Introduction
Despite the success of preventive vaccination and
the advances in the development of antivirals in the past decade,
hepatitis B virus (HBV) infection remains one of the leading causes
of chronic liver disease worldwide (1). However, the currently available
treatments with interferon (IFN) and nucleos(t) ide analog (NA) are
unsatisfactory, as IFNs are limited by multiple side-effects
(2), and NAs are compromised by
the potential resistance (3).
Furthermore, these drawbacks cannot be overcome completely by novel
agents within the same categories (4,5) or
optimization of current therapies (6). Thus, novel antivirals for HBV are
presently required.
In view of the lifecycle of HBV, various strategies
have been proposed experimentally to interfere with the key steps
of HBV replication: viral entry, nucleocapsid assembly, envelopment
of mature nucleocapsids, reverse transcription just targeted by NAs
(7–9), as well as covalent closed circle DNA
(cccDNA) at the level of transcription under hypothesis yet
(10). Among these, the assembly
of nucleocapsids has been studied extensively and appears
promising. This process is characterized by the complex formation
of pgRNA with HBV core (HBc) protein and polymerase and
self-assembly of an RNA-containing core particle in the cytoplasm
of hepatocytes, and the resultant nucleocapsids then provide a site
for following DNA replication (7). Small molecular compounds
phenopropenamides (11) and
dihydroarylpyrimidines, such as Bay 4-4109 (12), inhibit the maturation of HBV
nucleocapsids and subsequent replication, although challenged by
the potential hepatotoxicity due to fatty acid metabolism disorder
and mitochondrial inability (13). Both the internal fragment of HBc
(HBc78-117) (14) and the
intracellular single-chain variable fragment (scFv) antibody
against HBc, but not that against HBx, delivered by plasmid or
lentiviral vector were all capable of inhibiting HBV replication
in vitro by interfering with the function of HBc (15,16). Collectively, these data indicate
the prospects of encapsidation of HBV genome as an attractive
target for antiviral design, and, presumably, the scFv antibodies
against HBc are preferable alternatives for this purpose,
considering their well-known advantages of specificity for
antiviral therapeutics. However, together with the uncertainty in
clinical practice for small molecular compounds under study
(13), the aforementioned gene
therapies with plasmid or viral-based vectors are not only
challenged by transfection efficiency and cytotoxicity, but they
are also severely limited by the safety and ethical concerns
(17). Bioavailability on the
premise of safety is important in the design of any therapeutics
(18), and this applies to the
development of biologically anti-HBV agents including the scFv
antibodies.
This obstacle is expected to be overcome in part by
a new means of cell-penetrating peptides (CPPs), also known as
protein transduction domains (17,18). CPPs are a group of short cationic
sequences with generally negligible side-effects and are well known
by the efficient translocation of cargoes into a variety of cells
(19). These CPPs have
successfully delivered proteins, nucleic acids, and small molecule
therapeutics, thereby accomplishing the potential for diagnosing
and treating various diseases (19–21). As expected, this technique
provides a reasonable tool for intrabodies expressed within a
designated intracellular compartment, especially for the popular
engineered scFv with the merit of smaller size, as the safety or
ethical concerns associated with viral transfer system would be
eliminated if delivered as proteins (22). In the field of HBV infection, a
series of artificial synthetic peptides combining cell penetrating
sequence oligo-arginine R7 with several nucleocapsid binding
subunits were designed and evaluated in vitro. The results
showed that these recombinant peptides efficiently penetrated into
living cells and significantly inhibited nucleocapsid assembly and
HBV release (23). However, the
possible value of CPPs for those more specific peptides of scFvs
regarding the blocks of nucleocapsid assembly remains elusive. In
our previous study, we identified and purified a type of human scFv
specific to HBc protein with favorable affinity, however, its
cytoplasmic delivery in living cells was severely limited (24). Whether this scFv can be
internalized by CPPs and whether it can inhibit encapsidation, if
delivered by CPPs, remains to be explored.
Therefore, the present study purified the
aforementioned anti-HBc scFv (24) fused to cytoplasmic transduction
peptide (CTP), a type of common CPP with a higher level of liver
expression, less cytotoxicity and more cytoplasmic distribution
(19,25), and evaluated its effect on HBV
replication in vitro in HepG2.2.15 cells.
Materials and methods
Preparation of anti-HBc scFv fused to
CTP
A standard prokaryotic expression system with
Escherichia coli (E. coli) DH5α and BL21 (DE3) as
host strains and pET28a (Invitrogen) as basic plasmid was used for
the expression of target proteins anti-HBc scFv-CTP and anti-HBc
scFv. The sequence of scFv gene against HBc was described by Tang
et al (24), and the
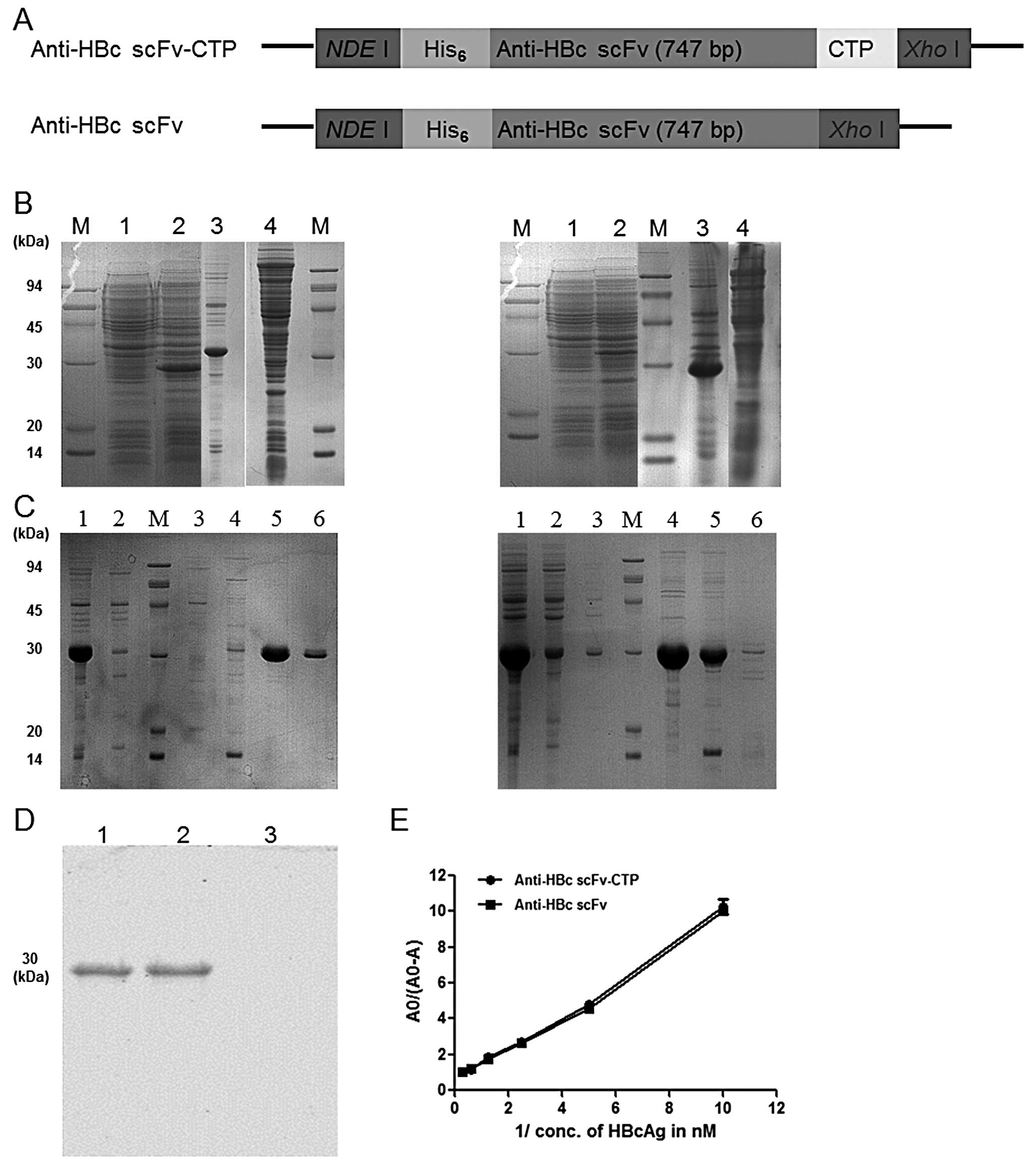
primers used for amplification are shown in Table I. As illustrated in Fig. 1A, the scFv gene alone or together
with a sequence of CTP at the C-terminus was amplified by PCR from
the pPNL6 and inserted into the NdeI/XhoI sites of
pET28a to yield the plasmid pET28a-scFv-CTP and pET28ascFv,
respectively, and a 6XHis tag located at the N-terminus of both
constructs was used for the following purification and detection.
Both recombinant plasmids were transiently transformed into the
E. coli BL21 bacteria and were induced to express by
isopropyl β-D-thiogalactopyranoside (IPTG) at a final concentration
of 1 mM at 37°C for 4 h. The recombinant proteins were purified
using nickel-nitrilotriacetic acid (Ni+-NTA) resin as
recommended by the manufacturer (HisTrap HP; Amersham Biosciences).
The purified proteins anti-HBc scFv-CTP and anti-HBc scFv were
identified with sodium dodecyl sulfate polyacrylamide gel
electrophoresis (SDS-PAGE) with Coomassie blue staining and further
identified with a standard protocol of western blotting by using
mouse anti-His monoclonal antibody (Abcam). The protein
concentration was determined by the Bradford assay (Bradford).
 | Table I.Primers for anti-HBc scFv and the
fusion protein. |
Table I.
Primers for anti-HBc scFv and the
fusion protein.
| Primer | Sequence
(5′-3′) |
|---|
| Anti-HBc scFv fused
to CTP | Forward:
CATATGaGCCCAGGTGAAGCTGCAGGAG |
| Reverse:
CTCGAGbGTGCACGGCGACCTCCCCGTTTGATTTCCAAC
TTAGCGACGCCGACGCCGGC |
| Anti-HBc scFv | Forward:
CATATGaGCCCAGGTGAAGCTGCAGGAG |
| Reverse:
CTCGAGbTTAGCGACGCCGACGCCGGC |
Analysis for the binding affinity of
anti-HBc scFv-CTP
The interaction between the purified protein
anti-HBc scFv-CTP and HBc was evaluated in solution phase by
enzyme-linked immunosorbent assay (ELISA) using the protocol
described by Friguet et al (26). Briefly, different dilutions of
antigen (0.1–10 nM) were incubated with diluted solutions of fusion
protein anti-HBc scFv-CTP or anti-HBc scFv for 16 h at 4°C, so that
equilibrium of the antigen-antibody reaction was reached. One
hundred microliters of such equilibrated solution was incubated in
antigen-coated ELISA plates (250 ng/well of antigen) for 90 min at
room temperature (RT) to capture free scFv, and was washed six
times with phosphate-buffered saline (PBS)-T (PBS plus 0.05%
Tween-20) to remove unbound proteins. The complex was incubated for
2 h with 100 μl of anti-His (1:1,000; Abcam) at RT, and
washed six times with PBS-T. Then, 100 μl of
o-phenylenediamine dihydrochloride (OPD; 1 mg/ml) in 1X stable
peroxide substrate buffer (Pierce) was added and the complex was
incubated for 15 min. The reaction was terminated by the addition
of 10 μl of 2.5 M sulfuric acid, and the absorbance at 450
nm was measured using a Synergy 2 spectrophotometer (BioTek).
Cell culture and administration with
agents
The HBV replicating cell line HepG2.2.15 used for
anti-HBV compound screening was maintained in Dulbecco’s modified
Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum
(FBS), 100 U/ml penicillin, 100 μg/ml streptomycin (Gibco),
and 380 μg/ml G418 (Sigma). Cells were cultured at 37°C in a
humidified atmosphere of 5% CO2.
Cells were seeded in 6 or 96-well plates as
necessary at a density of 2×105/ml. After 24 h, the
medium was exchanged to media containing different concentrations
of the fusion protein anti-HBc scFv-CTP (1.7, 5.0 and 15 μM,
respectively, dissolved in PBS), anti-HBc scFv (5 μM,
dissolved in PBS), or lamivudine (LMV; 1 μM, dissolved in
0.1% dimethyl sulf-oxide), every other day for three times, then
harvested at 48 h after the final administration, unless otherwise
indicated.
Cell viability assay for the
determination of cytotoxicity of the fusion protein
Cell viability was determined with CellTiter
96® AQueous Non-Radioactive Cell Proliferation Assay
(Promega), according to the manufacturer’s instructions. Briefly,
cells were seeded in a 96-well plate and treated as described
above, and CellTiter 96® Aqueous One Solution Reagent
(Promega) was added, followed by incubation for 2 h. Absorbance was
measured at 450 nm using a Synergy 2 spectrophotometer (BioTek),
and the 50% cytotoxicity concentration (CC50) was
calculated.
Immunofluorescent staining for the
detection of cytoplasmic translocation
Cells were seeded in a 6-well plate and treated with
different concentrations of proteins for 6 h. They were then fixed
with 4% paraformaldehyde in 0.1 M PBS and permeabilized with 0.1%
(v/v) Triton X-100 in PBS for 30 min. Mouse monoclonal IgG against
His (1:500; Abcam) was used as the primary antibody and
FITC-labeled IgG against mouse IgG (1:2,000; Jackson Immuno
Research) as the secondary antibody. The chromosome was stained
with propidium iodide (PI; Sigma) for nuclear indication. Images
were captured by a confocal laser scanning microscope (LSM 510;
Zeiss, Berlin, Germany).
Electrochemical illuminescence
quantification for HBsAg, HBeAg and real-time PCR for HBV DNA in
the culture supernatants
Cells were seeded in 6-well plates and treated with
different agents, and the culture supernatants were collected 48 h
after the final administration. Viral protein HBsAg and HBeAg
quantification was detected using the electrochemical illuminescent
immunoassay kits (Abbott Laboratories, Abbott Park, IL, USA) on an
ARCHITECT i2000 automatic immunoassay analyzer (Abbott
Laboratories). HBV DNA was quantified with a real-time fluorescence
quantitative polymerase chain reaction (PCR) kit (FQ-PCR; Daan,
Shenzhen, China) on an ABI 7500 Real-Time PCR System (Applied
Biosystems, USA), according to the manufacturer’s instructions.
Western blot analysis for intracellular
HBc, immunoprecipitation analysis for the scFv-CTP-HBc complex, and
agarose gel electrophoresis for nucleocapsids
For western blot analysis, cells in one well of a
6-well plate were washed twice with ice-cold PBS and lysed in 200
μl of lysis buffer containing 10 mM Tris-HCl (pH 8.0), 1 mM
ethylenediaminetetraacetic acid (EDTA), 50 mM NaCl, 1% NP-40, and
1X protease inhibitor cocktail (Qiagen). Twenty microliters of the
clarified cell lysate were mixed with 5 μl of 5X loading
buffer for SDS-PAGE (Invitrogen), denatured by boiling for 10 min,
then separated on a 12% SDS-PAGE gel and transferred onto
nitrocellulose membranes (Millipore). The membranes were blocked
with 5% fat-free milk and probed with the primary polyclonal
antibody against HBc (1:1,000; Dako). Bound antibodies were
detected by the HRP-conjugated secondary antibody against rabbit
IgG (1:5,000; Jackson Immuno Research) and visualized by enhanced
chemiluminescence (Pierce). Glyceraldehyde 3-phosphate
dehydrogenase (GAPDH) was used as a protein loading control.
For the scFv-CTP-HBc complex analysis, the
supernatants of cell lysates from one well of a 6-well plate were
incubated with anti-His antibody (1:500; Abcam) or rabbit
polyclonal anti-HBc (1:500; Dako) at 4°C under rotary agitation
overnight. Subsequently, 30 μl of pretreated protein A/G
sepharose/sample were added and incubated at 4°C under rotary
agitation for 4 h. The protein A/G sepharose beads were collected
by centrifugation and washed three times in PBS. After the final
wash, the bead pellet was re-suspended in 25 μl of 1X
SDS-PAGE sample loading buffer and boiled at 100°C for 5 min.
Approximately 20 μg of total protein were separated on 12%
SDS-PAGE and detected by immunoblotting with anti-HBc antibody
(Dako) or anti-His antibody (Abcam) as noted above.
For cytoplasmic nucleocapsid analysis, cells in one
well of a 6-well plate were lysed with lysis buffer (10 mM Tris pH
7.5, 1 mM EDTA, 50 mM NaCl, 8% sucrose, 1% NP-40) on ice for 10
min. Clarified supernatants were treated with 6 μM
MgCl2 and 200 μg/ml of DNase I (NEB). After 20
min of incubation at 37°C, the reaction was terminated by 1 mM
EDTA. Twenty microliters of the resultant sample were subjected to
1.2% agarose gel in 1X Tri-EDTA buffer (Sigma) at 50 V for 3 h.
Capsids were transferred directly to a nitrocellulose membrane
through capillary action in TNE (10 mM Tris-HCl, 150 mM NaCl, 1 mM
EDTA) buffer overnight, and were then blotted by anti-HBc antibody
(Dako).
Southern blot for replicative
intermediates of HBV
To extract HBV DNA from intracellular core
particles, cells were lysed in lysis buffer (10 mM Tris-HCl pH 8.0,
1 mM EDTA, 50 mM NaCl and 1% NP-40) at 37°C for 15 min. Following
centrifugation at 12,000 × g for 5 min, the supernatants were
treated with pronase (0.5 mg/ml; Sigma) in a buffer containing 150
mM NaCl and 0.5% SDS at 37°C for 1 h. After saturated phenol
extraction, viral DNA was precipitated out by ethanol at −20°C
overnight, dissolved in distilled deionized water and separated on
a 1.2% agarose gel at 60 V for 3 h. Gels were then subjected to
denaturalization in a solution containing 0.5 M NaOH and 1.5 M
NaCl, followed by neutralization in a buffer containing 1 M
Tris-HCl (pH 7.4) and 1.5 M NaCl. DNA was then blotted onto
Hybond-XL membrane (GE Healthcare) in 20X SSC buffer and hybridized
with 32P-labeled full-length HBV DNA probe, prepared by
using the Prime-a-Gene® labeling system (Promega). The
membranes were washed and signals were developed by a
phosphorimager screen (Fujinon).
Statistical analysis
All statistical analysis was performed using SPSS
software version 13.0 (SPSS Inc.). Continuous variables were
expressed as the means ± standard deviation, assessed using the
Student’s t-test or ANOVA analysis. All values are based on at
least three independent experiments. Two-sided P<0.05 was
considered to indicate a statistically significant difference.
Results
Fusion protein anti-HBc scFv-CTP
purification and its binding affinity
Briefly, soluble expression of the fusion protein
anti-HBc scFv-CTP and the control anti-HBc scFv without CTP were
both obtained by the standard prokaryotic expression system
(Fig. 1B). Then the target
proteins were purified successfully (Fig. 1C), and were blotted at the
expected binding site of 30 kDa by anti-His antibody (Fig. 1D). Finally, a >90% purity and a
≤1 mg/ml of concentration dissolved in water for the two purified
proteins was achieved, and a 1.012 nM (R2=0.98) and
1.142 nM (R2=0.99) of dissociation constant
KD was shown for both purified proteins, respectively,
as calculated from the slope of the straight lines in Fig. 1E.
CTP delivers the anti-HBc scFv into the
cytoplasm of HepG2.2.15 cells
To evaluate whether or not the aforementioned
purified fusion protein can enter cells, HepG2.2.15 cells were
treated with anti-HBc scFv-CTP for 6 h. Obvious FITC signals were
observed in the cytoplasm of HepG2.2.15 cells, in contrast to the
absence of signals in cells treated with the control protein
anti-HBc scFv (Fig. 2).
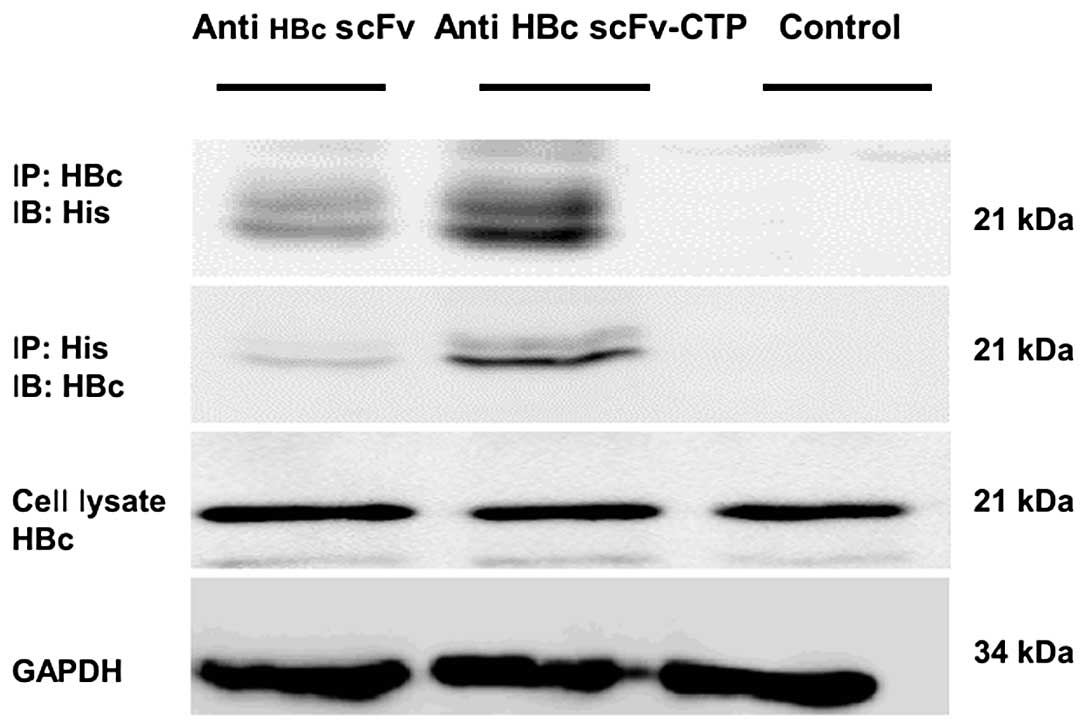
The fusion protein interacts with HBc in
HepG2.2.15 cells
As indicated from immunoprecipitation analysis using
cell lysates, the fusion protein anti-HBc scFv-CTP interacted with
HBc within HepG2.2.15 cells, but the anti-HBc scFv failed to be
precipitated by either anti-His or anti-HBc antibody (Fig. 3). Together with the results of
immunofluorescent staining, this suggests that CPP CTP provided
both the possibility of translocation across membranes and the
potential to interact with the target antigen HBc for anti-HBc scFv
in HepG2.2.15 cells.
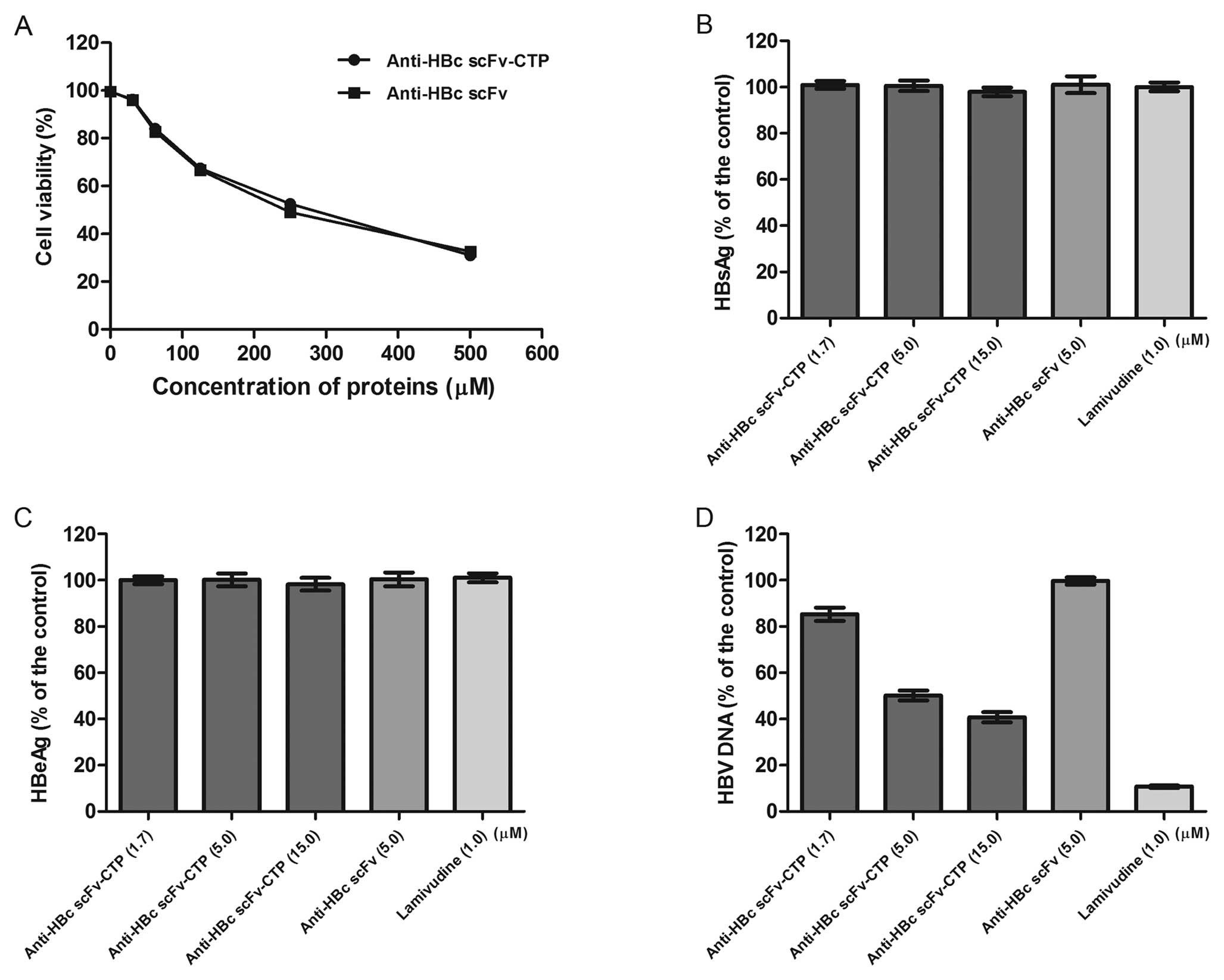
Cytotoxicity of the fusion protein
anti-HBc scFv-CTP
No obvious cytotoxicity was observed for the fusion
protein anti-HBc scFv-CTP and the control protein anti-HBc scFv.
The CC50 for the anti-HBc scFv-CTP and the anti-HBc scFv
was approximately 256 and 235 mM, respectively (Fig. 4A).
The fusion protein inhibits the
replication of HBV
Real-time PCR indicated that HBV DNA levels in
culture supernatants decreased significantly by the fusion protein
anti-HBc scFv-CTP, and a 5.1 μM of half maximal (50%) effect
concentration (EC50) for extracellular HBV DNA was
shown. Then, a lower dose of 1.7 μM and a higher dose of 15
μM (based on the ‘3-fold steps’) were chosen for the
subsequent evaluation for antiviral activity. Compared with the
result from the control protein anti-HBc scFv, our fusion protein
anti-HBc scFv-CTP inhibited the replication of HBV in a
dose-dependent manner, although the effect was less than that of
LMV (Fig. 4D). With respect to
the levels of HBsAg and HBeAg in culture supernatant, however, no
significant reduction was observed by the fusion protein (Fig. 4B and C).
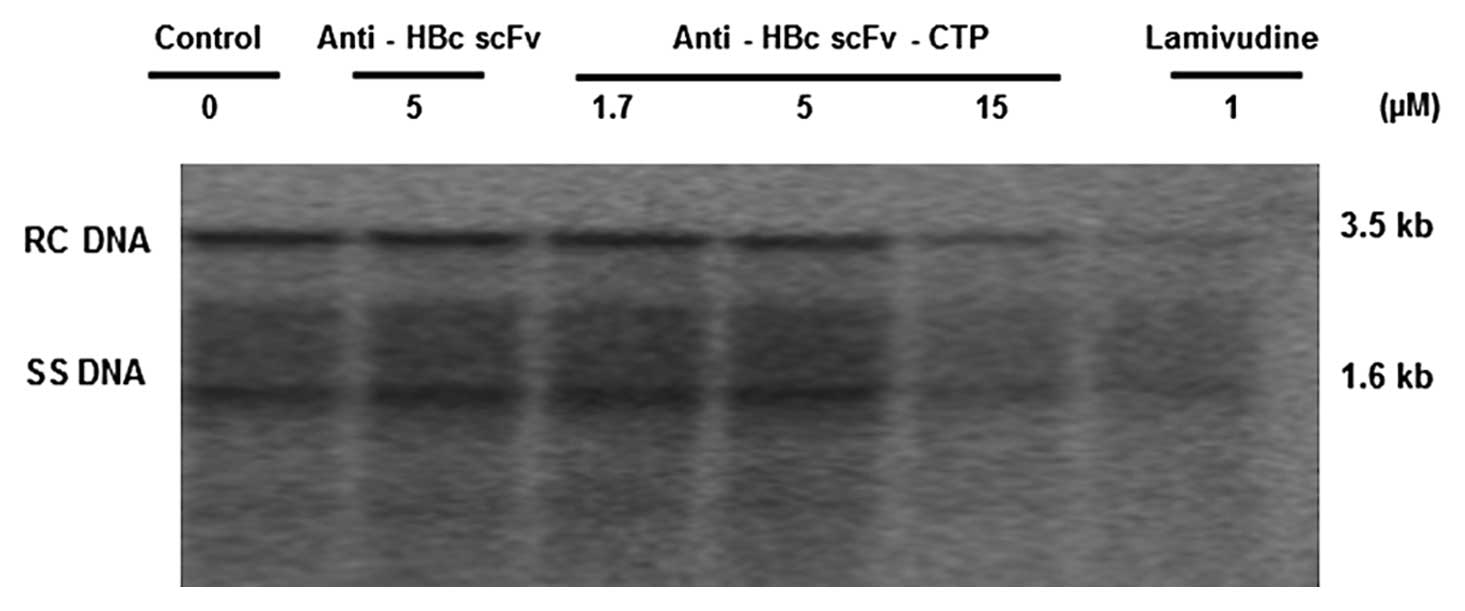
The fusion protein interferes with HBV
nucleocapsid assembly and decreases the intracellular HBV DNA
replication intermediates
To investigate the antiviral mechanism of fusion
protein anti-HBc scFv-CTP, we evaluated sequentially the
intracellular levels of HBc, nucleocapsids, and DNA replication
intermediates in HepG2.2.15 cells. The expression of HBc was not
affected by any of the agents involved, while the anti-HBc
scFv-CTP, but not the anti-HBc scFv, effectively reduced the level
of nucleocapsids, which was markedly different from that of LMV
(Fig. 5). Furthermore, southern
blotting indicated that the intracellular HBV DNA replication
intermediates decreased accordingly by the anti-HBc scFv fused to
CTP, and the magnitude of inhibition activity from the higher dose
was similar to that of LMV (Fig.
6).
Discussion
In the present study, we purified an anti-HBc scFv
fused to CTP using a previous screened sequence and identified its
efficacy in anti-HBV in vitro. Our results showed that the
fusion protein anti-HBc scFv-CTP was successfully translocated into
the cytoplasm of HepG2.2.15 cells, affecting nucleocapsid assembly
and markedly decreasing the replication of HBV in a micromolar
concentration.
One of the most attractive options for the
development of novel anti-HBV strategies, blocking of nucleocapsid
assembly was tested both by small molecular compounds (12) and by molecular-based approaches
(27) as potential candidates and
promising results have been obtained. However, these, including the
scFv against HBc developed by our team and others (15,24), have yet to be applied to clinical
practice. To overcome the major obstacle regarding the absence of
safe methods for delivering intracellular scFv to living target
cells, we investigated the possibility of CPPs as a means of
delivery for protein translocation by purifying a recombinant
protein derived from the previous scFv (24) and a high liver expression CPPs-CTP
(25). The fusion protein of
anti-HBc scFv-CTP was expressed correctly and purified successfully
using a prokaryotic expression system. Moreover, our protein showed
negligible cytotoxicity with a CC50 up to 256 mM and
retained a comparable binding affinity with the original
counterpart anti-HBc scFv (24).
Evaluation regarding the cell-penetrating activity
indicated that our fusion protein was translocated into the
cytoplasm of living HepG2.2.15 cells, persisting for at least 6 h.
This uptake across membrane and the stable intracellular expression
provided a premise for the function of the prior scFv with little
cytoplasmic activity (24). The
interaction between our fusion protein with intracellular HBc
further detected by immunoprecipitation suggested a direct impact
of the protein on its specific antigens in the cytoplasm of
HepG2.2.15 cells. This translocation was in marked contrast to the
original scFv alone, although the latter exhibited similar binding
affinity in the ex vivo context. Theoretically, this
valuable property in living cells should come from the fused
sequence of CTP.
Based on the results from the levels of HBV
replication, fusion protein, rather than the original one, worked
well in HepG2.2.15 cells, despite the absence of effect for viral
proteins HBsAg and HBeAg in culture supernatant. It was able to
inhibit the replication of HBV in a dose-dependent manner, although
the potency was less than LMV to some extent. This functional
improvement of anti-HBc scFv within living cells from CTP was
similar, in part, to the artificial oligoarginine carrying peptides
targeting the nucleocapsid binding subunits (23), thus making CPPs a suitable tool
for delivering macromolecules in the design of encapsidation
inhibitors. However, CTP may have certain advantages for future
clinical application among numerous CPPs, owing to its higher level
of liver expression and more cytoplasmic distribution confirmed in
an in vivo study (25).
Therefore, our fusion protein provides the possibility for future
therapeutic application for this purpose.
A mechanism study revealed that intracellular HBc
remained stable while HBV nucleocapsids were repressed
significantly, and HBV DNA replication intermediates, both
single-stranded and partially double-stranded DNA, were markedly
suppressed by the anti-HBc sFv-CTP in HepG2.2.15 cells. This
inhibition activity was in line with the intracellular scFv
delivered by expression vector (15) and the internal fragment derived
from HBc delivered by lentiviral vector (14). This suggests that assembly of
nucleocapsids is a pivotal process for HBV replication, and a small
interference on the premise of intracellular targeted expression
would, perhaps, affect its function of encapsidation, although
intracellular HBc protein as a whole is difficult to be neuralized
or decreased using current strategies. Our fusion protein anti-HBc
scFv-CTP is a possible alternative for future nucleocapsid
inhibitors. However, more powerful approaches capable of inhibiting
HBV replication and diminishing HBc are required.
In conclusion, several limitations of this study
need to be declared. Short peptide CTP alone was not purified as
another negative control, but its profiles regarding
bioavailability and safety have already been verified by Kim et
al (25), therefore, it is
negligible concerning the interferences of CTP. Second, northern
blotting for the detection of HBV RNA intermediates was not carried
out, however, nucleocapsid assembly targeted by our scFv occurred
after genome transcription thus no changes would be observed at
this level as revealed by the similar study with scFv against HBc
(15). Third, the clinical
application of therapeutic scFv, remains a significant challenge,
such as the inherent drawback of short half-life in serum is
impossible to be improved by CPPs. Nevertheless, another problem of
membrane translocation has to be resolved by this delivery system,
and prokaryotic expression systems provide opportunities both for
large-scale purification of scFv fused to CTP and for future
studies to overcome the other obstacles.
Abbreviations:
|
CHB
|
chronic hepatitis B;
|
|
HBV
|
hepatitis B virus;
|
|
IFN
|
interferon;
|
|
NA
|
nucleos(t)ide analog;
|
|
HBc
|
HBV core;
|
|
cccDNA
|
covalent closed circle DNA;
|
|
scFv
|
single-chain variable fragment;
|
|
CPP
|
cell-penetrating peptide;
|
|
CTP
|
cytoplasmic transduction peptide.
|
Acknowledgements
This study was funded by a grant from
the National Natural Science Foundation of China (general program,
no. 81070335/H0316 to Guoqing Zang).
References
|
1.
|
Zanetti AR, Van Damme P and Shouval D: The
global impact of vaccination against hepatitis B: a historical
overview. Vaccine. 26:6266–6273. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Perrillo R: Benefits and risks of
interferon therapy for hepatitis B. Hepatology. 49(Suppl 5):
S103–S111. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Dienstag JL: Benefits and risks of
nucleoside analog therapy for hepatitis B. Hepatology. 49(Suppl 5):
S112–S121. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Snow-Lampart A, Chappell B, Curtis M, et
al: No resistance to tenofovir disoproxil fumarate detected after
up to 144 weeks of therapy in patients monoinfected with chronic
hepatitis B virus. Hepatology. 53:763–773. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Bingfa X, Qinglin F, Hui H, Canjun W, Wei
W and Lihua S: Anti-hepatitis B virus activity and mechanisms of
recombinant human serum albumin-interferon-alpha-2b fusion protein
in vitro and in vivo. Pharmacology. 83:323–332. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Carey I and Harrison PM: Monotherapy
versus combination therapy for the treatment of chronic hepatitis
B. Expert Opin Investig Drugs. 18:1655–1666. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Urban S, Schulze A, Dandri M and Petersen
J: The replication cycle of hepatitis B virus. J Hepatol.
52:282–284. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Stein LL and Loomba R: Drug targets in
hepatitis B virus infection. Infect Disord Drug Targets. 9:105–116.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Grimm D, Thimme R and Blum HE: HBV life
cycle and novel drug targets. Hepatol Int. 5:644–653. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Levrero M, Pollicino T, Petersen J,
Belloni L, Raimondo G and Dandri M: Control of cccDNA function in
hepatitis B virus infection. J Hepatol. 51:581–592. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Katen SP, Chirapu SR, Finn MG and Zlotnick
A: Trapping of hepatitis B virus capsid assembly intermediates by
phenylpropenamide assembly accelerators. ACS Chem Biol.
5:1125–1136. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Deres K, Schroder CH, Paessens A, et al:
Inhibition of hepatitis B virus replication by drug-induced
depletion of nucleocapsids. Science. 299:893–896. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Shi C, Wu CQ, Cao AM, Sheng HZ, Yan XZ and
Liao MY: NMR-spectroscopy-based metabonomic approach to the
analysis of Bay41-4109, a novel anti-HBV compound, induced
hepatotoxicity in rats. Toxicol Lett. 173:161–167. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Han J, Pan X, Gao Y and Weil L: Inhibition
of hepatitis B virus replication by the internal fragment of
hepatitis B core protein. Virus Res. 150:129–134. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Yamamoto M, Hayashi N, Takehara T, et al:
Intracellular single-chain antibody against hepatitis B virus core
protein inhibits the replication of hepatitis B virus in cultured
cells. Hepatology. 30:300–307. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Jin YH, Hong SH, Kim K, Shin HJ and Park
S: Intracellular antibody fragment against hepatitis B virus X
protein does not inhibit viral replication. Yonsei Med J.
47:721–728. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Heng BC and Cao T: Making cell-permeable
recombinant telomerase (trans-telomerase) through fusion of its
catalytic subunit (hTERT) with protein transduction domains (PTD):
a possible strategy to overcome replicative senescence during ex
vivo culture of primary explanted cells. Med Hypotheses.
65:199–200. 2005.
|
|
18.
|
Gump JM and Dowdy SF: TAT transduction:
the molecular mechanism and therapeutic prospects. Trends Mol Med.
13:443–448. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Zorko M and Langel U: Cell-penetrating
peptides: mechanism and kinetics of cargo delivery. Adv Drug Deliv
Rev. 57:529–545. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Doorbar J and Griffin H: Intrabody
strategies for the treatment of human papillomavirus-associated
disease. Expert Opin Biol Ther. 7:677–689. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Fonseca SB, Pereira MP and Kelley SO:
Recent advances in the use of cell-penetrating peptides for medical
and biological applications. Adv Drug Deliv Rev. 61:953–964. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Chames P, Van Regenmortel M, Weiss E and
Baty D: Therapeutic antibodies: successes, limitations and hopes
for the future. Br J Pharmacol. 157:220–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Pan XB, Wei L, Han JC, Ma H, Deng K and
Cong X: Artificial recombinant cell-penetrating peptides interfere
with envelopment of hepatitis B virus nucleocapsid and viral
production. Antiviral Res. 89:109–114. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Tang ZH, Ma HH, Chen WS, Gu L, Li G and
Yao JL: Screening, identifying and sequencing of human single-chain
variable fragment specific to hepatitis B virus core protein.
Zhongguo Binglishengli Zazhi. 19:329–333. 2003.(In Chinese).
|
|
25.
|
Kim D, Jeon C, Kim JH, et al: Cytoplasmic
transduction peptide (CTP): new approach for the delivery of
biomolecules into cytoplasm in vitro and in vivo. Exp Cell Res.
312:1277–1288. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Friguet B, Chaffotte AF, Djavadi-Ohaniance
L and Goldberg ME: Measurements of the true affinity constant in
solution of antigen-antibody complexes by enzyme-linked
immunosorbent assay. J Immunol Methods. 77:305–319. 1985.
View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Butz K, Denk C, Fitscher B, et al: Peptide
aptamers targeting the hepatitis B virus core protein: a new class
of molecules with antiviral activity. Oncogene. 20:6579–6586. 2001.
View Article : Google Scholar : PubMed/NCBI
|