Introduction
Atrial fibrillation (AF) is the most common
arrhythmia in clinical practice, present in 2% of the general
population. The prevalence of AF increases markedly with age,
affecting as many as 9% of individuals 80 years or older (1). AF has several complications such as
thromboembolism and heart failure and is regarded as an independent
risk factor for cardiovascular mortality (2).
Numerous studies have focused on the pathogenesis of
AF. It is generally accepted that AF may be multifactorial and has
been linked to, among others, ectopy from the pulmonary veins,
catecholamine release and large atrial size. However, the
fundamental arrhythmia mechanisms of AF are rapid ectopic firing
and reentrant activity. Atrial remodeling may increase the risk of
ectopic or reentrant activity through a multitude of potential
mechanisms (3) and is considered
to be the new pathophysiological mechanism of AF (4).
In the past few years the relationship between
inflammation and AF has drawn significant attention. Inflammation
may be a cause of AF and this is supported by the high incidence of
AF in post-operative cardiac surgery, a state of severe
inflammatory process (5–7). However, evidence suggests that
inflammation may also play a prominent role in nonoperative onset
of AF (8).
Programmed cell death-1 (PD-1, Pdcd-1) is a
negative immunoreceptor belonging to the CD28/CTLA-4 family. PD-1
deficiency enhances T-cell activation and increases the
inflammation level. Previous observations indicated that the
PD-1−/− mice with different genetic backgrounds
presented different types of autoimmune diseases (9,10).
Another study found that PD-1−/− mice cleared adenovirus
infections more rapidly but developed more severe hepatocellular
injury compared with the control group (11). Meanwhile, the study of Carter
et al (12) indicated that
the PD-1−/− mice produced a significantly higher
concentration of inflammatory cytokines [interferon-γ (IFN-γ),
tumor necrosis factor (TNF), interleukin-6 (IL-6) and IL-17]. In
the past few years, as an inflammatory animal model,
PD-1−/− mice have been used to carry out extensive
research, however, none of these studies focused on AF.
In the present study, we compared the expression of
inflammatory cytokines, the level of atrial myocyte oxidative
stress, the atrial effective refractory period (AERP) and the
atrial myocardial fibrosis level of the C57BL/6 PD-1−/−
with the C57BL/6 mice. We observed the relationship between the
inflammation in the C57BL/6 PD-1−/− mice and the
pathogenesis of AF.
Materials and methods
Animals
Two groups of mice were used in our experiment: the
C57BL/6 and the C57BL/6-PD-1−/− group (15 male mice, 7–8
weeks old, in each group).
The C57BL/6-PD-1−/− mice were kindly
provided by Professor Tasuku Honjo of Kyoto University, Japan. All
experiments conformed to the guidelines for the care and use of
laboratory animals published by the US National Institutes of
Health (NIH Publication no. 85-23, revised 1996). The protocol was
approved by the Animal Care and Use Committee of Tangdu Hospital;
all animals were maintained under specific pathogen-free conditions
prior to the experiment.
Cytometric bead array (CBA) for cytokine
assessment
Cytokine assessment was carried out using a mouse
Th1/Th2/TH17 cytokine kit (BD Biosciences, San Jose, CA, USA) for
simultaneous detection of seven cytokines (IL-2, -4, -6, -10, -17A,
TNF and IFN-γ). The kit performance was optimized for analysis of
physiologically relevant concentrations (pg/ml level) of specific
cytokine proteins in serum samples.
The CBA technique utilizes micro particles or beads
dyed with discrete fluorescence intensity. The dye incorporated in
the beads fluoresces strongly at 650 nm (measured as FL4 signals in
the BD FACSCalibur flow cytometer) when excited with an argon
laser. Each particle population of a given intensity represents a
discrete population for constructing an immunoassay for a single
analyte and each particle is covalently coupled with an antibody
directed against a specific analyte. When these capture beads for
different analytes are used as a mixture, the level of individual
analyte within samples can be measured simultaneously. Detection is
mediated by the binding of specific detection antibodies that are
directly conjugated with phycoerythrin (PE), thus providing an FL2
fluorescent signal on the appropriate bead. This signal is
proportional to the concentration of the analyte (Fig. 1).
Mouse serum (50 μl) and PE detection antibody
were incubated with capture bead reagent for 2 h in the dark at
room temperature. All unbound antibodies were washed (1.0 ml wash
buffer), re-suspended in 250 μl before acquiring data on the
BD FACSCalibur bio-analyzer (BD Biosciences).
By dedicated CBA analysis software, seven individual
cytokine standard curves were used to determine the concentration
of each analyte in the test sample; detection was calculated from
curve estimation for an average of ten assays using power fit and
R2>0.99 for all cytokines (Fig. 2).
Cell isolation
The isolation of single atrial myocyte from mice was
performed as described by Cho et al (13). Mice were sacrificed by cervical
dislocation, and the heart was quickly removed. The heart was
cannulated by a 24-gauge needle and then retrogradely perfused via
the aorta on a Langendorff apparatus. During coronary perfusion,
all perfusates were maintained at 37°C and equilibrated with 100%
O2. Initially the heart was perfused with normal Tyrode
solution for 2–3 min to clear the blood. The heart was then
perfused with Ca2+-free solution for 2 min. Finally, the
heart was perfused with enzyme solution for 15 min. The enzyme
solution contained 0.15 mg/ml collagenase (Sigma-Aldrich) in
Ca2+-free solution. Following perfusion with enzyme
solution, the atria were separated from the ventricles and chopped
into small pieces. Single cells were dissociated in storage medium
from these small pieces using a blunt-tip glass pipette.
Generation of reactive oxygen species
(ROS)
Generation of myocardial cell intracellular ROS was
measured using the fluorescent dye 2′,7′-dichlorofluorescein
diacetate (DCFH-DA) (Sigma-Aldrich). DCFH-DA is a non-polar and
non-fluorescent compound that can diffuse into the cell where it is
deacetylated by cellular esterases into a non-fluorescent polar
derivative DCFH that is impermeable to the cell membrane. DCFH is
rapidly oxidized to the highly fluorescent dichlorofluorescein
(DCF) in the presence of intracellular ROS and can be analyzed with
excitation 495 nm/emission 525 nm (measured in the FL1
channel).
Atrial myocytes were seeded in 96-well black tissue
culture microplates. The cells received DCFH-DA (2 μmol/l)
for 20 min at 37°C. Following removal of the DCFH-DA, the
microplate was incubated at 37°C for 1 h (14), and data was acquired on a BD
FACSCalibur bioanalyzer (BD Biosciences).
Atrial effective refractory periods
(AERPs)
The electrophysiology research was performed
according to Etzion et al (15). All mice were anesthetized with
intraperitoneal injection of sodium pentobarbital (50 mg/kg).
Following intubation and mechanical ventilation, the chest was
opened through the right fourth intercostal space, and two bipolar
screws-in pacing lead were fixed in proper sequence to four sites
in the right atrial as follows: appendage, high lateral, low
lateral and anterior walls. During ventilation, the arterial blood
gases were adjusted to between pH 7.35 and 7.45.
Electrograms were recorded in bipolar mode at a
filter setting of 30–500 Hz and stored digitally on a custom
acquisition system (Quinton Electrophysiology, Canada). A
programmed stimulator (Fukuda Denshi BC02A, Japan) was used to
deliver square-wave impulses of 1-ms duration. The AERPs were
measured at each of two sites at basic cycle lengths (100 ms) after
30 sec of continuous pacing to achieve a steady state; an atrial
extra-stimulus was introduced after every six drive beat, and all
stimuli were twice the diastolic threshold. The initial
extra-stimulus coupling interval was set at 30 ms, and the coupling
interval was set in steps of 5 ms increasing after every six beat
until the extra-stimulus resulted in atrial capture. The coupling
interval was then reduced by 5 ms and increased in steps of 1 ms
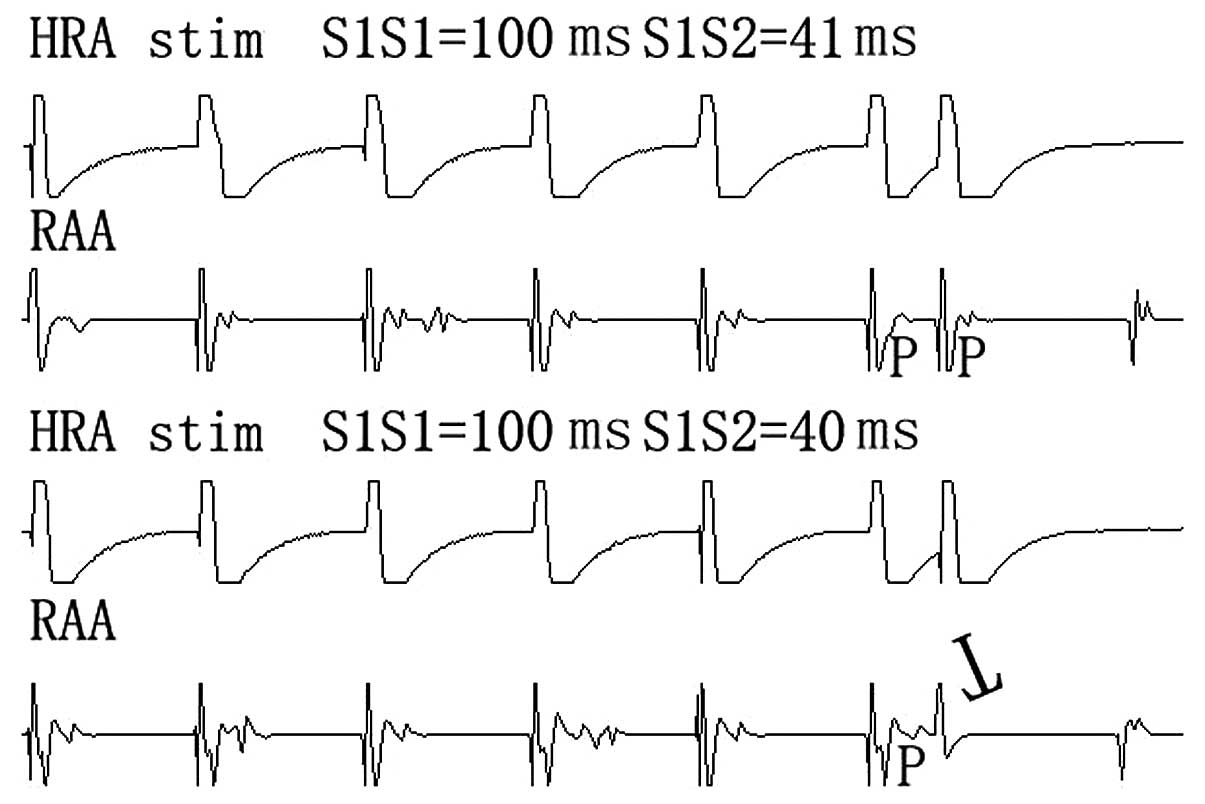
until atria captured again by extra-stimulus. AERP was defined as
the longest S1S2 coupling interval that failed to lead to atrial
capture (Fig. 3). Dispersion of
AERP (dAERP) equaled to maximum AERP minus minimum AERP among the
four sides.
Histological examination
Atrial cardiac muscle was dehydrated by storing in
70% ethanol, cleared and embedded in paraffin. Routine 4 μm
serial section was performed, and slides were dried in a 60°C oven.
For Masson staining, slides were submerged in Masson trichrome
solution (the Fourth Military Medical University Pathology
Laboratory, Xi’an, China) for 5 min, washed with 0.2% acetic acid
for 10 sec, followed by 5% phosphotungstic acid for 10 min, then
washed twice with 0.2% acetic acid solution, stained by 2% aniline
blue solution for 5 min, washed twice by 0.2% acetic acid,
dehydrated using gradient ethanol, cleared in xylene and sealed
using neutral Balsam.
Statistical analysis
All statistical analyses were performed using SPSS
software (version 18; SPSS Inc., Chicago, IL, USA). Data are
expressed as the means ± SD and compared by the two-sample
Student’s t-test. Differences were considered to be statistically
significant when P<0.05.
Results
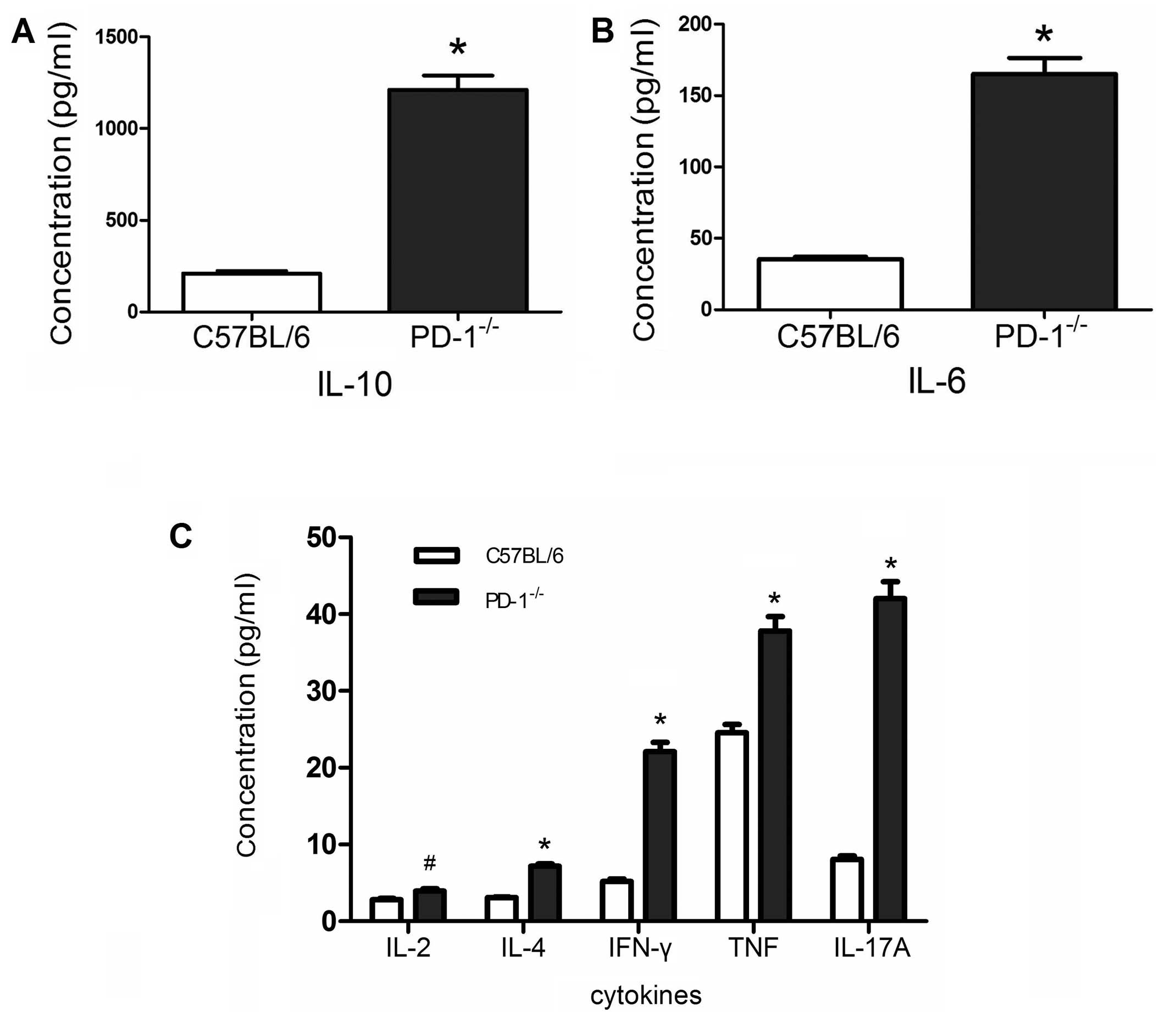
Inflammatory cytokines
There were significant differences in inflammatory
cytokine levels between the two groups. The inflammatory cytokine
levels of PD-1−/− mice were significantly higher than
those of the C57BL/6 mice. IL-10, 1212.35±300.42 vs. 208.8±57.25
pg/ml, P<0.001 (Fig. 4A);
IL-6, 165.21±42.49 vs. 35.47±6.04 pg/ml, P<0.001 (Fig. 4B); IL-2, 3.95±1.06 vs. 2.8±0.63
pg/ml, P<0.01; IL-4, 7.19±1.07 vs. 3.11±0.35 pg/ml, P<0.001;
IFN-γ, 22.1±4.72 vs. 5.17±1.27 pg/ml, P<0.001; TNF, 36.49±8.21
vs. 24.55±4.29 pg/ml, P<0.001; IL-17A, 42.03±8.53 vs. 8.05±1.76
pg/ml, P<0.001 (Fig. 4C).
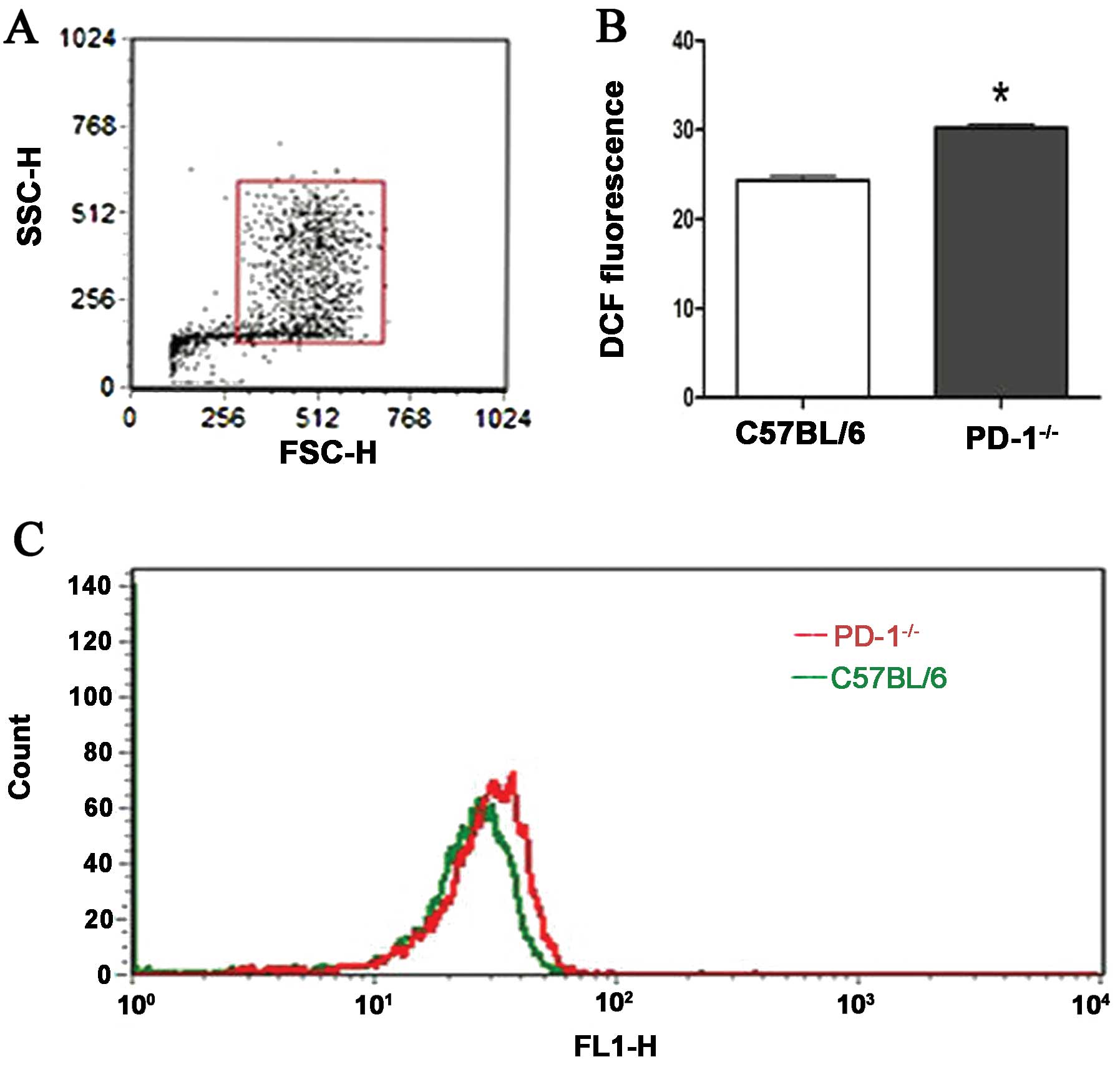
Oxidative stress
Intracellular ROS levels were measured by
fluorescent probe DCFH-DA (Fig.
5). There were significant differences in the mean DCF
fluorescence intensity between two groups (30.21±1.66 vs.
24.35±1.82, P<0.001).
AERPs
AERPs at each site were significantly shortened in
PD-1−/− mice compared with the C57BL/6 mice (Table I). We also found that the dAERP in
PD-1−/− mice was significantly increased, compared with
the C57BL/6 mice, 2.73±1.44 vs. 1.53±0.92 ms, P<0.05.
 | Table I.Comparison of AERP between the
C57BL/6 and the PD-1 deficient mice. |
Table I.
Comparison of AERP between the
C57BL/6 and the PD-1 deficient mice.
| AERPs BCL (ms) | C57BL/6 (n=15) | PD-1 deficient
(n=15) |
|---|
| RAA 100 | 42.47±0.74 | 41.73±1.03a |
| LRA 100 | 42.87±0.99 | 41.47±2.17a |
| HRA 100 | 42.80±0.94 | 41.87±1.19a |
| ARA 100 | 42.60±1.45 | 41.53±1.30a |
Pathological examination
Histological studies were performed to verify the
potential pathological substrate difference between the two groups.
Atrial myocardial fibrosis was detected in the PD-1−/−
group, but not in the C57BL/6 group (Fig. 6).
Discussion
PD-1−/− mice and
cardiovascular diseases
Programmed cell death-1 (PD-1) is an inhibitory
receptor in the CD28/CTLA-4 family and it can be inducibly
expressed on CD8 and CD4 T-cells, natural killer T-cells, B-cells
and activated monocytes (9,10,16). The ligands for PD-1 (PD-Ls) are
PD-L1 and PD-L2. PD-Ls pathways control the induction and
maintenance of peripheral T-cell tolerance (10). PD-1 deficiency enhances T-cell
activation and increases the inflammation level.
PD-1−/− mice were first reported by
Nishimura et al (17) in
1998. Experiments showed that the PD-1−/− mice with
different genetic backgrounds presented different types of diseases
and several were cardiovascular diseases. Nishimura et al
(18) found that some
2C-PD-1−/−H-2b/d mice died of a
Graft-Versus-Host-like (GVH-like) disease; the survivor mouse
myocardium presented inflammatory cell infiltration. Studies also
indicated that the heart of BALB/c-PD-1−/− mice showed
various degrees of inflammation with marked deposition of immune
complex on the surface of cardiomyocytes, and their sera contained
high titer auto-antibodies against cardiac troponin I (cTnI)
(19–21). In addition, MRL-PD-1−/−
mice developed fatal myocarditis (22), massive infiltration of
CD4+ and CD8+ T-cells and myeloid cells were
found in their hearts concomitant with the production of high-titer
auto-antibodies against cardiac myosin (22).
These studies indicated that the PD-1−/−
mice are closely associated with cardiovascular diseases; the basic
mechanism is the high system inflammation level resulting in the
inflammatory cell infiltration and/or the generation of
auto-antibodies against cardiac tissue.
In the present study, we found that the
PD-1−/− group presented atrial electricity remodeling
(shorter AERP and increased dAERP) and structural remodeling
(atrial myocardial fibrosis). It is generally accepted that AF
results from the presence of multiple reentrant circuits
originating in the atria (23),
and atrial remodeling increases the probability of generating
multiple atrial wavelets by dispersion of refractoriness and rapid
atrial activation (4). Our
findings strongly suggest that the PD-1−/− mice are more
likely to develop AF.
Inflammatory cytokines, reactive oxygen
species (ROS) and atrial remodeling
Previous studies indicated that inflammation may
lead to ‘atrial myocarditis’; atrial electrical and structural
remodeling resulting from it subsequently led to the initiation and
maintenance of AF (24,25).
Other studies indicated that T lymphocytes
participated in the cardiac remodeling (26,27) and Th1 and Th2 responses are
involved in the most fiber proliferative disorders. Th1 cytokines
(IFN-γ, TNF and IL-2) are considered to be involved in the
initiation phase of fibrosis and the Th2 cytokines (IL-4, -6 and
-10) during the latter stages (28). Th17 cells were characterized by
the production of IL-17 as signature cytokines and also participate
in the process of cardiac remodeling. Studies have shown that IL-17
could promote the production of collagen in cardiac fibroblasts
(29,30).
Inflammation is also involved in the process of
electrical remodeling. Atrial inflammation following cardiac
surgery could influence the electrical properties of atrial tissue,
and the level of atrial inflammation was associated with a
proportional increase in the inhomogeneity of atrial conduction and
AF duration (31). In this
process, inflammation cytokines may play an important role.
Previous studies found that overexpression of TNF could
downregulate connexin 40 (32),
Kv4.2, Kv4.3 and Kv1.5 and KChIP-2 (33,34). Downregulation of connexin 40 slows
down the conduction and increases the susceptibility of atrial
arrhythmias, decreases potassium channel-interacting protein,
reduces Ito (transient outward K current), IK,
slow1, and IK, slow2, which attenuate the effective
refractory period (ERP).
In the present study, although two groups of mice
lived in the same environment and were fed the same food and water,
the pathogen existing in the environment including microbe,
parasites and virus aggravated the inflammation process in
PD-1−/− mice. Therefore, the PD-1−/− group
presented higher levels of inflammatory cytokines.
Oxidative stress refers to the total burden of
potentially harmful ROS formed during cellular metabolism. High
oxidative stress levels may be closely related to atrial electrical
remodeling. Carnes et al (35) found that atrial tachy-pacing
provoked increased protein nitration indicating enhanced oxidative
stress in a dog model and the process was accompanied by a decrease
in the ERP. It also showed that pretreatment of the animals with
oral vitamin C (a water-soluble antioxidant) attenuated the ERP
shortening (36). In addition,
another study indicated that ROS also played an important role in
cardiac remodeling by angiotensin II (37).
In several pathophysiological conditions,
inflammation can augment oxidative stress and vice versa.
Therefore, some people speculate that inflammation and oxidative
stress cooperate at some level facilitating atrial remodeling
(25,38).
In the present study, we found that the
PD-1−/− group presented higher levels of inflammatory
cytokines compared with the C57BL/6 group. At the same time,
intracellular ROS of cardiac atrial myocytes were also higher. They
may be interrelated and result in shorter AERPs, increased dAERP
and myocardial fibrosis.
Our study is the first to prove that PD-1 deficiency
results in atrial electrical and structural remodeling, raising the
vulnerability of C57BL/6 mice to AF. However, it should be noted
that this was an animal study with a special gene background and
should not be over-interpreted. A clinical trial is required to
confirm that the systemic inflammation causes the onset of AF in
nonoperative settings. Sata et al (39) observed 15 patients with paroxysmal
AF and 11 patients with normal sinus rhythm. CRP, IL-6 and TNF-α
were measured at baseline, 24 h, and two weeks after cardioversion,
and they found that CRP, IL-6 and TNF-α were greater in the AF
group and did not normalize two weeks after cardioversion. The
study suggested that inflammation may be an independent risk factor
for AF, however their sample size was limited.
Tang et al (40) hypothesized that Chlamydia
pneumoniae infection was a possible cause of AF by initiating
inflammation response. They found that there was a close
epidemiological and pathological relationship between both sides,
but the hypothesis remains to be confirmed by clinical experiments.
Bigger sample clinical trials need to be performed to validate the
role of systemic inflammation in the onset of nonoperative AF.
In conclusion, PD-1 deficiency resulted in a
significant increase in inflammatory cytokines and intracellular
ROS levels in C57BL/6 mice. Higher inflammatory cytokines and
intracellular ROS levels led to atrial remodeling, and, due to
atrial remodeling, the C57BL/6 PD-1−/− mice may be more
susceptible to AF.
Acknowledgements
The authors thank Professor Tasuku
Honjo for kindly providing us with the C57BL/6 PD-1−/−
mice. This study was supported by the National Natural Science
Foundation of China (no. 81070161).
References
|
1.
|
Page RL: Clinical practice. Newly
diagnosed atrial fibrillation. N Engl J Med. 351:2408–2416. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Benjamin EJ, Wolf PA, D’Agostino RB,
Silbershatz H, Kannel WB and Levy D: Impact of atrial fibrillation
on the risk of death: the Framingham Heart Study. Circulation.
946–952. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Nattel S, Burstein B and Dobrev D: Atrial
remodeling and atrial fibrillation: mechanisms and implications.
Circ Arrhythm Electrophysiol. 1:62–73. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Sharma D, Li G, Xu G, Liu Y and Xu Y:
Atrial remodeling in atrial fibrillation and some related
microRNAs. Cardiology. 120:111–121. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Bruins P, te Velthuis H, Yazdanbakhsh AP,
et al: Activation of the complement system during and after
cardiopulmonary bypass surgery: postsurgery activation involves
C-reactive protein and is associated with postoperative arrhythmia.
Circulation. 96:3542–3548. 1997. View Article : Google Scholar
|
|
6.
|
Gabay C and Kushner I: Acute-phase
proteins and other systemic responses to inflammation. N Engl J
Med. 340:448–454. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Ommen SR, Odell JA and Stanton MS: Atrial
arrhythmias after cardiothoracic surgery. N Engl J Med.
336:1429–1434. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Engelmann MD and Svendsen JH: Inflammation
in the genesis and perpetuation of atrial fibrillation. Eur Heart
J. 26:2083–2092. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Greenwald RJ, Freeman GJ and Sharpe AH:
The B7 family revisited. Annu Rev Immunol. 23:515–548. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Okazaki T and Honjo T: The PD-1-PD-L
pathway in immunological tolerance. Trends Immunol. 27:195–201.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Iwai Y, Terawaki S, Ikegawa M, Okazaki T
and Honjo T: PD-1 inhibits antiviral immunity at the effector phase
in the liver. J Exp Med. 198:39–50. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Carter LL, Leach MW, Azoitei ML, et al:
PD-1/PD-L1, but not PD-1/PD-L2, interactions regulate the severity
of experimental autoimmune encephalomyelitis. J Neuroimmunol.
182:124–134. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Cho H, Nam GB, Lee SH, Earm YE and Ho WK:
Phosphatidylinositol 4,5-bisphosphate is acting as a signal
molecule in alpha(1)-adrenergic pathway via the modulation of
acetylcholine-activated K(+) channels in mouse atrial myocytes. J
Biol Chem. 276:159–164. 2001.PubMed/NCBI
|
|
14.
|
Golan O, Issan Y, Isak A, Leipziger J,
Robaye B and Shainberg A: Extracellular nucleotide derivatives
protect cardiomyocytes against hypoxic stress. Biochem Pharmacol.
81:1219–1227. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Etzion Y, Mor M, Shalev A, et al: New
insights into the atrial electrophysiology of rodents using a novel
modality: the miniature-bipolar hook electrode. Am J Physiol Heart
Circ Physiol. 295:H1460–H1469. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Chen L: Co-inhibitory molecules of the
B7-CD28 family in the control of T-cell immunity. Nat Rev Immunol.
4:336–347. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Nishimura H, Minato N, Nakano T and Honjo
T: Immunological studies on PD-1 deficient mice: implication of
PD-1 as a negative regulator for B cell responses. Int Immunol.
10:1563–1572. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Nishimura H, Nose M, Hiai H, Minato N and
Honjo T: Development of lupus-like autoimmune diseases by
disruption of the PD-1 gene encoding an ITIM motif-carrying
immunoreceptor. Immunity. 11:141–151. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Nishimura H, Okazaki T, Tanaka Y, et al:
Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice.
Science. 291:319–322. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Okazaki T and Honjo T: Pathogenic roles of
cardiac autoantibodies in dilated cardiomyopathy. Trends Mol Med.
11:322–326. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Okazaki T, Tanaka Y, Nishio R, et al:
Autoantibodies against cardiac troponin I are responsible for
dilated cardiomyopathy in PD-1-deficient mice. Nat Med.
9:1477–1483. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Wang J, Okazaki IM, Yoshida T, et al: PD-1
deficiency results in the development of fatal myocarditis in MRL
mice. Int Immunol. 22:443–452. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Falk RH: Atrial fibrillation. N Engl J
Med. 344:1067–1078. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Aviles RJ, Martin DO, Apperson-Hansen C,
et al: Inflammation as a risk factor for atrial fibrillation.
Circulation. 108:3006–3010. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Korantzopoulos P, Kolettis T, Siogas K and
Goudevenos J: Atrial fibrillation and electrical remodeling: the
potential role of inflammation and oxidative stress. Med Sci Monit.
9:225–229. 2003.PubMed/NCBI
|
|
26.
|
Jakubzick C, Choi ES, Kunkel SL, Joshi BH,
Puri RK and Hogaboam CM: Impact of interleukin-13 responsiveness on
the synthetic and proliferative properties of Th1- and Th2-type
pulmonary granuloma fibroblasts. Am J Pathol. 162:1475–1486. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Liu W, Feng W, Wang F, et al:
Adenovirus-mediated ICOSIg gene transfer alleviates cardiac
remodeling in experimental autoimmune myocarditis. Immunol Cell
Biol. 86:659–665. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Yu Q, Horak K and Larson DF: Role of T
lymphocytes in hypertension-induced cardiac extracellular matrix
remodeling. Hypertension. 48:98–104. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Cortez DM, Feldman MD, Mummidi S, et al:
IL-17 stimulates MMP-1 expression in primary human cardiac
fibroblasts via p38 MAPK- and ERK1/2-dependent C/EBP-beta,
NF-kappaB, and AP-1 activation. Am J Physiol Heart Circ Physiol.
293:H3356–H3365. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Feng W, Li W, Liu W, Wang F, Li Y and Yan
W: IL-17 induces myocardial fibrosis and enhances RANKL/OPG and
MMP/TIMP signaling in isoproterenol-induced heart failure. Exp Mol
Pathol. 87:212–218. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Ishii Y, Schuessler RB, Gaynor SL, et al:
Inflammation of atrium after cardiac surgery is associated with
inhomogeneity of atrial conduction and atrial fibrillation.
Circulation. 111:2881–2888. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Sawaya SE, Rajawat YS, Rami TG, et al:
Downregulation of connexin40 and increased prevalence of atrial
arrhythmias in transgenic mice with cardiac-restricted
overexpression of tumor necrosis factor. Am J Physiol Heart Circ
Physiol. 292:1561–1567. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Kawada H, Niwano S, Niwano H, et al: Tumor
necrosis factor-alpha downregulates the voltage gated outward
K+ current in cultured neonatal rat cardiomyocytes: a
possible cause of electrical remodeling in diseased hearts. Circ J.
70:605–609. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Petkova-Kirova PS, Gursoy E, Mehdi H,
McTiernan CF, London B and Salama G: Electrical remodeling of
cardiac myocytes from mice with heart failure due to the
overexpression of tumor necrosis factor-alpha. Am J Physiol Heart
Circ Physiol. 290:2098–2107. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Carnes CA, Chung MK, Nakayama T, et al:
Ascorbate attenuates atrial pacing-induced peroxynitrite formation
and electrical remodeling and decreases the incidence of
postoperative atrial fibrillation. Circ Res. 89:e32–e38. 2001.
View Article : Google Scholar
|
|
36.
|
Galaris D and Korantzopoulos P: On the
molecular mechanism of metmyoglobin-catalyzed reduction of hydrogen
peroxide by ascorbate. Free Radic Biol Med. 22:657–667. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Wu S, Gao J, Ohlemeyer C, et al:
Activation of AP-1 through reactive oxygen species by angiotensin
II in rat cardiomyocytes. Free Radic Biol Med. 39:1601–1610. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Korantzopoulos P, Galaris D, Papaioannides
D and Kokkoris S: C-reactive protein and oxidative stress in atrial
fibrillation. Int J Cardiol. 88:103–104. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Sata N, Hamada N, Horinouchi T, et al:
C-reactive protein and atrial fibrillation. Is inflammation a
consequence or a cause of atrial fibrillation? Jpn Heart J.
45:441–445. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Tang RB, Dong JZ, Liu XP and Ma CS:
Inflammation and atrial fibrillation: is Chlamydia
pneumoniae a candidate pathogen of atrial fibrillation. Med
Hypotheses. 67:462–466. 2006.PubMed/NCBI
|