Introduction
Thyroid cancer (THCA) ranks among the most prevalent
malignancies arising from the endocrine system (1), with a rising global incidence
(2). Although the five-year
survival rate for patients with THCA is notably high when treated
with radioiodine, mortality rates escalate significantly due to
lymph node and distant metastases (3). Consequently, elucidating the specific
molecular mechanisms underlying THCA metastasis is of paramount
importance.
Aurora kinase A (AURKA) is a member of the Aurora
family of serine/threonine kinases, involved in cell division
through the formation of bipolar mitotic spindles and the
maintenance of chromosomal stability (4). A previous study demonstrated that
AURKA promotes lymph node metastasis in papillary THCA by enhancing
cofilin-1 activity (5). In our
earlier research, the authors observed that AURKA reactivates
dormant laryngeal squamous cell carcinoma (LSCC) cells; however,
its role in THCA remains unclear (6).
Recent evidence indicated that
epithelial-mesenchymal transition (EMT) serves as a primary
mechanism for cancer metastasis in breast (7), gastric (8), pancreatic (9) and colorectal cancers (10), among others. EMT is the process by
which epithelial cells de-differentiate into mesenchymal-like
derivatives. A previous study revealed that ISL1 targeting AURKA
facilitates the development of neuroblastoma (NB), promoting EMT
and metastatic processes (11).
Similarly, the initiation of the EMT program in thyroid epithelial
cells contributes to recurrent and metastatic diseases (12). These findings suggest a link
between AURKA and EMT in the context of THCA metastasis.
Therefore, in the present study, reducing AURKA
expression decreased the proliferation and inhibited the transition
of BHT101 and BCPAP cells from the G0 phase to active division.
Furthermore, downregulating AURKA expression enhanced the movement,
migration and invasion capabilities of BHT101 and BCPAP cells.
Additionally, AURKA regulates proliferation-related molecules.
Specifically, the knockdown of AURKA expression increased the
expression of P130 and E2F4, while reducing the expression of P107.
Furthermore, upregulating AURKA expression promoted EMT, whereas
downregulation of AURKA expression resulted in the opposite effect.
In addition, the focal adhesion kinase (FAK) signaling pathway has
been shown to play a crucial role in THCA metastasis, with
inhibition of FAK hindering the movement, migration and invasion of
THCA cells. Therefore, AURKA promotes EMT by regulating P130 and
P107 to facilitate THCA metastasis.
Materials and methods
Cell culture
BCPAP and BHT101 cells were obtained from Shanghai
Institutes of Biological Sciences (Shanghai, China) and were
cultured in DMEM (YOBIBIO) supplemented with 10% fetal bovine serum
(YOBIBIO). The cells were maintained in a humidified incubator at
37˚C. In the TAE226 treatment protocol, THCA cells (including BCPAP
and BHT101) were treated with the FAK inhibitor TAE226 (2.1 µM/ml;
Selleck Chemicals) for 24 h at 37˚C. The control groups included
THCA cells treated with DMSO and THCA cells without any
treatment.
Plate colony formation assay
Cells, with or without treatment, were seeded into
6-well plates at a density of 1x103 cells per well.
After one month of culture, the cells were washed twice with PBS
and fixed with pure methanol for 15 min at 37˚C. The cells were
then stained with crystal violet for 30 min at 37˚C. Finally,
colonies consisting of >50 cells in each well were manually
counted.
Flow cytometry
The cells were seeded into 6-well plates and rinsed
three times with cold PBS. They were then fixed with precooled
anhydrous ethanol at 4˚C overnight. The fixed cells were removed
from 4˚C and equilibrated at room temperature for 15 min. The
samples were centrifuged at 3,162 x g for 5 min, and the
supernatant was discarded. A total of 10 ml of PBS was added, and
the mixture was well mixed before another centrifugation at 3,162 x
g for 5 min to remove the supernatant. After adding 2 ml of PBS and
mixing, the cells were transferred to a flow tube and were
centrifuged at 3,162 x g for 5 min to remove the supernatant. In
total, 40 µl of 50 µg/ml RNase A (Beyotime Institute of
Biotechnology) was added, and the mixture was well mixed before
incubation at 37˚C in the dark for 30 min with intermittent
shaking. A total of 20 µl of 50 µg/ml PI staining solution (BD
Pharmingen) was added, and the cells were incubated at room
temperature in the dark for 15 min. Cell cycle analysis was
performed by flow cytometry on a FACScan instrument (Beckman
Coulter, Inc.), and analyzed using FlowJo v10 (FlowJo LLC) and
GraphPad Prism software v6.0 (Dotmatics).
Immunofluorescence (IF)
Cells were seeded into Millicell EZ SLIDES
(MilliporeSigma) and fixed with 4% paraformaldehyde for 30 min at
37˚C. The slides were then rinsed three times with PBS, blocked
with 5% BSA for 1 h at 37˚C, and incubated overnight at 4˚C with
anti-AURKA (1:100; cat. no. 66757-1-Ig; Cell Signaling Technology,
Inc.), anti-P130 (1:100; cat. no. 13383; Cell Signaling Technology,
Inc.) and anti-P107 (1:100; cat. no. 13354-1-AP; Proteintech Group,
Inc.). The slides were incubated with Alexa Fluor® 488
goat anti-rabbit IgG (1:1,000; cat. no. 4412S; Cell Signaling
Technology, Inc.) and Alexa Fluor® 555 goat anti-rabbit
IgG (1:1,000; cat. no. 4413S; Cell Signaling Technology, Inc.) for
1 h at room temperature in the dark. DAPI (1:1,000; Beyotime
Institute of Biotechnology) was used to stain nuclei for 5 min in
the dark at 37˚C. The slides were rinsed three times with PBS and
analyzed by fluorescent microscopy at x10 magnification.
siRNA interference
The day before transfection, cells were seeded into
6-well plates at a density of 5x105 cells/ml (1.5 ml per
well). Transfection was performed 16 h later. For transfection,
siRNA was mixed with Opti-MEM to achieve a final concentration of
100 nM, with a total volume of 250 µl, and was left at room
temperature for 5 min. In a separate tube, 5 µl of
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) was added to 250 µl of Opti-MEM and incubated at
room temperature for 5 min. The diluted siRNA was then combined
with the transfection reagent and incubated at room temperature for
20 min. Finally, 500 µl of the RNA transfection mixture was added
to each well containing the cells. Subsequently, 6 h
post-transfection, the culture medium was replaced with 1.5 ml of
fresh complete medium. Cells were collected 48 h after transfection
for western blot analysis. The sequences were as follows: si-AURKA
forward, 5'-GAAGAGAGUUAUUCAUAGADTDT-3' and reverse,
5'-UCUAUGAAUAACUCUCUUCDTDT-3'); and siNC forward,
5'-UACGUACUAUCGCGCGGAUTT-3' and reverse,
5'-AUCCGCGCGATAGUACGUATT-3' were purchased from Shanghai Kaiji
Company.
Construction of overexpression
plasmids
The plasmid backbone used for the overexpression of
AURKA was pEX-3 (pGCMV/MCS/Neo), which was purchased from Shanghai
GenePharma Co., Ltd. The AURKA gene was amplified from genomic DNA
extracted from BCPAP and BHT101 cells using the Qiagen DNeasy Blood
& Tissue Kit (Qiagen GmbH) according to the manufacturer's
instructions. PCR amplification was performed using Taq DNA
polymerase (Thermo Fisher Scientific, Inc.) with the following
primers: Forward (EcoRI site),
5'-CCGGAATTCGCCACCATGCATATCACCATCACTCAACTCAGTGGAGCATTCAGATAG-3' and
reverse (BamHI site), 5'-CCGGGATCC
TCAGTTGGAGGTATCATGAACACTG-3'. The thermocycling conditions included
an initial denaturation at 95˚C for 5 min, followed by 35 cycles of
denaturation at 95˚C for 30 sec, annealing at 60˚C for 30 sec and
extension at 72˚C for 1 min per kb of template, with a final
extension at 72˚C for 10 min. PCR products were analyzed using 1.5%
agarose gel electrophoresis and visualized with SYBR Safe staining
(Invitrogen; Thermo Fisher Scientific, Inc.) under UV light using a
gel imaging system. The plasmid carrying the vector was extracted
from overnight bacterial cultures (3-5 ml) using the QIAGEN Plasmid
Mini Kit (Qiagen GmbH). The vector was then subjected to double
digestion with EcoRI and BamHI (1.5 µl of each
enzyme, 3 µl Green Buffer, 23 µl ddH2O and 1 µg vector) at 37˚C for
3 h. After digestion, the product was purified by 0.8% agarose gel
electrophoresis followed by gel extraction using QG buffer and
isopropanol precipitation. The target fragment was amplified using
PCR with 10 ng template DNA, 2 µl of each primer (10 µM), 25 µl PCR
mix and 21 µl double-distilled water. Following PCR, the product
was recovered and digested under similar conditions as the vector.
Both the vector and insert were ligated using T4 DNA Ligase (1 µl)
in a reaction containing 2 µl T4 DNA ligation buffer, 70 ng vector,
an equimolar amount of the insert (calculated based on vector
concentration) and double-distilled water to a final volume of 20
µl at 22˚C for 60 min. The ligation mixture (10 µl) was then used
to transform E. coli DH5α (EC0112; Thermo Fisher Scientific,
Inc.) competent BCPAP and BHT101 cells through heat shock (90 sec
at 42˚C), followed by incubation in SOC medium (cat. no. 15544034;
Thermo Fisher Scientific, Inc.) for 45 min at 17 x g at 37˚C.
Finally, the transformed cells were plated on selective agar plates
containing the appropriate antibiotic (ampicillin; 1 µg/ml) and
incubated overnight at 37˚C. Several single colonies were incubated
in small quantities, which were screened and confirmed by
sequencing (Sanger). The Sanger sequencing traces are shown
Fig. S1.
AURKA overexpression plasmid
transfection
For transfection, plasmid DNA (500 µg/µl) was
diluted in Opti-MEM and transfected using Lipofectamine®
2000 (Invitrogen; Thermo Fisher Scientific, Inc.). The mixture was
incubated at room temperature for 5-20 min to allow DNA-lipid
complexes to form before being added dropwise onto the cells. Cells
were incubated at 37˚C with 5% CO2 for 4-6 h before
replacing the transfection mixture with fresh growth medium. For
stable cell line selection, cells were cultured for 72 h
post-transfection before adding selection medium containing 1 mg/ml
G418. Selection was continued for 2 weeks until resistant colonies
emerged. Once stable cell lines were established, the cells were
maintained in selection medium containing 0.2 mg/ml G418 to
preserve stable knockdown efficiency. The effect of overexpression
was examined by western blotting. The time interval between
transfection and subsequent experiments was 24-72 h.
Western blotting
The procedures for the western blotting have been
previously described (6).
Antibodies used included Anti-P130 (1:2,000; rabbit mAb; 13383),
anti-E2F4 (1:2,000; rabbit mAb; 40291), anti-E-cad (1:2,000; mouse
mAb; 14472), anti-N-cad (1:2,000; mouse mAb; 14215), anti-Vimentin
(1:2,000; rabbit mAb; 5741), anti-Slug (1:2,000; rabbit mAb; 9585)
and anti-GAPDH (1:2,000; rabbit; 2118T), which were all obtained
from Cell Signaling Technology, Inc. Anti-P107 (1:2,000; rabbit;
13354-1-AP) and anti-AURKA (1:2,000; mouse; 66757-1-Ig) were
sourced from Proteintech Group, Inc. The membranes were incubated
with primary antibodies at 4˚C overnight. Proteins (40 µg/lane)
were separated by 10 or 12.5% SDS-PAGE for 2 h. Following the
addition of secondary antibodies (1:2,000; anti-rabbit IgG;
HRP-linked antibody; 7074; anti-mouse IgG; HRP-linked antibody;
7076; Cell Signaling Technology, Inc.), the membranes were
incubated with secondary antibodies at room temperature for 1 h.
The membranes were visualized using Thermo Pierce chemiluminescent
(ECL) Western Blotting Substrate (Thermo Fisher Scientific, Inc.)
and were detected with a Tanon 5200 system (Tanon Science and
Technology Co., Ltd.).
For the detection of the proteins of interest and
the loading control (GAPDH), it should be noted that the proteins
were not probed for on the same membranes due to the limitations of
the experimental setup. Despite this, the western blots were
conducted under consistent conditions to maintain the integrity of
the data. A total of 40 µg of protein was loaded into each lane to
ensure sample consistency across the experiments. Separate blots
were performed for each target protein to avoid cross-reactivity
and ensure reliable quantification.
Wound healing assay
Cells were seeded into six-well plates at a density
of 1x106 cells per well and cultured until they reached
confluence. The monolayer was then scratched using a 200-µl pipette
tip, and the medium was replaced with fresh medium containing 1%
FBS. Images of the cells were captured by fluorescence microscopy
at a magnification of x10 at 0 and 24 h post-scratch. To quantify
wound healing, the maximum width of the wound was measured and
analyzed using GraphPad Prism software v6.0 (Dotmatics).
Migration and invasion assays
For the migration assay, 2x105 cells
suspended in 200 µl of serum-free DMEM were added to the top
chamber of a Transwell insert (pore size, 8 µm; Corning, Inc.). The
lower chamber was filled with 600 µl of DMEM supplemented with 10%
FBS. After 24 h, the cells that had migrated to the lower surface
of the filter were fixed with 4% paraformaldehyde for 15 min at
37˚C and stained with 0.5% crystal violet solution for 20 min at
37˚C, and images were captured under an Olympus BX50 light
microscope (Olympus Corporation) at a magnification of x10.
For the invasion assay, the top chamber membrane was
first coated with Matrigel (Becton, Dickinson and Company) at 4˚C
overnight. After 24 h of culture at 37˚C, tumor cells remaining on
the upper side of the inserts were removed with cotton swabs. The
remaining steps were identical to those of the migration assay.
Statistical analysis
All experiments were repeated at least three times
and the data are presented as the mean ± SD. Significant
differences between groups were determined using an unpaired
Student's t-test (two groups) and one-way ANOVA (multiple groups;
GraphPad Prism software v6.0; Dotmatics), and Tukey's Honestly
Significant Difference test was used for post hoc comparisons.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Decreasing AURKA expression reduces
the proliferation of BHT101 and BCPAP cells
To investigate the specific regulatory mechanism by
which AURKA promotes THCA metastasis, AURKA expression was silenced
by transfecting AURKA-siRNA into BHT101 and BCPAP cells (designated
as BHT101/AURKA-siRNA and BCPAP/AURKA-siRNA, respectively) and were
compared with untreated controls (BHT101/AURKA-NC and
BCPAP/AURKA-NC). The silencing effect was confirmed by results
shown in Fig. 1A and B (P<0.01). The plate colony formation
assay demonstrated a reduction in the number of colonies formed by
BHT101/AURKA-siRNA (207±26.8) and BCPAP/AURKA-siRNA cells
(184±16.2) compared with BHT101/AURKA-NC (118±14.9) and
BCPAP/AURKA-NC cells (101±12.8; Fig.
1C and D; P<0.01). Flow
cytometric analysis revealed that AURKA inactivation induced G2/M
phase arrest in BHT101/AURKA-siRNA and BCPAP/AURKA-siRNA cells
(Fig. 1E-H; P<0.01). These
findings suggest that AURKA promotes proliferation by inhibiting
G2/M phase arrest in BHT101 and BCPAP cells.
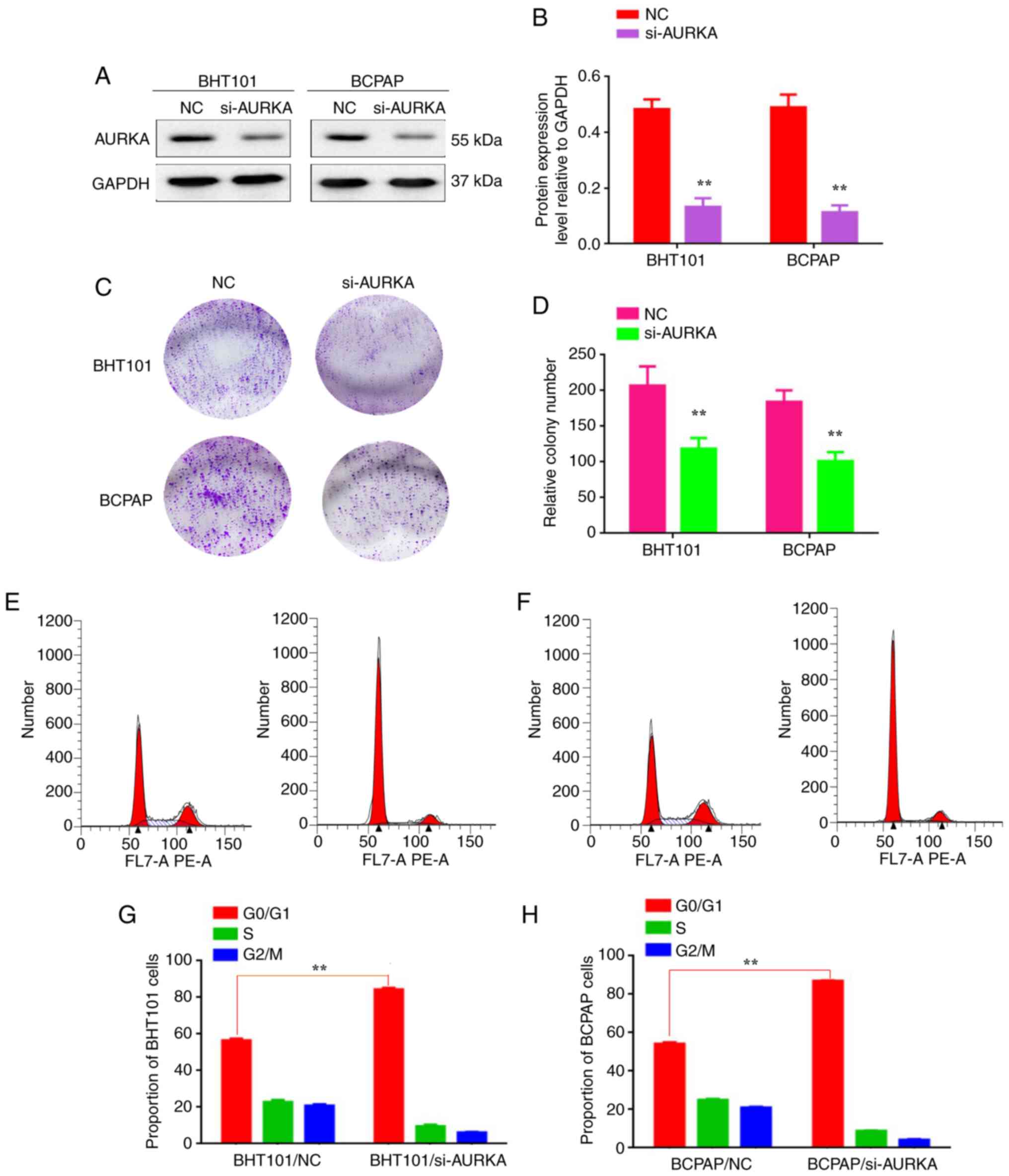 | Figure 1Decreasing AURKA expression reduces
the proliferation of BHT101 and BCPAP cells. (A) Western blot
analysis was used to detect the expression of AURKA in
BHT101/si-NC, BHT101/si-AURKA, BCPAP/si-NC and BCPAP/si-AURKA
cells. (B) The protein expression level relative to GAPDH in
BHT101/si-NC, BHT101/si-AURKA, BCPAP/si-NC and BCPAP/si-AURKA
cells. (C) Plate colony formation assay was used to detect the
proliferation of BHT101/si-NC, BHT101/si-AURKA, BCPAP/si-NC and
BCPAP/si-AURKA cells. (D) The relative colony number of
BHT101/si-NC, BHT101/si-AURKA, BCPAP/si-NC and BCPAP/si-AURKA
cells. (E-H) The proportion of G0/G1 cells of
BHT101/si-NC, BHT101/si-AURKA, BCPAP/si-NC and BCPAP/si-AURKA
cells. **P<0.01. AURKA, Aurora kinase A; siRNA, small
interfering RNA; NC, negative control. |
Decreasing AURKA expression inhibits
the migration and invasion of BHT101 and BCPAP cells
To further explore the true role of AURKA in THCA
metastasis, wound healing, migration and invasion assays were
conducted to assess the capabilities of BHT101 and BCPAP cells. The
results of the wound healing assay indicated that
BHT101/AURKA-siRNA and BCPAP/AURKA-siRNA cells exhibited reduced
motility at 24 h compared with BHT101/AURKA-NC and BCPAP/AURKA-NC
cells (Fig. 2A-D; P<0.01).
Additionally, it was observed that BHT101/AURKA-siRNA (64±2.6) and
BCPAP/AURKA-siRNA cells (70±1.76) migrated less than
BHT101/AURKA-NC cells (109±2.6) and BCPAP/AURKA-NC cells (129±2.6;
Fig. 2E-H; P<0.01).
Furthermore, BHT101/AURKA-siRNA cells (17±1.1) and
BCPAP/AURKA-siRNA cells (9±1.45) invaded less than BHT101/AURKA-NC
cells (56±3.5) and BCPAP/AURKA-NC cells (47±3.48; Fig. 2E-H; P<0.01). These results
suggest that reducing AURKA expression inhibited the migration and
invasion of BHT101 and BCPAP cells.
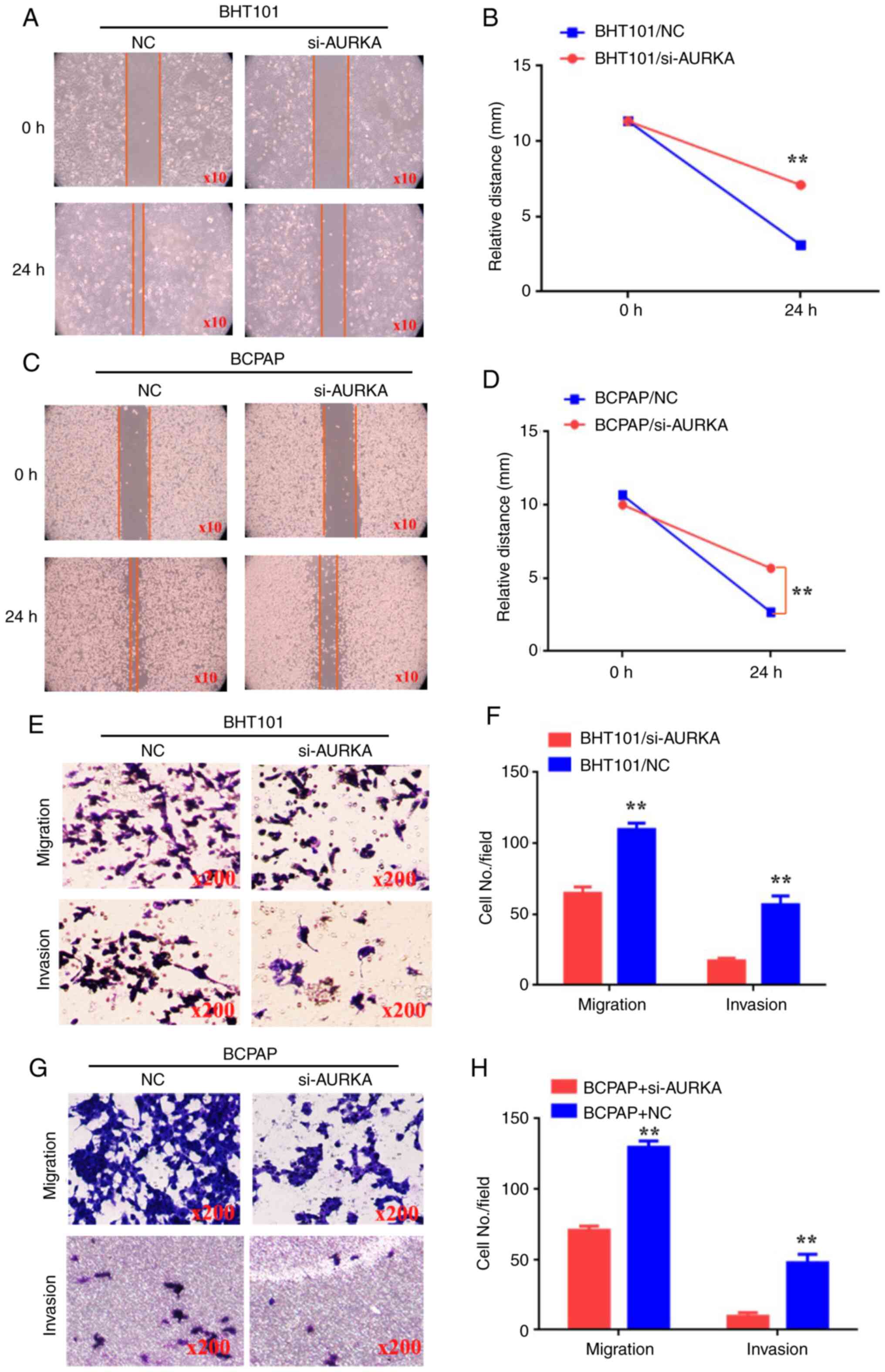 | Figure 2Decreasing AURKA expression inhibits
the migration and invasion of BHT101 and BCPAP cells. (A and C)
Wound healing assay was used to detect the ability of movement in
BHT101/si-NC, BHT101/si-AURKA, BCPAP/si-NC and BCPAP/si-AURKA
cells. (B and D) The relative distance (mm) of BHT101/si-NC,
BHT101/si-AURKA, BCPAP/si-NC and BCPAP/si-AURKA cells. (E and G)
Transwell and Matrigel assays were used to detect the ability of
migration and invasion in BHT101/si-NC, BHT101/si-AURKA,
BCPAP/si-NC and BCPAP/si-AURKA cells. (F and H) The cell number in
every field in BHT101/si-NC, BHT101/si-AURKA, BCPAP/si-NC and
BCPAP/si-AURKA cells. **P<0.01. AURKA, Aurora kinase
A; siRNA, small interfering RNA; NC, negative control. |
AURKA inhibition could reduce the
proliferation by regulating P130 and P107 molecules
Our previous study indicated that P130 and P107
molecules played crucial roles in cancer cell cycle and
proliferation (6). In the present
study, IF staining showed that P107 and P130 were localized in the
nucleus. P107 levels decreased, while P130 increased in
BHT101/AURKA-siRNA and BCPAP/AURKA-siRNA cells compared with
controls (BHT101/AURKA-NC cells and BCPAP/AURKA-NC cells; Fig. 3A). Similarly, western blotting
showed that proliferation-related molecule (P107) expression
decreased in BHT101/AURKA-siRNA and BCPAP/AURKA-siRNA cells.
Proliferation-related molecules (P130 and E2F4) increased in
BHT101/AURKA-siRNA and BCPAP/AURKA-siRNA cells (Fig. 3B and C; P<0.01). Furthermore, AURKA was
efficiently upregulated in BHT101 and BCPAP cells (Fig. 3D and E; P<0.01). Conversely, the
proliferation-related molecule (P107) increased in BHT101/AURKA and
BCPAP/AURKA cells. Proliferation-related molecules (P130 and E2F4)
decreased in BHT101/AURKA and BCPAP/AURKA cells (Fig. 3F and G; P<0.01). Therefore, it was concluded
that AURKA inhibition reduced proliferation by regulating P130 and
P107 molecules.
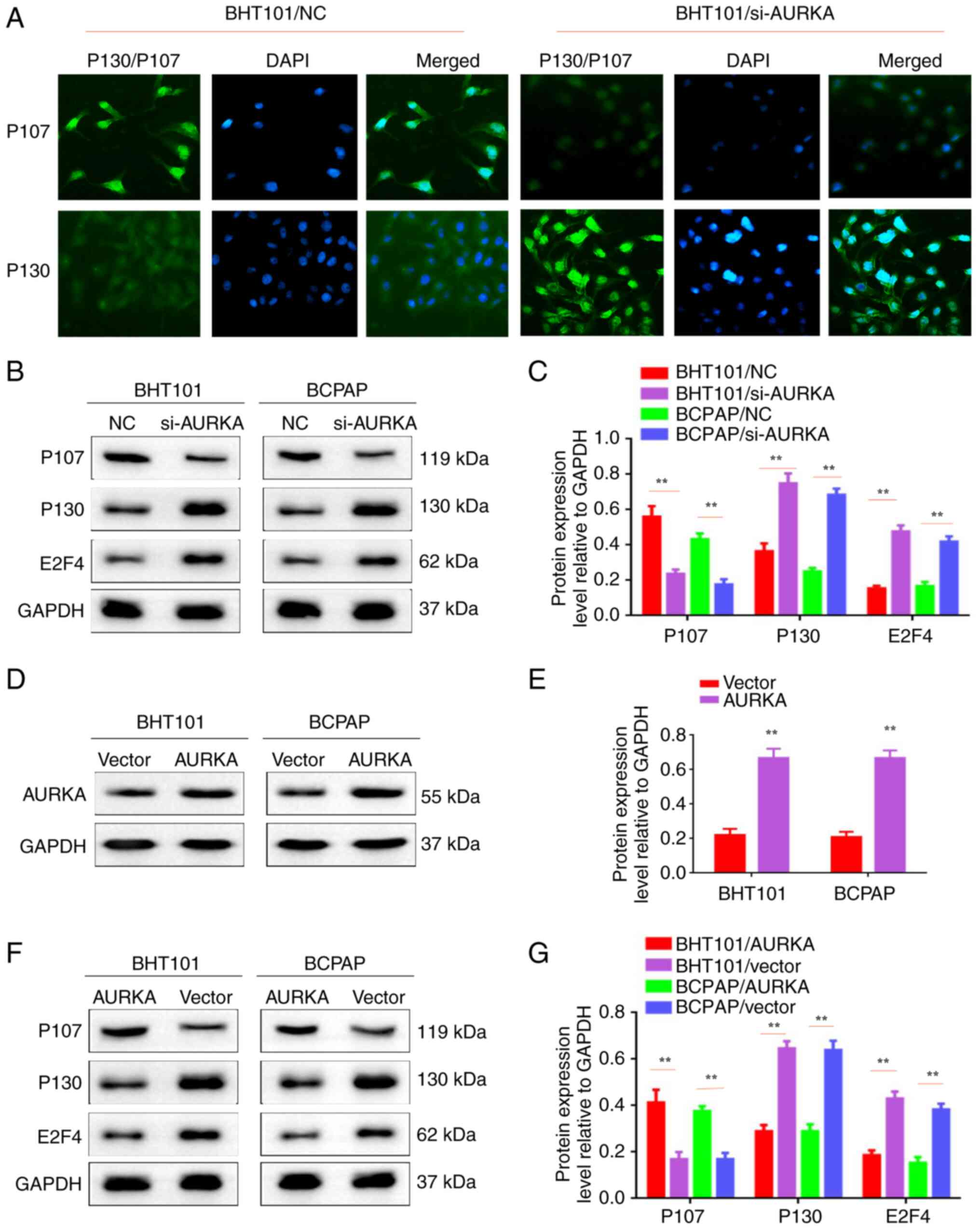 | Figure 3AURKA inhibition can reduce the
proliferation by regulating P130 and P107 molecules. (A)
Immunofluorescent staining assay was used to detect the expression
levels of P130 and P107 in BTH101/si-NC, BTH101/si-AURKA,
BCPAP/si-NC and BCPAP/si-AURKA cells. (B) Western blot analysis was
used to detect the expression levels of P107, P130, E2F4 and GAPDH
in BTH101/si-NC, BTH101/si-AURKA, BCPAP/si-NC and BCPAP/si-AURKA
cells. (C) The protein expression level relative to GAPDH in
BTH101/si-NC, BTH101/si-AURKA, BCPAP/si-NC and BCPAP/si-AURKA
cells. (D) Western blot analysis was used to detect the expression
of AURKA in BTH101/vector, BTH101/AURKA, BCPAP/vector and
BCPAP/AURKA cells. (E) The protein expression level relative to
GAPDH in BTH101/vector, BTH101/AURKA, BCPAP/vector and BCPAP/AURKA
cells. (F) Western blot analysis was used to detect the expression
levels of P107, P130, E2F4 and GAPDH in BTH101/vector,
BTH101/AURKA, BCPAP/vector and BCPAP/AURKA cells. (G) The protein
expression level relative to GAPDH in BTH101/vector, BTH101/AURKA,
BCPAP/vector and BCPAP/AURKA cells. **P<0.01. AURKA,
Aurora kinase A; siRNA, small interfering RNA; NC, negative
control. |
Inactivation of AURKA inhibits EMT in
BHT101 and BCPAP cells
EMT has been recognized to play a crucial role in
metastasis. In the present study, EMT-related proteins E-cadherin,
N-cadherin, vimentin and slug were assessed in BHT101/AURKA-siRNA,
BCPAP/AURKA-siRNA, BHT101/AURKA-NC and BCPAP/AURKA-NC cells. IF
staining results showed that E-cadherin expression was higher in
BHT101/AURKA-siRNA and BCPAP/AURKA-siRNA cells, while vimentin
expression was lower compared with controls (BHT101/AURKA-NC and
BCPAP/AURKA-NC cells). Western blot results similarly revealed
increased E-cadherin levels. However, other EMT-related molecules
(N-cadherin, vimentin and slug) were decreased (Fig. 4B-E; P<0.01). Thus, it was
concluded that AURKA inactivation inhibits EMT in BHT101 and BCPAP
cells.
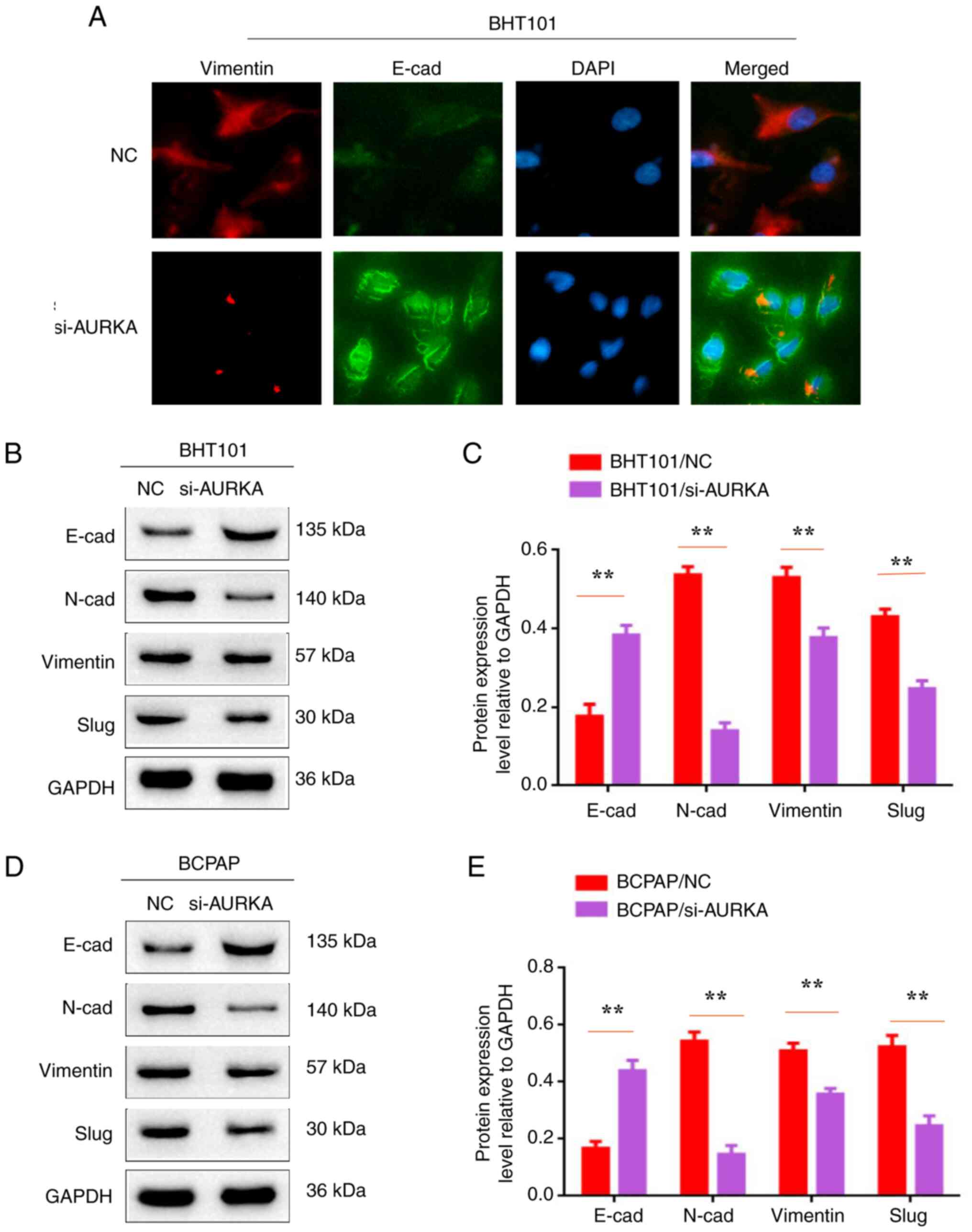 | Figure 4Inactivation of AURKA inhibits
epithelial-mesenchymal transition in BHT101 and BCPAP cells. (A)
Immunofluorescent staining assay was used to detect the expression
levels of Vimentin and E-cad in BTH101/si-NC, BTH101/si-AURKA,
BCPAP/si-NC and BCPAP/si-AURKA cells. (B and D) Western blot
analysis was used to detect the expression levels of E-cad, N-cad,
Vimentin, Slug and GAPDH in BTH101/si-NC, BTH101/si-AURKA,
BCPAP/si-NC and BCPAP/si-AURKA cells. (C and E) The protein
expression level relative to GAPDH in BTH101/si-NC,
BTH101/si-AURKA, BCPAP/si-NC and BCPAP/si-AURKA cells.
**P<0.01. AURKA, Aurora kinase A; siRNA, small
interfering RNA; NC, negative control; E-cad, E-cadherin; N-cad,
N-cadherin. |
Activation of AURKA promotes EMT in
BHT101 and BCPAP cells
To further explore AURKA-mediated EMT in THCA
metastasis, EMT-related proteins E-cadherin, N-cadherin, vimentin
and slug were assessed in BHT101/AURKA, BCPAP/AURKA, BHT101/vector
and BCPAP/vector cells. IF staining revealed that E-cadherin
expression was decreased and vimentin expression was increased in
BHT101/AURKA and BCPAP/AURKA cells compared with controls
(BHT101/vector and BCPAP/vector cells; Fig. 5A). Western blot analysis
demonstrated decreased E-cadherin levels and increased expression
of other EMT-related molecules (N-cadherin, vimentin and slug) in
AURKA-activated BHT101 and BCPAP cells (Fig. 5B-E; P<0.01). Therefore, it was
concluded that AURKA activation promotes EMT in BHT101 and BCPAP
cells.
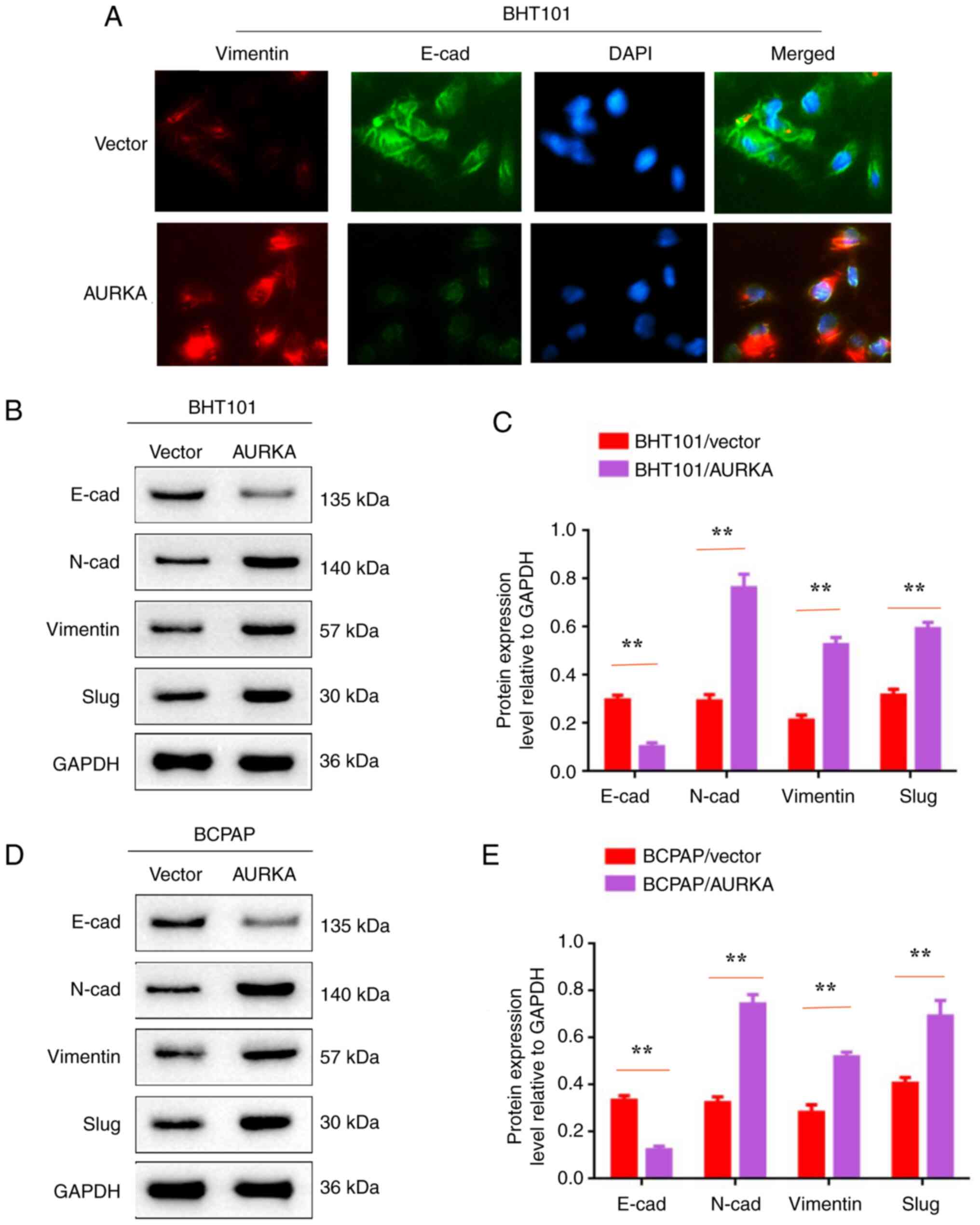 | Figure 5Activation of AURKA promotes
epithelial-mesenchymal transition in BHT101 and BCPAP cells. (A)
Immunofluorescent staining assay was used to detect the expression
levels of Vimentin and E-cad in BTH101/vector, BTH101/AURKA,
BCPAP/vector and BCPAP/AURKA cells. (B and D) Western blot analysis
was used to detect the expression levels of E-cad, N-cad, Vimentin,
Slug and GAPDH in BTH101/vector, BTH101/AURKA, BCPAP/vector and
BCPAP/AURKA cells. (C and E) The protein expression level relative
to GAPDH in BTH101/vector, BTH101/AURKA, BCPAP/vector and
BCPAP/AURKA cells. **P<0.01. AURKA, Aurora kinase A;
E-cad, E-cadherin; N-cad, N-cadherin. |
AURKA promotes EMT through the FAK
signaling pathway and FAK pathway inhibition impacts
proliferation-related proteins
The FAK signaling pathway has been recognized to
play a crucial role in cancer metastasis (13-16).
To explore the importance of FAK in THCA metastasis, protein
expression levels were determined using western blot analysis. FAK
protein levels decreased in BHT101/si-AURKA and BCPAP/si-AURKA
cells (Fig. 6A-D; P<0.01). To
further demonstrate that AURKA promotes EMT by regulating P130 and
P107 molecules through FAK signaling in THCA cells, P107, P130 and
E2F4 expression levels were explored in THCA cells treated with the
FAK inhibitor TAE226(17). Western
blot results revealed that P130 and E2F4 were increased and P107
decreased in BHT101/TAE226 and BCPAP/TAE226 cells (Fig. 6E-H, P<0.01). These findings
indicate that AURKA promotes EMT by regulating P130 and P107
molecules through the FAK signaling pathway in THCA cells.
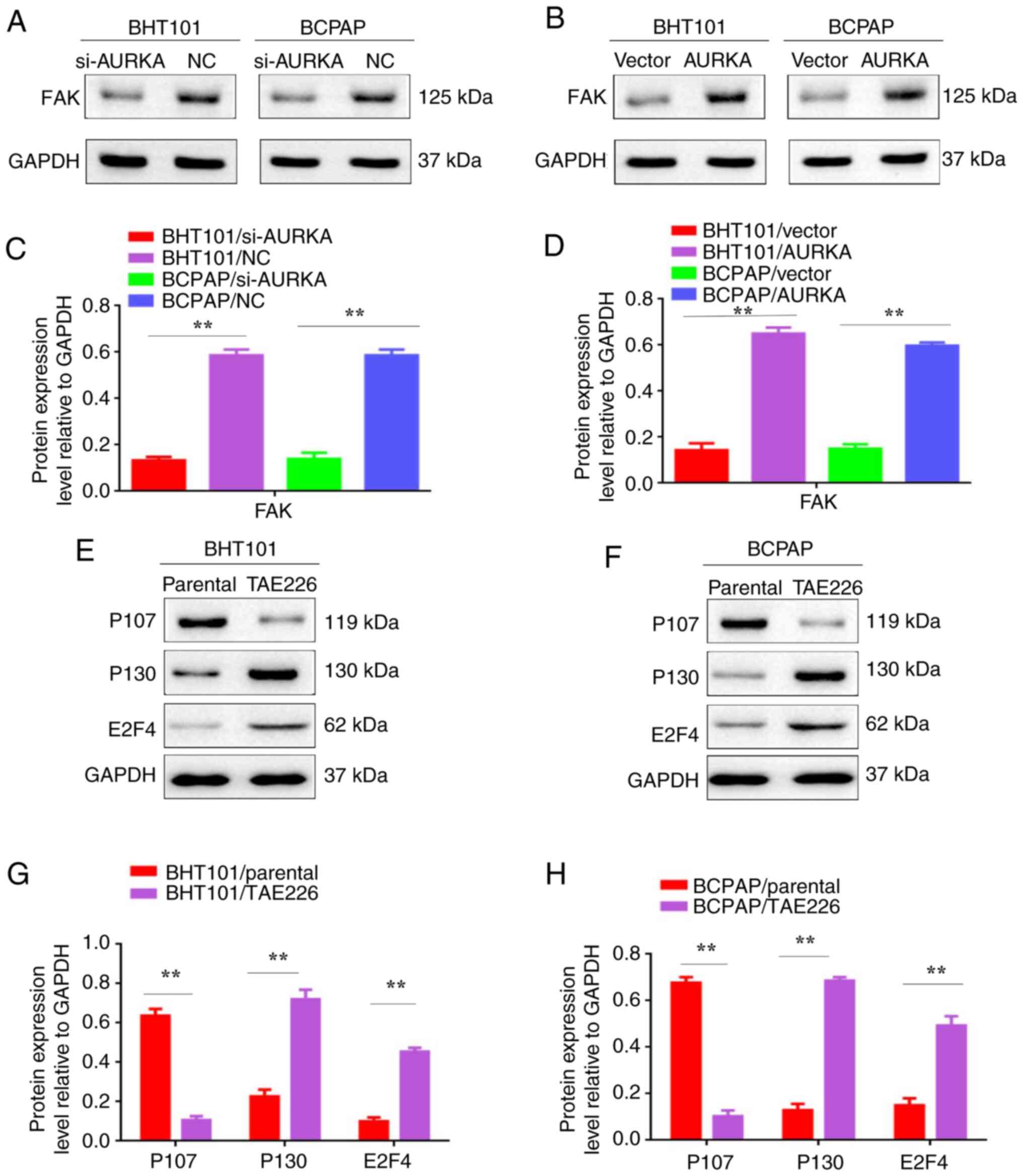 | Figure 6AURKA promotes epithelial-mesenchymal
transition through the FAK signaling pathway and FAK pathway
inhibition impacts proliferation-related proteins. (A) Western blot
analysis was used to detect the expression of FAK in BTH101/si-NC,
BTH101/si-AURKA, BCPAP/si-NC and BCPAP/si-AURKA cells. (B) Western
blot analysis was used to detect the expression of FAK in
BTH101/vector, BTH101/AURKA, BCPAP/vector and BCPAP/AURKA cells.
(C) The protein expression level relative to GAPDH in BTH101/si-NC,
BTH101/si-AURKA, BCPAP/si-NC and BCPAP/si-AURKA cells. (D) The
protein expression level relative to GAPDH in BTH101/vector,
BTH101/AURKA, BCPAP/vector and BCPAP/AURKA cells. (E) Western blot
analysis was used to detect the expression levels of P107, P130,
E2F4 and GAPDH in BTH101/si-NC, BTH101/si-AURKA, BCPAP/si-NC and
BCPAP/si-AURKA cells. (F) Western blot analysis was used to detect
the expression levels of P107, P130, E2F4 and GAPDH in
BTH101/vector, BTH101/AURKA, BCPAP/vector and BCPAP/AURKA cells.
(G) The protein expression level relative to GAPDH in BTH101/si-NC,
BTH101/si-AURKA, BCPAP/si-NC and BCPAP/si-AURKA cells. (H) The
protein expression level relative to GAPDH in BTH101/vector,
BTH101/AURKA, BCPAP/vector and BCPAP/AURKA cells.
**P<0.01. AURKA, Aurora kinase A; FAK, focal adhesion
kinase; siRNA, small interfering RNA; NC, negative control. |
Blocking FAK impairs the movement,
migration and invasion of THCA cells
To further investigate the potential of inhibiting
the tumor-promoting effects of THCA by blocking FAK expression,
wound-healing, migration and invasion assays were used to examine
the movement, migration and invasion abilities of THCA cells. THCA
cells were treated with the FAK inhibitor TAE226 (BCPAP/TAE226 and
BHT101/TAE226 cells). Control groups included THCA cells treated
with DMSO (BCPAP/parental and BHT101/parental cells) and THCA cells
without any treatment (BCPAP/ctrl and BHT101/ctrl cells). In the
wound-healing assay, BCPAP/TAE226 and BHT101/TAE226 cells
demonstrated reduced motility at 24 h compared with BCPAP/parental
and BHT101/parental cells (Fig.
7A-D; P<0.01). Moreover, the migration assay results
demonstrated that BCPAP/TAE226 (102±6.8) and BHT101/TAE226
(101±5.23) cells showed decreased movement through Matrigel
compared with BCPAP/parental (250±9.64), BCPAP/ctrl (248±13.96),
BHT101/parental (251±14.34) and BHT101/ctrl cells (252±17.61;
Fig. 7E and F; P<0.01). For the invasion assay, the
results identified that BCPAP/TAE226 (29±3.48) and BHT101/TAE226
(26±3.18) cells exhibited decreased movement through Matrigel
compared with BCPAP/parental (103±3.48), BCPAP/ctrl (102±9.20),
BHT101/parental (103±9.33) and BHT101/ctrl cells (109±6.93;
Fig. 7G and H; P<0.01). These findings indicated
that blocking FAK impairs the movement, migration and invasion of
THCA cells.
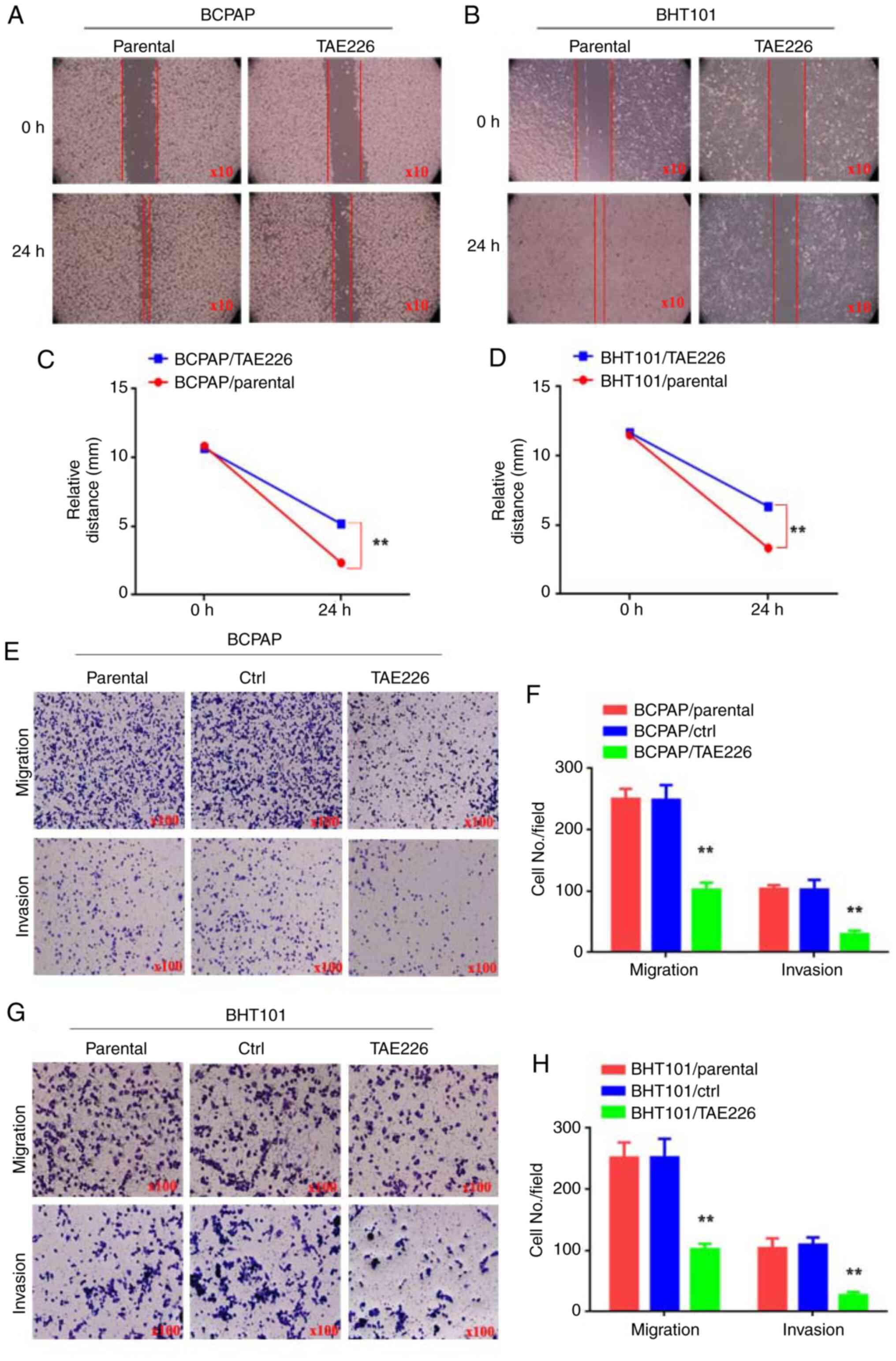 | Figure 7Blocking FAK impairs the movement,
migration and invasion of THCA cells. (A-D) The relative distance
(mm) of BTH101/parental, BTH101/TAE226, BCPAP/parental and
BCPAP/TAE226 cells. (E and G) Transwell assay was used to detect
the ability of migration and invasion in BTH101/parental,
BTH101/ctrl, BTH101/TAE226, BCPAP/parental, BCPAP/ctrl and
BCPAP/TAE226 cells. (F and H) The cell number in every field in
BTH101/parental, BTH101/ctrl, BTH101/TAE226, BCPAP/parental,
BCPAP/ctrl and BCPAP/TAE226 cells. **P<0.01. THCA,
thyroid cancer; FAK, focal adhesion kinase; ctrl, control. |
Discussion
THCA is a malignancy originating from thyroid
epithelial cells, characterized by a high incidence rate.
Metastasis remains the leading cause of mortality among patients
with THCA (18). However, the
precise molecular and cellular mechanisms governing THCA metastasis
are still not fully understood. Clarifying these mechanisms is
critical for identifying potential molecular targets aimed at
improving patient survival rates and quality of life.
Years following initial treatment, patients with
THCA might experience the development of local remnants or
disseminated tumors. This phenomenon can be attributed to tumor
dormancy-a phase where residual disease exists but remains
asymptomatic. The reactivation of dormant tumor cells could lead to
recurrence and metastasis. Therefore, exploring the specific
molecular mechanisms behind the activation of THCA dormancy holds
significant theoretical and practical value.
Building upon findings of previous studies that
highlighted the pivotal role of AURKA in LSCC tumorigenesis and
metastasis both in vitro (19) and in vivo (20), the authors examined the impact of
AURKA on cell proliferation, cell cycle progression and mobility in
LSCC cell lines. AURKA, an evolutionarily conserved Aurora
serine/threonine kinase, regulates the cell cycle (21) through processes such as centrosome
maturation, mitotic entry, centrosome separation, bipolar spindle
assembly, chromosome alignment, cytokinesis and mitotic exit
(22). Disruption during mitosis
can result in genetic instability and tumorigenesis. Colony
formation and flow cytometry assays demonstrated that AURKA
promoted proliferation, whereas the downregulation of AURKA
inhibited the transition from the G0 phase to active division in
BHT101 and BCPAP cells. Increased AURKA expression also enhanced
movement, migration and invasion capabilities in BHT101 and BCPAP
cells.
Research increasingly indicates that the
proliferation-associated proteins P130 and E2F4 are abundant in
quiescent cells (23,24), while P107 proteins are less
prevalent (25). E2F4, an E2F
transcription factor, mediates the expression of cell cycle
proteins (26). In the present
study, the inhibition of AURKA reduced proliferation by regulating
P130 and P107 molecules.
There is increasing evidence suggesting that EMT
plays a role in tumor invasion and metastasis (27). During tumor metastasis, cancer
cells acquire a mesenchymal phenotype and increased invasiveness
through EMT, allowing them to penetrate surrounding tissues. At
implantation sites, cancer cells eventually form metastases that
resemble the primary tumor through EMT (28), which is closely linked to tumor
biological characteristics and involves multiple regulatory
mechanisms. As research advances, various mechanisms have been
identified, including those involved in tumor growth, evasion and
dissemination, which hold significant clinical implications and may
provide new approaches for diagnosing and treating tumor
metastasis.
However, the molecular basis and regulatory
mechanisms of THCA EMT remain largely unexplored, presenting a
major challenge for research. In the present study, the
relationship between AURKA and EMT was explored in the context of
THCA metastasis. To the best of the authors' knowledge, no prior
studies have examined this aspect within THCA. Additionally,
upregulated AURKA expression promoted EMT, whereas downregulated
AURKA expression inhibited EMT.
While the present study provided valuable insights,
several limitations should be acknowledged. Primarily, the research
focuses on in vitro cell models without conducting in
vivo experiments to validate the observed phenomena, limiting
the applicability of the findings to clinical settings.
Additionally, the relatively small sample size may impact the
generalizability and statistical power of the results. The use of a
single or limited number of cell lines might not fully capture the
variability between different cell types. Long-term effects and
complex molecular mechanisms were also not thoroughly investigated,
and the techniques employed each have inherent limitations. Future
studies should aim to address these constraints to provide more
comprehensive and accurate information.
In conclusion, based on the aforementioned findings,
it was demonstrated that AURKA promotes EMT by regulating P130 and
P107 genes, thereby facilitating the metastasis of THCA.
Consequently, AURKA may represent a viable therapeutic target in
the treatment of THCA.
Supplementary Material
Verification of cloned colonies by
Sanger sequencing. Screening and confirmation were conducted using
Sanger sequencing to verify that the sequence of the inserted
fragment in the recombinant clones was completely consistent with
the target fragment sequence. AURKA, Aurora kinase A; CMV,
cytomegalovirus; PEX, plasmid vector.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by the Shanghai Healthy
Youth Project (grant no. 20234Y0055), the Natural Science
Foundation of Fujian (grant no. 2023J011342), Pudong the New Area
Clinical Characteristic Discipline (grant no. PWYts2021-15), the
Subject Construction Project of Pudong Health Commission of
Shanghai (grant no. PWZy2020-06), the Gongli Hospital National Fund
Cultivation Project (grant no. 2022GPY-B04), the Key Specialty
Construction Project of Health Bureau of Shanghai (grant no.
ZX2019C06) and the Pudong New Area Clinical Characteristic
Discipline (grant no. PWYts2021-15).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
LY and YG performed the experiments. LY and JL
analyzed the data. LY wrote the manuscript. GW revised the
manuscript. LY and GW designed the study. LY and GW interpreted the
data. LY and GW confirm the authenticity of all the raw data. All
authors read and approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Liao Y, Hua Y, Li Y, Zhang C, Yu W, Guo P,
Zou K, Li W, Sun Y, Wang R, et al: CRSP8 promotes thyroid cancer
progression by antagonizing IKKα-induced cell differentiation. Cell
Death Differ. 28:1347–1363. 2021.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Xu S, Mo S, Lin J, Yan Y, Liu X, Wu K,
Zhang H, Zhu Y, Chen L and Chen X: Loss of ID3 drives papillary
thyroid cancer metastasis by targeting E47-mediated epithelial to
mesenchymal transition. Cell Death Discov. 7(226)2021.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Zhan S, Wang T and Li J, Zhu H, Ge W and
Li J: Asporin interacts with HER2 to promote thyroid cancer
metastasis via the MAPK/EMT signaling pathway. Front Oncol.
12(762180)2022.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Asteriti IA, Polverino F, Stagni V,
Sterbini V, Ascanelli C, Naso FD, Mastrangelo A, Rosa A, Paiardini
A, Lindon C and Guarguaglini G: AurkA nuclear localization is
promoted by TPX2 and counteracted by protein degradation. Life Sci
Alliance. 6(e202201726)2023.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Maimaiti Y, Jie T, Jing Z, Changwen W, Pan
Y, Chen C and Tao H: Aurora kinase A induces papillary thyroid
cancer lymph node metastasis by promoting cofilin-1 activity.
Biochem Biophys Res Commun. 473:212–218. 2016.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Yang LY, He CY, Chen XH, Su LP, Liu BY and
Zhang H: Aurora kinase A revives dormant laryngeal squamous cell
carcinoma cells via FAK/PI3K/Akt pathway activation. Oncotarget.
7:48346–48359. 2016.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Ban Y, Zou Y, Liu Y, Lee SB, Bednarczyk
RB, Sheng J, Cao Y, Wong STC and Gao D: Targeting ribosome
biogenesis as a novel therapeutic approach to overcome EMT-related
chemoresistance in breast cancer bioRxiv. Preprint.
28(546927)2023.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Zhang Z, Li Y, Fan L, Wang B, Liu W, Cui J
and Tan B: LncRNA THUMPD3-AS1 promotes invasion and EMT in gastric
cancer by regulating the miR-1297/BCAT1 pathway. iScience.
26(108012)2023.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Meng F, Hua S, Chen X, Meng N and Lan T:
Lymph node metastasis related gene BICC1 promotes tumor progression
by promoting EMT and immune infiltration in pancreatic cancer. BMC
Med Genomics. 16(263)2023.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Koyama Y, Fujihara S, Chiyo T, Matsui T,
Hamaya S, Fujita K, Tani J, Morishita A, Kobara H, Ono M, et al:
Role of Mir-452-5p overexpression in epithelial-mesenchymal
transition (EMT) in early-stage colorectal cancer. In Vivo.
37:1980–1990. 2023.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Li M, Sun C, Bu X, Que Y, Zhang L, Zhang
Y, Zhang L, Lu S, Huang J, Zhu J, et al: ISL1 promoted
tumorigenesis and EMT via Aurora kinase A-induced activation of
PI3K/AKT signaling pathway in neuroblastoma. Cell Death Dis.
12(620)2021.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Miro C, Di Cicco E, Ambrosio R, Mancino G,
Di Girolamo D, Cicatiello AG, Sagliocchi S, Nappi A, De Stefano MA,
Luongo C, et al: Thyroid hormone induces progression and
invasiveness of squamous cell carcinomas by promoting a
ZEB-1/E-cadherin switch. Nat Commun. 10(5410)2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Jiang W, Cai F, Xu H, Lu Y, Chen J, Liu J,
Cao N, Zhang X, Chen X, Huang Q, et al: Extracellular signal
regulated kinase 5 promotes cell migration, invasion and lung
metastasis in a FAK-dependent manner. Protein Cell. 11:825–845.
2020.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Li H, Fu X, Zhao J, Li C, Li L, Xia P, Guo
J, Wei W, Zeng R, Wu J, et al: EXOC4 promotes diffuse-type gastric
cancer metastasis via activating FAK signal. Mol Cancer Res.
20:1021–1034. 2022.PubMed/NCBI View Article : Google Scholar
|
|
15
|
He X, Wang L, Li H, Liu Y, Tong C, Xie C,
Yan X, Luo D and Xiong X: CSF2 upregulates CXCL3 expression in
adipocytes to promote metastasis of breast cancer via the FAK
signaling pathway. J Mol Cell Biol. 15(mjad025)2023.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Liu P, Sun Y, Liu S, Niu J, Liu X and Chu
Q: SY-707, an ALK/FAK/IGF1R inhibitor, suppresses growth and
metastasis of breast cancer cells. Acta Biochim Biophys Sin
(Shanghai). 54:252–260. 2022.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Ghosh S and Cho SJ: Three-dimensional-QSAR
and relative binding affinity estimation of focal adhesion kinase
inhibitors. Molecules. 28(1464)2023.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Xiao Y, Tu Y and Li Y: Expression level of
long non-coding RNA colon adenocarcinoma hypermethylated serves as
a novel prognostic biomarker in patients with thyroid carcinoma.
Biosci Rep. 41(20210284)2021.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Zhang H, Chen X, Jin Y, Liu B and Zhou L:
Overexpression of Aurora-A promotes laryngeal cancer progression by
enhancing invasive ability and chromosomal instability. Eur Arch
Otorhinolaryngol. 269:607–614. 2012.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Zhang H, Chen X, Liu B and Zhou L: Effects
of stable knockdown of Aurora kinase A on proliferation, migration,
chromosomal instability, and expression of focal adhesion kinase
and matrix metalloproteinase-2 in HEp-2 cells. Mol Cell Biochem.
357:95–106. 2011.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Niu NK, Wang ZL, Pan ST, Ding HQ, Au GH,
He ZX, Zhou ZW, Xiao G, Yang YX, Zhang X, et al: Pro-apoptotic and
pro-autophagic effects of the Aurora kinase A inhibitor alisertib
(MLN8237) on human osteosarcoma U-2 OS and MG-63 cells through the
activation of mitochondria-mediated pathway and inhibition of p38
MAPK/PI3K/Akt/mTOR signaling pathway. Drug Des Devel Ther.
9:1555–1584. 2015.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Dar AA, Goff LW, Majid S, Berlin J and
El-Rifai W: Aurora kinase inhibitors - rising stars in cancer
therapeutics? Mol Cancer Ther. 9:268–278. 2010.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Correa RJ, Peart T, Valdes YR, DiMattia GE
and Shepherd TG: Modulation of AKT activity is associated with
reversible dormancy in ascites-derived epithelial ovarian cancer
spheroids. Carcinogenesis. 33:49–58. 2012.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Ruppender N, Larson S, Lakely B, Kollath
L, Brown L, Coleman I, Coleman R, Nguyen H, Nelson PS, Corey E, et
al: Cellular adhesion promotes prostate cancer cells escape from
dormancy. PLoS One. 10(130565)2015.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Spiliotaki M, Mavroudis D, Kapranou K,
Markomanolaki H, Kallergi G, Koinis F, Kalbakis K, Georgoulias V
and Agelaki S: Evaluation of proliferation and apoptosis markers in
circulating tumor cells of women with early breast cancer who are
candidates for tumor dormancy. Breast Cancer Res.
16(485)2014.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Iyirhiaro GO, Zhang Y, Estey C, O'Hare MJ,
Safarpour F, Parsanejad M, Wang S, Abdel-Messih E, Callaghan SM,
During MJ, et al: Regulation of ischemic neuronal death by
E2F4-p130 protein complexes. J Biol Chem. 289(18202)2014.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Huang Y, Hong W and Wei X: The molecular
mechanisms and therapeutic strategies of EMT in tumor progression
and metastasis. J Hematol Oncol. 15(129)2022.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Subbalakshmi AR, Sahoo S, McMullen I,
Saxena AN, Venugopal SK, Somarelli JA and Jolly MK: KLF4 induces
mesenchymal-epithelial transition (MET) by suppressing multiple
emt-inducing transcription factors. Cancers (Basel).
13(5135)2021.PubMed/NCBI View Article : Google Scholar
|














