Introduction
Human prion diseases, also known as transmissible
spongiform encephalopathies (TSEs), are fatal neurodegenerative
disorders including Kuru, Creutzfeldt-Jakob disease (CJD),
Gerstmann-Sträussler-Scheinker disease (GSS) and fatal familial
insomnia (FFI). As an autosomal dominant prion disease, FFI
patients carry the D178N mutation coupled with a methionine at the
polymorphic site 129 of the prion protein gene (PRNP) on
chromosome 20 (1,2). Clinically, FFI is characterized by
rapidly progressive insomnia, prominent autonomic alterations and
behavioral disturbance (3,4).
The prominent pathology of FFI occurs mainly in the thalamus,
showing severe astrogliosis and loss of neurons, however, the
lesion profiles may vary in different patients and different brain
areas (3,5,6).
Additionally, deposits of PrPSc, the pathological
isoform of PrP, in brain tissues of FFI are usually infrequent
(2,7).
More than ten FFI patients have been diagnosed
genetically and/or pathologically since conducting CJD surveillance
in mainland China, starting in 2006. To further investigate the
neuropathologic and pathogenic features of Chinese FFI patients,
postmortem brains from three FFI patients were comparatively
analyzed based on PrPSc, the formation of astrogliosis
and the loss of neurons in various brain regions. Meanwhile, the
transcriptions of glial fibrillary acidic protein (GFAP) and
neuron-specific enolase (NSE) specific mRNA in different regions
were evaluated. Despite the different status of PrPSc
deposits in brains among the three patients, all cases showed more
severe reactive astrogliosis in the region of the thalamus than in
the cortex region. Neuronal loss among different regions was highly
comparable. We also observed that the transcriptional levels of
GFAP and NSE specific mRNAs were coincident with the expressions of
respective proteins in brain tissues of the FFI patients.
Materials and methods
Patients
The postmortem brains of three FFI cases that were
diagnosed by the experts from the Chinese National Surveillance
Network for CJD (CNSNC) were enrolled in this study. The clinical
presentation and pedigree of the family for 2 of the 3 cases, a
48-year-old male (Case 1) and a 26-year-old female (Case 2), were
described previously (8).
Case 3 was a 55-year-old man who was referred to the
first affiliated hospital of Zhengzhou University in 2009. The
patient was hospitalized with complaints of sleep disturbance and
restlessness for more than 1 month. During the initial admission,
the patient complained of general tiredness and weakness,
accompanied by belching and sour regurgitation. Neurological
examination showed mutism, generalized hyperreflexia of the limbs
and bilateral Babinski signs (+). During hospitalization, physical
movements rapidly declined and the patient later became bedridden.
EEG examination did not suggest CJD-related electroencephalogram
periodic sharp wave complexes (PWSC) and MRI scanning did not
reveal any obvious abnormalities. CSF 14-3-3 protein was positive.
His general condition deteriorated progressively and he was
discharged 4 months later. Severe akinetic mutism marked the end of
his clinical course and the patient succumbed to the disease 2
months after discharge. The total clinical course was approximately
8 months. His peripheral blood was obtained when he was in
hospital, and PRNP gene sequencing revealed a D178N mutation
with M129M homozygote.
Interviews with this patient’s family members
revealed that an elder brother had also developed similar symptoms
at the age of 55. The symptoms included initial insomnia and
restlessness, followed by progressive dementia, severe myoclonus,
pyramidal and extrapyramidal signs. Akinetic mutism, sleepiness,
lethargy marked the terminus of the clinical course which lasted 14
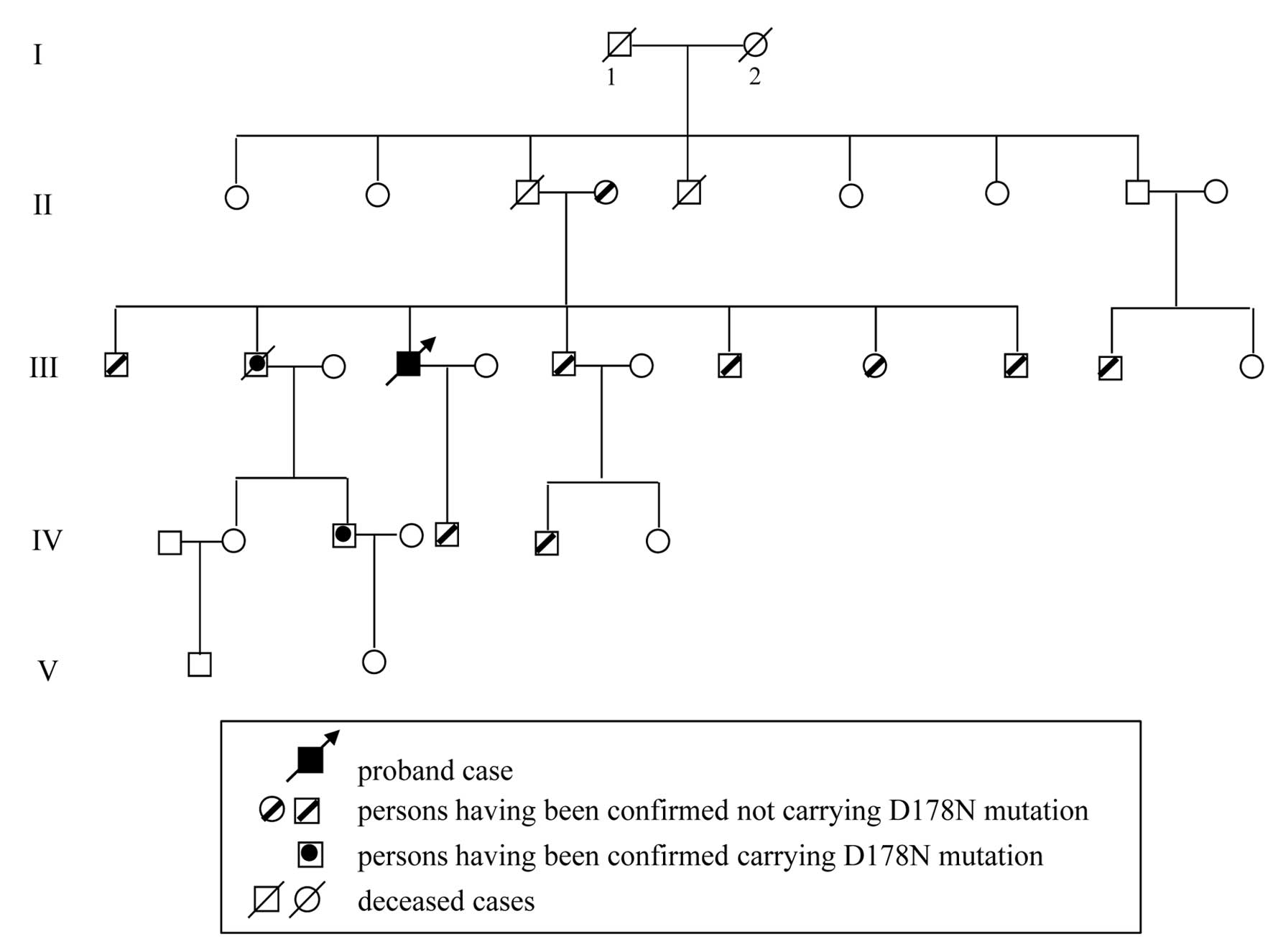
months. Eleven family members of the Case 3 patient were screened
for PRNP. D178N mutations were observed in his second elder
brother and the son of this brother who was still healthy. The
families of the other patients did not consent to genetic analysis.
The pedigree of the family of Case 3 is shown in Fig 1. The main clinical manifestations
of the 3 FFI cases are summarized in Table I.
 | Table IGeneral features and clinical
manifestations of three FFI cases. |
Table I
General features and clinical
manifestations of three FFI cases.
| | | | | | | Progressive
sympathetic symptoms | | | | |
|---|
| | | | | | |
| | | | |
|---|
| No. of Case | Gender | Age at onset (y) | Family history | Foremost
symptoms | Other signs and
symptoms | Blood pressure
(mmHg) | Excessive
sweating | Salivation | Minor evening
pyrexia | Weight loss | CSF 14-3-3 | EEG (PSW) | MRI | Duration of illness
(m) |
|---|
| 1 | M | 48 | Yes | Sleep disturbance,
sympathetic symptoms | Insomnia,
dysautonomia, motor, abnormalities | 145/95 | Yes | Yes | Yes | Yes | ND | − | Normal | 10 |
| 2 | F | 26 | Yes | Sleep
disturbance | Insomnia,
dysautonomia, myoclonus | 142/98 | Yes | Yes | Yes | Yes | ND | − | Normal | 14 |
| 3 | M | 55 | Yes | Sleep disturbance,
fidget | Insomnia,
diaphoresis, hyperthermia, memory loss | 145/95 | Yes | Yes | No | Yes | + | − | Normal | 8 |
Preparation of brain homogenates and
western blot analyses
Human brain tissues from the regions of the thalamus
(mediodorsal thalamic nuclei), the cingulate gyrus, the frontal
cortex, the parietal cortex, the occipital cortex and the temporal
cortex of each patient, brain tissues of a definitely diagnosed
G114V genetic CJD (gCJD) patient and a sporadic CJD (sCJD) patient,
as well as normal C57BL/6 mouse brains, were individually
collected. Brain homogenates [10% (w/v)] were prepared based on the
protocol previously described (8). Aliquots of brain homogenates were
separated on 12% SDS-PAGE and electroblotted onto a nitrocellulose
membrane. Membranes were probed with various primary antibodies at
4°C overnight, including 1:2,000-diluted anti-neuron specific
enolase mAb (NSE; Abcam, UK), 1:1,000-diluted anti-glial fibrillary
acidic protein mAb (GFAP; Santa Cruz Biotechnology, Inc., Santa
Cruz, CA, USA), 1:5,000-diluted anti-PrP mAb (3F4; Chemicon) and
1:1,000-diluted anti-β-actin mAb (Santa Cruz Biotechnology, Inc.),
respectively. After incubating with 1:5,000-diluted horseradish
peroxidase (HRP)-conjugated goat anti-mouse IgG (Santa Cruz
Biotechnology, Inc.) at room temperature for 2 h, the blots were
developed using Enhanced Chemiluminescence system (ECL; Amersham
Life Sciences) and visualized on autoradiography films. Images were
captured by ChemiDoc™ XRS+ Imager (Bio-Rad) and analyzed with
ImageJ software.
To detect the presence of proteinase K (PK)
resistant PrP in brain tissues, the brain homogenates were digested
with a final concentration of 20 μg/ml PK at 37°C for 60 min prior
to western blot analyses. To compare the PK-resistance of PrP among
the 3 brains, tissue homogenates from the thalamus were subjected
to treatment of various amounts of PK, i.e. 0, 5, 10, 15, 20 and 25
μg/ml PK.
Conformational stability assay
One hundred microliters of tissue homogenates of the
thalamus region from each FFI case, a G114V gCJD patient (9) and an sCJD case, were mixed with 5
volumes of cold methanol at −20°C for 2 h. Following centrifugation
at 20,000 × g for 30 min, the pellets were resuspended in 100 μl
GdnHCl (1, 2, 3, 4, 5 and 6 M) and incubated at 37°C for 12 h as
described elsewhere (10).
Subsequently, five volumes of cold methanol was added into each
preparation for protein precipitation and the pellets were
resuspended with 100 μl TN buffer (10 mM Tris, 130 mM NaCl, pH
7.0). Half the section was employed directly into SDS-PAGE and the
rest was subjected to PK-digestion (20 μg/ml) prior to
SDS-PAGE.
Nissl staining and immunohistochemical
(IHC) assays
Brain paraffin sections (5 μm) of 6 brain regions
were prepared. For Nissl staining, slices were stained with Nissl
(1% toluidine blue) for 30 min based on the protocol described
elsewhere (11). IHC assays were
performed according to published protocol (12). Briefly, the sections were
incubated overnight at 4°C with 1:500-diluted mAb for NSE, or
1:500-diluted mAb for GFAP. Then, the sections were incubated for 1
h with 1:250-diluted HRP-conjugated goat anti-mouse secondary
antibody (Vector Laboratories, USA) and visualized by incubation
with 3,3-diaminobenzidine tetrahydrochloride (DAB). GFAP or
NSE-positive staining regions were separately assessed based on
integral optical density (IOD) representing the total intensity of
all positive signals in an optical field (13). The OD analyses were performed in 3
individual sections by 2 investigators under double blindness.
Nonspecific background correction in each section was carried out
by subtracting the OD value of the blank area obtained from the
same section.
Quantitative real-time PCR (qRT-PCR)
Real-time PCR in this study was performed in an ABI
7900HT Fast sequence detector (Applied Biosystems). Total tissue
RNAs were extracted from different brain regions with a commercial
RNeasy Lipid Tissue mini kit (Qiagen), and were subjected to
first-strand cDNA synthesis with Reverse Transcription System
(Promega) according to the manufacturer’s protocols. The specific
primers were designed based on the sequences of human GFAP, NSE and
GAPDH in GenBank (NM_183079.1, NM_001975.2 and NM_002046.3),
including: 5′-GGAGA GGAAGATTGAGTCGC-3′ and 5′-CGGTAGTCGTTGG
CTTCG-3′, 5′-CGACAAGGCTGGCTACAC-3′ and 5′-CAG GTCATCACCCACAATC-3′,
and 5′-ACCACAGTCCAT GCCATCAC-3′ and 5′-TCCACCACCCTGTTGCTGTA-3′,
respectively. PCR amplification was performed in triplicate with a
total of 40 cycles (15 sec at 95°C and 30 sec at 56°C). The
comparative Ct (the fractional cycle number at which the amount of
amplified target reached a fixed threshold) method was used for the
relative quantitative detection of the expressions of the targeting
genes. The relative Ct for the target gene was subtracted from the
Ct for the GAPDH gene (14).
Statistical analysis
Statistical analyses were performed using SPSS 17.0
statistical package. All values in figures were expressed as means
± SD. Quantitative analysis of immunoblot images was carried out
using software ImageJ. Differences between means were assessed with
the one-way ANOVA test. P-value <0.05 was considered to indicate
statistically significant differences.
Ethics statement
Written consent for further investigation and
publication was obtained from the relatives of the patients. Usage
of the stored human brain samples in this study was approved by the
Ethics Committee of the National Institute for Viral Disease
Prevention and Control, China CDC.
Results
Features of PrPSc in FFI
cases
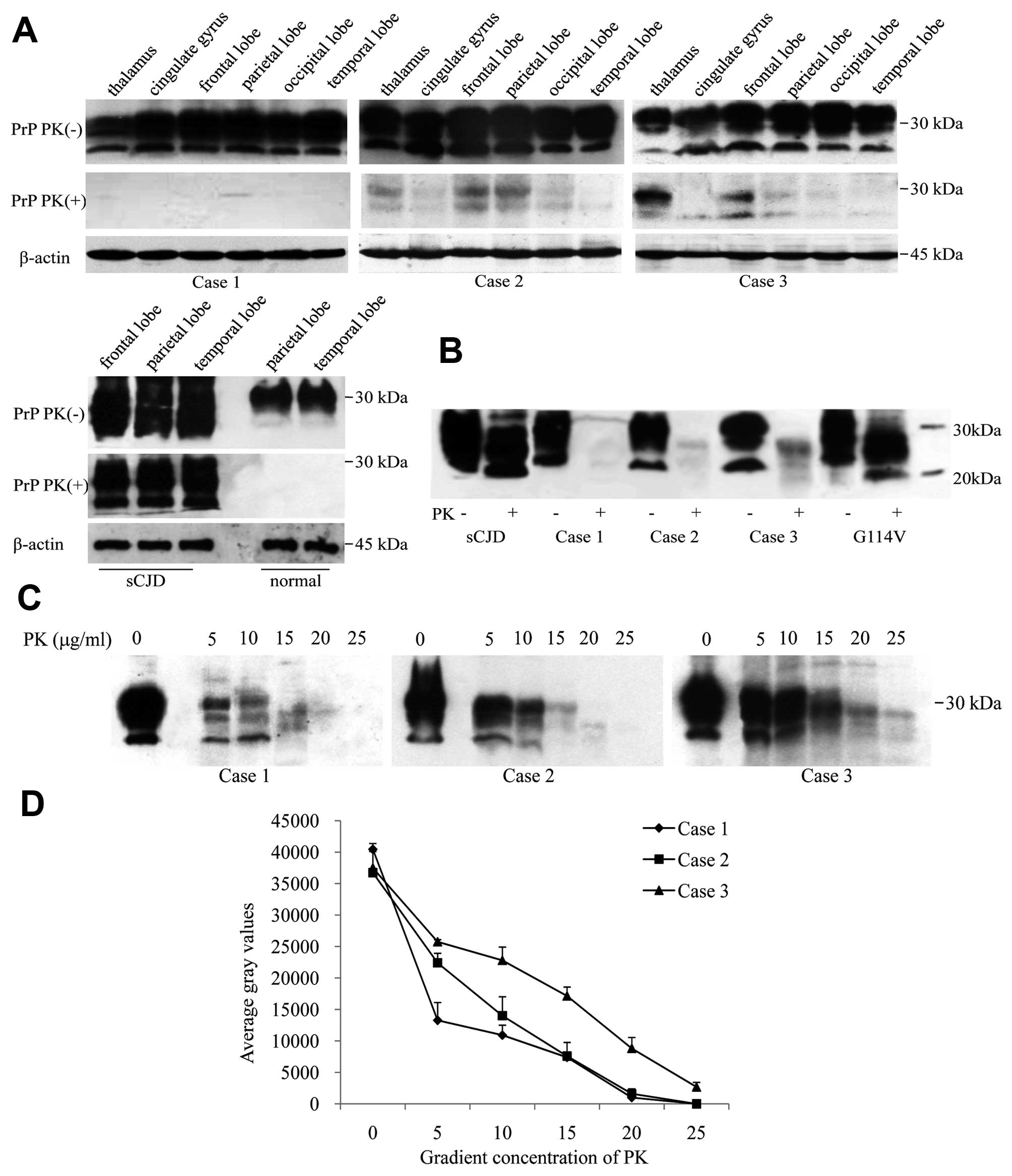
Six brain regions of each patient were comparatively
analyzed with PrP-specific western blot analyses. The signals of
three typical glycosylated isoforms of PrP were observed in all
preparations without PK digestion, in which the signal intensities
in the thalamus were slightly weaker than in the cortex regions
(Fig. 2A, upper row). To address
the deposits of PK-resistant PrP (PrPSc) in the brains,
various tissue homogenates were treated with 20 μg/ml PK. Clear
PrPSc signals were detected in the brains of Cases 2 and
3, particularly in the regions of the thalamus, frontal and
parietal lobes, while almost no PrPSc was found in the
brain tissues of Case 1 (Fig. 2A,
middle row). In contrast to the profiles of type-1 PrPSc
in a G114V gCJD and an sCJD with M129 homozygote which contained
predominant monoglycosylated PrPSc, both detectable
PrPSc signals in Cases 2 and 3 showed predominant
diglycosylated PrPSc patterns (Fig. 2B), which is more like that of
type-2B PrPSc (8).
To assess the PK-resistance of PrPSc in
the brains, the homogenates of the thalamus region of the three
cases were subjected to treatments with a serial concentration of
PK. Western blot analyses showed very weak PrPSc signals
in the preparations of Case 1 below 20 μg/ml PK, but obvious
PrPSc signals in the preparations of Cases 2 and 3
(Fig. 2C). Analyses of the
intensities of PrPSc signals revealed that the curve of
PrPSc in Case 1 dropped gradually in the reactions from
5 to 10 μg/ml PK and reached an undetectable level in that of 20
μg/ml PK, while in Cases 2 and 3 the data maintained relative high
levels in the preparations with low PK concentrations (from 5 to 15
μg/ml) and dropped down to almost baseline in 25 μg/ml PK (Fig. 2D).
Conformational stabilities of
PrPSc from FFI, gCJD and sCJD
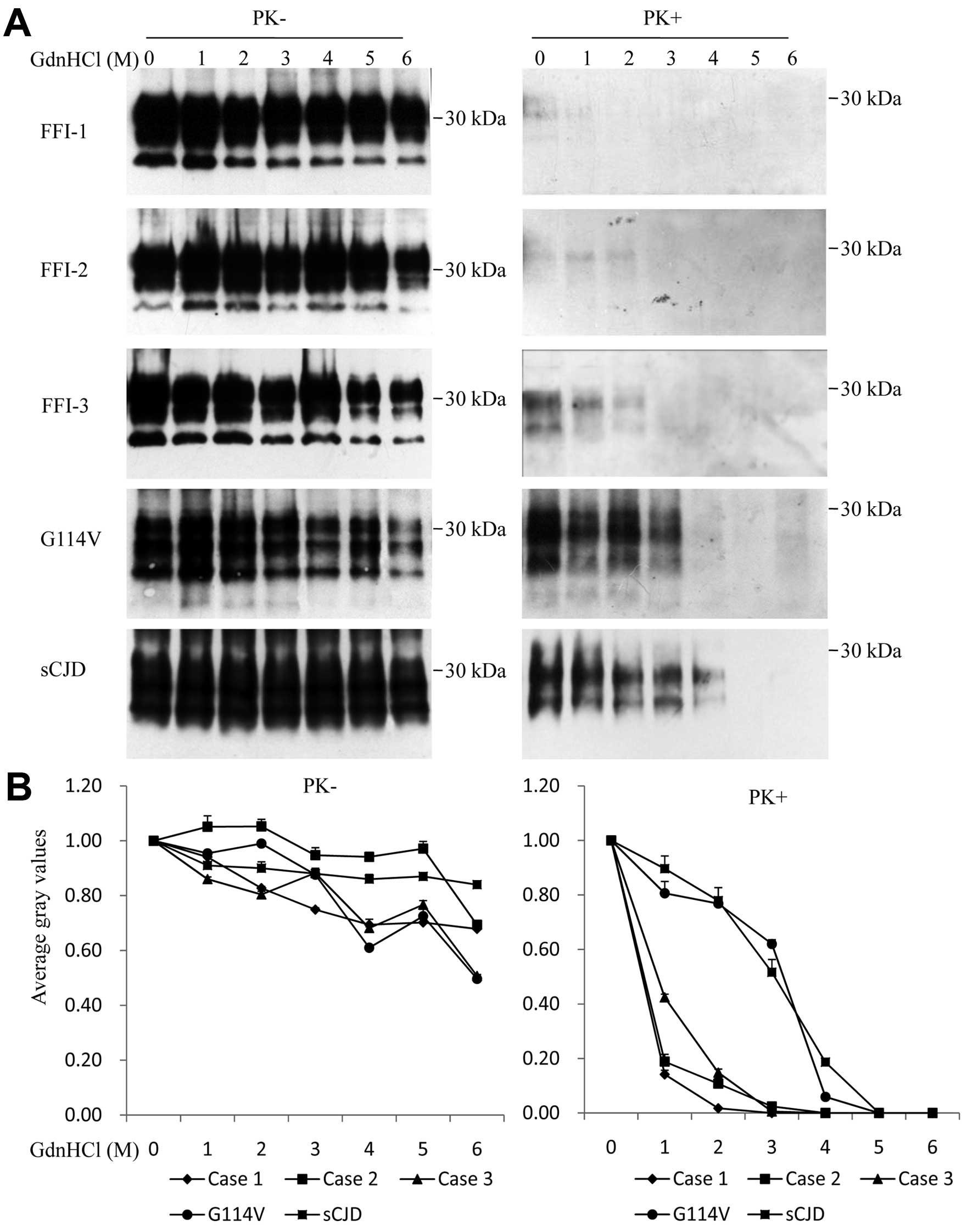
To test the conformational stabilities of
PrPSc in the brains of FFI cases following exposure to
GdnHCl, equal amounts of homogenates from the thalamus region of
FFI cases, a G114V gCJD patient and an sCJD patient, were incubated
with increasing concentrations of GdnHCl. Western blot analyses
revealed that the total PrP signals (without treatment of PK) were
comparable among five tested samples and decreased slightly along
with the increase of GdnHCl (Fig. 3A
and B, left). When the GdnHCl-treated brain samples were
subsequently exposed to 20 μg/ml PK, the PrPSc signals
in the FFI samples were generally markedly weaker than those shown
for the G114V gCJD and sCJD samples. Extremely weak
PrPSc signals were observed in the preparations of FFI
Case 1 treated with 0 and 1 M GdnHCl, while weak but repeatedly
observed PrPSc signals were seen in FFI Cases 2 and 3
treated with 0, 1 and 2 M GdnHCl (Fig. 3A, right). Analyses of the curves
of the PrPSc gray values along with the increase of
GdnHCl showed the obviously different patterns among those CJD
subtypes, that PrPSc signals of G114V gCJD and sCJD
cases possessed stronger conformational stabilities in GdnHCl,
while the PrPSc in the brains of the FFI cases possessed
less stable conformational topology (Fig. 3B, right).
Marked astrogliosis in the thalamus of
FFI cases
To compare the astrogliosis in various brain regions
of the 3 FFI cases, the brain slides from six regions of each
patient were immunohistochemically stained with a GFAP-specific
antibody. Obvious GFAP-positive stained astrocyte-like cells and
fibril-like structures were observed in all tested slides, and the
thalamus regions of the 3 patients appeared more severely affected
(Fig. 4A). The IOD values of GFAP
staining for all three FFI cases were the highest in the thalamus.
Comparatively, Case 3 displayed overall lower IOD values in each
brain region when compared with the other 2 cases (Fig. 4B). Furthermore, various brain
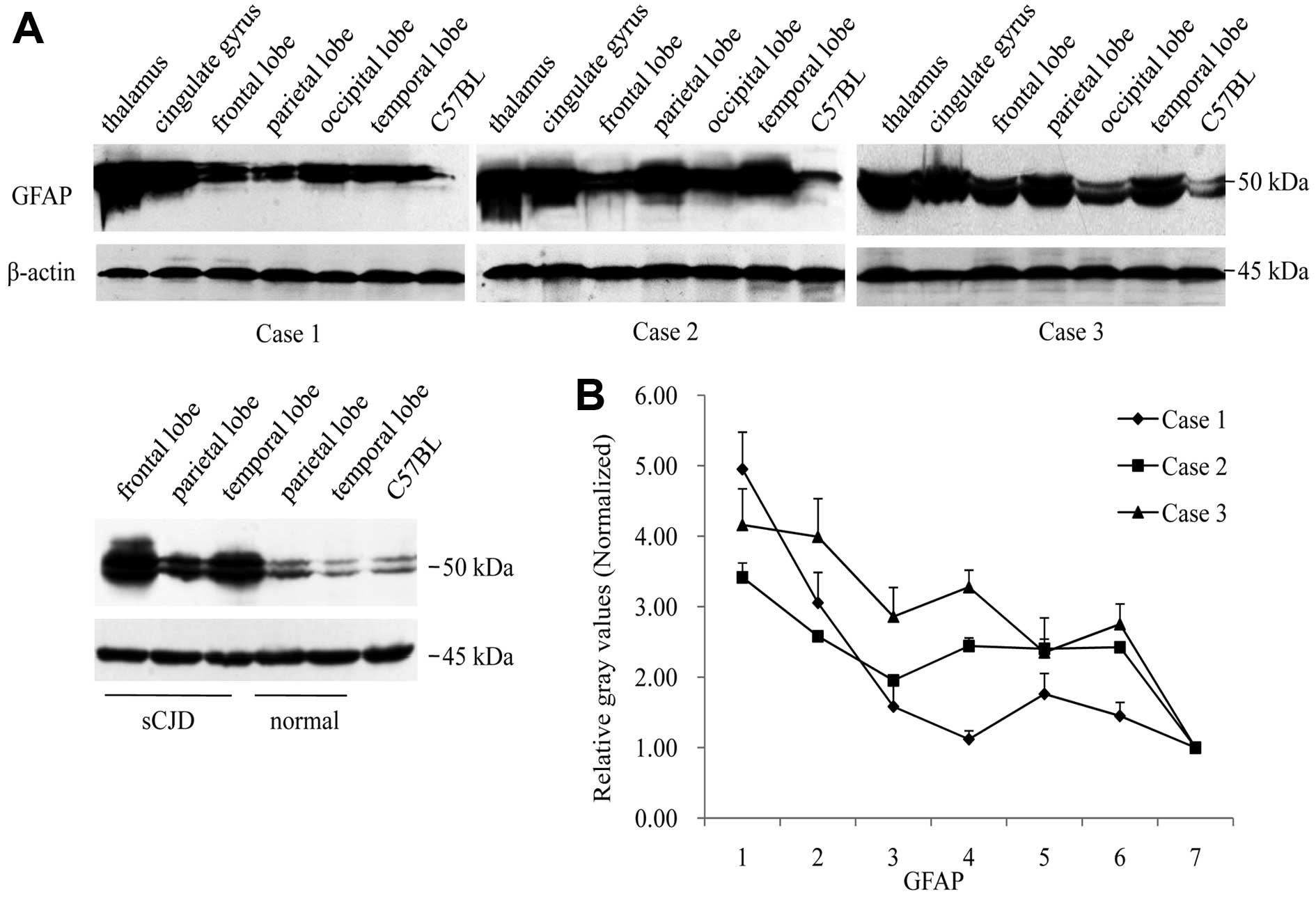
homogenates were employed into GFAP-specific western blot analyses.
Fig. 5A illustrates the
GFAP-specific signals in all preparations at the position of
Mr. Fifty GFAP signals in the thalamus of the three cases
were markedly stronger than those of the individual cortex regions.
Analyses of the relative gray values of GFAP signals following
normalization with those of the controls of the normal human brain
and the pooled mice brain homogenates revealed similar patterns of
the GFAP distributions to those the IHC assay showed in the brain
regions (Fig. 5B), indicating
more astrogliosis in the regions of the thalamus of the 3 FFI
cases.
Neuron loss in the different brain
regions of FFI cases
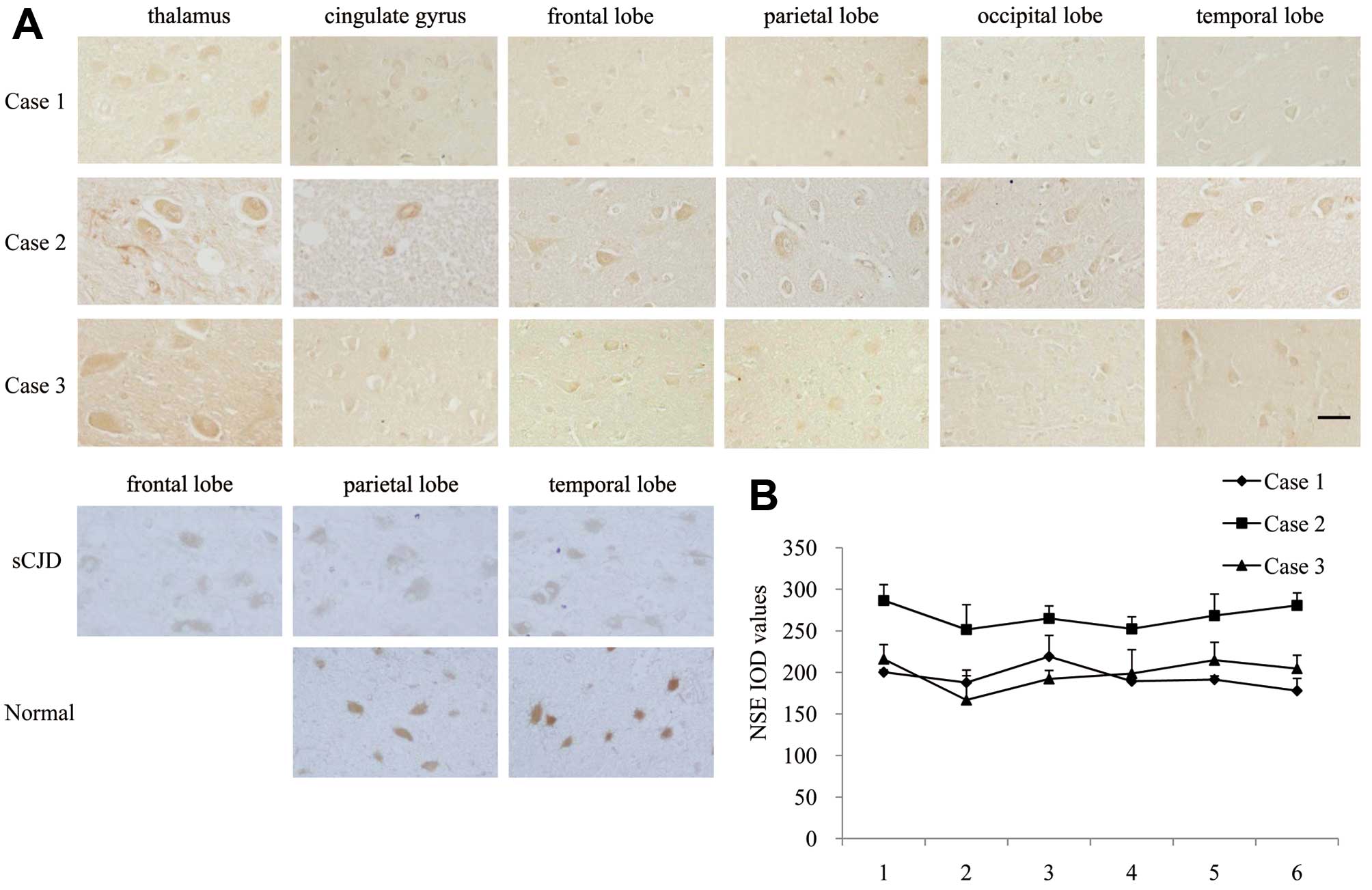
Slides of 6 brain regions from 3 FFI cases were
immunohistologically stained with an NSE-specific mAb. Large
elliptic positive-stained cells were observed in all tested slides
(Fig. 6A). Quantification of
histochemical staining for NSE showed almost identical levels among
the 6 tested regions for each patient, while Case 2 was relatively
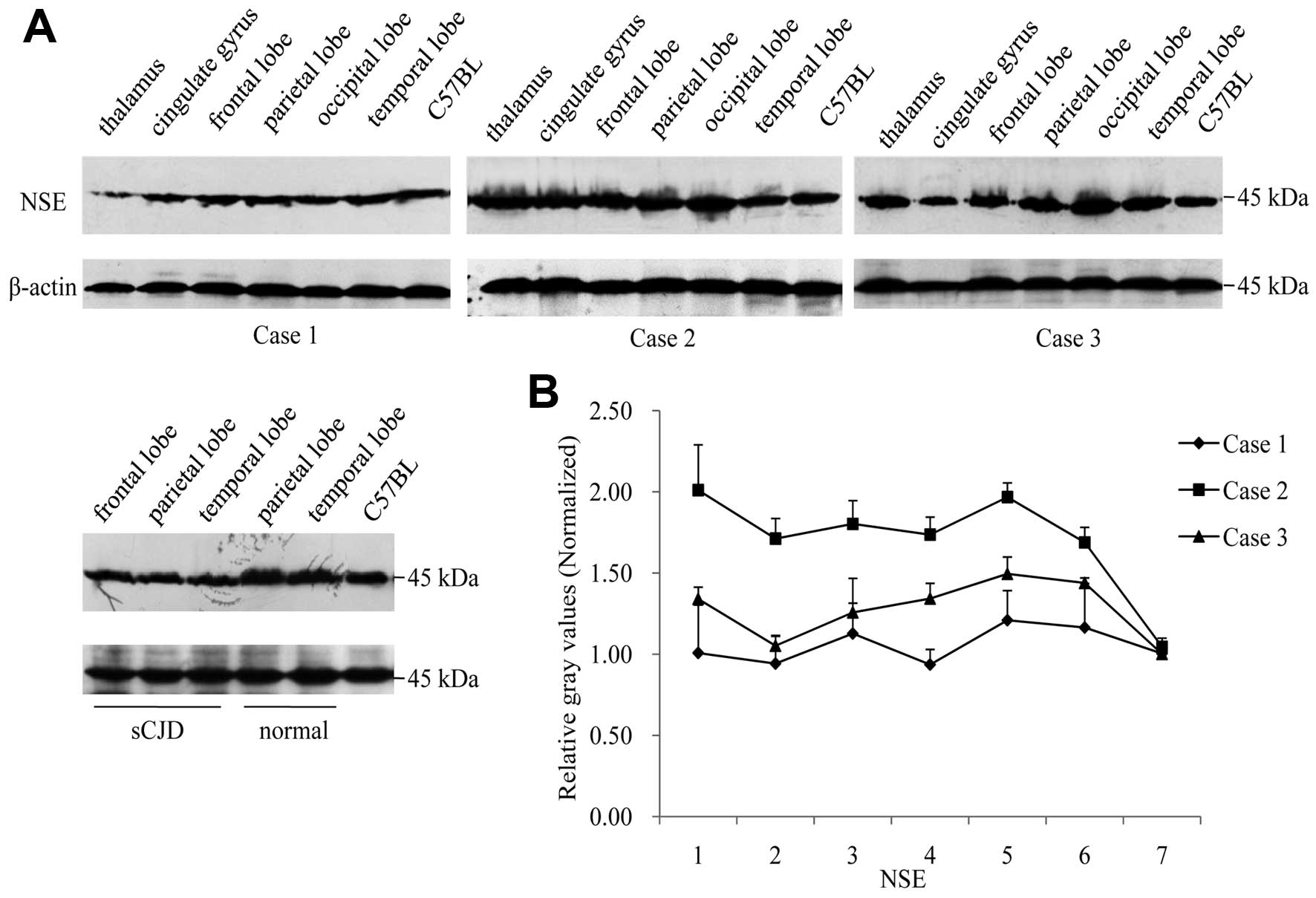
higher (Fig. 6B). In NSE-specific
western blot analyses, the 45 kDa-large band was detected in all
tested samples, showing similar signal intensities among various
FFI cases (Fig. 7A). Relative
gray values of NSE signals in each preparation following
equilibration with those of the normal human and mice brain
homogenates illustrated comparable data among various brain
regions, in which the data of Case 2 were higher (Fig. 7B).
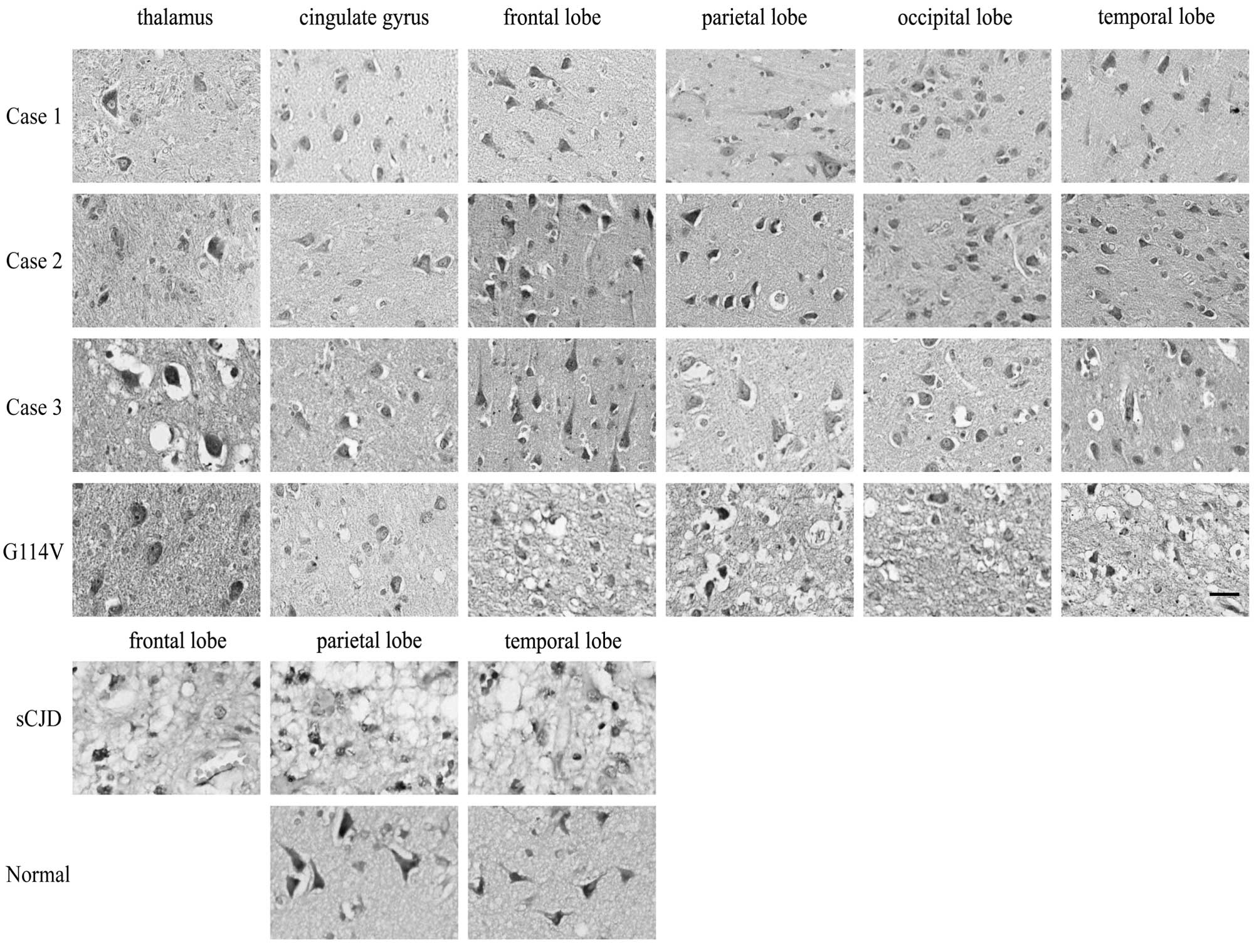
Slides of 6 brain regions from three FFI cases and a
G114V gCJD were assayed by Nissl staining. It revealed less
purple-blue stained Nissl bodies in the regions of the thalamus of
the three FFI cases accompanied by vacuoles, compared with the
observations in the same region of G114V gCJD (Fig. 8). By contrast, more severe
spongiform degenerations were observed in the 4 cortex lobes of the
G114V gCJD case than in the 3 FFI cases, accompanied by obvious
pyknotic nuclei (Fig. 8). This
finding suggests more severe neuronal damage in the thalamus region
of FFI.
Transcriptions of GFAP and NSE in brains
of FFI cases
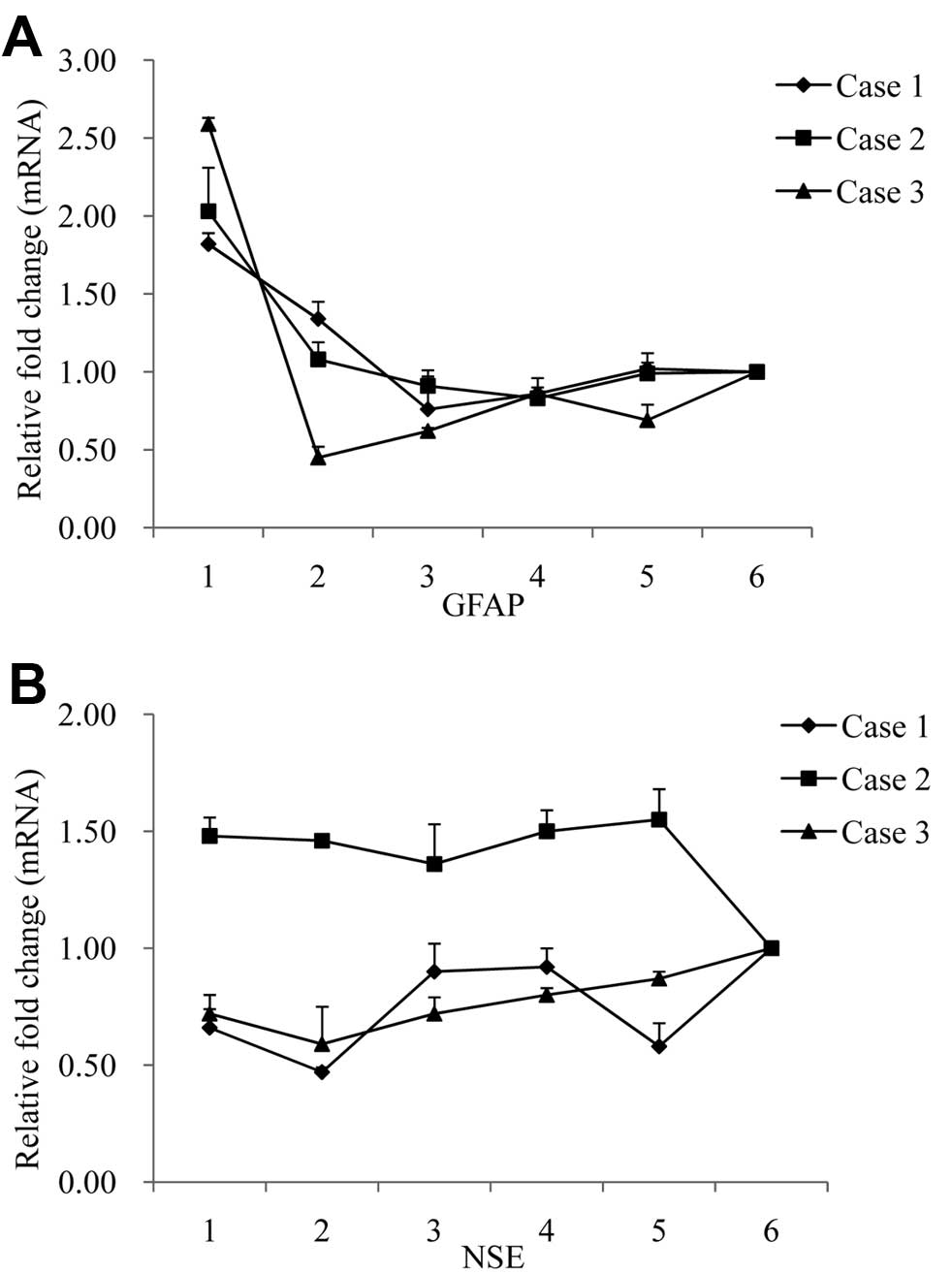
To evaluate the transcriptional levels of GFAP and
NSE-specific mRNAs in various brain regions, total RNAs extracted
from 6 brain regions of 3 FFI cases were subjected to qRT-PCRs for
GFAP and NSE, respectively, using the amplification of GAPDH mRNA
as the endogenous control. Compared to the 4 cortex regions, the
thalamus expressed by far the highest relative quantities of
GFAP-specific mRNA (2−ΔΔCt) (Fig. 9A). There was little difference in
the relative expression of NSE specific mRNAs (2−ΔΔCt)
among various brain regions of each patient, although the values in
Case 2 appeared higher than in the other 2 cases (Fig. 9B). The data suggested that the
transcriptions of GFAP and NSE in brain tissues of FFI patients
correlated positively with the levels of the proteins.
Discussion
In this study, we investigated evidence for
astrogliosis and neuronal loss in various brain regions from
postmortem samples obtained from 3 Chinese FFI cases. Based on the
semi-quantitative methodologies of IHC and western blot analyses
for GFAP and NSE, the brains of the 3 FFI cases displayed similar
neuropathological patterns, whereby a more active gliosis in the
regions of the thalamus was found compared to the cortex
regions.
GFAP is restrictively expressed by astrocyte, and is
commonly used as a special biomarker for astrocyte. Astrogliosis is
a basic pathogenic sign often associated with CNS injuries
(15), showing increased
expression of GFAP in brain tissues. Severe astrogliosis in the
thalamus is significant for the FFI-related pathologic pattern,
which is distinct from other types of human prion diseases
(9,16). Our data here provide evidence that
not only higher numbers of GFAP-stained cells, but also more deeply
GFAP-stained and larger astrocytes are observed in the thalamus of
FFI patients. Our observations emphasize again the unique
pathogenesis of FFI, caused by the existence of the PRNP
allele of the mutation of D178N with M129M homozygote.
Neuronal loss is another pathological feature of
prion diseases, although it is not the specific one (3,4,6,17).
From the Nissl staining, which presents the situation of the
neuronal structural destruction, it appears that more severe
neuronal damage occurs in the thalamus of the FFI cases than in the
G114V gCJD case, who had sCJD-like clinical presentations and
neuropathological changes (9).
However, we were unable to find significant differences in the
number of NSE-stained cells or degree of NSE gene/protein
expression among the various brain regions of individual FFI
patients. Furthermore, since the amount of NSE-positive cells or
signals in the brain of the Case 2 patient, who was 26 years old at
onset, tested repeatedly higher than in the other 2 patients, who
were in their 50’s at onset, an age-related factor is possible.
Our study here illustrates again the common
characteristics of FFI, presence of little or no PrPSc
in brain tissues with less PK-resistance and unstable in
conformation when exposed to GdnHCl, possibly indicating a special
topology of PrP with D178N mutation and M129M homozygous. In line
with several other studies (2,4,18),
our data reveals whether or not deposition of PrPSc
seems to significantly affect the neuropathological changes and the
clinical manifestations of FFI; it may also highlight that the
large plaques or severe aggregations of PrPSc are not
the major component for neurotoxicity in prion diseases (19).
In CJD patients, some disease-related genes are
usually continually transcribed in the CNS tissues, even at the
final stage of illness. We previously demonstrated that PrP mRNAs
are continuously transcribed in the brain tissues of a G114V gCJD,
which is speculated to supply PrPC as the substrates for
PrPSc replication (9).
Active transcription of GFAP in brains, especially in the thalamus,
may reflect an intense reactive gliosis against the progressively
severe brain injury. Identification of NSE-related mRNAs in FFI
brains during their terminal stage may indicate the presence of
non-functional neurons or neuron networks.
To date, more than 56 mutations in PrP have been
identified as being involved in the development of human genetic
TSEs with a variety of geographical distributions and frequencies
(16). Although the genetic
tendencies, the pathologic involvements, the clinical
manifestations and the laboratory features may vary, no other TSE
induces such prominent thalamic dysfunction characterized by
insomnia and dysautonomia as do TSEs containing a D178N mutation
with M129M polymorphism. Why FFI-associated PrP mutant has tropisms
for the thalamus remains unknown. Other human genetic prion
diseases also show region-specific damage, such as GSS in the
cerebellum (20). Further studies
into the role of PrP, as well the local microenvironment, are
required in order to better understand the pathogenesis of this
mysterious disease.
Acknowledgements
We thank Dr Dennis J. Grab (Johns Hopkins
University) for reading our manuscript prior to submission. This
study was supported by the China Mega-Project for Infectious
Disease (2011ZX10004-101), the Young Scholar Scientific Research
Foundation of China CDC (2012A102), and the SKLID Development Grant
(2012SKLID102 and 2011SKLID211).
References
|
1
|
Goldfarb LG, Petersen RB, Tabaton M, et
al: Fatal familial insomnia and familial Creutzfeldt-Jakob disease:
disease phenotype determined by a DNA polymorphism. Science.
258:806–808. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Medori R, Montagna P, Tritschler HJ, et
al: Fatal familial insomnia: a second kindred with mutation of
prion protein gene at codon 178. Neurology. 42:669–670. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lugaresi E, Medori R, Montagna P, et al:
Fatal familial insomnia and dysautonomia with selective
degeneration of thalamic nuclei. N Engl J Med. 315:997–1003. 1986.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Manetto V, Medori R, Cortelli P, et al:
Fatal familial insomnia: clinical and pathologic study of five new
cases. Neurology. 42:312–319. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ayuso Blanco T, Urriza Mena J, Caballero
Martinez C, Iriarte Franco J, Munoz R and Garcia-Bragado F: Fatal
familiar insomnia: clinical, neurophysiological and
histopathological study of two cases. Neurologia. 21:414–420.
2006.(In Spanish).
|
|
6
|
Parchi P, Petersen RB, Chen SG, et al:
Molecular pathology of fatal familial insomnia. Brain Pathol.
8:539–548. 1998. View Article : Google Scholar
|
|
7
|
Brown P, Kenney K, Little B, et al:
Intracerebral distribution of infectious amyloid protein in
spongiform encephalopathy. Ann Neurol. 38:245–253. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shi XH, Han J, Zhang J, et al: Clinical,
histopathological and genetic studies in a family with fatal
familial insomnia. Infect Genet Evol. 10:292–297. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shi Q, Zhang BY, Gao C, et al: The
diversities of PrP(Sc) distributions and pathologic changes in
various brain regions from a Chinese patient with G114V genetic
CJD. Neuropathology. 32:51–59. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Thackray AM, Hopkins L, Spiropoulos J and
Bujdoso R: Molecular and transmission characteristics of
primary-passaged ovine scrapie isolates in conventional and ovine
PrP transgenic mice. J Virol. 82:11197–11207. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zheng Q, Zhu D, Bai Y, Wu Y, Jia J and Hu
Y: Exercise improves recovery after ischemic brain injury by
inducing the expression of angiopoietin-1 and Tie-2 in rats. Tohoku
J Exp Med. 224:221–228. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang J, Chen L, Zhang BY, et al:
Comparison study on clinical and neuropathological characteristics
of hamsters inoculated with scrapie strain 263K in different
challenging pathways. Biomed Environ Sci. 17:65–78. 2004.
|
|
13
|
Ruifrok AC, Katz RL and Johnston DA:
Comparison of quantification of histochemical staining by
hue-saturation-intensity (HSI) transformation and
color-deconvolution. Appl Immunohistochem Mol Morphol. 11:85–91.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Abdulmawjood A, Schonenbrucher H and Bulte
M: Novel molecular method for detection of bovine-specific central
nervous system tissues as bovine spongiform encephalopathy risk
material in meat and meat products. J Mol Diagn. 7:368–374. 2005.
View Article : Google Scholar
|
|
15
|
Eng LF and Ghirnikar RS: GFAP and
astrogliosis. Brain Pathol. 4:229–237. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Pastore M, Chin SS, Bell KL, et al:
Creutzfeldt-Jakob disease (CJD) with a mutation at codon 148 of
prion protein gene: relationship with sporadic CJD. Am J Pathol.
167:1729–1738. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Spacey SD, Pastore M, McGillivray B,
Fleming J, Gambetti P and Feldman H: Fatal familial insomnia: the
first account in a family of Chinese descent. Arch Neurol.
61:122–125. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Almer G, Hainfellner JA, Brucke T, et al:
Fatal familial insomnia: a new Austrian family. Brain. 122:5–16.
1999. View Article : Google Scholar
|
|
19
|
Faucheux BA, Morain E, Diouron V, et al:
Quantification of surviving cerebellar granule neurones and
abnormal prion protein (PrPSc) deposition in sporadic
Creutzfeldt-Jakob disease supports a pathogenic role for small
PrPSc deposits common to the various molecular subtypes.
Neuropathol Appl Neurobiol. 37:500–512. 2011.PubMed/NCBI
|
|
20
|
Kretzschmar HA, Kufer P, Riethmuller G,
DeArmond S, Prusiner SB and Schiffer D: Prion protein mutation at
codon 102 in an Italian family with Gerstmann-Straussler-Scheinker
syndrome. Neurology. 42:809–810. 1992. View Article : Google Scholar : PubMed/NCBI
|