Introduction
microRNAs (miRNAs) are a class of RNA molecules,
typically 19–25 nucleotides (nt) long, comprising highly conserved
families of non-coding RNA that have gained recognition as
important regulators of diverse cell processes, such as
proliferation, differentiation, development, and cell death
(1). miRNAs are negative
regulators of gene expression that inhibit the translation or
promote the degradation of target mRNAs, with an estimated 30% of
transcribed mRNAs thought to be susceptible to miRNA-mediated
regulation (2).
miRNAs play an important role in regulating normal
organ physiology and development (3). The elucidation of the spatial and
temporal patterns of their expression is important for
understanding the precise role of miRNAs in organogenesis (4). To gain a more complete understanding
of miRNA functions, investigations were conducted into global
patterns of miRNA expression in mammalian tissues, and a number of
miRNAs enriched in specific tissues were identified (5). Although the spatial and temporal
expression patterns of some developing mouse tissues, such as,
retina, bladder and brain, have been analyzed, those of the heart
remain to be investiagated (6–8).
Through this process, specific miRNAs at critical stages of organ
development have been identified and quantified, providing valuable
insight into their role during organogenesis.
Findings of previous studies suggested that miRNAs
play an essential role in the maintenance of cardiac development
and disease (9). A series of
studies, profiling miRNA expression in rodent and human hearts
under various pathological conditions, including cardiac
hypertrophy, heart failure and myocardial infarction demonstrated
that miRNAs are involved in cardiac pathophysiology (10–12). The global patterns of miRNA
expression of normal human and mouse heart have been profiled in
adults (3). Furthermore, some
cardiac-specific miRNAs, including miR-1, miR-133a and miR-208a,
involved in maintaining cardiac development and function have been
identified (13–15).
However, the heart, more than any other organ, has
to maintain a high level of function throughout the lifespan of the
organism, starting from the early primitive heart tube, to
formation of the heart chambers, and throughout life (16). It is known that many miRNAs show
spatially and/or temporally restricted expression patterns
(17). Thus, by characterizing
the spatial and temporal expression profiles of miRNAs in the
developing heart, we can improve our understanding of heart
development and gene regulation. The differentially expressed
miRNAs of mouse ventricular chambers in 3 distinct developmental
stages [embryonic day (E)12.5, E15.5 and E18.5] have been profiled
(18). Most of the differentially
expressed miRNAs exhibited a relatively discrete peak of expression
at ventricular developmental stages; however, spatial and temporal
expression profiles of miRNAs in heart have not been examined in
detail.
The mouse heart shows great similarity to the human
heart, with respect to anatomy, growth and development, making the
mouse an important experimental model for biomedical research
(19). The heart is the first
functional organ during mouse embryonic development. During this
stage, the primitive heart tube, in which the heart begins to beat
at approximately the E9.0, begins to form. The form of the heart
starts to take shape at approximately E10.0, and at E12.5–18.5, the
tube undergoes a complex series of movements and tissue remodeling
events that lead to the formation of the 4-chambered heart
(20). Based on this
developmental timeline, we selected 4 key time-points (E12.5,
E14.5, E16.5 and E18.5) representing the process of normal heart
development, to perform a miRNA screening by next-generation
sequencing in C57BL/6 mice. The aim of this study was to explore
the mechanisms of miRNAs in embryonic heart development, and offer
a foundation for future functional analyses.
Materials and methods
Experimental animals
The Nanjing Medical University Animal Care and Use
Committee approved the experimental protocols used in this study.
Pathogen-free male and female C57BL/6J mice were obtained from the
animal center of the Nanjing Medical University. The animals were
housed in individual cases in a temperature-controlled room with a
12-h light/dark cycle. At the age of 6 months, the males and
females were mated. Pregnancy was detected by visual inspection of
a distended abdomen. At E12.5, E14.5, E16.5 and E18.5, pregnant
mice were sacrificed with CO2, embryos were collected
and fetal hearts dissected and pooled within each age group for
further analysis. The 4 experimental groups were designated as: M1
(E18.5), M2 (E16.5), M3 (E14.5) and M4 (E12.5).
Hematoxylin and eosin (H&E)
staining
Collected fetal hearts were washed with cold PBS and
then fixed in formalin overnight at 4°C. Sections (7 μm) of
paraformaldehyde-fixed heart tissue were obtained and stained with
H&E for morphological analysis. H&E sections were viewed
under a light microscope at magnifications of ×40 to observe
changes in fetal heart development at the 4 experimental
time-points.
Isolation of miRNA, and sequencing by
oligonucleotide ligation and detection (SOLiD) sequencing and
analysis
At each time point, fetal cardiac tissue was
removed, snap-frozen in liquid nitrogen and stored at −80°C for
later analysis. Total miRNA was extracted from cardiac tissue of
fetal mice using the mirVana miRNA Isolation kit (Applied
Biosystems-Life Technologies Co., Grand Island, NY, USA) according
to the manufacturer’s instructions.
The methodological details of sample processing, RNA
extraction, library construction, and SOLiD sequencing were
described in our previous study (21). Samples of miRNA (100 ng) isolated
from cardiac tissue of fetal mouse were processed into sequencing
libraries using the Small RNA Expression kit (Applied Biosystems).
Briefly, RNA was ligated overnight with the adapters from the kit,
reverse-transcribed, RNAse H-treated and PCR amplified before
agarose gel electrophoresis for size selection of miRNAs containing
inserted sequences of 16–61 nt. Libraries were amplified onto beads
using emulsion PCR, deposited on slides and sequenced using the
SOLiD v2 sequencing system (Applied Biosystems) at the State Key
Laboratory of Bioelectronics, Southeast University, China. Data
were analyzed with the SOLiD System Small RNA Analysis Pipeline
Tool (RNA2MAP). Acceptable sequences were compared with sequences
in the mouse miRBase database (release 14.0, http://www.mirbase.org; Sanger). The threshold for
selection was set conservatively to include beads sampled a minimum
of 10 times in any of the libraries.
Quantitative real-time PCR
The methodological details of quantitative real-time
PCR (qRT-PCR) were described in our previous study (21), which was performed to confirm the
differential expression of miRNAs identified by SOLiD sequencing.
Briefly, total RNA was isolated from cardiac tissue of fetal mice
using TRIzol reagent (Invitrogen, Carlsbad, CA, USA). Single-strand
cDNA was synthesized as follows: the reverse transcription mixture
contained 2 μl total RNA, 1 μl mmu-miRNA reverse primer (Table I), 1 μl ReverTra Ace, 4 μl 5X
buffer, 2 μl dNTP mix (10 mM), 1 μl RNasin, 1 μl random primer and
8 μl RNase-Free H2O (20 μl total volume). The reaction
was performed according to the manufacturer’s instructions using
the Applied Biosystems 7300 real-time PCR system (Applied
Biosystems) (Table II). Data were
analyzed using an iCycler™ iQ Optical System Software, Version 3.0a
(Bio-Rad Laboratories, Hercules, CA, USA). The relative level of
mmu-miRNA was calculated relative to U6 RNA (internal control)
using the 2−ΔΔCt method.
 | Table I.RT primer sequences. |
Table I.
RT primer sequences.
| Gene name | RT primers |
|---|
| let-7a |
5′-CGTCGCGGCATCGAGTGGAGCAGACCGACAGCGCGACGGATTAGGAAAGA-3′ |
| let-7d |
5′-CGTCGCGGCATCGAGTGGAGCAGACCGACAGCGCGACGGATAAGAAAGGC-3′ |
| let-7e |
5′-CGTCGCGGCATCGAGTGGAGCAGACCGACAGCGCGATATACAACCTCC-3′ |
| let-7f |
5′-CGTCGCGGCATCGAGTGGAGCAGACCGACAGCGCGACGGATATACAATCTA-3′ |
| miR-206 |
5′-CGTCGCGGCATCGAGTGGAGCAGACCGACAGCGCGACGGCCACATGC-3′ |
| miR-184 |
5′-CGTCGCGGCATCGAGTGGAGCAGACCGACAGCGCGACGACCTACCCTT-3′ |
| miR-146b |
5′-CGTCGCGGCATCGAGTGGAGCAGACCGACAGCGCGACGGAGAACTTTG-3′ |
| Table II.Primers for real-time RT-PCR. |
Table II.
Primers for real-time RT-PCR.
| Gene name | Forward primer | Reverse primer |
|---|
| let-7a |
5′-GCTACTGTCTTTCCTAAG-3′ |
5′-GCATCGAGTGGAGCAGAC-3′ |
| let-7d |
5′-TTAACTATACGACCTGCTGC-3′ |
5′-GCATCGAGTGGAGCAGAC-3′ |
| let-7e |
5′-GGGTGAGGTAGGAGGTTGTATA-3′ |
5′-GCATCGAGTGGAGCAGAC-3′ |
| let-7f |
5′-GGTGAGGTAGTAGATTGTATA-3′ |
5′-GCATCGAGTGGAGCAGAC-3′ |
| miR-206 |
5′-GGATATAAAGAAGCATGT-3′ |
5′-GCATCGAGTGGAGCAGACC-3′ |
| miR-184 |
5′-GAACTGATAAGGGTAGGA-3′ |
5′-GCATCGAGTGGAGCAGAC-3′ |
| miR-146b |
5′-GGTGGCCAAAGTTCTCTCA-3′ |
5′-GCATCGAGTGGAGCAGAC-3′ |
| U6 |
5′-CAGGGGCCATGCTAAATCTTC-3′ |
5′-CTTCGGCAGCACATATACTAAAAT-3′ |
Target gene ontology and network analysis
of target gene-miRNAs
The methodological details of target gene ontology
and network analysis of target gene-miRNAs were described in our
previous study (21). In brief,
target genes were analyzed by gene ontology (http://www.babelomics.bioinfo.cipf.es/). A graphical
representation of the network between the miRNAs and their
predicted targets involved in cardiac development was identified by
IPA analysis (http://www.ingenuity.com/).
Statistical analysis
Putative miRNA candidates were selected according to
the following criteria: i) at least 10 copies by SOLiD sequencing;
ii) fold-change >2, based on the normalized counts between
different time-points (M1 vs. M2, M1 vs. M3, M1 vs. M4, M2 vs. M3,
M2 vs. M4 and M3 vs. M4), as well as between the later development
group (M1+M2) and early development group (M3+M4); iii) the data of
the qRT-PCR were presented as the mean ± SEM. P<0.05 was
considered statistically significant.
Results
Overview of the SOLiD sequencing
data
After SOLiD sequencing, raw reads were obtained from
the small RNA library. Low-quality reads were removed, and the 39
adaptor sequences were trimmed. Small RNA sequences ranging in size
from 18 to 28 nt were retrieved from the raw data set. The size
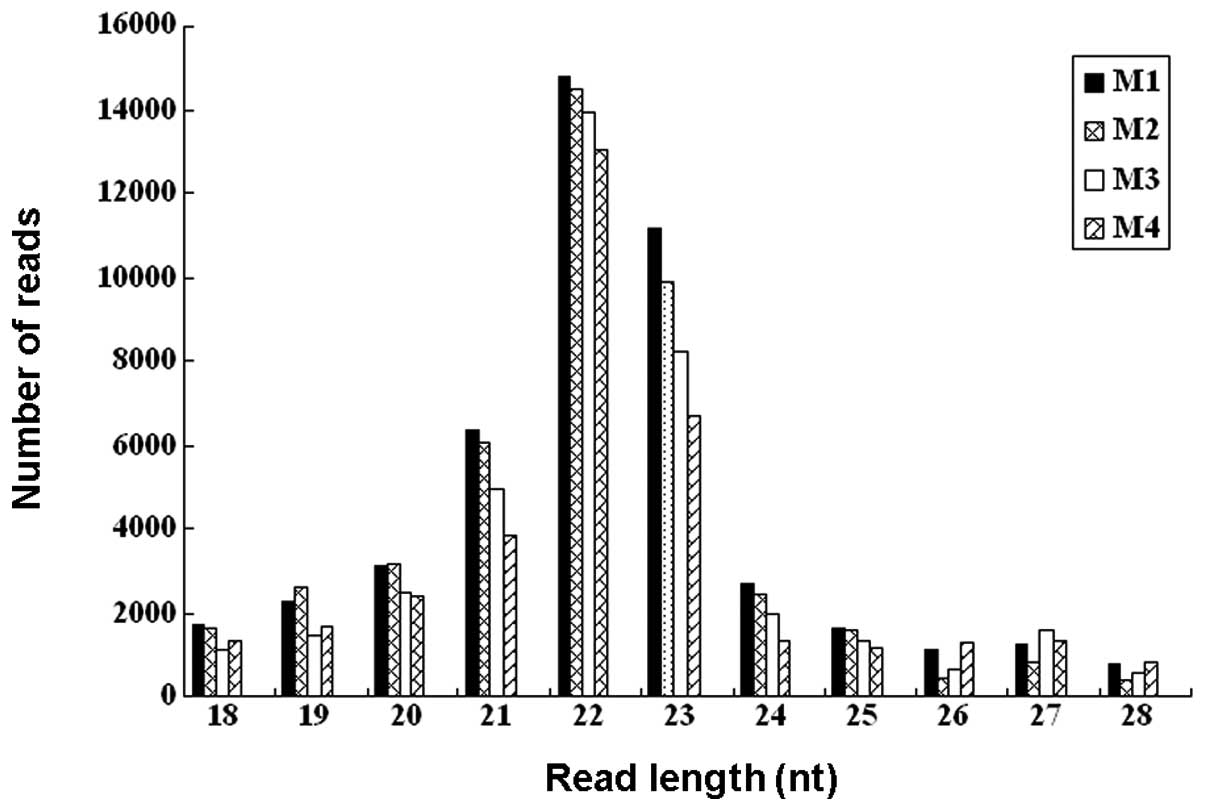
distributions of the reads are shown in Fig. 1. The majority of the small RNAs
were between 21 and 23 nt in size. Sequences of 22 nt accounted for
31.5–37.3% of total sequence reads in the 4 samples, which is the
typical size range for Dicer-derived products. The 23 nt size class
was also dominant.
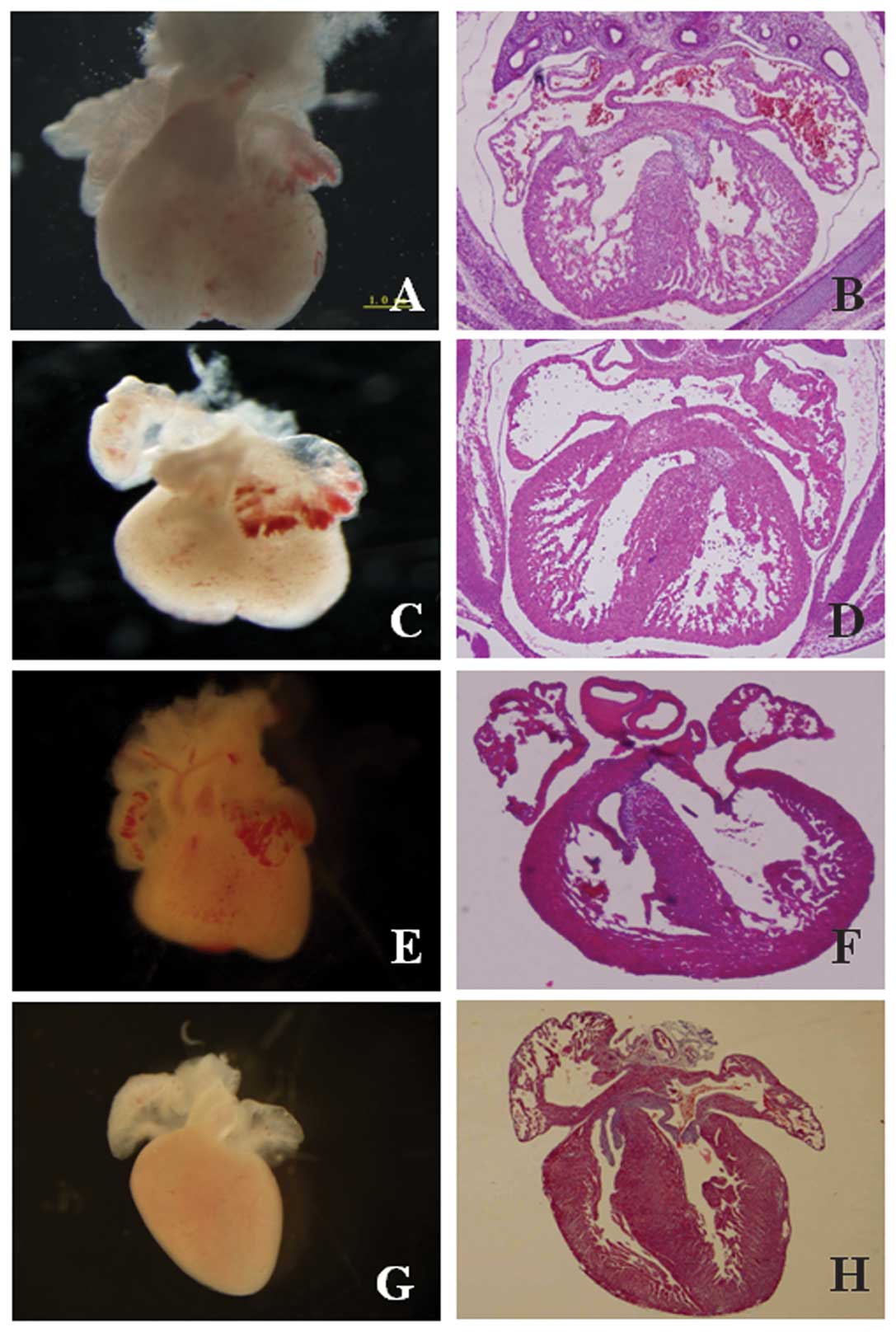
Histology
A series of hearts were collected at each time point
for histological analysis using H&E (Fig. 2). The typical features of the
developing heart were observed at each time point: in the M4 group,
the endocardial cushions appeared to fuse, and the sepals, aorta
and tracheal were also visible. In the M3 group, aortic and
pulmonary arterial structures were found to be well-developed, and
the left ventricular outflow tract, the inflow tract and mitral
valve were all visible. In the M2 group, the myocardium had
completed development. In the M1 group, the endocardial, myocardial
and epicardial layers had matured.
miRNA expression profile
The expression profiles of miRNAs in the cardiac
tissue of fetal mice were analyzed in 4 samples. The top expressed
miRNAs of the 4 groups are shown in Table III, including miRNAs present at
>1% of total read counts. The types of the top expressed miRNAs
gradually increased along with the increasing gestational age from
E12.5 to E18.5. The 10 miRNAs included in the top expressed miRNAs
of the 4 groups are: mmu-miR-23b, mmu-miR-24, mmu-miR-23a,
mmu-miR-375, mmu-miR-29a, mmu-miR-93, mmu-miR-21, mmu-miR-25,
mmu-let-7b and mmu-miR-27b.
| Table III.The top expressed miRNAs in the
developing heart. |
Table III.
The top expressed miRNAs in the
developing heart.
M1 (E18.5)
| M2 (E16.5)
| M3 (E14.5)
| M4 (E12.5)
|
|---|
| miRNA | % | miRNA | % | miRNA | % | miRNA | % |
|---|
| mmu-miR-23b | 22.07 | mmu-miR-23b | 22.89 | mmu-miR-23b | 32.09 | mmu-miR-23b | 30.65 |
| mmu-miR-23a | 13.99 | mmu-miR-24 | 17.07 | mmu-miR-24 | 18.42 | mmu-miR-24 | 18.11 |
| mmu-miR-24 | 10.94 | mmu-miR-23a | 8.79 | mmu-miR-23a | 11.41 | mmu-miR-23a | 10.77 |
| mmu-miR-221 | 5.47 | mmu-miR-375 | 7.36 | mmu-miR-375 | 5.30 | mmu-miR-375 | 7.54 |
| mmu-miR-375 | 4.37 | mmu-miR-93 | 3.08 | mmu-miR-29a | 2.96 | mmu-miR-29a | 2.41 |
| mmu-miR-29a | 4.25 | mmu-miR-29a | 2.91 | mmu-miR-93 | 2.68 | mmu-miR-93 | 2.28 |
| mmu-miR-93 | 2.30 | mmu-let-7a | 2.17 | mmu-miR-21 | 1.26 | mmu-miR-21 | 1.58 |
| mmu-miR-21 | 2.16 | mmu-miR-351 | 1.60 | mmu-miR-27b | 1.22 | mmu-miR-25 | 1.58 |
| mmu-miR-222 | 2.03 | mmu-miR-25 | 1.54 | mmu-miR-25 | 1.22 | mmu-let-7b | 1.52 |
| mmu-let-7b | 1.80 | mmu-let-7f | 1.48 | mmu-let-7b | 1.16 | mmu-miR-27b | 1.39 |
| mmu-let-7a | 1.76 | mmu-let-7b | 1.43 | mmu-miR-320 | 1.02 | | |
| mmu-miR-27b | 1.23 | mmu-miR-26a | 1.37 | | | | |
| mmu-miR-25 | 1.23 | mmu-miR-181d | 1.37 | | | | |
| mmu-miR-26a | 1.21 | mmu-miR-27b | 1.31 | | | | |
| mmu-let-7f | 1.20 | mmu-miR-181b | 1.20 | | | | |
| mmu-miR-22 | 1.06 | | | | | | |
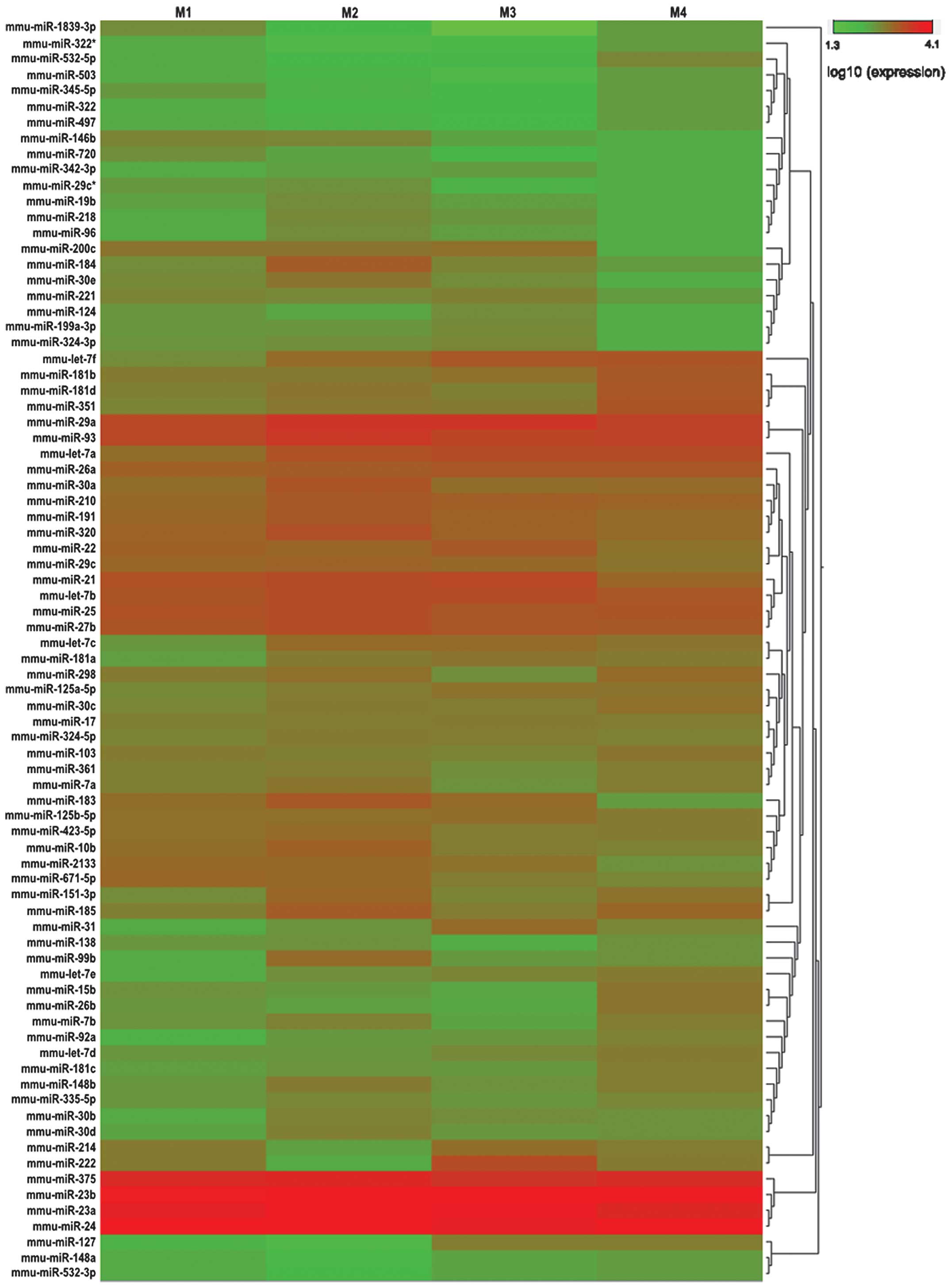
We used a threshold of at least 10 copies by SOLiD
sequencing in the 4 groups, and found 80 miRNAs that met those
requirements. These miRNAs were used for further hierarchical
cluster analysis (Fig. 3). As a
result, 77 differentially expressed miRNAs in the 4 groups (M1, M2,
M3 and M4) passed our fold-change filter (fold-change >2.0),
including 51 differentially upregulated miRNAs and 26
differentially downregulated miRNAs (Tables IV and V). Among the differentially expressed
miRNAs, we identified 8 downregulated and 6 upregulated miRNAs in
M1/M2; 2 downregulated and 8 upregulated miRNAs in M1/M3; 5
downregulated and 7 upregulated miRNAs in M1/M4; 3 downregulated
and 13 upregulated miRNAs in M2/M3; 3 downregulated and 13
upregulated miRNAs in M2/M4; 5 downregulated and 4 upregulated
miRNAs in M3/M4, all with a fold-change >2 (Tables IV and V). From this list of differentially
expressed miRNAs, we aimed to identify miRNAs that were
consistently downregulated or upregulated during heart development,
however, no miRNA met these requirements.
| Table IV.Differentially expressed miRNAs
upregulated in the developing heart. |
Table IV.
Differentially expressed miRNAs
upregulated in the developing heart.
M1 vs. M2
| M1 vs. M3
| M1 vs. M4
| M2 vs. M3
| M2 vs. M4
| M3 vs. M4
|
|---|
| miRNA | Ratio | miRNA | Ratio | miRNA | Ratio | miRNA | Ratio | miRNA | Ratio | miRNA | Ratio |
|---|
| mmu-miR-221 | 47.88 | mmu-miR-222 | 53.42 | mmu-miR-221 | 28.77 | mmu-miR-222 | 10.52 | mmu-let-7f | 7.81 | mmu-miR-184 | 3.51 |
| mmu-miR-222 | 5.08 | mmu-miR-221 | 43.50 | mmu-let-7f | 6.31 | mmu-miR-351 | 6.77 | mmu-miR-351 | 6.31 | mmu-let-7c | 2.94 |
| mmu-miR-181b | 4.84 | mmu-miR-214 | 9.83 | mmu-miR-222 | 5.34 | mmu-miR-214 | 6.39 | mmu-miR-181d | 4.33 | mmu-miR-151-3p | 2.42 |
| mmu-miR-21 | 2.70 | mmu-let-7f | 3.20 | mmu-let-7c | 4.65 | mmu-miR-15b | 6.24 | mmu-let-7a | 3.81 | mmu-miR-185 | 2.18 |
| mmu-miR-2133 | 2.51 | mmu-let-7d | 2.51 | mmu-miR-22 | 3.35 | mmu-miR-181b | 5.71 | mmu-miR-15b | 3.61 | | |
| mmu-miR-22 | 2.06 | mmu-miR-181b | 2.34 | mmu-let-7a | 3.09 | mmu-let-7d | 5.07 | mmu-let-7c | 3.61 | | |
| | mmu-miR-22 | 2.20 | mmu-miR-185 | 2.25 | mmu-miR-181d | 5.05 | mmu-miR-181b | 3.15 | | |
| |
mmu-miR-125a-5p | 2.05 | | | mmu-miR-181c | 4.06 | mmu-let-7d | 3.15 | | |
| | | | | | mmu-let-7f | 3.96 | mmu-miR-181a | 3.15 | | |
| | | | | | mmu-miR-30c | 2.68 | mmu-miR-7b | 2.70 | | |
| | | | | | mmu-let-7a | 2.29 | mmu-miR-148b | 2.70 | | |
| | | | | | mmu-miR-298 | 2.23 | mmu-miR-151-3p | 2.70 | | |
| | | | | |
mmu-miR-125a-5p | 2.20 | mmu-miR-185 | 2.52 | | |
| | | | | | mmu-miR-7b | 2.01 |
mmu-miR-125a-5p | 2.40 | | |
| Table V.Differentially expressed miRNAs
downregulated in the developing heart. |
Table V.
Differentially expressed miRNAs
downregulated in the developing heart.
M1 vs. M2
| M1 vs. M3
| M1 vs. M4
| M2 vs. M3
| M2 vs. M4
| M3 vs. M4
|
|---|
| miRNA | Ratio | miRNA | Ratio | miRNA | Ratio | miRNA | Ratio | miRNA | Ratio | miRNA | Ratio |
|---|
| mmu-miR-361 | 0.45 | mmu-miR-185 | 0.44 | mmu-miR-361 | 0.49 | mmu-miR-2133 | 0.41 | mmu-miR-671-5p | 0.30 | mmu-miR-103 | 0.49 |
| mmu-miR-148b | 0.42 | mmu-miR-26b | 0.35 | mmu-miR-26b | 0.49 | mmu-miR-184 | 0.17 | mmu-miR-103 | 0.25 | mmu-miR-181c | 0.44 |
| mmu-miR-181b | 0.41 | | | mmu-miR-298 | 0.42 | mmu-miR-183 | 0.16 | mmu-miR-183 | 0.20 | mmu-miR-26b | 0.44 |
| mmu-miR-185 | 0.38 | | | mmu-miR-671-5p | 0.41 | | | | | mmu-miR-214 | 0.14 |
| mmu-miR-181c | 0.32 | | | mmu-miR-103 | 0.41 | | | | | mmu-miR-222 | 0.10 |
| mmu-miR-138 | 0.30 | | | | | | | | | | |
| mmu-miR-15b | 0.15 | | | | | | | | | | |
| mmu-miR-26b | 0.14 | | | | | | | | | | |
Since there was no miRNA showing obvious
fold-changes consistently between the 4 groups, we re-analyzed our
data to profile the differentially expressed miRNAs between the
later development group (M1+M2) and the early development group
(M3+M4). Using this analysis 16 differentially expressed miRNAs
located in 11 chromosomes were identified (Table VI), 3 of which were downregulated
and 13 of which were upregulated, with a fold-change >2.
| Table VI.Differentially expressed miRNAs of
the fetal mouse heart the between late development and early
development groups. |
Table VI.
Differentially expressed miRNAs of
the fetal mouse heart the between late development and early
development groups.
| miRNA | (M1+M2) vs.
(M3+M4) | Log2
[(M1+M2)/(M3+M4)] | Chromosomal
localization | Start locus | Stop locus |
|---|
| mmu-miR-127 | 8.90 | 3.15 | 12 | 110040653 | 110040722 |
| mmu-miR-31 | 5.18 | 2.37 | 4 | 88381788 | 88381893 |
| mmu-let-7f | 4.75 | 2.25 | 13 | 48633198 | 48633286 |
| mmu-let-7e | 3.78 | 1.92 | 17 | 17534970 | 17535062 |
| mmu-miR-532-5p | 3.09 | 1.63 | X | 6405361 | 6405456 |
| mmu-miR-92a | 3.03 | 1.60 | 14 | 115443649 | 115443728 |
| mmu-let-7d | 2.91 | 1.54 | 13 | 48631381 | 48631483 |
| mmu-miR-15b | 2.63 | 1.40 | 3 | 68813694 | 68813757 |
| mmu-let-7a | 2.59 | 1.37 | 13 | 48633548 | 48633641 |
| mmu-miR-148a | 2.41 | 1.27 | 6 | 51219811 | 51219909 |
| mmu-miR-181a | 2.36 | 1.24 | 1 | 139863032 | 139863118 |
| mmu-miR-532-3p | 2.23 | 1.16 | X | 6825528 | 6825623 |
|
mmu-miR-125a-5p | 2.22 | 1.15 | 17 | 17967776 | 17967843 |
| mmu-miR-206 | 0.46 | −1.12 | 1 | 20669091 | 20669163 |
| mmu-miR-184 | 0.41 | −1.29 | 9 | 89697098 | 89697166 |
| mmu-miR-146b | 0.34 | −1.56 | 19 | 46417252 | 46417360 |
Validation of differentially expressed
miRNAs
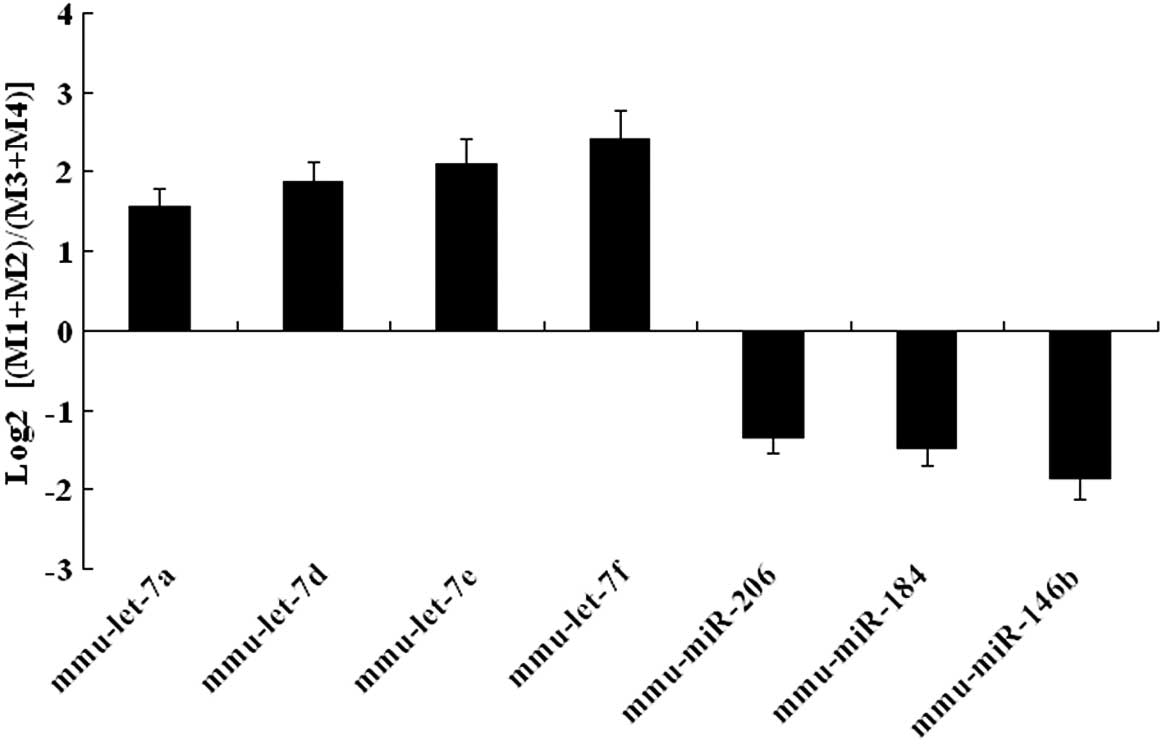
SOLiD sequencing results were validated by qRT-PCR
expression analysis in the later development and early development
groups. Upregulated miRNAs (mmu-let-7a, mmu-let-7d, mmu-let-7e and
mmu-let-7f) and downregulated miRNAs (mmu-miR-206, mmu-miR-184 and
mmu-miR-146b) were selected for additional analysis. This
confirmatory process showed that our expression data obtained by
qRT-PCR analysis were comparable with the sequencing data (Fig. 4).
Target gene ontology and target
gene-miRNA network analysis
Of the miRNAs identified for additional analysis, a
subset (mmu-let-7a, mmu-let-7d, mmu-let-7e and mmu-let-7f) was
found to be from the same polycistronic miRNA cluster (let-7), and
were upregulated when comparing the later development group with
the early development group. Predicted targets of the 4
differentially expressed miRNAs of the let-7 cluster were grouped
into different categories. The top 13 gene ontology (GO) terms are
shown in Table VII. The majority
of targets were classified according to developmental processes,
cell organization and biogenesis, thus reflecting the profound
biological changes occurring in the developing mouse heart. A
network relationship between the predicted targets of the 4
differentially expressed miRNAs of the let-7 cluster and these
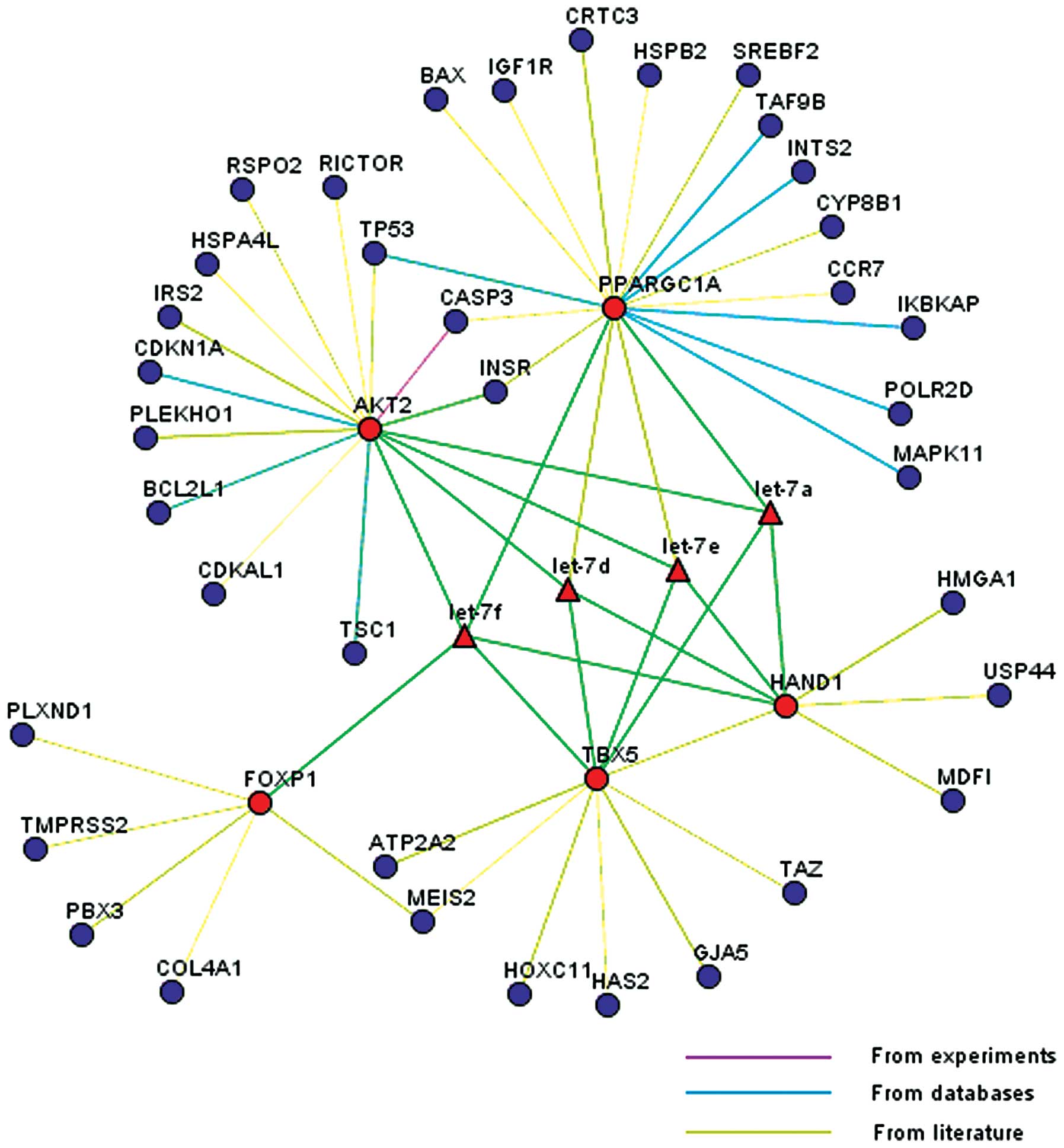
miRNAs was analyzed. Five candidate genes (FOXP1, TBX5, HAND1, AKT2
and PPARGC1A), known to be involved in cardiac development were
profiled. These five genes were located in the center of the
network, whereas the remaining 37 genes, which were associated with
the 5 candidate genes, were located on the edge of the network
(Fig. 5).
| Table VII.Gene ontology analysis of targets of
mmu-let-7a/7d/7e/7f. |
Table VII.
Gene ontology analysis of targets of
mmu-let-7a/7d/7e/7f.
| Biological process
category, n | Targets of
let-7a | Targets of
let-7d | Targets of
let-7e | Targets of
let-7f |
|---|
| Cell cycle and
proliferation | 49 | 51 | 49 | 52 |
| Stress
response | 48 | 47 | 47 | 48 |
| Transport | 97 | 107 | 101 | 96 |
| Developmental
processes | 112 | 111 | 113 | 115 |
| RNA metabolism | 101 | 97 | 101 | 104 |
| DNA metabolism | 18 | 15 | 18 | 20 |
| Other metabolic
processes | 98 | 97 | 104 | 100 |
| Cell organization
and biogenesis | 77 | 77 | 76 | 81 |
| Cell-cell
signaling | 14 | 16 | 15 | 13 |
| Signal
transduction | 102 | 100 | 101 | 101 |
| Cell adhesion | 21 | 19 | 21 | 20 |
| Protein
metabolism | 128 | 122 | 132 | 127 |
| Death | 35 | 35 | 35 | 34 |
Discussion
In the present study, we have characterized the
miRNA expression profile in the developing mouse heart from E12.5
to E18.5 using next-generation sequencing. Currently, the most
well-studied miRNAs are generally those that are expressed at the
highest levels in tissues (22).
The 10 top expressed miRNAs identified in the developing heart in
the M1, M2, M3 and M4 groups are: mmu-miR-23b, mmu-miR-24,
mmu-miR-23a, mmu-miR-375, mmu-miR-29a, mmu-miR-93, mmu-miR-21,
mmu-miR-25, mmu-let-7b and mmu-miR-27b. This dataset has some
overlap with previously published data on miRNA expression in the
adult mouse heart (23–25); specifically, there are 3 miRNAs
(mmu-miR-27b, mmu-miR-23a and mmu-miR-24) which appear to be highly
abundant in both embryonic and adult hearts (Table VIII). These miRNAs have previously
been investigated to assess their function in the heart, including
the creation of cardiac-specific miRNA transgenic mice. For
example, transgenic mice with cardiomyocyte-specific
over-expression of miR-27b are able to induce cardiac hypertrophy
and dysfunction (26), miR-23a
transgenic mice also exhibit exaggerated cardiac hypertrophy
(27), and transgenic mice with
cardiac overexpression of miR-24 results in embryonic lethality
(28). Given the insightful
phenotypes observed in these mice, other miRNAs abundant in the
developing heart should be investigated to gain a better
understanding of the regulatory mechanisms of cardiac
development.
| Table VIII.Top expressed miRNAs from the
reported literature in adult mouse heart. |
Table VIII.
Top expressed miRNAs from the
reported literature in adult mouse heart.
Landgraf et
al (23)
| Takada et al
(25)
| Rao et al
(24)
|
|---|
| miRNA | % | miRNA | % | miRNA |
|---|
| mmu-miR-1 | 30.16 |
mmu-mir-126-3p/-5p | 20.84 | mmu-miR-1 |
| mmu-miR-208 | 8.73 | mmu-mir-1 | 17.37 | mmu-miR-29a |
| mmu-miR-126 | 7.14 | mmu-mir-189/24 | 13.40 | mmu-let-7c |
| mmu-miR-143 | 3.17 |
mmu-mir-30e/30e* | 10.17 | mmu-let-7d |
| mmu-miR-26a | 3.17 | mmu-mir-191 | 6.70 | mmu-miR-378 |
| mmu-let-7d | 3.17 | mmu-mir-143 | 5.21 | mmu-let-7f |
| mmu-miR-144 | 3.17 | mmu-mir-124a | 4.22 | mmu-miR-26a |
| mmu-miR-451 | 3.17 | mmu-mir-144 | 2.48 | mmu-miR-143 |
| mmu-miR-133a | 3.17 | mmu-mir-145 | 1.49 | mmu-miR-24 |
| mmu-miR-16 | 2.38 | mmu-let-7a | 1.49 | mmu-miR-30c |
| mmu-miR-22 | 2.38 | mmu-mir-29a | 1.24 | mmu-miR-133a |
| mmu-miR-27a | 2.38 | | | mmu-let-7a |
| mmu-miR-29b | 1.59 | | | mmu-miR-126 |
| mmu-let-7c | 1.59 | | | mmu-miR-30d |
| mmu-miR-30a | 1.59 | | | mmu-miR-22 |
| mmu-let-7a | 1.59 | | | mmu-miR-29c |
| mmu-let-7b | 1.59 | | | mmu-miR-125b |
| mmu-let-7f | 1.59 | | | mmu-miR-30a |
| mmu-miR-146a | 1.59 | | | mmu-miR-30e |
| mmu-miR-23a | 1.59 | | | mmu-miR-27b |
| | | | mmu-let-7b |
| | | | mmu-miR-26b |
Although some of the miRNAs identified as being
highly abundant in the developing heart (i.e., mmu-miR-21,
mmu-miR-25, mmu-miR-93 and mmu-miR-375) in this study, were not the
top expressed miRNAs of the adult mouse heart (Table VIII), some of these miRNAs have
previously been associated with heart function. For example,
mmu-miR-21 is known as a differentiation-state-related miRNA
(29), and as a cardiac muscle
marker. Overexpression of miR-21 in a transgenic mouse heart
resulted in suppression of the ischemia-induced upregulation of
PTEN and FasL expression, a smaller infarct size, and ameliorated
heart failure (30). Mmu-miR-25
and mmu-miR-93, both of which are in the miR-106b-25 cluster, also
seem to have some significance for heart function. Transfection of
miR-25 is sufficient to decrease the collagen gene expression in
isolated cardiac fibroblasts in vitro (31), and miR-93 is downregulated during
cardiac hypertrophy (32). It has
also been postulated that miR-375 is a potential biomarker of acute
ST-segment elevation myocardial infarction (33).
A third group of miRNAs to be considered are those
that were highly abundant in the adult mouse heart (i.e.,
mmu-miR-1, mmu-miR-126 and mmu-miR-133a) (Table VIII), but were not in the top
expressed miRNAs of the present study. Previous studies have shown
that both miR-1 and miR-133 are important in the remodeling of the
heart that occurs during cardiogenesis (34), and miR-126 has been shown to be
sufficient to regulate vascular integrity and angiogenesis
(35).
miRNAs may be expressed preferably during particular
developmental time-points, or within certain tissues. Moreover,
within a developmental framework, miRNAs may exhibit dynamic
expression patterns (36). A
major focus of our study was to define the repertoire of miRNAs
expressed at different time-points of heart development. In terms
of broad classes with expression that changed during development,
two major expression profiles were identified: miRNAs expressed
predominantly early in heart development and those expressed
predominantly in the mature and developed heart. Our comparative
clustering analyses have shown that there is 1 upregulated miRNA
(mmu-miR-185) and 1 downregulated miRNA (mmu-miR-103) when we
compared M1, M2, M3 and M4 (E12.5, early in heart development),
respectively. This showed that the 2 highly conserved miRNA
expression patterns at this point of heart development may be
important, but no mechanistic studies investigating the
relationship between these 2 miRNAs and heart development and
function are currently available.
There are 3 upregulated miRNAs (mmu-miR-221,
mmumiR-222, and mmu-miR-22) and 1 downregulated miRNA (mmu-miR-26b)
differentially expressed when we compare M1 (E18.5, mature and
developed heart) and M2, M3, M4, respectively. This showed that the
4 highly conserved miRNA expression patterns at this point of
development may be important in heart and vascular development.
miR-221 and miR-222 are known to be novel regulators for vascular
smooth muscle cell proliferation (37), while miR-22-deficient mice show
evidence of cardiac decompensation and left ventricular dilation
(38). Overexpression of miR-26b
in the heart was shown to inhibit upregulation of its targets and
the development of hypertrophy (39).
For comparative analysis, we also profiled the
differentially expressed microRNAs between the 4 different
experimental time-points. Hierarchical cluster analysis showed a
number of miRNAs that were differentially expressed during the
investigated time frame of development, although no miRNAs were
consistently downregulated or upregulated in the different groups.
As such, we profiled the differentially expressed miRNAs between
the later development (M1+M2) and early development (M3+M4) groups,
and 3 downregulated and 13 upregulated miRNAs were identified. From
this group of 16 differentially expressed miRNAs, let-7a/7d/7e/7f
were all upregulated miRNAs that are known to be abundantly
expressed in adult mouse heart tissue (24). Predicted targets of
let-7a/7d/7e/7f are associated with developmental processes, cell
organization and biogenesis, indicating profound biological changes
occurring in the developing mouse heart. A network analysis of the
predicted targets of let-7a/7d/7e/7f has shown that 5 target genes
(FOXP1, TBX5, HAND1, AKT2 and PPARGC1A) are known to be involved in
cardiac development (40). It is
also known that these miRNAs, which are involved in mouse embryonic
hearts, are able to regulate the expression of the 5 target genes
by binding to a highly conserved target site in their 3′UTR.
In conclusion, our experiments identified a series
of miRNAs abundantly expressed in the developing heart, as well as
several differentially expressed miRNAs between late and early
heart development. We believe that these miRNAs likely play an
important role in heart development, thus additional studies may
clarify the mechanism(s) of normal heart development, and provide a
physiological basis for future investigations on congenital heart
disease.
Acknowledgements
This study was supported by grants
from the National Natural Science Foundation of China (Grant no.
81070500), the Key Medical Personnel Foundation of Jiangsu Province
(Grant no. RC2011021), the Nanjing Medical Science and Technique
Development Foundation, and the Science and Technology Development
Foundation of Nanjing Medical University (Grant no.
2010NJMUZ15).
References
|
1.
|
DP BartelMicroRNAs: genomics, biogenesis,
mechanism, and
functionCell116281297200410.1016/S0092-8674(04)00045-514744438
|
|
2.
|
BP LewisCB BurgeDP BartelConserved seed
pairing, often flanked by adenosines, indicates that thousands of
human genes are microRNA
targetsCell1201520200510.1016/j.cell.2004.12.03515652477
|
|
3.
|
Y LiangD RidzonL WongC
ChenCharacterization of microRNA expression profiles in normal
human tissuesBMC
Genomics8166200710.1186/1471-2164-8-16617565689
|
|
4.
|
E WienholdsWP KloostermanE MiskaE
Alvarez-SaavedraE BerezikovE de BruijnHR HorvitzS KauppinenRH
PlasterkMicroRNA expression in zebrafish embryonic
developmentScience309310311200510.1126/science.1114519
|
|
5.
|
M Lagos-QuintanaR RauhutA YalcinJ MeyerW
LendeckelT TuschlIdentification of tissue-specific microRNAs from
mouseCurr Biol12735739200210.1016/S0960-9822(02)00809-612007417
|
|
6.
|
L Hackler JrJ WanA SwaroopJ QianDJ
ZackMicroRNA profile of the developing mouse retinaInvest
Ophthalmol Vis Sci5118231831201010.1167/iovs.09-465719933188
|
|
7.
|
KH LingPJ BrautiganCN HahnT DaishJR
RaynerPS CheahJM RaisonS PiltzJR MannDM MattiskeDeep sequencing
analysis of the developing mouse brain reveals a novel microRNABMC
Genomics12176201110.1186/1471-2164-12-17621466694
|
|
8.
|
B LiuGR CunhaLS BaskinDifferential
expression of microRNAs in mouse embryonic bladderBiochem Biophys
Res Commun385528533200910.1016/j.bbrc.2009.05.08819470377
|
|
9.
|
T ThumD CatalucciJ BauersachsMicroRNAs:
novel regulators in cardiac development and diseaseCardiovasc
Res79562570200810.1093/cvr/cvn13718511432
|
|
10.
|
D SayedC HongIY ChenJ LypowyM
AbdellatifMicroRNAs play an essential role in the development of
cardiac hypertrophyCirc
Res100416424200710.1161/01.RES.0000257913.42552.2317234972
|
|
11.
|
C SucharovMR BristowJD PortmiRNA
expression in the failing human heart: functional correlatesJ Mol
Cell Cardiol45185192200810.1016/j.yjmcc.2008.04.01418582896
|
|
12.
|
RE vanLB SutherlandJE ThatcherJM DiMaioRH
NaseemWS MarshallJA HillEN OlsonDysregulation of microRNAs after
myocardial infarction reveals a role of miR-29 in cardiac
fibrosisProc Natl Acad Sci
USA1051302713032200810.1073/pnas.080503810518723672
|
|
13.
|
A CareD CatalucciF FelicettiD BonciA
AddarioP GalloML BangP SegnaliniY GuND DaltonMicroRNA-133 controls
cardiac hypertrophyNat Med13613618200710.1038/nm158217468766
|
|
14.
|
RE vanLB SutherlandX QiJA RichardsonJ
HillEN OlsonControl of stress-dependent cardiac growth and gene
expression by a
microRNAScience316575579200710.1126/science.113908917379774
|
|
15.
|
Y ZhaoJF RansomA LiV VedanthamM von
DrehleAN MuthT TsuchihashiMT McManusRJ SchwartzD
SrivastavaDysregulation of cardiogenesis, cardiac conduction, and
cell cycle in mice lacking
miRNA-1-2Cell129303317200710.1016/j.cell.2007.03.03017397913
|
|
16.
|
N LiuEN OlsonMicroRNA regulatory networks
in cardiovascular developmentDev
Cell18510525201010.1016/j.devcel.2010.03.01020412767
|
|
17.
|
A StarkJ BrenneckeN BushatiRB RussellSM
CohenAnimal MicroRNAs confer robustness to gene expression and have
a significant impact on 3′UTR
evolutionCell12311331146200516337999
|
|
18.
|
A ChinchillaE LozanoH DaimiFJ EstebanC
CristAE AranegaD FrancoMicroRNA profiling during mouse ventricular
maturation: a role for miR-27 modulating Mef2c expressionCardiovasc
Res8998108201110.1093/cvr/cvq26420736237
|
|
19.
|
A WesselsD SedmeraDevelopmental anatomy of
the heart: a tale of mice and manPhysiol
Genomics15165176200310.1152/physiolgenomics.00033.200314612588
|
|
20.
|
SM SavolainenJF FoleySA ElmoreHistology
atlas of the developing mouse heart with emphasis on E11.5 to
E18.5Toxicol Pathol37395414200910.1177/019262330933506019359541
|
|
21.
|
ZB YuSP HanYF BaiC ZhuY PanXR GuomicroRNA
expression profiling in fetal single ventricle malformation
identified by deep sequencingInt J Mol Med295360201221935567
|
|
22.
|
X WangX WangSystematic identification of
microRNA functions by combining target prediction and expression
profilingNucleic Acids
Res3416461652200610.1093/nar/gkl06816549876
|
|
23.
|
P LandgrafM RusuR SheridanA SewerN IovinoA
AravinS PfefferA RiceAO KamphorstM LandthalerA mammalian microRNA
expression atlas based on small RNA library
sequencingCell12914011414200710.1016/j.cell.2007.04.04017604727
|
|
24.
|
PK RaoY ToyamaHR ChiangS GuptaM BauerR
MedvidF ReinhardtR LiaoM KriegerR JaenischLoss of cardiac
microRNA-mediated regulation leads to dilated cardiomyopathy and
heart failureCirc
Res105585594200910.1161/CIRCRESAHA.109.20045119679836
|
|
25.
|
S TakadaE BerezikovY YamashitaM
Lagos-QuintanaWP KloostermanM EnomotoH HatanakaS FujiwaraH
WatanabeM SodaMouse microRNA profiles determined with a new and
sensitive cloning methodNucleic Acids
Res34e115200610.1093/nar/gkl65316973894
|
|
26.
|
J WangY SongY ZhangH XiaoQ SunN HouS GuoY
WangK FanD ZhanCardiomyocyte overexpression of miR-27b induces
cardiac hypertrophy and dysfunction in miceCell
Res22516527201210.1038/cr.2011.13221844895
|
|
27.
|
K WangZQ LinB LongJH LiJ ZhouPF LiCardiac
hypertrophy is positively regulated by MicroRNA miR-23aJ Biol
Chem287589599201210.1074/jbc.M111.26694022084234
|
|
28.
|
RE vanLB SutherlandN LiuAH WilliamsJ
McAnallyRD GerardJA RichardsonEN OlsonA signature pattern of
stress-responsive microRNAs that can evoke cardiac hypertrophy and
heart failureProc Natl Acad Sci
USA1031825518260200610.1073/pnas.060879110317108080
|
|
29.
|
MR SuhY LeeJY KimSK KimSH MoonJY LeeKY
ChaHM ChungHS YoonSY MoonHuman embryonic stem cells express a
unique set of microRNAsDev
Biol270488498200410.1016/j.ydbio.2004.02.01915183728
|
|
30.
|
D SayedM HeC HongS GaoS RaneZ YangM
AbdellatifMicroRNA-21 is a downstream effector of AKT that mediates
its antiapoptotic effects via suppression of Fas ligandJ Biol
Chem2852028120290201010.1074/jbc.M110.10920720404348
|
|
31.
|
V DivakaranJ AdrogueM IshiyamaML EntmanS
HaudekN SivasubramanianDL MannAdaptive and maladptive effects of
SMAD3 signaling in the adult heart after hemodynamic pressure
overloadingCirc Heart
Fail2633642200910.1161/CIRCHEARTFAILURE.108.82307019919989
|
|
32.
|
PA Da Costa MartinsLJ De WindtMicroRNAs in
control of cardiac hypertrophyCardiovasc
Res93563572201222266752
|
|
33.
|
Y D’AlessandraP DevannaF LimanaS StrainoCA
DiPG BrambillaM RubinoMC CarenaL SpazzafumoM De SimoneCirculating
microRNAs are new and sensitive biomarkers of myocardial
infarctionEur Heart J3127652773201020534597
|
|
34.
|
V DivakaranDL MannThe emerging role of
microRNAs in cardiac remodeling and heart failureCirc
Res10310721083200810.1161/CIRCRESAHA.108.18308718988904
|
|
35.
|
S WangAB AuroraBA JohnsonX QiJ McAnallyJA
HillJA RichardsonR Bassel-DubyEN OlsonThe endothelial-specific
microRNA miR-126 governs vascular integrity and angiogenesisDev
Cell15261271200810.1016/j.devcel.2008.07.00218694565
|
|
36.
|
F FaziC NerviMicroRNA: basic mechanisms
and transcriptional regulatory networks for cell fate
determinationCardiovasc
Res79553561200810.1093/cvr/cvn15118539629
|
|
37.
|
X LiuY ChengS ZhangY LinJ YangC ZhangA
necessary role of miR-221 and miR-222 in vascular smooth muscle
cell proliferation and neointimal hyperplasiaCirc
Res104476487200910.1161/CIRCRESAHA.108.18536319150885
|
|
38.
|
P GurhaC Abreu-GoodgerT WangMO RamirezAL
DrumondS van DongenY ChenN BartonicekAJ EnrightB LeeTargeted
deletion of microRNA-22 promotes stress induced cardiac dilation
and contractile
dysfunctionCirculation12527512761201210.1161/CIRCULATIONAHA.111.04435422570371
|
|
39.
|
M HanZ YangD SayedM HeS GaoL LinS YoonM
AbdellatifGATA4 expression is primarily regulated via a
miR-26b-dependent post-transcriptional mechanism during cardiac
hypertrophyCardiovasc Res93645654201210.1093/cvr/cvs00122219180
|
|
40.
|
VK KhodiyarDP HillD HoweTZ BerardiniS
TweediePJ TalmudR BreckenridgeS BhattarcharyaP RileyP ScamblerRC
LoveringThe representation of heart development in the gene
ontologyDev Biol354917201110.1016/j.ydbio.2011.03.01121419760
|