKC activity can antagonize cisplatin cytotoxicity (3).
XPA. NER is an important DNA repair pathway that promotes ICL repair by mediating lesion recognition and incision through >30 DNA repair-associated proteins. Among these, the XPA protein has a pivotal role in DNA damage verification and assembly of the NER incision complex (66). One related study has shown that XPA expression is inversely associated with the prognosis of patients with TGCT and that increased co-expression of ERCC1, XPF and XPA is also associated with unfavorable overall survival. In primary TGCTs, increased XPA expression, and, to a lesser extent, NER co-expression, may promote increased DNA repair capacity; therefore, XPA may be a new promising therapeutic target in TGCTs (67).
REV7. REV7 (also known as MAD2L2, MAD2B or FANCV) is a HORMA protein and is regulated through stable structural rearrangement (68). Under physiological conditions, endogenous and exogenous agent-induced DNA damage is commonly repaired using the DNA repair system (DDR). If the damage remains unrepaired, the damage-tolerance system is activated. Translesion synthesis (TLS) is a major mechanism underlying DNA damage tolerance (69). REV7 is highly expressed in human TGCT tissues (70) and is a multifunctional protein involved in TLS, DNA repair, cell cycle regulation, gene expression and histone modification through protein-protein interactions. REV7 plays a critical role in the DSB repair pathway. Mechanistically, REV7 and 53BP1 promote non-homologous terminal junctions at DSB sites, thereby promoting DNA damage repair. Knockdown of REV7 could lead to the downregulation of DNA repair-associated genes, as well as cell cycle checkpoint genes in TCGT cells, while promoting the transcription of pro-apoptotic genes. Therefore, REV7 inactivation antagonizes the cisplatin resistance induced by DNA damage-associated DSB accumulation. Collectively, REV7 loss contributes to the effective cytotoxicity of cisplatin in cancer cells, allowing drug-resistant TGCTs to regain their sensitivity to cisplatin, suggesting that targeting REV7 could be a potential treatment for patients with elevated REV7 or cisplatin resistance (71).
Neddylation
Neddylation refers to the process by which the small ubiquitin-like molecule neuronal precursor cell expression and development down-regulated protein 8 (NEDD8) is coupled to a target protein to alter its function, stability or subcellular position. The activation of neddylation increases the degradation of tumor suppressor proteins 21, p27 and p53, thereby promoting tumor progression. It has been observed in lung, breast and pancreatic cancer. A recent study found that the overexpression of neddylation-related protein NEDD8-activating enzyme E1, which can lead to cisplatin resistance, and the neddylation inhibitor MLN4924 and cisplatin co-induced apoptosis and cell cycle arrest, effectively reducing the viability of TCGTs. Therefore, MLN4924 in combination with cisplatin could be a potential treatment for resistant TGCTs (72).
Other mechanisms
Enhancer of zeste homolog 2 (EZH2) is an epigenetic transcriptional suppressor involved in cell cycle control and cell fate determination (73). Genetic, transcriptional and post-transcriptional dysregulation of EZH2 are frequently observed in numerous cancer types (74). They play different roles in different types of tumor cells. Studies have shown that EZH2 upregulation increases cisplatin resistance in lung, ovarian and breast cancers, and the combination of EZH2 inhibitors and cisplatin may be beneficial for oncotherapy (75-77). However, at 833 K, in the NT2/D1 and 2102EP cell lines of TGCTs, decreased EZH2 expression resulted in cisplatin resistance. EZH2 is a negative regulator of cisplatin resistance in TGCTs. In addition, cisplatin resistance in TGCTs is associated with decreased H3K27 methylation (78), ubiquitination of H2A-K119 and expression of (B cell-specific Moloney murine leukemia virus integration site-1) BMI1(79). However, DNA methyltransferase 3b knockdown leads to the induction of histone H3 lysine 27 (H3K27m3), EZH2 and BMI1 expression, thereby enhancing the sensitivity of TGCT cells to cisplatin (80). Gain of chromosome 3p25.3 was detected in all cisplatin-resistant TGCT cell lines, and the copy number in this region was associated with the level of cisplatin resistance. Gains in this region were detected at a low frequency in primary tumors and at a higher frequency in relapsed and/or cisplatin-resistant tumors. Increased chromosome 3p25.3 is associated with shorter progression-free and overall survival (81).
Mitotic arrest deficient-2 (MAD2) is a key activator of the mitotic spindle assembly checkpoint (SAC) (82), which defers the start of anaphase during mitosis when microtubules are broken or centromeres are mislocated or unattached. SAC damage causes cells to enter anaphase prematurely, resulting in chromosomal missegregation, aneuploidy or chromosomal instability (83). Abnormal MAD2 expression has a role in chromosomal abnormalities in different types of cancer cells, including TCs. Mad2γ is a new isomer of MAD2 derived from the alternative splicing of MAD2 pre-mRNA without including exons 2 and 3. Studies have shown that the expression of MAD2γ is markedly increased in patients with TGCT who are resistant to cisplatin chemotherapy, suggesting that MAD2γ overexpression is related to cisplatin resistance. Furthermore, the expression of MAD2γ increased after cisplatin treatment. Research has shown that SAC destruction is associated with resistance to DNA-damaging chemotherapy drugs. The N-terminal domain of MAD2a can interact with proteins that participate in the DNA damage response and is thought to be important for chemotherapy resistance. Similarly, MAD2γ retains the N-terminal domain of MAD2a and is related to chemotherapy drug resistance; its overexpression promotes cisplatin resistance (84). It was found that the mRNA levels of the meiosis-related gene Testis-expressed gene 11 (TEX11) and mobility-group nucleosome-binding gene High-mobility group nucleosome-binding domain 5 (HMGN5) are significantly upregulated in TGCTs, and knockdown of HMGN5 or TEX11 in cisplatin-resistant TGCTs significantly reduces the activity of cisplatin-resistant TGCT cells (85).
4. Potential therapeutic drugs
In the past 50 years, significant progress has been made in the treatment of TGCTs, particularly advanced diseases, and the cure rate has increased from 25% in the 1970s to nearly 80% (86). Radical orchiectomy combined with chemotherapy is currently considered the standard treatment (87,88). As TGCT is highly sensitive to chemotherapy, particularly cisplatin chemotherapy, the combination of cisplatin, etoposide and bleomycin or ifosfamide remains the first-line treatment (10,89,90). These combination chemotherapy regimens have shown high efficacy in the treatment of early and advanced TGCT, resulting in a significant increase in the 5-year survival rate to >95% (91,92). However, owing to the long-term side effects of platinum chemotherapy, patients still experience long-term toxicity after being cured (93,94). Approximately 20-30% of patients cannot be cured by standard treatment, particularly those who develop cisplatin resistance, and the prognosis is still unfavorable (95,96). Patients with recurrent TGCT are usually treated with high-dose chemotherapy, but this treatment results in severe adverse effects and cytotoxicity (97-99). Therefore, there is an urgent need to develop new, less toxic drugs to overcome this challenge and to improve the health status and quality of life of patients.
Numerous studies have shown that epigenetic drugs can induce tumor suppressor gene expression, making cancer sensitive to chemotherapeutic drugs again (100-102). Of note, TGCT tumorigenesis is closely associated with epigenetic and drug resistance mechanisms. Therefore, epigenetic drugs may be a viable treatment option for TGCT treatment (103-105). Numerous agents have been tested in TGCT cell lines and animal models with promising results (103,106-108). DNA methylation is a key epigenetic regulatory mechanism that silences gene expression by adding methyl groups to promoter regions of genes through the action of DNA methyltransferase (DNMT). This phenomenon often leads to loss of tumor suppressor gene function (109-112). DNA methylation is mainly mediated by DNMT1, whereas DNA remethylation is mainly mediated by DNMT3A and DNMT3B (113). Studies have shown that chemical DNA demethylation can restore the expression of silencing genes, e.g., using a first-generation DNMT inhibitor (DNMTI) 5-aza. The cytotoxic activity of 5-aza as a ribonucleoside is based on its wide range of effects on DNA methylation patterns, cellular transcriptomes and gene expression profiles. It can bind to DNA and RNA and is primarily used to treat myelodysplastic syndromes (114,115). A previous study demonstrated that 5-aza could contribute to decreased cisplatin resistance in a seminoma cell line (116). Oing et al (107) investigated the effects of 5-aza on two different EC cell lines (NCCIT carrying TP53 mutations and 2102Ep carrying wild-type TP53), using isogenic resistant sublines 2102EP-R and NCCIT-R to establish an acquired cisplatin resistance model system in vivo and in vitro. They found that nanomolar doses of EC cells are highly sensitive to 5-aza regardless of cisplatin sensitivity and that 5-aza at nanomolar concentrations can overcome cisplatin resistance and induce an intense and long-lasting apoptotic response in EC cells (107). DNMTIs such as 5-aza and decitabine (DAC) have been approved by the US Food and Drug Administration (FDA), and breakthroughs have been made in the treatment of hematological cancers by blocking the activity of DNMT (117). DNMTIs have also shown potential in preclinical experimental studies for the treatment of TGCT (106,118). Beyrouthy et al (118) in their experiments with cisplatin-resistant embryonic cancer cells pretreated with 5-aza, found that the drug not only affected resistant cells, but also through the induction of p53 target genes or through corresponding promoter methylation to the expression of other genes (such as MGMT, RASSF1A and HOXA-9), to restore sensitivity to cisplatin chemotherapy. Collectively, these results suggest that 5-aza can promote apoptosis and overcome cisplatin resistance in non-cysteine germ cell tumor cells. As mentioned previously, 5-aza is a first-generation DNMTI. Notably, SGI-110, a second-generation DNMTI, has been used to treat refractory TGCTs (106). Issa et al (119) evaluated the effects of SGI-110 (a demethylating agent) on EC cells and in cisplatin-resistant non-seminoma testicular cancer animal models and found that cisplatin-resistant cells and tumors derived from EC were highly sensitive to SGI-110, which showed potential antitumor effects in EC (106). Notably, very low doses of SGI-110 as a single agent eliminated the progression of cisplatin-resistant EC tumors (106). In addition, they conducted a Phase I trial combining SGI-110 with cisplatin in patients with relapsed cisplatin-resistant TGCT (120), indicating that the combination of SGI-110 and cisplatin displayed good efficacy in patients with platinum-refractory germ cell cancer (120). Furthermore, Lobo et al (105) tested a newly synthesized flavonoid-derived compound, MLo1302, in TGCT cell lines and found that it significantly reduced tumor cell activity and induced apoptosis and cell cycle arrest by reducing DNMT expression.
In addition to DNMTIs, histone deacetylase inhibitors (HDACIs) related to histone modifications have also shown promising results in preclinical studies (103,121,122). Lobo et al (103) used FDA-approved HDACIs, Belinostat and Panobinostat, and observed that these agents reduced cell viability and induced cell cycle arrest and apoptosis in cisplatin-sensitive and cisplatin-resistant TGCT cell lines. In addition, Steinemann et al (122,123) used a novel dual-mode compound called animacroxam, which was conjugated to an HDAC-inhibiting component and a cytoskeletal-disrupting component, to show significant anti-proliferation, cell cycle arrest and apoptosis-inducing effects in TGCT cell lines, and no nonspecific cytotoxicity was observed. Of note, animacroxam also reduced glucose uptake in TGCT and inhibited the expression of glycolytic enzymes, leading to a collapse in glycolytic energy production (122,123). Nettersheim et al (124) used the HDACI romidepsin and found that it was highly toxic to cisplatin-resistant TGCT cells. They also showed that combined treatment with the glucocorticoid dexamethasone further enhanced the expression of romidepsin effector factors and reduced TGCT cell activity more significantly than monotherapy (124). Notably, the bromodomain inhibitor JQ1, which acts by interfering with the function of bromodomain and extra terminal (BET) (125), increased apoptosis and growth arrest in TGCT cells, reduced tumor size and vascular density in mice and was more effective in combination with romidepsin (108). Burmeister et al (126) treated cisplatin-resistant TGCT cells with dual HDAC and BET inhibitors, resulting in decreased cell viability and impairment of the cell cycle. Furthermore, in the TGCT xenograft model, the dual inhibitor significantly reduced the tumor burden.
Other targeted agents have also shown potential for the treatment of cisplatin-resistant TGCT. TLS protects cells against DNA damage. Recently, Lengert et al (127) found that TLS inhibitors may overcome the resistance of TGCTs to cisplatin, and the targets can be diverse. MG-132 (carbobenzoxyl-L-leucyl-L-leucyl-L-leucine) is a peptide aldehyde that effectively blocks the proteolytic activity of the 26S proteasome complex and prevents TLS in human cancer cells but not in normal cells (128). However, the exact mechanism through which MG-132 inhibits TLS remains elusive. However, MG-132 showed significant cytotoxicity in cisplatin-resistant TGCT cell lines in the nanomolar range (127). In addition, Hasibeder et al (129) demonstrated the potential of the phytoestrogens Belamcanda chinensis extract and tectorigenin to inhibit TGCT.
Combination therapy is a promising therapeutic strategy. For cisplatin-resistant TGCTs, triple therapy with gemcitabine, oxaliplatin and paclitaxel is the standard of care (130). In addition, the combination of palbociclib and cisplatin greatly reduced TGCT cell viability in vitro and in zebrafish embryos and the drug combination also played a positive role in cell recovery after toxic damage (51). In addition, it has been reported that inhibitors specifically targeting PARP, MDM2 or AKT/mTOR combined with cisplatin have been shown to successfully overcome cisplatin resistance, which was also demonstrated in patient-derived xenograft models (10). Therefore, these agents may be promising candidates for the treatment of TGCT, particularly cisplatin-resistant TGCT; however, their application in patients with TGCT still requires further clinical studies and validation (Table II).
5. Conclusions and future perspectives
Cisplatin is the first-line drug for TGCT; therefore, it is necessary to study the mechanisms of its sensitivity and resistance.
In the mechanism of TGCT sensitivity to cisplatin, the expression of the NER essential proteins XPA, XPF and ERCC1 was significantly associated with TGCT sensitivity to cisplatin. In addition, defects in DSB homologous recombination repair also lead to high sensitivity of TGCT cell lines to cisplatin. Furthermore, the expression of the apoptosis-related factor p53 and its related proteins such as NOXA and OCT4 is also closely related to cisplatin sensitivity.
In the mechanism of TGCT resistance to cisplatin, studies have shown that cisplatin-resistant cells are mainly associated with the apoptotic pathway (24), DNA methylation, tumor cell cycle regulation-related factors and DNA damage repair pathways.
In the apoptotic signaling pathway, MDM2 can mediate the ubiquitination and further degradation of p53, thereby inhibiting apoptosis and cell cycle arrest and promoting cisplatin resistance (24). PDGF phosphorylates PDGFR, thereby promoting the PDGFR/PI3K/AKT signaling pathway, resulting in enhanced AKT phosphorylation. Enhanced phosphorylation of p21 and MDM2 by AKT promotes the degradation of p53. In addition, decreased expression of OCT4 could result in a decrease in NOXA, which inhibits apoptosis and promotes cisplatin resistance (23). By contrast, inhibition of FOXO3 expression leads to the suppression of PUMA and FAS, thereby inhibiting the process of programmed death (35-37). Similarly, upregulation of IGFR promotes AKT phosphorylation and inhibits apoptosis (49). Regarding the mechanism of tumor cell cycle-related regulatory factors, p-AKT could lead to the enhancement of p21 phosphorylation, which leads to an increase in cytoplasmic p21, and high expression of cytoplasmic p21 could inhibit cell apoptosis and promote drug resistance. Cytoplasmic p21 is a new therapeutic target or biomarker that also plays an important role in chemotherapy resistance in other tumor cells. For instance, nuclear protein 1 contributes to chemotherapy resistance in breast cancer by inducing Akt-mediated phosphorylation and the subsequent cytoplasmic relocalization of p21(131). In addition, cytoplasmic p21 is a potential predictor of cisplatin sensitivity in ovarian cancer (132). Therefore, whether it is possible to regain sensitivity to chemotherapy by inhibiting the expression of p21 in the cytoplasm or altering its localization needs to be further investigated.
In addition, studies have shown that increased methylation can promote cisplatin resistance, and VIRMA, METTL3 and IGF2BP1 can promote the upregulation of m6A and thus promote drug resistance, whereas DNMT1 inhibitors 5-aza and SGI-110 can inhibit methylation and lead to cisplatin sensitivity, which can be used as a potential treatment for resistant TGCTs. However, its practicability requires further investigation. In the DNA repair system, 53BP1 was far from HR. Upregulation of HR and downregulation of NHEJ were positively correlated with cisplatin resistance, and the downregulation of 53BP1 and DNA-PKCs may promote cisplatin resistance (3,133).
In the TCGT NT2/D1 cell line, the CDK4/6 inhibitor palbocinib enhanced CDK4/6 protein expression in NT2/D1 cells, thereby inhibiting the cytotoxic effect of cisplatin and inducing cisplatin resistance by influencing tumor cell cycle regulation-related factors (51). However, the specific mechanisms involved require further investigation. For instance, whether an increase in CDK4/6 protein expression directly leads to cisplatin resistance or whether other factors lead to cisplatin resistance requires further exploration (51). In addition, palbocinb reduced RB phosphorylation in TGCTs, thus promoting apoptosis and inhibiting cisplatin resistance. Whether RB phosphorylation is inhibited in NT2/D1 cells to promote cisplatin resistance should be investigated in the future (134). MG-132 has been proven to promote apoptosis in refractory TGCTs and it can treat resistant TGCTs alone or in combination with cisplatin; however, its mechanism of action remains elusive (127). Study has shown that p53 promotes apoptosis in cisplatin-resistant TGCTs. Notably, MDM2-mediated ubiquitination and degradation of p53 can result in the downregulation of p53 and inhibition of apoptosis, contributing to cisplatin resistance (135). Therefore, it is speculated that MG-132, a proteasome inhibitor, promotes apoptosis by inhibiting the ubiquitination and degradation of p53, thereby inhibiting cisplatin resistance, which requires further investigation (136).
In DNA repair systems, REV7 can bind to 53BP1 to promote NHEJ, and depletion of REV7 leads to the accumulation of DNA double-strand breaks and activation of cell apoptosis, thereby inhibiting cisplatin resistance (137). However, study has shown that cisplatin resistance is negatively related to NHEJ and that REV7 upregulation could promote NHEJ, which inhibits cisplatin resistance (71). However, study has shown that REV7 depletion can also inhibit cisplatin resistance, and whether this is a contradiction requires further discussion. In addition, the DNA repair-related factor XPA is highly expressed in TGCTs, and its association with cisplatin resistance in TGCTs should be further studied (138). In cisplatin-sensitive and drug-resistant TGCT cells, there were differential expressions of the L1TD1, GAL, NTF3, NANOG, WNT6, POU5F1, IGFBP2, IGFBP7, PCP4, ZFP42, ID2, TRIB3 and SLC40A1 genes (139). However, whether their corresponding proteins are differentially expressed has not yet been studied, and whether these proteins can be used as potential therapeutic targets needs to be studied (134).
In cisplatin-resistant TGCT therapy, studies have shown that methylation is associated with cisplatin-resistant TGCT; therefore, DNMTIs such as 5-aza and DAC SGI-110 can restore sensitivity to cisplatin-resistant chemotherapy by inhibiting methylation. In addition, drugs such as HDACIs, belinostat and panobinostat can exert therapeutic effects by inducing apoptosis. Furthermore, TLS inhibitors such as MG-132 and lomidoxin can enhance the toxic effects on cisplatin-resistant TGCT cells. Although the efficacy of certain drugs has been demonstrated in patient-derived xenotransplantation models, their use in patients with TGCT still requires further clinical research and validation.
In addition, similar to the mechanism of cisplatin resistance in other tumors, such as in ovarian yolk sac tumor (oYST), long-term cisplatin exposure resulted in a 7-fold increase in the IC50 concentration in resistant cells. Drug-resistant cells showed significantly increased expression of prominin-1 (CD133), ATP-binding cassette subfamily G member 2 and aldehyde dehydrogenase 3 subtype A1 (ALDH3A1), decreased gene and promoter methylation, increased expression of ALDH1A3 and increased overall ALDH enzyme activity (140). The study showed that high expression of ALDH1A3 and elevated activity of ALDH were detected in cisplatin-resistant TCGT cells, which is identical to oYST (141). However, in TGCTs, reduced methylation promotes cisplatin resistance, in contrast to that in oYST. However, the role of other factors in the cisplatin resistance mechanism of TGCTs remains elusive and may be considered a potential factor. In medulloblastoma, anti-miR-31 enhances cisplatin resistance through the PI3K/AKT and NF-κB pathways (142). Similarly, in glioblastoma, circPTN (a newly discovered circular RNA) contributes to cisplatin resistance through PI3K/AKT signaling via the miR-3-542p/PIK3R3 pathway (143). The PI3K/AKT pathway also affects cisplatin resistance in TGCTs; therefore, it is worth exploring whether miR-31 and circPTN also play a role in cisplatin resistance in TGCTs. A previous study showed that PD-L1 is highly expressed in TGCTs (144). Studies have shown that in non-small cell lung cancer (NSCLC), NSCLC cell-derived programmed death-ligand 1 (PD-L1)-containing exosomes promote cell stemness and increase the resistance of NSCLC cells to cisplatin (145), and high expression of PD-L1 in cancer cells drives immune-independent, cell-intrinsic functions, leading to resistance to DNA-damaging therapies, such as cisplatin (146). In NSCLC, the increase in CDK5 could lead to the downregulation of F-box only protein 22, which leads to the downregulation of PD-L1 ubiquitination, and the increase in PD-L1 levels leads to cisplatin resistance (146). In medulloblastoma, CDK5 and PD-L1 also play a role in cisplatin resistance; therefore, the role of CDK5 in cisplatin resistance of TGCTs is also worth discussing (146).
Acknowledgements
The figures were created using BioRender.
Funding
Funding: This work was supported by the Zhejiang Provincial Natural Science Foundation of China (grant no. LY24C050001), Youth Science and Technology Leader of Ningbo (grant no. 2023QL052), the National Natural Science Foundation of China (grant no. 32270821) and the Natural Science Foundation of Ningbo (grant no. 2021J065).
Availability of data and materials
Not applicable.
Authors' contributions
ZZ, XL, JS and LC conceived the study, performed the investigation, and wrote and edited the original draft. XJ and MY wrote, reviewed and edited the manuscript. All authors read and approved the final manuscript. Data authentication is not applicable.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
|
Gilligan T, Lin DW, Aggarwal R, Chism D, Cost N, Derweesh IH, Emamekhoo H, Feldman DR, Geynisman DM, Hancock SL, et al: Testicular cancer, version 2.2020, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. 17:1529–1554. 2019.PubMed/NCBI View Article : Google Scholar | |
|
Winter C and Albers P: Testicular germ cell tumors: Pathogenesis, diagnosis and treatment. Nat Rev Endocrinol. 7:43–53. 2011.PubMed/NCBI View Article : Google Scholar | |
|
Caggiano C, Cavallo F, Giannattasio T, Cappelletti G, Rossi P, Grimaldi P, Feldman DR, Jasin M and Barchi M: Testicular germ cell tumors acquire cisplatin resistance by rebalancing the usage of DNA repair pathways. Cancers (Basel). 13(787)2021.PubMed/NCBI View Article : Google Scholar | |
|
Shen H, Shih J, Hollern DP, Wang L, Bowlby R, Tickoo SK, Thorsson V, Mungall AJ, Newton Y, Hegde AM, et al: Integrated molecular characterization of testicular germ cell tumors. Cell Rep. 23:3392–3406. 2018.PubMed/NCBI View Article : Google Scholar | |
|
Batool A, Liu XM, Zhang CL, Hao CF, Chen SR and Liu YX: Recent advances in the regulation of testicular germ cell tumors by microRNAs. Front Biosci (Landmark Ed). 24:765–776. 2019.PubMed/NCBI View Article : Google Scholar | |
|
Batool A, Karimi N, Wu XN, Chen SR and Liu YX: Testicular germ cell tumor: A comprehensive review. Cell Mol Life Sci. 76:1713–1727. 2019.PubMed/NCBI View Article : Google Scholar | |
|
Fukawa T and Kanayama HO: Current knowledge of risk factors for testicular germ cell tumors. Int J Urol. 25:337–344. 2018.PubMed/NCBI View Article : Google Scholar | |
|
Voutsadakis IA: The chemosensitivity of testicular germ cell tumors. Cell Oncol (Dordr). 37:79–94. 2014.PubMed/NCBI View Article : Google Scholar | |
|
Brown A, Kumar S and Tchounwou PB: Cisplatin-based chemotherapy of human cancers. J Cancer Sci Ther. 11(97)2019.PubMed/NCBI | |
|
de Vries G, Rosas-Plaza X, van Vugt MATM, Gietema JA and de Jong S: Testicular cancer: Determinants of cisplatin sensitivity and novel therapeutic opportunities. Cancer Treat Rev. 88(102054)2020.PubMed/NCBI View Article : Google Scholar | |
|
Yang Y, Yang C, Li T, Yu S, Gan T, Hu J, Cui J and Zheng X: The deubiquitinase USP38 promotes NHEJ repair through regulation of HDAC1 activity and regulates cancer cell response to genotoxic insults. Cancer Res. 80:719–731. 2020.PubMed/NCBI View Article : Google Scholar | |
|
Hoeijmakers JH: DNA damage, aging, and cancer. N Engl J Med. 361:1475–1485. 2009.PubMed/NCBI View Article : Google Scholar | |
|
Groelly FJ, Fawkes M, Dagg RA, Blackford AN and Tarsounas M: Targeting DNA damage response pathways in cancer. Nat Rev Cancer. 23:78–94. 2023.PubMed/NCBI View Article : Google Scholar | |
|
Jackson SP and Bartek J: The DNA-damage response in human biology and disease. Nature. 461:1071–1078. 2009.PubMed/NCBI View Article : Google Scholar | |
|
Yang D and Wang AH: Structural studies of interactions between anticancer platinum drugs and DNA. Prog Biophys Mol Biol. 66:81–111. 1996.PubMed/NCBI View Article : Google Scholar | |
|
Eastman A: Characterization of the adducts produced in DNA by cis-diamminedichloroplatinum(II) and cis-dichloro(ethylenediamine)platinum(II). Biochemistry. 22:3927–3933. 1983.PubMed/NCBI View Article : Google Scholar | |
|
Jordan P and Carmo-Fonseca M: Molecular mechanisms involved in cisplatin cytotoxicity. Cell Mol Life Sci. 57:1229–1235. 2000.PubMed/NCBI View Article : Google Scholar | |
|
Országhová Z, Kalavska K, Mego M and Chovanec M: Overcoming chemotherapy resistance in germ cell tumors. Biomedicines. 10(972)2022.PubMed/NCBI View Article : Google Scholar | |
|
Welsh C, Day R, McGurk C, Masters JR, Wood RD and Köberle B: Reduced levels of XPA, ERCC1 and XPF DNA repair proteins in testis tumor cell lines. Int J Cancer. 110:352–361. 2004.PubMed/NCBI View Article : Google Scholar | |
|
Usanova S, Piée-Staffa A, Sied U, Thomale J, Schneider A, Kaina B and Köberle B: Cisplatin sensitivity of testis tumour cells is due to deficiency in interstrand-crosslink repair and low ERCC1-XPF expression. Mol Cancer. 9(248)2010.PubMed/NCBI View Article : Google Scholar | |
|
Mego M, Cierna Z, Svetlovska D, Macak D, Machalekova K, Miskovska V, Chovanec M, Usakova V, Obertova J, Babal P and Mardiak J: PARP expression in germ cell tumours. J Clin Pathol. 66:607–612. 2013.PubMed/NCBI View Article : Google Scholar | |
|
Cavallo F, Graziani G, Antinozzi C, Feldman DR, Houldsworth J, Bosl GJ, Chaganti RS, Moynahan ME, Jasin M and Barchi M: Reduced proficiency in homologous recombination underlies the high sensitivity of embryonal carcinoma testicular germ cell tumors to cisplatin and poly (adp-ribose) polymerase inhibition. PLoS One. 7(e51563)2012.PubMed/NCBI View Article : Google Scholar | |
|
Gutekunst M, Mueller T, Weilbacher A, Dengler MA, Bedke J, Kruck S, Oren M, Aulitzky WE and van der Kuip H: Cisplatin hypersensitivity of testicular germ cell tumors is determined by high constitutive Noxa levels mediated by Oct-4. Cancer Res. 73:1460–1469. 2013.PubMed/NCBI View Article : Google Scholar | |
|
Koster R, Timmer-Bosscha H, Bischoff R, Gietema JA and de Jong S: Disruption of the MDM2-p53 interaction strongly potentiates p53-dependent apoptosis in cisplatin-resistant human testicular carcinoma cells via the Fas/FasL pathway. Cell Death Dis. 2(e148)2011.PubMed/NCBI View Article : Google Scholar | |
|
Spierings DCJ, de Vries EGE, Vellenga E and de Jong S: Loss of drug-induced activation of the CD95 apoptotic pathway in a cisplatin-resistant testicular germ cell tumor cell line. Cell Death Differ. 10:808–822. 2003.PubMed/NCBI View Article : Google Scholar | |
|
Gutekunst M, Oren M, Weilbacher A, Dengler MA, Markwardt C, Thomale J, Aulitzky WE and van der Kuip H: p53 hypersensitivity is the predominant mechanism of the unique responsiveness of testicular germ cell tumor (TGCT) cells to cisplatin. PLoS One. 6(e19198)2011.PubMed/NCBI View Article : Google Scholar | |
|
Morsi RZ, Hage-Sleiman R, Kobeissy H and Dbaibo G: Noxa: Role in cancer pathogenesis and treatment. Curr Cancer Drug Targets. 18:914–928. 2018.PubMed/NCBI View Article : Google Scholar | |
|
Schmidtova S, Kalavska K and Kucerova L: Molecular mechanisms of cisplatin chemoresistance and its circumventing in testicular germ cell tumors. Curr Oncol Rep. 20(88)2018.PubMed/NCBI View Article : Google Scholar | |
|
Woldu SL, Amatruda JF and Bagrodia A: Testicular germ cell tumor genomics. Curr Opin Urol. 27:41–47. 2017.PubMed/NCBI View Article : Google Scholar | |
|
Lobo J, Alzamora MA, Guimarães R, Cantante M, Lopes P, Braga I, Maurício J, Jerónimo C and Henrique R: p53 and MDM2 expression in primary and metastatic testicular germ cell tumors: Association with clinical outcome. Andrology. 8:1233–1242. 2020.PubMed/NCBI View Article : Google Scholar | |
|
Han MH, Park SW, Do HJ, Chung HJ, Song H and Kim JH, Kim NH, Park KH and Kim JH: Growth and differentiation factor 3 is transcriptionally regulated by OCT4 in human embryonic carcinoma cells. Biol Pharm Bull. 39:1802–1808. 2016.PubMed/NCBI View Article : Google Scholar | |
|
Cheng CJ, Wu YC, Shu JA, Ling TY, Kuo HC, Wu JY, Chang EE, Chang SC and Huang YH: Aberrant expression and distribution of the OCT-4 transcription factor in seminomas. J Biomed Sci. 14:797–807. 2007.PubMed/NCBI View Article : Google Scholar | |
|
Mohiuddin IS, Wei SJ and Kang MH: Role of OCT4 in cancer stem-like cells and chemotherapy resistance. Biochim Biophys Acta Mol Basis Dis. 1866(165432)2020.PubMed/NCBI View Article : Google Scholar | |
|
Wu YC, Ling TY, Lu SH, Kuo HC, Ho HN, Yeh SD, Shen CN and Huang YH: Chemotherapeutic sensitivity of testicular germ cell tumors under hypoxic conditions is negatively regulated by SENP1-controlled sumoylation of OCT4. Cancer Res. 72:4963–4973. 2012.PubMed/NCBI View Article : Google Scholar | |
|
Li L, Xu M, Li X, Lv C, Zhang X, Yu H, Zhang M, Fu Y, Meng H and Zhou J: Platelet-derived growth factor-B (PDGF-B) induced by hypoxia promotes the survival of pulmonary arterial endothelial cells through the PI3K/Akt/Stat3 pathway. Cell Physiol Biochem. 35:441–451. 2015.PubMed/NCBI View Article : Google Scholar | |
|
Pullamsetti SS, Berghausen EM, Dabral S, Tretyn A, Butrous E, Savai R, Butrous G, Dahal BK, Brandes RP, Ghofrani HA, et al: Role of Src tyrosine kinases in experimental pulmonary hypertension. Arterioscler Thromb Vasc Biol. 32:1354–1365. 2012.PubMed/NCBI View Article : Google Scholar | |
|
Noskovičová N, Petřek M, Eickelberg O and Heinzelmann K: Platelet-derived growth factor signaling in the lung. From lung development and disease to clinical studies. Am J Respir Cell Mol Biol. 52:263–284. 2015.PubMed/NCBI View Article : Google Scholar | |
|
Rieg AD, Suleiman S, Anker C, Verjans E, Rossaint R, Uhlig S and Martin C: PDGF-BB regulates the pulmonary vascular tone: Impact of prostaglandins, calcium, MAPK- and PI3K/AKT/mTOR signalling and actin polymerisation in pulmonary veins of guinea pigs. Respir Res. 19(120)2018.PubMed/NCBI View Article : Google Scholar | |
|
Liu P, Cheng H, Roberts TM and Zhao JJ: Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 8:627–644. 2009.PubMed/NCBI View Article : Google Scholar | |
|
Zhao L and Vogt PK: Class I PI3K in oncogenic cellular transformation. Oncogene. 27:5486–5496. 2008.PubMed/NCBI View Article : Google Scholar | |
|
Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC and Abraham RT: The PI3K pathway in human disease. Cell. 170:605–635. 2017.PubMed/NCBI View Article : Google Scholar | |
|
Papa A and Pandolfi PP: The PTEN-PI3K axis in cancer. Biomolecules. 9(153)2019.PubMed/NCBI View Article : Google Scholar | |
|
Di Vizio D, Cito L, Boccia A, Chieffi P, Insabato L, Pettinato G, Motti ML, Schepis F, D'Amico W, Fabiani F, et al: Loss of the tumor suppressor gene PTEN marks the transition from intratubular germ cell neoplasias (ITGCN) to invasive germ cell tumors. Oncogene. 24:1882–1894. 2005.PubMed/NCBI View Article : Google Scholar | |
|
Juliachs M, Muñoz C, Moutinho CA, Vidal A, Condom E, Esteller M, Graupera M, Casanovas O, Germà JR, Villanueva A and Viñals F: The PDGFRβ-AKT pathway contributes to CDDP-acquired resistance in testicular germ cell tumors. Clin Cancer Res. 20:658–667. 2014.PubMed/NCBI View Article : Google Scholar | |
|
Koster R, di Pietro A, Timmer-Bosscha H, Gibcus JH, van den Berg A, Suurmeijer AJ, Bischoff R, Gietema JA and de Jong S: Cytoplasmic p21 expression levels determine cisplatin resistance in human testicular cancer. J Clin Invest. 120:3594–3605. 2010.PubMed/NCBI View Article : Google Scholar | |
|
Zhang X, Tang N, Hadden TJ and Rishi AK: Akt, FoxO and regulation of apoptosis. Biochim Biophys Acta. 1813:1978–1986. 2011.PubMed/NCBI View Article : Google Scholar | |
|
Selfe J, Goddard NC, McIntyre A, Taylor KR, Renshaw J, Popov SD, Thway K, Summersgill B, Huddart RA, Gilbert DC and Shipley JM: IGF1R signalling in testicular germ cell tumour cells impacts on cell survival and acquired cisplatin resistance. J Pathol. 244:242–253. 2018.PubMed/NCBI View Article : Google Scholar | |
|
Gilbert D, Rapley E and Shipley J: Testicular germ cell tumours: Predisposition genes and the male germ cell niche. Nat Rev Cancer. 11:278–288. 2011.PubMed/NCBI View Article : Google Scholar | |
|
Selfe J and Shipley JM: IGF signalling in germ cells and testicular germ cell tumours: Roles and therapeutic approaches. Andrology. 7:536–544. 2019.PubMed/NCBI View Article : Google Scholar | |
|
Schneeweiss-Gleixner M, Byrgazov K, Stefanzl G, Berger D, Eisenwort G, Lucini CB, Herndlhofer S, Preuner S, Obrova K, Pusic P, et al: CDK4/CDK6 inhibition as a novel strategy to suppress the growth and survival of BCR-ABL1T315I+ clones in TKI-resistant CML. EBioMedicine. 50:111–121. 2019.PubMed/NCBI View Article : Google Scholar | |
|
Rossini E, Bosatta V, Abate A, Fragni M, Salvi V, Basnet RM, Zizioli D, Bosisio D, Piovani G, Valcamonico F, et al: Cisplatin cytotoxicity in human testicular germ cell tumor cell lines is enhanced by the CDK4/6 inhibitor palbociclib. Clin Genitourin Cancer. 19:316–324. 2021.PubMed/NCBI View Article : Google Scholar | |
|
Mayer F, Stoop H, Scheffer GL, Scheper R, Oosterhuis JW, Looijenga LH and Bokemeyer C: Molecular determinants of treatment response in human germ cell tumors. Clin Cancer Res. 9:767–773. 2003.PubMed/NCBI | |
|
Jacobsen C and Honecker F: Cisplatin resistance in germ cell tumours: Models and mechanisms. Andrology. 3:111–121. 2015.PubMed/NCBI View Article : Google Scholar | |
|
Oosterhuis JW and Looijenga LHJ: Human germ cell tumours from a developmental perspective. Nat Rev Cancer. 19:522–537. 2019.PubMed/NCBI View Article : Google Scholar | |
|
Sonnenburg D, Spinella MJ and Albany C: Epigenetic targeting of platinum resistant testicular cancer. Curr Cancer Drug Targets. 16:789–795. 2016.PubMed/NCBI View Article : Google Scholar | |
|
Fazal Z, Singh R, Fang F, Bikorimana E, Baldwin H, Corbet A, Tomlin M, Yerby C, Adra N, Albany C, et al: Hypermethylation and global remodelling of DNA methylation is associated with acquired cisplatin resistance in testicular germ cell tumours. Epigenetics. 16:1071–1084. 2021.PubMed/NCBI View Article : Google Scholar | |
|
Liu ZX, Li LM, Sun HL and Liu SM: Link between m6A modification and cancers. Front Bioeng Biotechnol. 6(89)2018.PubMed/NCBI View Article : Google Scholar | |
|
Miranda-Gonçalves V, Lobo J, Guimarães-Teixeira C, Barros-Silva D, Guimarães R, Cantante M, Braga I, Maurício J, Oing C, Honecker F, et al: The component of the m6A writer complex VIRMA is implicated in aggressive tumor phenotype, DNA damage response and cisplatin resistance in germ cell tumors. J Exp Clin Cancer Res. 40(268)2021.PubMed/NCBI View Article : Google Scholar | |
|
Wei J, Yin Y, Zhou J, Chen H, Peng J, Yang J and Tang Y: METTL3 potentiates resistance to cisplatin through m6A modification of TFAP2C in seminoma. J Cell Mol Med. 24:11366–11380. 2020.PubMed/NCBI View Article : Google Scholar | |
|
Doghish AS, Moustafa HAM, Elballal MS, Sallam AM, El-Dakroury WA, Abdel Mageed SS, Elesawy AE, Abdelmaksoud NM, Shahin RK, Midan HM, et al: The potential role of miRNAs in the pathogenesis of testicular germ cell tumors-A Focus on signaling pathways interplay. Pathol Res Pract. 248(154611)2023.PubMed/NCBI View Article : Google Scholar | |
|
Sawant A, Floyd AM, Dangeti M, Lei W, Sobol RW and Patrick SM: Differential role of base excision repair proteins in mediating cisplatin cytotoxicity. DNA Repair (Amst). 51:46–59. 2017.PubMed/NCBI View Article : Google Scholar | |
|
Cavallo F, Caggiano C, Jasin M and Barchi M: Assessing homologous recombination and interstrand cross-link repair in embryonal carcinoma testicular germ cell tumor cell lines. Methods Mol Biol. 2195:113–123. 2021.PubMed/NCBI View Article : Google Scholar | |
|
Pannunzio NR, Watanabe G and Lieber MR: Nonhomologous DNA end-joining for repair of DNA double-strand breaks. J Biol Chem. 293:10512–10523. 2018.PubMed/NCBI View Article : Google Scholar | |
|
Scully R, Panday A, Elango R and Willis NA: DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat Rev Mol Cell Biol. 20:698–714. 2019.PubMed/NCBI View Article : Google Scholar | |
|
Chang HHY, Pannunzio NR, Adachi N and Lieber MR: Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat Rev Mol Cell Biol. 18:495–506. 2017.PubMed/NCBI View Article : Google Scholar | |
|
Sugitani N, Sivley RM, Perry KE, Capra JA and Chazin WJ: XPA: A key scaffold for human nucleotide excision repair. DNA Repair (Amst). 44:123–135. 2016.PubMed/NCBI View Article : Google Scholar | |
|
Cierna Z, Miskovska V, Roska J, Jurkovicova D, Pulzova LB, Sestakova Z, Hurbanova L, Machalekova K, Chovanec M, Rejlekova K, et al: Increased levels of XPA might be the basis of cisplatin resistance in germ cell tumours. BMC Cancer. 20(17)2020.PubMed/NCBI View Article : Google Scholar | |
|
Clairmont CS and D'Andrea AD: REV7 directs DNA repair pathway choice. Trends Cell Biol. 31:965–978. 2021.PubMed/NCBI View Article : Google Scholar | |
|
Dash RC and Hadden K: Protein-protein interactions in translesion synthesis. Molecules. 26(5544)2021.PubMed/NCBI View Article : Google Scholar | |
|
Shimada Y, Kato T, Sakurai Y, Watanabe H, Nonaka M, Nanaura N, Ichinoe M and Murakumo Y: Identification of the promoter region regulating the transcription of the REV7 gene. Biochem Biophys Res Commun. 662:8–17. 2023.PubMed/NCBI View Article : Google Scholar | |
|
Sakurai Y, Ichinoe M, Yoshida K, Nakazato Y, Saito S, Satoh M, Nakada N, Sanoyama I, Umezawa A, Numata Y, et al: Inactivation of REV7 enhances chemosensitivity and overcomes acquired chemoresistance in testicular germ cell tumors. Cancer Lett. 489:100–110. 2020.PubMed/NCBI View Article : Google Scholar | |
|
Funke K, Einsfelder U, Hansen A, Arévalo L, Schneider S, Nettersheim D, Stein V and Schorle H: Genome-scale CRISPR screen reveals neddylation to contribute to cisplatin resistance of testicular germ cell tumours. Br J Cancer. 128:2270–2282. 2023.PubMed/NCBI View Article : Google Scholar | |
|
Hinz S, Magheli A, Weikert S, Schulze W, Krause H, Schrader M, Miller K and Kempkensteffen C: Deregulation of EZH2 expression in human spermatogenic disorders and testicular germ cell tumors. World J Urol. 28:631–635. 2010.PubMed/NCBI View Article : Google Scholar | |
|
Yamagishi M and Uchimaru K: Targeting EZH2 in cancer therapy. Curr Opin Oncol. 29:375–381. 2017.PubMed/NCBI View Article : Google Scholar | |
|
Sun S, Zhao S, Yang Q, Wang W, Cai E, Wen Y, Yu L, Wang Z and Cai J: Enhancer of zeste homolog 2 promotes cisplatin resistance by reducing cellular platinum accumulation. Cancer Sci. 109:1853–1864. 2018.PubMed/NCBI View Article : Google Scholar | |
|
Dou D, Ge X, Wang X, Xu X, Zhang Z, Seng J, Cao Z, Gu Y and Han M: EZH2 contributes to cisplatin resistance in breast cancer by epigenetically suppressing miR-381 expression. Onco Targets Ther. 12:9627–9637. 2019.PubMed/NCBI View Article : Google Scholar | |
|
Xu C, Hao K, Hu H, Sheng Z, Yan J, Wang Q and Yu L: Expression of the enhancer of zeste homolog 2 in biopsy specimen predicts chemoresistance and survival in advanced non-small cell lung cancer receiving first-line platinum-based chemotherapy. Lung Cancer. 86:268–273. 2014.PubMed/NCBI View Article : Google Scholar | |
|
Singh R, Fazal Z, Corbet AK, Bikorimana E, Rodriguez JC, Khan EM, Shahid K, Freemantle SJ and Spinella MJ: Epigenetic remodeling through downregulation of polycomb repressive complex 2 mediates chemotherapy resistance in testicular germ cell tumors. Cancers (Basel). 11(796)2019.PubMed/NCBI View Article : Google Scholar | |
|
Samaržija I, Tomljanović M, Novak Kujundžić R and Trošelj KG: EZH2 inhibition and cisplatin as a combination anticancer therapy: An overview of preclinical studies. Cancers (Basel). 14(4761)2022.PubMed/NCBI View Article : Google Scholar | |
|
Singh R, Fazal Z, Bikorimana E, Boyd RI, Yerby C, Tomlin M, Baldwin H, Shokry D, Corbet AK, Shahid K, et al: Reciprocal epigenetic remodeling controls testicular cancer hypersensitivity to hypomethylating agents and chemotherapy. Mol Oncol. 16:683–698. 2022.PubMed/NCBI View Article : Google Scholar | |
|
Timmerman DM, Eleveld TF, Sriram S, Dorssers LCJ, Gillis AJM, Schmidtova S, Kalavska K, van de Werken HJG, Oing C, Honecker F, et al: Chromosome 3p25.3 gain is associated with cisplatin resistance and is an independent predictor of poor outcome in male malignant germ cell tumors. J Clin Oncol. 40:3077–3087. 2022.PubMed/NCBI View Article : Google Scholar | |
|
Yu H: Structural activation of Mad2 in the mitotic spindle checkpoint: The two-state Mad2 model versus the Mad2 template model. J Cell Biol. 173:153–157. 2006.PubMed/NCBI View Article : Google Scholar | |
|
Henriques AC, Silva PMA, Sarmento B and Bousbaa H: The Mad2-binding protein p31comet as a potential target for human cancer therapy. Curr Cancer Drug Targets. 21:401–415. 2021.PubMed/NCBI View Article : Google Scholar | |
|
López-Saavedra A, Ramírez-Otero M, Díaz-Chávez J, Cáceres-Gutiérrez R, Justo-Garrido M, Andonegui MA, Mendoza J, Downie-Ruíz Á, Cortés-González C, Reynoso N, et al: MAD2γ, a novel MAD2 isoform, reduces mitotic arrest and is associated with resistance in testicular germ cell tumors. Cell Cycle. 15:2066–2076. 2016.PubMed/NCBI View Article : Google Scholar | |
|
Kitayama S, Ikeda K, Sato W, Takeshita H, Kawakami S, Inoue S and Horie K: Testis-expressed gene 11 inhibits cisplatin-induced DNA damage and contributes to chemoresistance in testicular germ cell tumor. Sci Rep. 12(18423)2022.PubMed/NCBI View Article : Google Scholar | |
|
McHugh DJ, Gleeson JP and Feldman DR: Testicular cancer in 2023: Current status and recent progress. CA Cancer J Clin. 74:167–186. 2024.PubMed/NCBI View Article : Google Scholar | |
|
Paffenholz P, Pfister D and Heidenreich A: Testis-preserving strategies in testicular germ cell tumors and germ cell neoplasia in situ. Transl Androl Urol. 9 (Suppl 1):S24–S30. 2020.PubMed/NCBI View Article : Google Scholar | |
|
Chang MM, Pan BS, Wang CY and Huang BM: Cordycepin-induced unfolded protein response-dependent cell death, and AKT/MAPK-mediated drug resistance in mouse testicular tumor cells. Cancer Med. 8:3949–3964. 2019.PubMed/NCBI View Article : Google Scholar | |
|
Hu Z, Yu J, Gui G, Chen Y, Huang R, Jiang L, Kwong JSW, Li Y and Zhang L: Cisplatin for testicular germ cell tumors: A rapid review. J Evid Based Med. 9:144–151. 2016.PubMed/NCBI View Article : Google Scholar | |
|
Chovanec M, Hanna N, Cary KC, Einhorn L and Albany C: Management of stage I testicular germ cell tumours. Nat Rev Urol. 13:663–673. 2016.PubMed/NCBI View Article : Google Scholar | |
|
Sadek KM, AbdEllatief HY, Mahmoud SFE, Alexiou A, Papadakis M, Al-Hajeili M, Saad HM and Batiha GE: New insights on testicular cancer prevalence with novel diagnostic biomarkers and therapeutic approaches. Cancer Rep (Hoboken). 7(e2052)2024.PubMed/NCBI View Article : Google Scholar | |
|
Curreri SA, Fung C and Beard CJ: Secondary malignant neoplasms in testicular cancer survivors. Urol Oncol. 33:392–398. 2015.PubMed/NCBI View Article : Google Scholar | |
|
Beitzen-Heineke A, Rolling CC, Seidel C, Erley J, Molwitz I, Muellerleile K, Saering D, Senftinger J, Börschel N, Engel NW, et al: Long-term cardiotoxicity in germ cell cancer survivors after platinum-based chemotherapy: Cardiac MR shows impaired systolic function and tissue alterations. Eur Radiol. 34:4102–4112. 2024.PubMed/NCBI View Article : Google Scholar | |
|
Chovanec M, Abu Zaid M, Hanna N, El-Kouri N, Einhorn LH and Albany C: Long-term toxicity of cisplatin in germ-cell tumor survivors. Ann Oncol. 28:2670–2679. 2017.PubMed/NCBI View Article : Google Scholar | |
|
Porcu P, Bhatia S, Sharma M and Einhorn LH: Results of treatment after relapse from high-dose chemotherapy in germ cell tumors. J Clin Oncol. 18:1181–1186. 2000.PubMed/NCBI View Article : Google Scholar | |
|
Chieffi P, De Martino M and Esposito F: Further insights into testicular germ cell tumor oncogenesis: Potential therapeutic targets. Expert Rev Anticancer Ther. 20:189–195. 2020.PubMed/NCBI View Article : Google Scholar | |
|
Einhorn LH, Williams SD, Chamness A, Brames MJ, Perkins SM and Abonour R: High-dose chemotherapy and stem-cell rescue for metastatic germ-cell tumors. N Engl J Med. 357:340–348. 2007.PubMed/NCBI View Article : Google Scholar | |
|
Lorch A, Bascoul-Mollevi C, Kramar A, Einhorn L, Necchi A, Massard C, De Giorgi U, Fléchon A, Margolin K, Lotz JP, et al: Conventional-dose versus high-dose chemotherapy as first salvage treatment in male patients with metastatic germ cell tumors: Evidence from a large international database. J Clin Oncol. 29:2178–2184. 2011.PubMed/NCBI View Article : Google Scholar | |
|
Nichols CR, Tricot G, Williams SD, van Besien K, Loehrer PJ, Roth BJ, Akard L, Hoffman R, Goulet R, Wolff SN, et al: Dose-intensive chemotherapy in refractory germ cell cancer-a phase I/II trial of high-dose carboplatin and etoposide with autologous bone marrow transplantation. J Clin Oncol. 7:932–939. 1989.PubMed/NCBI View Article : Google Scholar | |
|
Su X, Wang Z, Li L, Zheng M, Zheng C, Gong P, Zhao P, Ma Y, Tao Q and Cai L: Lipid-polymer nanoparticles encapsulating doxorubicin and 2'-deoxy-5-azacytidine enhance the sensitivity of cancer cells to chemical therapeutics. Mol Pharm. 10:1901–1909. 2013.PubMed/NCBI View Article : Google Scholar | |
|
Ohtani H, Ørskov AD, Helbo AS, Gillberg L, Liu M, Zhou W, Ungerstedt J, Hellström-Lindberg E, Sun W, Liang G, et al: Activation of a subset of evolutionarily young transposable elements and innate immunity are linked to clinical responses to 5-azacytidine. Cancer Res. 80:2441–2450. 2020.PubMed/NCBI View Article : Google Scholar | |
|
Stahl M, Kohrman N, Gore SD, Kim TK, Zeidan AM and Prebet T: Epigenetics in cancer: A hematological perspective. PLoS Genet. 12(e1006193)2016.PubMed/NCBI View Article : Google Scholar | |
|
Lobo J, Guimarães-Teixeira C, Barros-Silva D, Miranda-Gonçalves V, Camilo V, Guimarães R, Cantante M, Braga I, Maurício J, Oing C, et al: Efficacy of HDAC inhibitors belinostat and panobinostat against cisplatin-sensitive and cisplatin-resistant testicular germ cell tumors. Cancers (Basel). 12(2903)2020.PubMed/NCBI View Article : Google Scholar | |
|
Cardoso AR, Lobo J, Miranda-Gonçalves V, Henrique R and Jerónimo C: Epigenetic alterations as therapeutic targets in testicular germ cell tumours: Current and future application of ‘epidrugs’. Epigenetics. 16:353–372. 2021.PubMed/NCBI View Article : Google Scholar | |
|
Lobo J, Cardoso AR, Miranda-Gonçalves V, Looijenga LHJ, Lopez M, Arimondo PB, Henrique R and Jerónimo C: Targeting germ cell tumors with the newly synthesized flavanone-derived compound MLo1302 efficiently reduces tumor cell viability and induces apoptosis and cell cycle arrest. Pharmaceutics. 13(73)2021.PubMed/NCBI View Article : Google Scholar | |
|
Albany C, Hever-Jardine MP, von Herrmann KM, Yim CY, Tam J, Warzecha JM, Shin L, Bock SE, Curran BS, Chaudhry AS, et al: Refractory testicular germ cell tumors are highly sensitive to the second generation DNA methylation inhibitor guadecitabine. Oncotarget. 8:2949–2959. 2017.PubMed/NCBI View Article : Google Scholar | |
|
Oing C, Verem I, Mansour WY, Bokemeyer C, Dyshlovoy S and Honecker F: 5-Azacitidine exerts prolonged pro-apoptotic effects and overcomes cisplatin-resistance in non-seminomatous germ cell tumor cells. Int J Mol Sci. 20(21)2018.PubMed/NCBI View Article : Google Scholar | |
|
Jostes S, Nettersheim D, Fellermeyer M, Schneider S, Hafezi F, Honecker F, Schumacher V, Geyer M, Kristiansen G and Schorle H: The bromodomain inhibitor JQ1 triggers growth arrest and apoptosis in testicular germ cell tumours in vitro and in vivo. J Cell Mol Med. 21:1300–1314. 2017.PubMed/NCBI View Article : Google Scholar | |
|
Zhou W, Chen H, Hong X, Niu X and Lu Q: Knockdown of DNA methyltransferase-1 inhibits proliferation and derepresses tumor suppressor genes in myeloma cells. Oncol Lett. 8:2130–2134. 2014.PubMed/NCBI View Article : Google Scholar | |
|
Tulsyan S, Aftab M, Sisodiya S, Khan A, Chikara A, Tanwar P and Hussain S: Molecular basis of epigenetic regulation in cancer diagnosis and treatment. Front Genet. 13(885635)2022.PubMed/NCBI View Article : Google Scholar | |
|
Lecomte S, Demay F, Ferrière F and Pakdel F: Phytochemicals targeting estrogen receptors: Beneficial rather than adverse effects? Int J Mol Sci. 18(1381)2017.PubMed/NCBI View Article : Google Scholar | |
|
Van Neste L, Bigley J, Toll A, Otto G, Clark J, Delrée P, Van Criekinge W and Epstein JI: A tissue biopsy-based epigenetic multiplex PCR assay for prostate cancer detection. BMC Urol. 12(16)2012.PubMed/NCBI View Article : Google Scholar | |
|
Jin B and Robertson KD: DNA methyltransferases, DNA damage repair, and cancer. Adv Exp Med Biol. 754:3–29. 2013.PubMed/NCBI View Article : Google Scholar | |
|
Stresemann C and Lyko F: Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int J Cancer. 123:8–13. 2008.PubMed/NCBI View Article : Google Scholar | |
|
Hollenbach PW, Nguyen AN, Brady H, Williams M, Ning Y, Richard N, Krushel L, Aukerman SL, Heise C and MacBeth KJ: A comparison of azacitidine and decitabine activities in acute myeloid leukemia cell lines. PLoS One. 5(e9001)2010.PubMed/NCBI View Article : Google Scholar | |
|
Wermann H, Stoop H, Gillis AJM, Honecker F, van Gurp RJ, Ammerpohl O, Richter J, Oosterhuis JW, Bokemeyer C and Looijenga LH: Global DNA methylation in fetal human germ cells and germ cell tumours: Association with differentiation and cisplatin resistance. J Pathol. 221:433–442. 2010.PubMed/NCBI View Article : Google Scholar | |
|
Rüter B, Wijermans PW and Lübbert M: DNA methylation as a therapeutic target in hematologic disorders: Recent results in older patients with myelodysplasia and acute myeloid leukemia. Int J Hematol. 80:128–135. 2004.PubMed/NCBI View Article : Google Scholar | |
|
Beyrouthy MJ, Garner KM, Hever MP, Freemantle SJ, Eastman A, Dmitrovsky E and Spinella MJ: High DNA methyltransferase 3B expression mediates 5-aza-deoxycytidine hypersensitivity in testicular germ cell tumors. Cancer Res. 69:9360–9366. 2009.PubMed/NCBI View Article : Google Scholar | |
|
Issa JJ, Roboz G, Rizzieri D, Jabbour E, Stock W, O'Connell C, Yee K, Tibes R, Griffiths EA, Walsh K, et al: Safety and tolerability of guadecitabine (SGI-110) in patients with myelodysplastic syndrome and acute myeloid leukaemia: A multicentre, randomised, dose-escalation phase 1 study. Lancet Oncol. 16:1099–1110. 2015.PubMed/NCBI View Article : Google Scholar | |
|
Albany C, Fazal Z, Singh R, Bikorimana E, Adra N, Hanna NH, Einhorn LH, Perkins SM, Sandusky GE, Christensen BC, et al: A phase 1 study of combined guadecitabine and cisplatin in platinum refractory germ cell cancer. Cancer Med. 10:156–163. 2021.PubMed/NCBI View Article : Google Scholar | |
|
Kurz L, Miklyaeva A, Skowron MA, Overbeck N, Poschmann G, Becker T, Eul K, Kurz T, Schönberger S, Calaminus G, et al: ARID1A regulates transcription and the epigenetic landscape via POLE and DMAP1 while ARID1A deficiency or pharmacological inhibition sensitizes germ cell tumor cells to ATR inhibition. Cancers (Basel). 12(905)2020.PubMed/NCBI View Article : Google Scholar | |
|
Steinemann G, Dittmer A, Kuzyniak W, Hoffmann B, Schrader M, Schobert R, Biersack B, Nitzsche B and Höpfner M: Animacroxam, a novel dual-mode compound targeting histone deacetylases and cytoskeletal integrity of testicular germ cell cancer cells. Mol Cancer Ther. 16:2364–2374. 2017.PubMed/NCBI View Article : Google Scholar | |
|
Steinemann G, Dittmer A, Schmidt J, Josuttis D, Fähling M, Biersack B, Beindorff N, Jolante Koziolek E, Schobert R, Brenner W, et al: Antitumor and antiangiogenic activity of the novel chimeric inhibitor animacroxam in testicular germ cell cancer. Mol Oncol. 13:2679–2696. 2019.PubMed/NCBI View Article : Google Scholar | |
|
Nettersheim D, Berger D, Jostes S, Skowron M and Schorle H: Deciphering the molecular effects of romidepsin on germ cell tumours: DHRS2 is involved in cell cycle arrest but not apoptosis or induction of romidepsin effectors. J Cell Mol Med. 23:670–679. 2019.PubMed/NCBI View Article : Google Scholar | |
|
Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, et al: BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 146:904–917. 2011.PubMed/NCBI View Article : Google Scholar | |
|
Burmeister A, Stephan A, Alves Avelar LA, Müller MR, Seiwert A, Höfmann S, Fischer F, Torres-Gomez H, Hoffmann MJ, Niegisch G, et al: Establishment and evaluation of dual HDAC/BET inhibitors as therapeutic options for germ cell tumors and other urological malignancies. Mol Cancer Ther. 21:1674–1688. 2022.PubMed/NCBI View Article : Google Scholar | |
|
Lengert AVH, Pereira LDNB, Cabral ERM, Gomes INF, Jesus LM, Gonçalves MFS, Rocha AOD, Tassinari TA, Silva LSD, Laus AC, et al: Potential new therapeutic approaches for cisplatin-resistant testicular germ cell tumors. Front Biosci (Landmark Ed). 27(245)2022.PubMed/NCBI View Article : Google Scholar | |
|
Huang TT, Nijman SMB, Mirchandani KD, Galardy PJ, Cohn MA, Haas W, Gygi SP, Ploegh HL, Bernards R and D'Andrea AD: Regulation of monoubiquitinated PCNA by DUB autocleavage. Nat Cell Biol. 8:339–347. 2006.PubMed/NCBI View Article : Google Scholar | |
|
Hasibeder A, Venkataramani V, Thelen P, Radzun HJ and Schweyer S: Phytoestrogens regulate the proliferation and expression of stem cell factors in cell lines of malignant testicular germ cell tumors. Int J Oncol. 43:1385–1394. 2013.PubMed/NCBI View Article : Google Scholar | |
|
Kalavska K, Schmidtova S, Chovanec M and Mego M: Immunotherapy in testicular germ cell tumors. Front Oncol. 10(573977)2020.PubMed/NCBI View Article : Google Scholar | |
|
Vincent AJ, Ren S, Harris LG, Devine DJ, Samant RS, Fodstad O and Shevde LA: Cytoplasmic translocation of p21 mediates NUPR1-induced chemoresistance: NUPR1 and p21 in chemoresistance. FEBS Lett. 586:3429–3434. 2012.PubMed/NCBI View Article : Google Scholar | |
|
Xia X, Ma Q, Li X, Ji T, Chen P, Xu H, Li K, Fang Y, Weng D, Weng Y, et al: Cytoplasmic p21 is a potential predictor for cisplatin sensitivity in ovarian cancer. BMC Cancer. 11(399)2011.PubMed/NCBI View Article : Google Scholar | |
|
Wang X, Wang Y, Gou S and Chen F: A trifunctional Pt(II) complex alleviates the NHEJ/HR-related DSBs repairs to evade cisplatin-resistance in NSCLC. Bioorg Chem. 104(104210)2020.PubMed/NCBI View Article : Google Scholar | |
|
Costa A, Forte IM, Pentimalli F, Iannuzzi CA, Alfano L, Capone F, Camerlingo R, Calabrese A, von Arx C, Benot Dominguez R, et al: Pharmacological inhibition of CDK4/6 impairs diffuse pleural mesothelioma 3D spheroid growth and reduces viability of cisplatin-resistant cells. Front Oncol. 14(1418951)2024.PubMed/NCBI View Article : Google Scholar | |
|
Romano FJ, Rossetti S, Conteduca V, Schepisi G, Cavaliere C, Di Franco R, La Mantia E, Castaldo L, Nocerino F, Ametrano G, et al: Role of DNA repair machinery and p53 in the testicular germ cell cancer: A review. Oncotarget. 7:85641–85649. 2016.PubMed/NCBI View Article : Google Scholar | |
|
Spierings DC, de Vries EG, Stel AJ, te Rietstap N, Vellenga E and de Jong S: Low p21Waf1/Cip1 protein level sensitizes testicular germ cell tumor cells to Fas-mediated apoptosis. Oncogene. 23:4862–4872. 2004.PubMed/NCBI View Article : Google Scholar | |
|
Tomida J, Takata KI, Bhetawal S, Person MD, Chao HP, Tang DG and Wood RD: FAM35A associates with REV7 and modulates DNA damage responses of normal and BRCA1-defective cells. EMBO J. 37(e99543)2018.PubMed/NCBI View Article : Google Scholar | |
|
Lind GE, Skotheim RI, Fraga MF, Abeler VM, Esteller M and Lothe RA: Novel epigenetically deregulated genes in testicular cancer include homeobox genes and SCGB3A1 (HIN-1). J Pathol. 210:441–449. 2006.PubMed/NCBI View Article : Google Scholar | |
|
Roška J, Wachsmannová L, Hurbanová L, Šestáková Z, Mueller T, Jurkovičová D and Chovanec M: Differential gene expression in cisplatin-resistant and -sensitive testicular germ cell tumor cell lines. Oncotarget. 11:4735–4753. 2020.PubMed/NCBI View Article : Google Scholar | |
|
Schmidtova S, Dorssers LCJ, Kalavska K, Gillis AJM, Oosterhuis JW, Stoop H, Miklikova S, Kozovska Z, Burikova M, Gercakova K, et al: Napabucasin overcomes cisplatin resistance in ovarian germ cell tumor-derived cell line by inhibiting cancer stemness. Cancer Cell Int. 20(364)2020.PubMed/NCBI View Article : Google Scholar | |
|
Schmidtova S, Kalavska K, Gercakova K, Cierna Z, Miklikova S, Smolkova B, Buocikova V, Miskovska V, Durinikova E, Burikova M, et al: Disulfiram overcomes cisplatin resistance in human embryonal carcinoma cells. Cancers (Basel). 11(1224)2019.PubMed/NCBI View Article : Google Scholar | |
|
Ma H, Cao W and Ding M: MicroRNA-31 weakens cisplatin resistance of medulloblastoma cells via NF-κB and PI3K/AKT pathways. Biofactors. 46:831–838. 2020.PubMed/NCBI View Article : Google Scholar | |
|
Luo H, Yi T, Huang D, Chen X, Li X, Wan Q, Huang H, Huang H, Wei H, Song Y, et al: circ_PTN contributes to -cisplatin resistance in glioblastoma via PI3K/AKT signaling through the miR-542-3p/PIK3R3 pathway. Mol Ther Nucleic Acids. 26:1255–1269. 2021.PubMed/NCBI View Article : Google Scholar | |
|
Fankhauser CD, Curioni-Fontecedro A, Allmann V, Beyer J, Tischler V, Sulser T, Moch H and Bode PK: Frequent PD-L1 expression in testicular germ cell tumors. Br J Cancer. 113:411–413. 2015.PubMed/NCBI View Article : Google Scholar | |
|
Hong W, Xue M, Jiang J, Zhang Y and Gao X: Circular RNA circ-CPA4/let-7 miRNA/PD-L1 axis regulates cell growth, stemness, drug resistance and immune evasion in non-small cell lung cancer (NSCLC). J Exp Clin Cancer Res. 39(149)2020.PubMed/NCBI View Article : Google Scholar | |
|
De S, Holvey-Bates EG, Mahen K, Willard B and Stark GR: The ubiquitin E3 ligase FBXO22 degrades PD-L1 and sensitizes cancer cells to DNA damage. Proc Natl Acad Sci USA. 118(e2112674118)2021.PubMed/NCBI View Article : Google Scholar | |
|
Liu L, Lian J, Zhang H, Tian H, Liang M, Yin M and Sun F: MicroRNA-302a sensitizes testicular embryonal carcinoma cells to cisplatin-induced cell death. J Cell Physiol. 228:2294–2304. 2013.PubMed/NCBI View Article : Google Scholar | |
|
Wei J, Gan Y, Peng D, Jiang X, Kitazawa R, Xiang Y, Dai Y, Tang Y and Yang J: Long non-coding RNA H19 promotes TDRG1 expression and cisplatin resistance by sequestering miRNA-106b-5p in seminoma. Cancer Med. 7:6247–6257. 2018.PubMed/NCBI View Article : Google Scholar | |
|
Huang H, Tian H, Duan Z, Cao Y, Zhang XS and Sun F: microRNA-383 impairs phosphorylation of H2AX by targeting PNUTS and inducing cell cycle arrest in testicular embryonal carcinoma cells. Cell Signal. 26:903–911. 2014.PubMed/NCBI View Article : Google Scholar | |
|
Elesawy AE, Abulsoud AI, Moustafa HAM, Elballal MS, Sallam AM, Elazazy O, El-Dakroury WA, Abdel Mageed SS, Abdelmaksoud NM, Midan HM, et al: miRNAs orchestration of testicular germ cell tumors-particular emphasis on diagnosis, progression and drug resistance. Pathol Res Pract. 248(154612)2023.PubMed/NCBI View Article : Google Scholar | |
|
Özata DM, Li X, Lee L, Liu J, Warsito D, Hajeri P, Hultman I, Fotouhi O, Marklund S, Ährlund-Richter L, et al: Loss of miR-514a-3p regulation of PEG3 activates the NF-kappa B pathway in human testicular germ cell tumors. Cell Death Dis. 8(e2759)2017.PubMed/NCBI View Article : Google Scholar | |
|
Roška J, Lobo J, Ivovič D, Wachsmannová L, Mueller T, Henrique R, Jerónimo C, Chovanec M and Jurkovičová D: Integrated microarray-based data analysis of miRNA expression profiles: Identification of novel biomarkers of cisplatin-resistance in testicular germ cell tumours. Int J Mol Sci. 24(2495)2023.PubMed/NCBI View Article : Google Scholar |