1. Introduction
Almost 2% of the people of Western industrialized
countries are affected by Alzheimer’s disease (AD) (1,2).
But what is this ailment that threatens a growing number people in
our aging populations? It is a very slowly expanding
neurodegenerative process that betrays its presence by
disconnecting and ultimately destroying neural networks in the
hippocampus, the brain’s ancient memory-recording and accessing
‘machine’ (3,4). The by far most common late onset AD
(LOAD) cases account for over 70% of dementia cases in individuals
>70 years of age (5). The
incidence of AD increases exponentially with age and doubles every
5 years after the age of 65 (1).
In the rare early-onset familiar AD (EOFAD) cases genetic mutations
support an Aβ peptide overproduction (1,2).
The LOAD pathogenesis is still controversial; it begins 30–40 years
before the phenotypic emergence of clinical symptoms, in the
entorhinal cortex and the dentate gyrus, where the
aggregation-prone Aβ1–42 peptides (Aβs), which derive
from the sequential activity of two proteases, BACE1 and
γ-secretase, on the amyloid precursor protein (APP), progressively
disrupt the neuronal networks (3–8).
In normal brains, neurons release at synapses
nontoxic Aβ42 monomers, the physiological levels of
which are kept at low, safe levels by various clearance mechanisms
involving the activation of proteases, phagocytosis by microglia
and dumping into the blood by transporters such as LRP1 (4). But in the aging brains of
susceptible persons the Aβ42 clearing mechanisms start
to fail and the accumulating Aβ42 monomers will
aggregate into toxic soluble oligomers and protofibrils driving the
brain into the onset of the pathology (3,4).
Thus, AD starts stealthily maybe as early as during childhood in a
subcortical region such as the locus coeruleus, from which a
prion-like tau mutant would progressively spread and/or maybe in
the brain’s default mode network (DMN), which includes the medial
temporal memory-recording region (3,4–10).
Aβs overproduction associates with the dangerous spread of
phosphorylated tau protein (3,4,6).
Ultimately, the functionally disturbed neurons cause a lethal
accrual of the toxic prion-like pE Aβ3–34 (pyroglutamyl
Aβ3–34) along with the Aβs oligomers with which it
associates (11–15). Indeed pE Aβ3–34 is
likely ‘the AD’s hatchet man’ as it has been called by Jawhar et
al (12).
In the present study we aim at updating some of the
mechanisms supporting AD development and progression, i.e. i) the
interactions of two nerve cell membrane receptors with Aβs and
their effects; ii) the complex involvement of a perhaps unduly
overlooked class of glial cells, the astrocytes, in AD; and iii)
the involvement of the primary cilia of neurons and astrocytes
alike in AD. Here we shall briefly review such topics in their own
contexts.
2. Nerve cell membrane receptors in AD
p75 neurotrophin receptor
(p75NTR)
The p75NTR is a TNF-family low-affinity
receptor for neurotrophins such as nerve growth factor (NGF),
neurotrophin (NT)-3, NT-4 and brain-derived neurotrophic factor
(BDNF). The interest in p75NTR role, if any, in AD
development and progression was triggered by the studies of Yaar
et al (16,17) and Kuner et al (18), who showed that Aβs could bind to
both p75NTR monomers and trimers, thereby activating its
intracellular signalling and inducing apoptosis in human
neuroblastoma cells. At about such early times, we employed
neuroblastoma cell clones that did not express any of the
neurotrophin receptors or had been engineered to express
full-length or various truncated forms of the p75NTR to
demonstrate that p75NTR binds Aβs via its extracellular
domain and, as a consequence, via its death domain directly signals
cell death. In fact, this signaling led to caspase-8 and caspase-3
activation and to reactive oxygen species (ROS) production and
cellular oxidative stress (19,20). Moreover, the direct and indirect
(inflammatory) mechanisms of neuronal damage by Aβs could interact
synergistically, since cytokines released from an activated
microglia, like TNF-α and IL-1β, remarkably potentiated the
neurotoxic actions of the Aβs/p75NTR signaling (19,20). Altogether, these findings
indicated that the privileged targets of the cytotoxic actions of
Aβs in AD might be p75NTR-expressing neurons endowed
with receptors for proinflammatory cytokines (19,20).
Concurrently, by means of the same human
neuroblastoma cell clones either devoid of all the neurotrophin
receptors or expressing the full-length or variously truncated
forms of p75NTR, we could prove that the neuronal death
induced by the prion protein fragment PrP106–126 is
mediated through its binding to the extracellular domain of
p75NTR and the subsequent signaling of its death domain
causing the downstream activation of caspase-8 and production of
ROS (20,21). Since then other laboratories have
corroborated the idea that the Aβs/p75NTR binding
engenders a signaling causing neuronal apoptosis (22,23).
More recently, we demonstrated that, besides binding
and activating p75NTR receptors, Aβ1–42 and
its surrogate active peptide Aβ25–35, but not the
reverse sequence Aβ42-1 peptide, at least double the
membrane complement of p75NTR receptors in SH-SY5Y human
neuroblastoma cells (24). We
concurrently established that p75NTR is overexpressed
above the level of corresponding wild-type mice in the hippocampal
membranes of two strains of AD transgenic mice, i) in
12–15-month-old AD-triple transgenic (Tg) mice
(3xTg-AD) harboring PS1 (M146V), AβPP (Swe) and tau
(P301L) and ii) in 7-month-old B6.Cg Tg-AD mice harboring
PSEN1dE9 and AβPP (Swe). Importantly, this increase correlated with
the age-dependent rise in Aβ1–42 levels in the AD mice
(24). Evidence was also gained
that the Aβ42 oligomers known as Aβ-derived diffusible
ligands (ADDLs) induced the expression of p75NTR protein
via the phosphorylation of insulin-like growth factor-1 receptor
(IGF-1R) in SH-SY5Y human neuroblastoma cells (25). An in vivo microinjection of
ADDLs also increased the p75NTR protein expression by
1.4-fold in the ipsilateral hippocampus as compared to the
non-injected contralateral hippocampus. Moreover, in the
ADDLs-microinjected mouse hippocampi IGF-1R phosphorylation surged
within 30 min, while the co-administration of picropodophyllin, an
IGF-1R kinase inhibitor, prevented any ADDLs-induced
p75NTR expression from occurring (25). In addition, in the hippocampi of
6-month-old AβPPswe/PS1dE9 Tg-AD model mice that had
accumulated significant amounts of Aβ1–42 a higher
p75NTR protein expression together with higher levels of
IGF-1R phosphorylation were detected with respect to the hippocampi
from age-matched wild-type mice (25). Hence, Aβ42
oligomer-mediated IGF-1R activation may trigger an increase in
p75NTR protein expression in the hippocampus of a
Tg-AD mouse model brain during the early stages of disease
development.
Notably, these findings raised an important
question, i.e. whether the Aβ42’s accumulation is also
coupled with an increased hippocampal membrane-associated
p75NTR expression in human AD brains. Indeed, the
mechanisms controlling the hippocampal expression of
p75NTR are poorly known. It is a commonly held view that
the p75NTR proteins are not expressed by the hippocampal
nerve cells, but are carried to the hippocampus via the afferent
axons of basal forebrain cholinergic neurons (BFCNs). Yet, BFCNs
are selectively killed in the early phases of AD, which would
entail a p75NTR fall in the hippocampi of AD brains
(26). On the other hand, a high
concentration of p75NTR receptors is detectable in the
membranes of the primary cilia of dentate gyrus granule
cells in the mouse hippocampus (27). Others have reported
p75NTR protein expression in normal mice granule cell
precursors up to the early postmitotic maturation of neuroblasts
(28) and in dendritic spines and
afferent terminals of hippocampal CA1 pyramidal neurons of normal
C57BL/6 mice (29). To solve this
question, we used polyclonal and monoclonal antibodies against the
p75NTR receptor’s intra- and extracellular domains.
Thus, we were able to show that the mean level of
membrane-associated p75NTR in the hippocampal formation
is significantly higher (~2-fold, p<0.03) in human AD brains
than in identical samples of hippocampal formation in age-matched
non-AD human brains (30). As
yet, we do not know whether the same types of nerve cells express
p75NTR receptors in murine and human hippocampi,
respectively. Nevertheless, an elevated membrane-bound
p75NTR in the human hippocampus could be another
characteristic of AD. It remains to be determined whether and/or
how such an increased expression of membrane-bound
p75NTR might be a cause of the hippocampal destruction
causing the cognitive decay in AD patients.
Calcium-Sensing Receptor (CaSR)
The highly conserved CaSR gene encodes the CaSR
protein, which belongs to family C of G-protein-coupled receptors
(GPCRs), whose members have no sequence homology with those of
other GPCR families (31). The
CaSR’s huge (>600 amino acids) bilobed extracellular N-terminus
domain looks like a Venus Flytrap (VFT), whose lobes are joined via
a three-strand hinge to 7 transmembrane α-helices (TM1–TM7) ending
with the intracellular C-terminus (32). A cysteine-rich region (CRR) links
the VFT to the 7TM region and is important for signal transmission
from the VFT-like domain to the TM1–TM7 (33). CaSR’s intracellular tail includes
two regions essential for its cell surface expression and
biological activity (34). By
rearranging the two 7TM regions, ligand binding permits the
intracellular CaSRs C-tails to bind various G proteins
(Gqα, Giα and G11α) (35). CaSRs form homodimers (CaSR/CaSR)
or heterodimers (CaSR/mGluR) in their membrane-bound form, although
they can function even as monomers (32). Dimers are assembled at the ER to
allow CaSR transport to the plasmalemma (36). The huge VFT lobes of CaSR
homodimers cooperatively bind ligands, e.g. Ca2+
(35). The CaSR detects changes
in [Ca2+]e (35), but is a non-selective receptor
(37). Ca2+, di- and
tri-valent cations, antibiotics and polyamines are the
CasR-activating orthosteric agonists, whereas endogenous ligands or
factors, like pH, ionic strength, [Na+]e and
aromatic L-α-amino acids are the allosteric CaSR modulators
(37). Intracellular signaling,
mediated via Ca2+ influx, has been connected to MAPK
(MEK/ERK and JNK) activation, ion channel function, gene
expression, cell proliferation and cell death (35). Most significantly, even Aβs do
bind and activate the CaSR (38).
Hence, CaSR-expressing neurons and glial cells of all types are
susceptible to the cytotoxic effects of the CasR-activating
Aβ42 oligomers and fibrillar aggregates (39). The interest in CaSR’s role in AD
pathogenesis has been increasing since the first evidence was
gained of Aβ42/CaSR interactions in hippocampal
pyramidal neurons (40). But CaSR
expression by the astrocytes entails deep neuropathologic
implications since they play significant roles in inflammatory and
degenerative brain diseases (39–41). Using cultured phenotypically
stable normal adult human astrocytes freshly isolated from the
temporal lobe cerebral cortex we could show that exogenous
Aβ-stimulated CaSR signaling triggers i) the expression of nitric
oxide synthase-2 (NOS-2); ii) the expression and activity of GTP
cyclohydrolase 1 (GCH1), which produces tetrahydrobiopterin (BH4);
and iii) the synthesis and release of large amounts of NO on the
part of the BH4-dimerized/activated NOS-2 (41–44). In its turn, the overproduced NO
can be fairly damaging to neurons and glial cells (see also
below).
Moreover, using the same cultured phenotypically
locked-in normal adult human astrocytes exposed to normoxic
conditions we could demonstrate that the Aβs/CaSR interaction also
induces within 18–24 hours the nuclear translocation of the
hypoxia-inducible HIF1α/HIF1β transcription complex that drives the
expression of three VEGF-A splice variants (VEGF-A121,
VEGF-A165 and VEGF-A189) and the increased
synthesis and secretion especially of VEGF-A165 (45 and
unpublished results).
Finally, and perhaps most interesting of all, the
Aβ-activated CaSR signalling also stimulates the normal human adult
astrocytes in culture to make significant amounts of their own
Aβ42 oligomers (46),
which accumulate inside the cells but are also released into the
medium (our unpublished results). Thus the Aβ/CaSR-evoked
signalling can simultaneously modulate the expression/production of
NO, VEGF-A and Aβ42 in human astrocytes.
3. Astrocytes in AD
Neurons have attracted the most attention from
people trying to understand AD pathophysiologic mechanisms
(3,4). Obviously they are very important in
the AD story and with them die the person’s cognition and
ultimately other functions. Concerning glial cells, undeniably
there are more astrocytes than neurons in the human brain, although
there are arguments about the size of this majority that ranges
from 1.4- to 10-fold the actual neurons’ numbers (47). Until recently, astrocytes have
been relegated to simple janitorial roles: they have not been
believed to be able to make Aβs, but only to sweep them up and then
die if and when they accumulate too much of Aβs (48), as Aβs are supposedly only made and
released by the neurons. There is an increasing realization that
astrocytes are much more important than previously thought; they
actually protect neurons and are in fact the neuron partners in
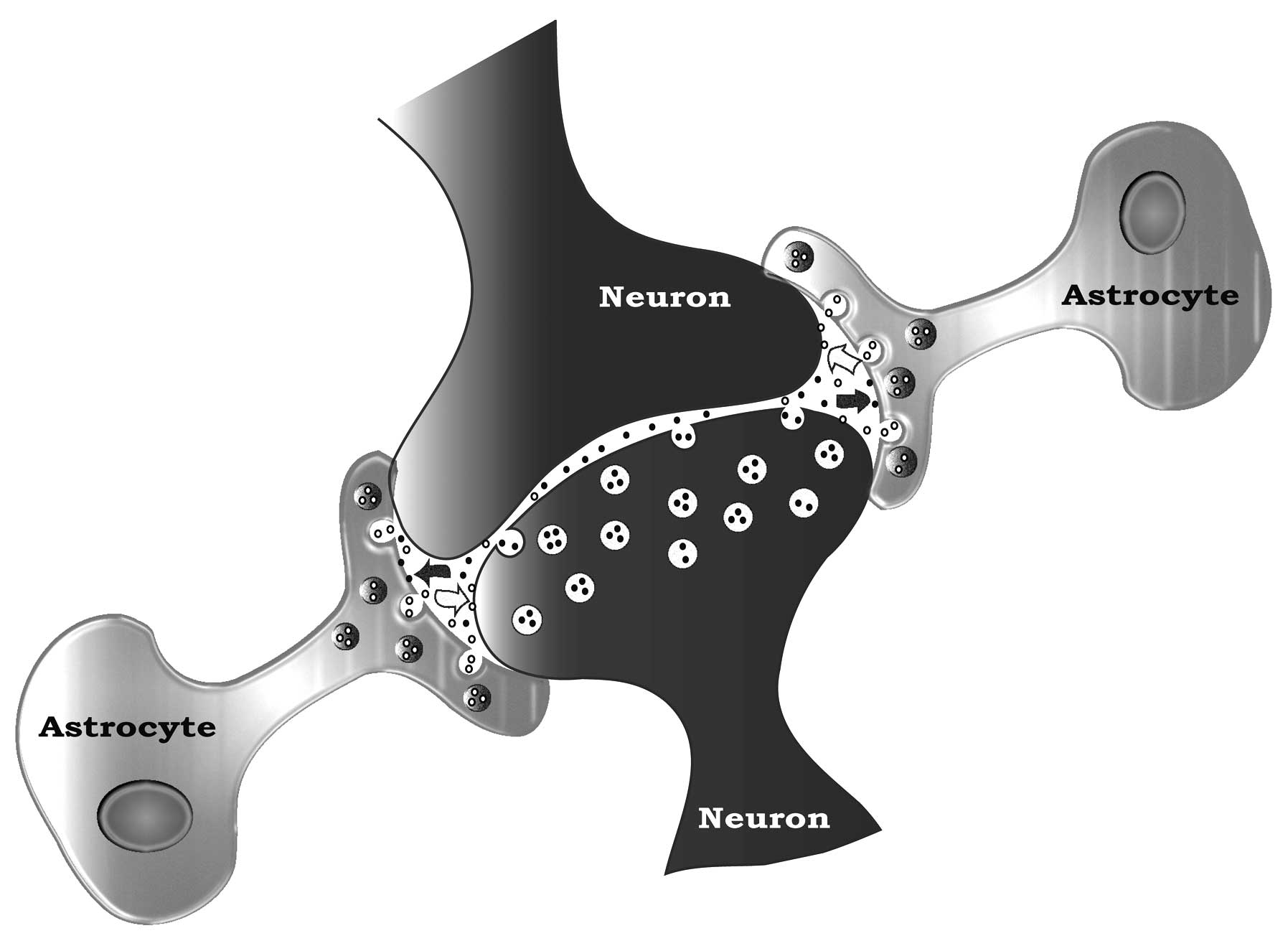
synaptic formation and function. They physically cradle or embrace
neurons (Fig. 1) (49), shielding them from the signaling
noise of neighboring neurons. They keep their neuron synapses
optimally functional by regulating synaptic K+, by
sweeping up secreted neurotransmitters (e.g., glutamate) from the
synaptic space and removing transmitters spilled into the nearby
space by neighbouring neurons. Astrocytes also collaborate in
neuronal signaling with their own gliotransmitters, and they can
stimulate synapse formation (47–50).
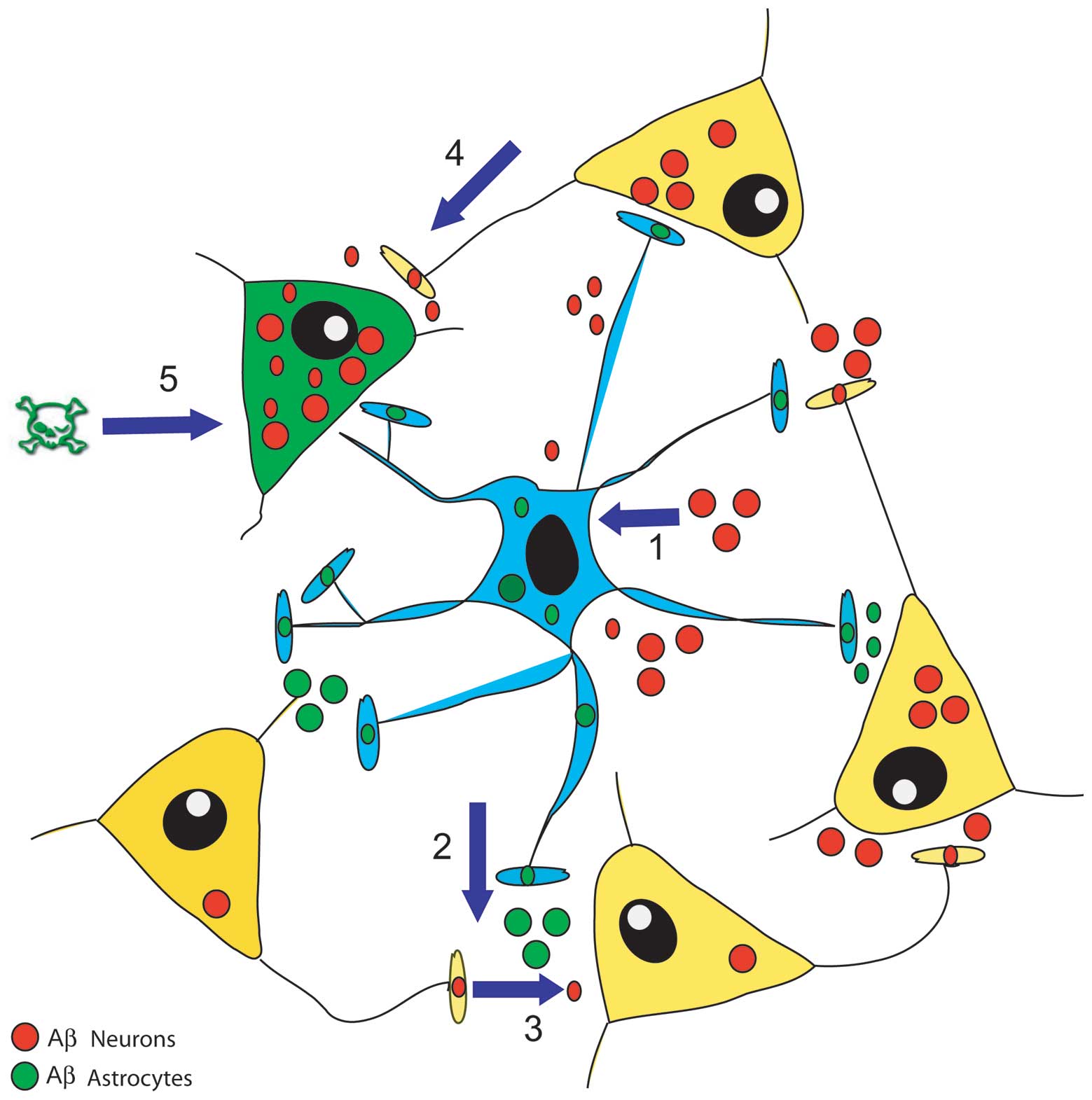
Recent findings have added a novel facet to this
picture (Fig. 2). Astrocytes are
no longer just by-standing synapse blankets that only clean up the
Aβs released from shattered neurons in, for example, the
increasingly cognitively disabled AD brains. Astrocytes are
actually stimulated by their neuron Aβs to make and secrete their
own Aβs (46 and unpublished results). This means that as
astrocyte-contacting neurons in key regions of the brain enter the
covert early stages of AD and start over-secreting Aβs, they
directly spray the astrocytes with their Aβs. The exciting finding
is that this causes the same astrocytes to make and release their
own Aβs and spread them to other neurons in local networks,
stimulating such neurons to join and enlarge the pool of cells
making Aβs. In this way, Aβs-exposed astrocytes act as vectors of a
contagious, self-sustaining and Aβs-spreading disease. But this
might not last, as the accumulating Aβs released from both neurons
and astrocytes reach a level that stimulates the latter cells to
start making large amounts of nitric oxide (NO), from which highly
toxic peroxynitrites (ONOO-) can be engendered (41–44). Both these diffusible agents damage
neurons and astrocytes to the point of inducing cell death.
Obviously, the progressive loss of astrocytes besides neurons
impairs synaptic function and the provision of supplies from the
circulation (47,49,51).
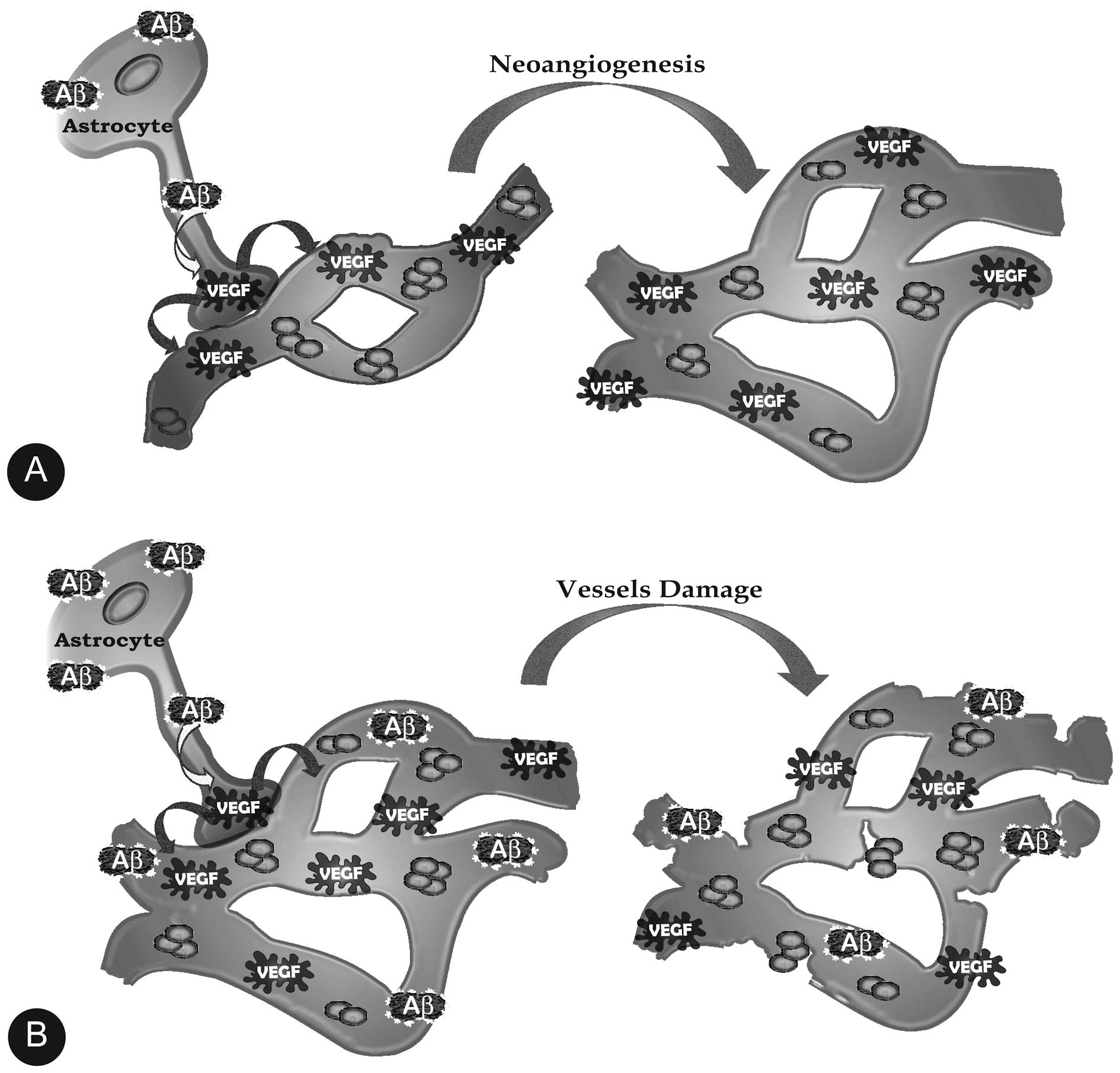
Amongst other activities, astrocytes send
information about the activities of the neurons they cradle to
their end-feet on local blood vessels, which adjusts the blood flow
to provide the glucose and oxygen needed to feed the busy neurons.
However, as in vitro (45), the accumulating Aβs in the key
regions of the pre-AD brain such as the dentate gyrus and
the CA3 subregions also stimulate astrocytes to make VEGF-A
(52–54), which as expected stimulates the
growth of blood vessels (Fig. 3).
This increase in the local vascular density magnifies the blood
flow and the blood oxygen-dependent (BOLD) functional magnetic
resonance imaging (fMRI) signaling from an active region such as a
hippocampus trying to respond to a dentate
gyrus/CA3-directed task (55,56). This has been wrongly interpreted
as if the already declining hippocampi of AD-bound people with
pre-AD mild cognitive impairment (MCI) are hyperactive, which they
are not (57–59). Again, in the early stages the
VEGF-expanded vascular networks are intact and the blood vessels
are functional, but this ends when the accumulating Aβs stimulate
the astrocytes (and microglia) to make huge, damaging amounts of
NO, which contributes to the perforation and severing of the blood
vessels and with this the breaching of the blood-brain barrier and
its disastrous consequences for brain function (60,61) (Fig.
3).
4. Nerve cell primary cilia in AD
Contrary to the old neurological tenet, in the brain
of adult humans and rodents there are two principal areas in which
neurogenesis does occur, the subgranular zone (SGZ) of the dentate
gyrus and the subventricular zone (SVZ) (62–64). In the hippocampal SGZ a pool of
neural stem cells, the astrocyte-like type 1 radial glial cells,
are able to produce new granule cells when they are needed for
memory encoding (65,66). These cells express some properties
of the astrocytes, including glial fibrillary acidic protein
(GFAP), the typical astrocytic marker, are endowed with vascular
end-feet and occupy their special SGZ niche: the upshot is a blood
vessel-associated gap-junctionally interconnected astrocytic
syncytium. Most of these cells possess non-motile, 4–8 μm-long
sensory antennas protruding from their bodies, the primary cilia,
wrapped in a plasma membrane that is stuffed with various kinds of
receptors. Among the receptors found in the rodent granule cell
cilia are the p75NTR receptor (10,24,39), the somatostatin type 3 receptor
(SSTR3) (67,68) and the Sonic hedgehog (Shh)
system’s smoothened (SMO) and Patched (Ptch) proteins (65,69–72). Conversely, the neurotrophin
tyrosine kinase receptor TrkA does not co-localize in the primary
cilia membrane. Signals from the receptors on these cilia are
believed to drive several fundamental activities such as
neurogenesis, neuroblast maturation and memory encoding.
Neurotrophin (e.g., BDNF)-induced p75NTR signalling from
the primary cilia drives the proliferation of granule cells
precursors in the SVZ of the dentate gyrus, as preventing
this signalling severely reduces neurogenesis (65,66,73).
How these primary cilia might be involved in
AD-linked cognitive deterioration? It appears that the accumulating
toxic Aβ42 oligomers in AD brains at first stimulate the
proliferation of GC progenitors. But later, when such oligomers are
caught in the fibrillary plaques, the newly formed neuroblasts
cannot mature or ultimately survive (74–76). The failure of the newly generated
neurons to mature and the resulting granule cell layer shrinkage
and memory failure is likely, at least partly, to be due to the
characteristic decline of somatostatin in AD and with it of the
cilial SSTR3 signalling needed for memory encoding (65,66). These notions enticed us to surmise
that primary cilium damage may cause the crippling decline of new
memory formation in AD (77).
This view is supported by the striking shortening of dentate
gyrus granule cell primary cilia linked to the strongly
reduced neurogenesis in AD Tg mice accumulating both
Aβ42 and tau protein (78,79).
Moreover, cilial p75NTR can be bound and
activated by Aβ42 (19,22). This would elicit an initially
increased neural progenitor cell proliferation in the early stages
of AD (80–82), meanwhile, the hippocampal supply
of acetylcholine (Ach) is progressively reduced by the accumulating
Aβ42 that kills Ach-producing basal forebrain
cholinergic septal neurons (BFCSNs) (83). Thus, despite the increased
Aβ42/p75NTR-stimulated progenitor cell
proliferation, neurogenesis is not actually increased because fewer
progenitor cells survive in the lack of Ach and cilial SSTR3
receptor signalling essential for neuroblasts maturation and de
novo memory encoding (65) is
silenced by the absence of SST in AD brains (66,84).
In this context, a mention is deserved by the
Leptin-induced signalling from Leptin b-receptors located in the
cell (not primary cilium) membrane, which may also stimulate, via
the Shh Smo and the release of cilium-located Gli-A nuclear
transcription factor, the primary cilium-dependent proliferation of
transit-amplifying progenitors in the dentate gyrus of the
adult hippocampal formation (85). It follows that daily doses of
Leptin might halt AD development if given perhaps in the pre-MCI or
MCI stage of the ailment (86).
However, some caution is at present warranted. These
tiny primary cilia are a technical challenge to isolate and
directly analyse. In addition, we presently do not know whether
human dentate gyral granule cells are ciliated or whether human
neuroblast maturation and integration into the granule cell layer
of SVZ are also driven from primary cilia. On an encouraging note,
we have indeed found ciliated cells in samples of hippocampi from
octogenarian normal and AD humans and in phenotypically normal
astrocytes isolated from adult human cerebral cortices (82).
5. Conclusions
It is quite clear from the foregoing discussion that
AD will not be understood by only considering neurons (3,4)
and microglia (87). We must take
into account the intimate collaboration between neurons and their
astrocyte cradlers and trans-network communications. In other words
we must pay serious attention to the astrocytes’ roles in AD.
Moreover, at the subcellular level, important protagonists are
emerging such as primary cilia and receptors such as the
p75NTR and the CaSR (88), as their interactions with Aβs can
modulate or alter fundamental cellular functions like
Aβ42, NO, VEGF-A and proinflammatory cytokine production
and release, proliferative responses and/or damage and
malfunctioning of cerebral blood vessels, neurogenesis and cell
death. Although as for now the AD picture seems to be more
intricate than ever, we hope that these and other new acquisitions
on the pathophysiologic mechanisms of this ailment will help pave
the way to novel, hopefully effective therapeutic approaches.
Acknowledgements
The authors are deeply indebted to Drs I. Dal Prà,
A. Chiarini, C. Gaudet, M. Ménard, T. Atkinson and L. Brown whose
dedicated and intense collaboration was indispensable to achieve
many of the scientific results mentioned in this study.
References
|
1
|
Alzheimer’s Association. Alzheimer’s
disease facts and figures. Alzheimers Dement. 4:110–133. 2008.
|
|
2
|
Kukull WA, Higdon R, Bowen JD, McCormick
WC, Teri L, Schellenberg GD, van Belle G, Jolley L and Larson EB:
Dementia and Alzheimer disease incidence: a prospective cohort
study. Arch Neurol. 59:1737–1746. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Querfurth HW and LaFerla FM: Alzheimer’s
disease. N Engl J Med. 362:329–344. 2010.
|
|
4
|
Selkoe DJ, Mandelkow E and Holtzman DM:
The Biology of Alzheimer Disease. Cold Spring Harbor Laboratory
Press; Cold Spring Harbor, NY: 2012
|
|
5
|
Choy RW, Cheng Z and Schekman R: Amyloid
precursor protein (APP) traffics from the cell surface via
endosomes for amyloid β (Aβ) production in the trans-Golgi network.
Proc Natl Acad Sci USA. 109:E2077–E2082. 2012.PubMed/NCBI
|
|
6
|
LaFerla FM, Green KN and Oddo S:
Intracellular amyloid-beta in Alzheimer’s disease. Nat Rev
Neurosci. 8:499–509. 2007.
|
|
7
|
Li S, Shankar GM and Selkoe DJ: How do
soluble oligomers of amyloid beta-protein impair hippocampal
synaptic plasticity? Front Cell Neurosci. 4:52010.PubMed/NCBI
|
|
8
|
Siegenthaler BM and Rajendran L: Retromers
in Alzheimer’s disease. Neurodegener Dis. 10:116–121. 2012.
|
|
9
|
Whitfield JF: The road to LOAD (late-onset
Alzheimer’s disease) and possible ways to block it. Expert Opin
Ther Targets. 11:1257–1260. 2007.
|
|
10
|
Dal Prà I, Chiarini A, Pacchiana R,
Chakravarthy B, Whitfield JF and Armato U: Emerging concepts of how
β-amyloid proteins and pro-inflammatory cytokines might collaborate
to produce an ‘Alzheimer brain’ (Review). Mol Med Rep. 1:173–178.
2008.
|
|
11
|
Hartlage-Rübsamen M, Morawski M, Waniek A,
Jäger C, Zeitschel U, Koch B, Cynis H, Schilling S, Schliebs R,
Demuth HU and Rossner S: Glutaminyl cyclase contributes to the
formation of focal and diffuse pyroglutamate (pGlu)-Aβ deposits in
hippocampus via distinct cellular mechanisms. Acta Neuropathol.
121:705–719. 2011.PubMed/NCBI
|
|
12
|
Jawhar S, Wirth O and Bayer TA:
Pyroglutamate amyloid-β (Aβ): a hatchet man in Alzheimer disease. J
Biol Chem. 286:38825–38832. 2011.
|
|
13
|
Nussbaum JM, Schilling S, Cynis H, Silva
A, Swanson E, Wangsanut T, Tayler K, Wiltgen B, Hatami A, Rönicke
R, Reymann K, Hutter-Paier B, Alexandru A, Jagla W, Graubner S,
Glabe CG, Demuth HU and Bloom GS: Prion-like behaviour and
tau-dependent cytotoxicity of pyroglutamylated amyloid-β. Nature.
485:651–655. 2012.PubMed/NCBI
|
|
14
|
Prusiner SB: Cell biology. A unifying role
for prions in neurodegenerative diseases. Science. 336:1511–1513.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Stöhr J, Watts JC, Mensinger ZL, Oehler A,
Grillo SK, DeArmond SJ, Prusiner SB and Giles K: Purified and
synthetic Alzheimer’s amyloid beta (Aβ) prions. Proc Natl Acad Sci
USA. 109:11025–11030. 2012.
|
|
16
|
Yaar M, Zhai S, Pilch PF, Doyle SM,
Eisenhauer PB, Fine RE and Gilchrest BA: Binding of β-amyloid to
the p75 neurotrophin receptor induces apoptosis. A possible
mechanism for Alzheimer’s disease. J Clin Invest. 100:2333–2340.
1997.
|
|
17
|
Yaar M, Zhai S, Fine RE, Eisenhauer PB,
Arbie BL, Stewart KB and Gilchrest BA: Amyloid-β binds trimers as
well as monomers of the 75-kDa neurotrophin receptor and activates
receptor signaling. J Biol Chem. 277:7720–7725. 2001.
|
|
18
|
Kuner P, Schubenel R and Hertel C:
β-amyloid binds to p75NTR and activates NF-kappaB in human
neuroblastoma cells. J Neurosci Res. 54:798–804. 1998.
|
|
19
|
Perini G, Della-Bianca V, Politi V, Della
Valle G, Dal Prà I, Rossi F and Armato U: Role of p75 neurotrophin
receptor in the neurotoxicity by β-amyloid peptides and synergistic
effect of inflammatory cytokines. J Exp Med. 195:907–918. 2002.
|
|
20
|
Chiarini A, Dal Prà I, Whitfield JF and
Armato U: The killing of neurons by beta-amyloid peptides, prions
and pro-inflammatory cytokines. Ital J Anat Embryol. 111:221–246.
2006.PubMed/NCBI
|
|
21
|
Della-Bianca V, Rossi F, Armato U, Dal Prà
I, Costantini C, Perini G, Politi V and Della Valle G: Neurotrophin
p75 receptor is involved in neuronal damage by prion
peptide-(106-126). J Biol Chem. 276:38929–38933. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sotthibundhu A, Li QX, Thangnipon W and
Coulson EJ: Abeta(1–42) stimulates adult SVZ neurogenesis through
the p75 neurotrophin receptor. Neurobiol Aging. 30:1975–1985.
2009.
|
|
23
|
Bai M, Trivedi S and Brown EM:
Dimerization of the extracellular calcium-sensing receptor (CaR) on
the cell surface of CaR-transfected HEK293 cells. J Biol Chem.
273:23605–23610. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chakravarthy B, Gaudet C, Ménard M,
Atkinson T, Brown L, Laferla FM, Armato U and Whitfield J:
Amyloid-beta peptides stimulate the expression of the p75(NTR)
neurotrophin receptor in SHSY5Y human neuroblastoma cells and AD
transgenic mice. J Alzheimers Dis. 19:915–925. 2010.PubMed/NCBI
|
|
25
|
Ito S, Ménard M, Atkinson T, Gaudet C,
Brown L, Whitfield J and Chakravarthy B: Involvement of
insulin-like growth factor 1 receptor signaling in the amyloid-β
peptide oligomers-induced p75 neurotrophin receptor protein
expression in mouse hippocampus. J Alzheimers Dis. 31:493–506.
2012.
|
|
26
|
Mufson EJ, Ma SY, Dills J, Cochran EJ,
Leurgans S, Wuu J, Bennett DA, Jaffar S, Gilmor ML, Levey AI and
Kordower JH: Loss of basal forebrain P75 (NTR) immunoreactivity in
subjects with mild cognitive impairment and Alzheimer’s disease. J
Comp Neurol. 443:136–153. 2002.PubMed/NCBI
|
|
27
|
Chakravarthy B, Gaudet C, Ménard M,
Atkinson T, Chiarini A, Dal Prà I and Whitfield J: The p75
neurotrophin receptor is localized to primary cilia in adult mouse
hippocampal dentate gyrus granule cells. Biochem Biophys Res
Commun. 401:458–462. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Woo NH, Teng HK, Siao CJ, Chiaruttini C,
Pang PT, Milner TA, Hempstead BL and Lu B: Activation of p75NTR by
proBDNF facilitates hippocampal long-term depression. Nat Neurosci.
8:1069–1077. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bernabeu RO and Longo FM: The p75
neurotrophin receptor is expressed by adult mouse dentate
progenitor cells and regulates neuronal and non-neuronal cell
genesis. BMC Neurosci. 11:1362010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chakravarthy B, Ménard M, Ito S, Gaudet C,
Dal Prà I, Armato U and Whitfield J: Hippocampal
membrane-associated p75NTR levels are increased in Alzheimer’s
disease. J Alzheimers Dis. 30:675–684. 2012.PubMed/NCBI
|
|
31
|
Brown EM and MacLeod RJ: Extracellular
calcium sensing and extracellular calcium signaling. Physiol Rev.
81:239–297. 2001.PubMed/NCBI
|
|
32
|
Msaouel P, Nixon AM, Bramos AP, Baiba E
and Kentarchos NE: Extracellular calcium-sensing receptor: an
overview of physiology, pathophysiology and clinical perspectives.
In Vivo. 18:739–753. 2004.PubMed/NCBI
|
|
33
|
Jensen AA and Bräuner-Osborne H:
Allosteric modulation of the calcium-sensing receptor. Curr
Neuropharmacol. 5:180–186. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Magno AL, Ward BK and Ratajczak T: The
calcium-sensing receptor: a molecular perspective. Endocr Rev.
32:3–30. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hofer AM and Brown EM: Extracellular
calcium sensing and signalling. Nat Rev Mol Cell Biol. 4:530–538.
2003. View Article : Google Scholar
|
|
36
|
Pidasheva S, Grant M, Canaff L, Ercan O,
Kumar U and Hendy GN: Calcium-sensing receptor dimerizes in the
endoplasmic reticulum: biochemical and biophysical characterization
of CaSR mutants retained intracellularly. Hum Mol Genet.
15:2200–2209. 2006. View Article : Google Scholar
|
|
37
|
Chang W and Shoback D: Extracellular
Ca2+-sensing receptors-an overview. Cell Calcium.
35:183–196. 2004.
|
|
38
|
Ye C, Ho-Pao CL, Kanazirska M, Quinn S,
Rogers K, Seidman CE, Seidman JG, Brown EM and Vassilev PM:
Amyloid-beta proteins activate Ca(2+)-permeable channels through
calcium-sensing receptors. J Neurosci Res. 47:547–554. 1997.
|
|
39
|
Chiarini A, Dal Prà I, Marconi M,
Chakravarthy B, Whitfield JF and Armato U: Calcium-sensing receptor
(CaSR) in human brain’s pathophysiology: roles in late-onset
Alzheimer’s disease (LOAD). Curr Pharm Biotechnol. 10:317–326.
2009.
|
|
40
|
Conley YP, Mukherjee A, Kammerer C,
DeKosky ST, Kamboh MI, Finegold DN and Ferrel RE: Evidence
supporting a role for the calcium-sensing receptor in Alzheimer
disease. Am J Med Genet B Neuropsychiatr Genet. 150B:703–709. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Dal Prà I, Chiarini A, Nemeth EF, Armato U
and Whitfield JF: Roles of Ca2+ and the
Ca2+-sensing receptor (CaSR) in the expression of
inducible NOS (nitric oxide synthase)-2 and its BH4
(tetrahydrobiopterin)-dependent activation in cytokine-stimulated
adult human astrocytes. J Cell Biochem. 96:428–438. 2005.
|
|
42
|
Chiarini A, Dal Prà I, Gottardo R,
Bortolotti F, Whitfield JF and Armato U: The BH4
(tetrahydrobiopterin)-dependent activation, but not the expression,
of inducible NOS (nitric oxide synthase)-2 in proinflammatory
cytokine-stimulated, cultured normal human astrocytes is mediated
by MEK-ERK kinases. J Cell Biochem. 94:731–743. 2005.
|
|
43
|
Chiarini A, Dal Prà I, Menapace L,
Pacchiana R, Whitfield JF and Armato U: Soluble amyloid β-peptide
and myelin basic protein strongly stimulate, alone and in synergism
with combined proinflammatory cytokines, the expression of
functional nitric oxide synthase-2 in normal adult human
astrocytes. Int J Mol Med. 16:801–807. 2005.
|
|
44
|
Chiarini A, Armato U, Pacchiana R and Dal
Prà I: Proteomic analysis of GTP cyclohydrolase 1 multiprotein
complexes in cultured normal adult human astrocytes under both
basal and cytokine-activated conditions. Proteomics. 9:1850–1860.
2009. View Article : Google Scholar
|
|
45
|
Chiarini A, Whitfield J, Bonafini C,
Chakravarthy B, Armato U and Dal Prà I: Amyloid-β(25–35), an
amyloid-β(1–42) surrogate, and proinflammatory cytokines stimulate
VEGF-A secretion by cultured, early passage, normoxic adult human
cerebral astrocytes. J Alzheimers Dis. 21:915–926. 2010.
|
|
46
|
Dal Prà I, Whitfileld JF, Pacchiana R,
Bonafini C, Talacchi A, Chakravarthy B, Armato U and Chiarini A:
The amyloid-β42 proxy, amyloid-β25–35, induces normal human
cerebral astrocytes to produce amyloid-β42. J Alzheimers Dis.
24:335–347. 2011.
|
|
47
|
Nedergaard M, Ransom B and Goldman SA: New
roles for astrocytes: redefining the functional architecture of the
brain. Trends Neurosci. 26:523–530. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Nagele RG and Wegiel J, Venkataraman V,
Imaki H, Wang KC and Wegiel J: Contribution of glial cells to the
development of amyloid plaques in Alzheimer’s disease. Neurobiol
Aging. 25:663–674. 2004.
|
|
49
|
Nedergaard M and Verkhratsky A: Artefact
versus reality-how astrocytes contribute to synaptic events. Glia.
60:1013–1023. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Theodosis DT, Poulain DA and Oliet SH:
Activity-dependent structural and functional plasticity of
astrocyte-neuron interactions. Physiol Rev. 88:983–1008. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Guenette SY: Astrocytes: a cellular player
in Abeta clearance and degradation. Trends Mol Med. 9:279–280.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Biron KE, Dickstein DL, Gopaul R and
Jefferies WA: Amyloid triggers extensive cerebral angiogenesis
causing blood brain barrier permeability and hypervascularity in
Alzheimer’s disease. PLoS One. 6:e237892011.PubMed/NCBI
|
|
53
|
Pogue AI and Lukiw WJ: Angiogenic
signaling in Alzheimer’s disease. Neuroreport. 15:1507–1510.
2004.
|
|
54
|
Zand L, Ryu JK and McLarnon JG: Induction
of angiogenesis in the beta-amyloid peptide-injected rat
hippocampus. Neuroreport. 16:129–132. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Bell RD and Zlokovic BV: Neurovascular
mechanisms and blood-brain barrier disorder in Alzheimer’s disease.
Acta Neuropatol. 118:103–113. 2008.
|
|
56
|
Bakker A, Krauss GL, Albert MS, Speck CL,
Jones LR, Stark CE, Yassa MA, Bassett SS, Shelton AL and Gallagher
M: Reduction of hippocampal hyperactivity improves cognition in
anamnestic mild cognitive impairment. Neuron. 74:467–474. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Putcha D, Brickhouse M, O’Keefe K,
Sullivan C, Rentz D, Marshall G, Dickerson B and Sperling R:
Hippocampal hyperactivation associated with cortical thinning in
Alzheimer’s disease signature regions in non-demented elderly
adults. J Neurosci. 31:17680–17688. 2011.PubMed/NCBI
|
|
58
|
Sperling R: Potential of functional MRI as
a biomarker in early Alzheimer’s disease. Neurobiol Aging. 32(Suppl
1): S37–S43. 2011.
|
|
59
|
Yassa MA, Stark SM, Bakker A, Albert MS,
Gallagher M and Stark CE: High-resolution structural and functional
MRI of hippocampal CA3 and dentate gyrus in patients with
anamnestic mild cognitive impairment. Neuroimage. 51:1242–1252.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Jantaratnotai N, Ryu JK, Schwab C, McGeer
PL and McLarnon JG: Comparison of vascular perturbations in an
Aβ-injected animal model and in AD brain. Int J Alzheimers Dis.
2011:9182802011.PubMed/NCBI
|
|
61
|
Meyer-Luehmann M, Spires-Jones TL, Prada
C, Garcia-Alloza M, de Calignon A, Rozkalne A, Koenigsknecht-Talboo
J, Holtzman DM, Bacskai BJ and Hyman BT: Rapid appearance and local
toxicity of amyloid-beta plaques in a mouse model of Alzheimer’s
disease. Nature. 451:720–724. 2008.PubMed/NCBI
|
|
62
|
Altman J and Das GD: Postnatal
neurogenesis in the guinea-pig. Nature. 214:1098–1101. 1967.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Nottebohm F: Testosterone triggers growth
of brain vocal control nuclei in adult female canaries. Brain Res.
189:429–436. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Kempermann G: Adult Neurogenesis. 2.
Oxford University Press; New York: 2011
|
|
65
|
Einstein EB, Patterson CA, Hon BJ, Regan
KA, Reddi J, Melnikoff DE, Mateer MJ, Schulz S, Johnson BN and
Tallent MK: Somatostatin signaling in neuronal cilia is critical
for object recognition memory. J Neurosci. 30:4306–4314. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Burgos-Ramos E, Hervás-Aguilar A,
Aguado-Liera D, Puebla-Jiménez L, Hernández-Pinto AM, Barrios V and
Arilla-Ferreiro E: Somatostatin and Alzheimer’s disease. Mol Cell
Endocrinol. 286:104–111. 2008.
|
|
67
|
Händel M, Schultz S, Stanarius A, Schreff
M, Erdtmann-Vourliotis M, Schmidt H, Wolf G and Höllt V: Selective
targeting of somatostatin receptor 3 to neuronal cilia.
Neuroscience. 89:909–926. 1999.PubMed/NCBI
|
|
68
|
Stanić D, Malmgren H, He H, Scott L,
Aperia A and Hökfelt T: Developmental changes in frequency of the
ciliary somatostatin receptor 3 protein. Brain Res. 1249:101–112.
2009.PubMed/NCBI
|
|
69
|
Berbari NF, Johnson AD, Lewis JS, Askwith
CC and Mykytyn K: Identification of ciliary localization sequences
within the third intracellular loop of G protein-coupled receptors.
Mol Biol Cell. 19:1540–1547. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Goetz SC, Ocbina PJ and Anderson KV: The
primary cilium as a hedgehog signal transduction machine. Methods
Cell Biol. 94:199–222. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Corbit KC, Aanstad P, Singla V, Norman AR,
Stainier DY and Reiter JF: Vertebrate smoothened functions at the
primary cilium. Nature. 437:1018–1021. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Han YG, Spassky N, Romaguera-Ros M,
Garcia-Verdugo JM, Aguilar A, Schneider-Maunoury S and
Alvarez-Buylla A: Hedgehog signaling and primary cilia are required
for the formation of adult neural stem cells. Nat Neurosci.
11:277–284. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
73
|
Amador-Arjona A, Elliott J, Miller A,
Ginbey A, Pazour GJ, Enikolopov G, Roberts AJ and Terskikh AV:
Primary cilia regulate proliferation of amplifying progenitors in
adult hippocampus: implications for learning and memory. J
Neurosci. 31:9933–9944. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Schaeffer EL, Novaes BA, Da Silva ER, Skaf
HD and Mendes-Neto AG: Strategies to promote differentiation of
newborn neurons into mature functional cells in Alzheimer brain.
Prog Neuropsychopharmacol Biol Psychiatry. 33:1087–1102. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
van Tijn P, Kamphuis W, Marlatt MW, Hol EM
and Lucassen PJ: Presenilin mouse and zebrafish models for
dementia: focus on neurogenesis. Prog Neurobiol. 93:149–164.
2011.PubMed/NCBI
|
|
76
|
Waldau B and Shetty AK: Behavior of neural
stem cells in the Alzheimer brain. Cell Mol Life Sci. 65:2372–2384.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Armato U, Chakravarthy B, Chiarini A, Dal
Prà I and Whitfield JF: Is Alzheimer’s disease at least partly a
ciliopathy? J Alzheimers Dis. 1:101e2011. View Article : Google Scholar
|
|
78
|
Gaudet C, Ménard M, Brown L, Atkinson T,
LaFerla FM, Ito S, Armato U, Dal Prà I, Whitfield J and
Chakravarthy B: Reduction of the immunostainable length of the
hippocampal dentate granule cells’ primary cilia in 3xAD-transgenic
mice producing human Aβ1–42 and tau. Biochem Biophys Res Commun.
September 17–2012.(Epub ahead of print).
|
|
79
|
Rodríguez JJ, Jones VC, Tabuchi M, Allan
SM, Knight EM, LaFerla FM, Oddo S and Verkhratsky A: Impaired adult
neurogenesis in the dentate gyrus of a triple transgenic mouse
model of Alzheimer’s disease. PLoS One. 3:e29352008.PubMed/NCBI
|
|
80
|
Avila J, Insausti R and Del Rio J: Memory
and neurogenesis in aging and Alzheimer’s disease. Aging Dis.
1:30–36. 2010.
|
|
81
|
Shetty AK: Reelin signaling, hippocampal
neurogenesis and efficacy of aspirin intake and stem cell
transplantation in aging and Alzheimer’s disease. Aging Dis.
1:2–11. 2010.PubMed/NCBI
|
|
82
|
Whitfield JF, Chakravarthy B, Chiarini A
and Dal Prà I: The primary cilium: The tiny driver of dentate gyral
neurogenesis. Neurogenesis Research. Clark GJ and Anderson WT:
Chapter V. Nova Science Publishers Inc; Hauppauge, NY: pp. 137–159.
2012, (In press). ISBN: 9781620817230
|
|
83
|
Fortress AM, Buhusi M, Helke KL and
Granholm AC: Cholinergic degeneration and alterations in the TrkA
and p75NTR balance as a result of pro-NGF injection into aged rats.
J Aging Res. 2011:4605432011. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Armato U, Chakravarthy B, Chiarini A, Dal
Prà I and Whitfield JF: A Paradigm-changing surprise from dentate
gyrus granule cells-cilium-localized p75NTR may drive their
progenitor cell proliferation. J Alzheimers Dis. 1:e1042011.
View Article : Google Scholar
|
|
85
|
Pérez-González R, Antequera D, Vargas T,
Spuch C, Bolos M and Carro E: Leptin induces the proliferation of
neuronal progenitors and neuroprotection in a mouse model of
Alzheimer’s disease. J Alzheimers Dis. 24:17–25. 2011.PubMed/NCBI
|
|
86
|
Armato U, Chakravarthy B, Chiarini A,
Chioffi F, Dal Prà I and Whitfield JF: Leptin, sonic hedgehogs and
neurogenesis-a primary cilium’s tale. J Alzheimers Dis. 1:e1052012.
View Article : Google Scholar
|
|
87
|
Bianca VD, Dusi S, Bianchini E, Dal Prà I
and Rossi F: Beta-amyloid activates the O2-forming NADPH
oxidase in microglia, monocytes and neutrophils. A possible
inflammatory mechanism of neuronal damage in Alzheimer’s disease. J
Biol Chem. 274:15493–15499. 1999.PubMed/NCBI
|
|
88
|
Armato U, Bonafini C, Chakravarthy B,
Pacchiana R, Chiarini A, Whitfield JF and Dal Prà I: The
calcium-sensing receptor: A novel Alzheimer’s disease crucial
target? J Neurol Sci. July 27–2012.(Epub ahead of print).
View Article : Google Scholar
|