Introduction
Obesity is a common metabolic disorder that is
rapidly becoming a global public health problem. It is associated
with an increased risk of several life-threatening diseases such as
type 2 diabetes, cardiovascular diseases, and multiple types of
cancer and may represent a leading preventable cause of death
(1,2). Obesity is characterized by
pathologic growth of adipose tissues to accommodate excess energy
intake through an increase in adipocyte number as a result of
increased proliferation and differentiation (hyperplasia) and
adipocyte size (hypertrophy) (3).
Thus, targeting adipocyte biology via inhibition of adipocyte
differentiation (adipogenesis) from fibroblastic preadipocytes into
mature adipocytes and induction of apoptosis in adipose tissues, as
well as identifying potential factors that regulate these processes
are of great importance in the prevention and treatment of obesity
(4).
The 3T3-L1 cell line is a well-established and
widely used in vitro model of obesity for studying adipocyte
differentiation. The differentiation process follows a precise
ordered and temporal series of events regulated by a number of
transcription factors and multiple signaling pathways for the
development of the adipocyte phenotype (5,6).
The hormonal cocktail consisting of insulin, dexamethasone, and
3-isobutyl-1-methylxanthine, activates an adipogenic program, which
occurs in well-defined phases. The stimulated cells immediately
re-enter the cell cycle and progress through at least two
cell-cycle divisions, a phase often referred to as mitotic clonal
expansion (MCE), a process essential for adipocyte differentiation
(7). After MCE, the cells
permanently withdraw from the cell cycle, begin to accumulate
lipid, and undergo terminal differentiation into mature adipocytes.
Induction of adipogenesis by hormones leads to the expression of
specific adipogenic transcription factors CCAAT/enhancer binding
proteins (C/EBPs), such as C/EBPβ, C/EBPδ, adipocyte determination
and differentiation-dependent factor 1/sterol regulatory
element-binding protein (ADD1/SREBP1), as well as cell cycle
regulators that together facilitate expression and activity of
peroxisome proliferator-activated receptor-γ (PPARγ) and C/EBPα
(5,6,8,9).
C/EBPα and PPARγ then coordinately drive expression of
adipocyte-specific genes such as adipocyte fatty acid-binding
protein 2 (aP2), fatty acid synthase (FAS), acetyl-CoA carboxylase
(ACC), lipoprotein lipase (LPL) and ATP citrate lyase (ACL) and
cluster of differentiation (CD) 36, many of which characterize the
final stages of differentiation (5,6,8,9).
5′-AMP-activated protein kinase (AMPK) is a major
regulator of cellular energy homeostasis which coordinates
metabolic pathways in order to balance nutrient supply with energy
demand. AMPK is activated by a variety of cellular stresses that
decrease ATP generation including metabolic poisons as well as
pathologic cues such as nutrient starvation, ischemia and hypoxia
(10–12). Under these conditions, the
activated AMPK phosphorylates many substrates that turn on
alternative catabolic pathways to generate more ATP. Overall, AMPK
activation leads to energy preservation for cell survival at the
expense of growth and proliferation via long term transcriptional
control of key players of various metabolic pathways.
Interestingly, knockout of AMPKα1 or AMPKα2 subunits led to the
development of obesity and insulin resistance in mice (13,14). In addition, AMPK was proposed to
be involved in the process of adipocyte differentiation, although
its role in adipogenesis is not entirely understood. Several
AMPK-activating compounds were found to have anti-adipogenic
effects via MCE inhibition and downregulation of the adipogenic
transcriptional pathways (15–18). Since AMPK is involved in the
regulation of a variety of metabolic processes and plays a key role
in glucose and lipid homeostasis, it is a promising target of
anti-obesity agents.
Recently, our laboratory has identified several
novel small-molecule compounds with anticancer properties. The lead
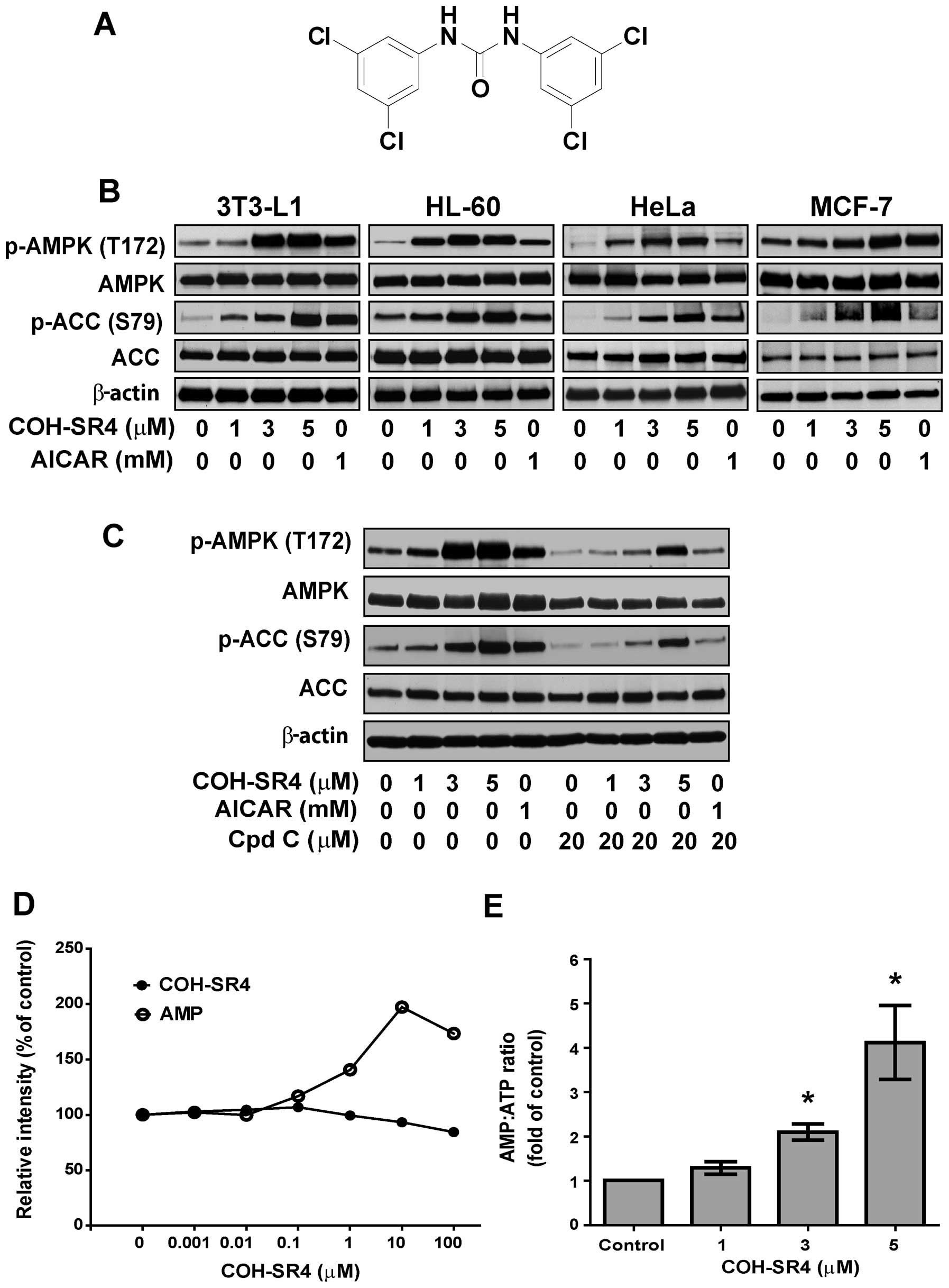
compound COH-SR4 (Fig. 1A) showed
potent anti-proliferative activities against leukemia, melanoma,
breast and lung cancers both in vitro and in vivo
(19,20, unpublished data). Since cancer and
obesity share several key metabolic regulators, we hypothesized
that COH-SR4 would also exhibit anti-adipogenic properties. In this
study, we investigated whether COH-SR4 prevents adipocyte
differentiation, and examined its inhibitory mechanisms on 3T3-L1
adipogenesis, specifically AMPK activation, and associated
downstream pathways.
Materials and methods
Chemicals, antibodies and reagents
COH-SR4 was synthesized according to a previously
validated protocol by Dr Christopher Lincoln at the Chemical GMP
Synthesis Facility, Translational Medicinal Chemistry Laboratory,
Beckman Research Institute of the City of Hope (20). The purity of the compound was
confirmed by 1H-NMR, 13C-NMR and HRMS-ESI
analyses. COH-SR4 was dissolved in DMSO at 25 mM stock solution.
Antibodies against CDK2, cyclin E1, p27Kip1, FAS, ACL,
aP2, PPARγ, AMPKα, phospho-AMPKα (T172), ACC, phospho-ACC (S79),
raptor, phospho-raptor (S792), TCS2, phospho-TSC2 (S1387),
phospho-Akt (S473), Akt, S6K1, phospho-S6K1 (T389), phospho-4E-BP1
(S65) and 4E-BP were obtained from Cell Signaling Technology
(Danvers, MA, USA). C/EBPα (p42) and SREBP1 antibodies were from
EMD Millipore (Billerica, MA, USA), while S6K was obtained Santa
Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). The AdipoRed™
Adipogenesis Assay, XTT Proliferation Assay, CycLex AMPK Kinase
Assay, and the colorimetric LDH Cytotoxicity Assay Kits were
purchased from Lonza (Walkersville, MD, USA), American Type Culture
Collection (ATCC, Manassas, VA, USA), CycLex Co., Ltd., (Nagano,
Japan), and BioVision (Milpitas, CA, USA), respectively. The
recombinant active human AMPK (α1β1γ1) was from SignalChem
(Richmond, BC, Canada). AMPKα1, AMPKα2 and control siRNA were
obtained from Invitrogen (San Diego, CA, USA) and Santa Cruz
Biotechnology, Inc. All other chemicals and reagents were purchased
from Sigma-Aldrich (St. Louis, MO, USA).
Cell culture and adipocyte
differentiation
3T3-L1 preadipocytes were obtained from Zen-Bio,
Inc. (Research Triangle Park, NC, USA) and were cultured in
Dulbecco’s modified Eagle’s medium (DMEM) containing 10% (v/v)
fetal bovine serum (FBS) (ATCC) and antibiotics at 37°C in a
humidified atmosphere of 5% CO2. Cells were subcultured
every 2–3 days at ~80–90% confluency. Depending on experiments,
3T3-L1 preadipocytes were plated in 6-, 12-, 24- or 48-well culture
plates with the culture medium at a density that allowed them to
reach confluence in 2 days. Two days after confluency (day 0),
growth-arrested 3T3-L1 preadipocytes were incubated in
differentiation medium (DM) cocktail containing 10 μg/ml insulin, 1
mM dexamethasone and 0.5 mM isobutyl-methylxanthine together with
or without COH-SR4. On day 2, the medium was replaced with DMEM
containing 10 μg/ml insulin only. Cells were then cultured in
normal medium from day 4 to 7. COH-SR4 was added in every
replacement of the medium except on experiments where it was added
only on days 0–2, 2–4 or 4–7. On day 7, fully differentiated cells
were harvested for protein analysis or stained with Oil Red O dye
or AdipoRed reagent.
Oil Red O staining
Cells on day 7 were washed twice with PBS, fixed
with 10% neutral buffered formalin for 1 h at room temperature,
washed with PBS and then dried completely. The fixed cells were
stained with Oil Red O in an isopropanol/distilled water (6:4)
solution for 30 min at room temperature and then washed twice with
distilled water. The stained lipid droplets were observed, and
microscopic images were obtained from randomly selected fields
under a phase contrast microscope (AX70; Olympus, Tokyo, Japan)
equipped with a digital camera and processed using ImagePro Plus
software (Media Cybernetics, Silver Spring, MD, USA).
Triglyceride assay
Quantification of intracellular triglyceride content
was measured on day 7 using a commercially available kit (AdipoRed
Assay Reagent; Lonza) according to the manufacturer’s directions.
Fluorescence was measured with an excitation wavelength of 485 nm
and emission wavelength of 572 nm.
AMPK activity assay
AMPK activity was determined using the CycLex AMPK
Kinase Assay kit according to the manufacturer’s instructions.
Briefly, recombinant active human AMPK (α1β1γ1) was incubated with
the indicated concentrations of AMP or COH-SR4 for 30 min at 30°C
in a pre-coated plate with a substrate peptide corresponding to
mouse insulin receptor substrate-1 (IRS-1). AMPK activity was
measured by monitoring the phosphorylation of Ser-789 in IRS-1
using an anti-mouse phospho-Ser-789 IRS-1 monoclonal antibody and
peroxidase-coupled anti-mouse IgG antibody. Conversion of the
chromogenic substrate tetramethylbenzidine was quantified by
absorbance measurement at 450 nm.
Cell proliferation assay
Cell proliferation was determined using
2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide
(XTT) assay according to the manufacturer’s instructions (ATCC).
Briefly, cells were seeded on 48-well plates and incubated with
test compounds for the indicated time periods. Activated XTT
solution was then added to each well of the plate. After a 4-h
incubation at 37°C, the absorbance was measured at 475 nm using a
reference wavelength of 630 nm. In addition to the XTT assay, cell
numbers were also estimated. Confluent 3T3-L1 preadipocytes were
grown in 6-well culture plate and incubated with either DMSO
vehicle or COH-SR4 (1–5 μM) in DM cocktail medium for 1–2 days. The
cells were trypsinized, harvested and counted using a cell counter
(Z1 Coulter Counter; Beckman Coulter, Inc., Brea, CA, USA).
Cytotoxicity assay
3T3-L1 preadipocytes were grown in a 48-well culture
plate in DMEM with 10% FBS and antibiotics and incubated with
either DMSO vehicle or COH-SR4 (1–5 μM). After 48 h, the released
lactate dehydrogenase (LDH) into the culture supernatant was
measured by the colorimetric LDH Cytotoxicity Assay (BioVision)
according to the manufacturer’s instructions.
Hoechst 33258 staining
After experimental treatment, cell cultures were
fixed with 4% paraformaldehyde, washed twice with PBS, and stained
with Hoechst 33258 (5 μg/ml) for 5 min in the dark, then followed
by extensive PBS washes. Nuclear staining was examined under a
fluorescence microscope and images were captured using ImagePro
Plus software. Cells that exhibited reduced nuclear size, chromatin
condensation, intense fluorescence, and nuclear fragmentation were
considered as apoptotic.
Nucleotide extraction and
measurement
Cellular levels of AMP, ATP and ADP were measured by
UV-HPLC. In brief, 3T3-L1 preadipocytes were treated with COH-SR4
for 60 min. After the incubation period, cells were washed once
with ice cold PBS, centrifuged, and 50 μl of PBS and 50 μl of 6%
TCA was added to the cell pellet. After thorough mixing, the sample
was placed in a 4° sonicator for 10 min and then centrifuged at
14,000 rpm for 10 min. A 100 μl aliquot of supernatant was removed
and mixed with 100 μl of deionized water. Finally, 18.5 μl of 1 M
KOH, 30 μl of 150 mM KH2PO4/150 mM KCl
solution (pH 6.0) and 0.5% acetonitrile was added. The final
mixture was centrifuged at 14,000 rpm for 5 min. The supernatant
was then transferred to an auto-injector vial and 10 μl was
injected into a Thermo ODS Hypersil C18 column (3 μm, 150×4.6 mm)
(Thermo Scientific, Rockford, IL, USA). Isocratic separation was
achieved using a mobile phase consisting of 150 mM
KH2PO4, 150 mM KCl (pH 6.0), and 0.5%
acetonitrile. The flow rate was set to 0.8 ml/min and a total run
time of 20 min. UV-HPLC analysis of ATP, ADP and AMP was performed
using a Shimadzu SCL-10AVP system (Shimadzu Scientific Instruments,
Columbia, MD, USA). Nucleotides were detected by their absorbance
at 260 nm and compared with the elution position of standards.
Retention times were 3.27, 4.68 and 4.87 min for ADP, AMP and ATP,
respectively.
Cell cycle analysis by flow
cytometry
Cells were harvested and fixed with 70% ethanol at
4°C for 24 h. After removal of ethanol by washing with ice cold
PBS, cells were resuspended in 1 ml propidium iodide (PI) staining
solution (4% PI, 2 μg/ml RNase in PBS) and incubated for 30 min at
37°C. Fluorescence-activated cell sorting (FACS) analysis was
performed using a CyAn ADP cytometer (Beckman Coulter, Inc.) as
previously described (19).
Transient transfection with small
interfering RNA (siRNA)
To knockdown the endogenous AMPK, 3T3-L1 cells were
transiently transfected with 100 nM mouse siRNAs targeting AMPKα1
and AMPKα2 or with the non-silencing control siRNA using
Lipofectamine™ RNAiMAX (Invitrogen) in culture medium without
antibiotics according to the manufacturer’s recommendations. After
24 h, the medium was replaced with DM with or without COH-SR4, and
cell lysates were prepared 24 h or 7 days after drug treatment. In
separate experiments, AMPKα1/α2 siRNA-treated cells were incubated
with or without COH-SR4 in DM for 48 h (day 0–2). The cells were
then incubated in DM+ insulin for 2 days and normal DMEM/10% FBS
for 3 days. At day 7, differentiated cells were assayed for Oil Red
O and AdipoRed staining. siRNA sequences used in the present study
are available upon request.
Protein extraction and western
blotting
3T3-L1 cells were harvested and washed twice with
ice cold PBS. Total cytosolic proteins were extracted with cell
lysis buffer (Cell Signaling Technology), and the protein
concentration was determined using the DC Protein Assay Kit
(Bio-Rad, Hercules, CA, USA). Protein electrophoresis and western
blotting were performed as described previously (19). Equal loading of proteins was
confirmed by stripping and restaining the membranes with β-actin
antibodies.
Statistical analyses
Statistical analyses were performed using Prism
software (GraphPad, San Diego, CA, USA). Data are presented as
means ± SEM. Comparison of two groups was analyzed by the Student’s
t-test. For multiple group comparison, data were first analyzed by
one-way ANOVA followed by Bonferonni’s test. Values of P<0.05
were considered to indicate statistically significant results.
Results
COH-SR4 activates AMPK
First, we investigated whether COH-SR4 activates
AMPK in different types of cells. COH-SR4 treatment resulted in a
dose-dependent increase in the phosphorylation of AMPK and its
substrate ACC in 3T3-L1 preadipocytes, as well as in cancer cells
such as HL-60, HeLa, MCF-7 (Fig.
1B), A-549, H-358 and H-520 (data not shown), showing that this
compound can activate AMPK in a wide variety of cells, similarly
with AICAR, a known AMPK agonist. Pre-treatment with the AMPK
inhibitor compound C blocked the ability of COH-SR4 to activate
AMPK in 3T3-L1 cells, as indicated by the reduction in AMPK and ACC
phosphorylation (Fig. 1C). In
vitro assays with purified AMPK revealed that COH-SR4 is not a
direct activator of the kinase (Fig.
1D), implying that this compound acts upstream from AMPK
activation. Indeed, we observed that COH-SR4 significantly
decreased the intracellular ATP and increased the AMP:ATP ratio
(Fig. 1E) in 3T3-L1 cells,
further indicating that this compound indirectly activates
AMPK.
COH-SR4 inhibits 3T3-L1 adipocyte
differentiation
We next examined the effects of COH-SR4 on adipocyte
differentiation. 3T3-L1 preadipocytes were stimulated to
differentiate in the presence of increasing concentrations of
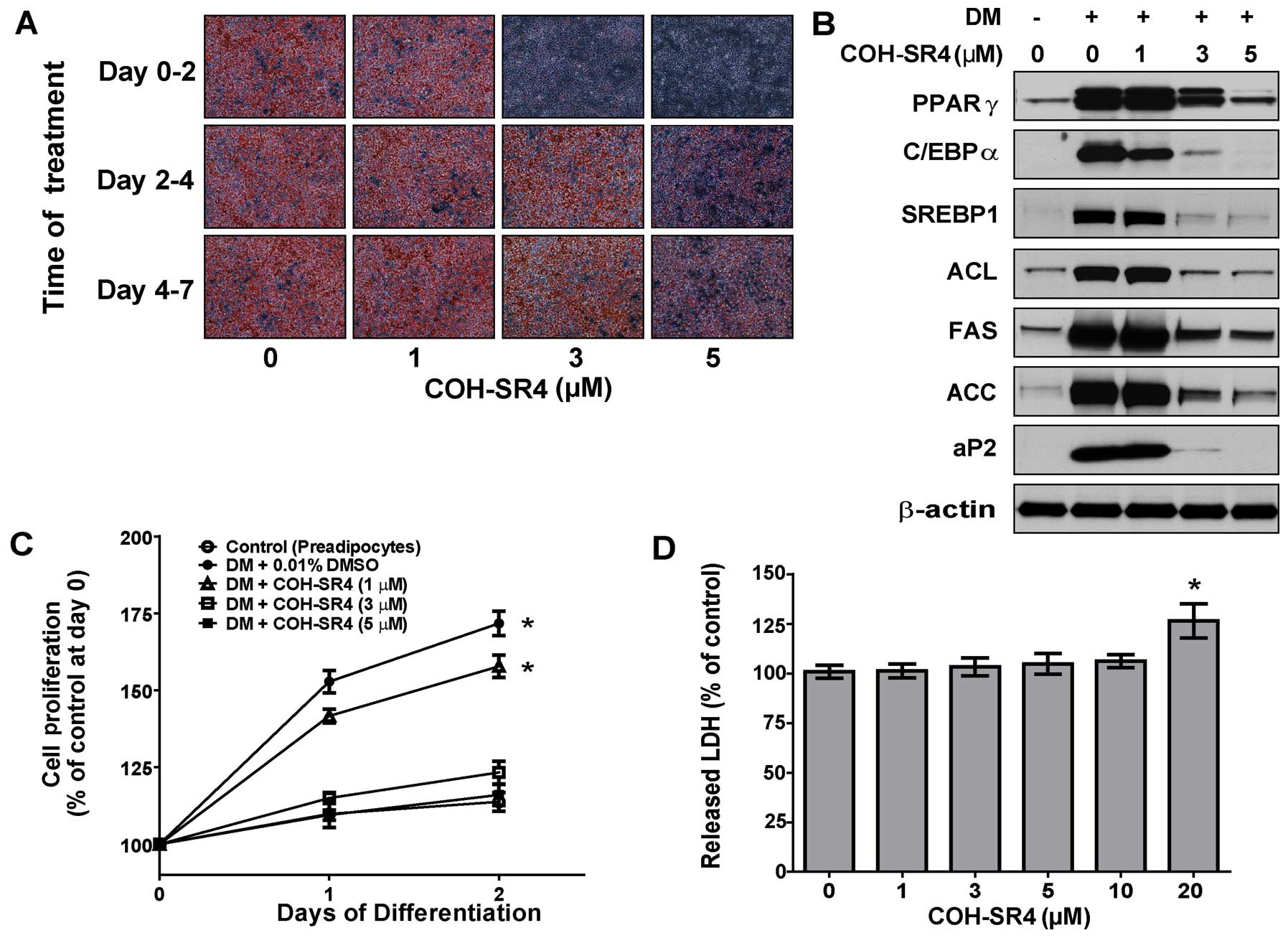
COH-SR4 (1–5 μM). By day 7 of differentiation, COH-SR4
dose-dependently inhibited lipid accumulation as revealed by Oil
Red O staining (Fig. 2A). In
particular, treatment with either 3 or 5 μM COH-SR4 almost
completely inhibited the formation of lipid droplets in the
adipocytes and the cells appeared morphologically similar to the
untreated preadipocyte control. The results of the Oil Red O
staining were confirmed by quantitative analyses of intracellular
triglyceride content. We estimated that COH-SR4 inhibited lipid
accumulation in 3T3-L1 adipocytes with an apparent IC50
of ~1.5 μM (Fig. 2B). These
results demonstrated that COH-SR4 is a potent inhibitor of
adipocyte differentiation.
Adipogenesis is a highly regulated process requiring
coordinated expression and activation of key transcription factors
which include C/EBPs, PPARγ, SREBPs, as well as adipogenic genes
such as FAS and aP2 (6,9,10).
Using western blotting, we confirmed the elevated expression of
these adipogenic effectors when 3T3-L1 cells were allowed to
differentiate for 7 days. Treatments with COH-SR4 dose-dependently
decreased the protein levels of PPARγ, C/EBPα, SREBP1, FAS and aP2,
as well as ACL and ACC, key proteins involved in fatty acid
synthesis (Fig. 2C). Taken
together, these data indicate that COH-SR4 effectively inhibited
the expression of key adipogenesis-related proteins, thereby
confirming its anti-adipogenic properties.
Since we established that COH-SR4 activates AMPK in
3T3-L1 preadipocytes, we next investigated whether AMPK is
activated by this compound during 3T3-L1 differentiation. Confluent
3T3-L1 preadipocytes were allowed to differentiate in the presence
of various concentrations of COH-SR4 for 7 days, and the proteins
were analyzed by western blotting (Fig. 2D). Treatment with COH-SR4 resulted
in a time- and dose-dependent increase in phosphorylated AMPK. AMPK
was activated as early as 1 h (data not shown) but the strongest
activation mainly occurred during days 1 to 3. Even though AMPK
activation increased gradually during the differentiation in the
absence of the test compound, COH-SR4 treatment rendered AMPK able
to be in a fully activated state during the early differentiation
phase, which was detrimental to adipocyte differentiation.
COH-SR4 inhibits MCE during the early
stage of adipogenesis without inducing cytotoxicity and
apoptosis
3T3-L1 cells were treated at three different phases
during the differentiation process: the proliferation phase (day
0–2), the differentiation phase (day 2–4) and the terminal
differentiation phase (day 4–7). The effects of COH-SR4 on
adipocyte differentiation were assessed on day 7 by Oil Red O
staining. Treatment with COH-SR4 during the proliferation phase
(days 0–2) resulted in a nearly complete inhibition of lipid
accumulation almost similar to that of a continuous (0–7 day)
treatment, whereas the compound had lesser inhibitory effects when
the cells were treated on days 2–4 only or 4–7 only (Fig. 3A). Moreover, the expression of
adipogenic transcription factors and protein markers associated
with adipogenesis were also suppressed when COH-SR4 treatment was
carried out on days 0–2 of differentiation (Fig. 3B). These observations were almost
similar to what was previously observed using day 0–7 treatment
(Fig. 2C). Therefore, these data
showed that the effects of COH-SR4 that caused inhibition of
adipocyte differentiation acted during the proliferation phase and
were less effective at the later phases of differentiation.
Since MCE is crucial for adipocyte differentiation,
we next determined whether COH-SR4 affects preadipocyte MCE during
the proliferation phase. 3T3-L1 cells were treated with increasing
concentrations of COH-SR4 in DM cocktail medium and after 1–2 days,
cell proliferation was estimated using a cell counter. While the
number of DM-treated control cells increased 175% from day 0 to 2,
the number of 3T3-L1 cells treated with COH-SR4 decreased
significantly in a dose-dependent fashion (Fig. 3C). However, the LDH cytotoxicity
assay in proliferating preadipocytes revealed no noticeable
toxicity associated with COH-SR4 at concentrations up to 20 μM
(Fig. 3D). In addition, COH-SR4
did not induce apoptosis in these differentiating adipocytes (data
not shown). These results demonstrated that COH-SR4 inhibited
adipocyte differentiation at least partly via inhibition of MCE
during the proliferation phase without interfering with cell
viability or inducing apoptosis.
COH-SR4 promotes cell cycle arrest
COH-SR4 treatment for 24 h of post-confluent 3T3
preadipocytes in DM cocktail led to a dose-dependent cell cycle
arrest at the G1 phase (Fig. 4A,
upper panel). The DNA content cytogram profile of 5 μM of COH-SR4
was comparable to that of the normal growth-arrested preadipocytes,
suggesting that this compound inhibited clonal expansion of 3T3-L1
cells by inducing cell cycle arrest (Fig. 4A, lower panel). Western blot
analysis of cell cycle regulator proteins confirmed that COH-SR4
induces cell cycle arrest in differentiating 3T3-L1 cells. After a
24-h treatment, COH-SR4 dose-dependently decreased the protein
levels of cyclin A, cyclin B1 and CDK2, but not cyclin E1 (Fig. 4B). In addition, the protein level
of p27, a potent CDK inhibitor involved in G1 arrest (21), was upregulated by COH-SR4. Based
on these results, COH-SR4 treatment modulated the level of proteins
active during S and G2 phases of the cell cycle, confirming the
results of FACS analysis indicating G1 arrest induced by
COH-SR4.
 | Figure 4COH-SR4 causes cell cycle arrest and
inhibits mTOR in differentiating adipocytes. (A) 3T3-L1 cells were
exposed to DM with or without COH-SR4 for 24 h. Cells were stained
with PI solution followed by analyses of cell cycle distribution
using flow cytometry. The percentage of the cell population at each
stage of the cell cycle was determined using the ModFit software.
Results were from three independent experiments with three
replicates each (n=9). Representative western blot analyses showing
the effects of COH-SR4 compound on (B) cell cycle regulatory
proteins and (C) mTOR signaling. Total cell lysates from 3T3-L1
cells treated with DM with or without COH-SR4 for 24 h were
resolved under electrophoresis and immunoblotted with antibodies
against cyclin A, cyclin B1, cyclin E1, CDK2, p27Kip1,
total and phosphorylated raptor, total and phosphorylated TSC2,
total and phosphorylated p70SK6 (p-S6K), total and phosphorylated
4E-BP, total and phosphorylated Akt, and β-actin, which served as
an internal control. |
COH-SR4 modulates mTORC1 but not mTORC2
function
To further elucidate the mechanism through which
COH-SR4 modulates the cell cycle and cell proliferation, we also
analyzed proteins involved in mammalian target of rapamycin (mTOR)
signaling, a multicomplex (mTORC1 and mTORC2) pathway that controls
cell cycle progression and cell proliferation in many types of
cells (22–25). Previous studies showed that AMPK
inhibits mTOR by phosphorylating either tuberous sclerosis protein
2 (TSC2) or the mTORC1 subunit raptor directly (26,27). Western blot analyses revealed that
COH-SR4 treatment dose-dependently increased the phosphorylation of
both TCS2 and raptor (Fig. 4C).
The increased phosphorylations coincided well with the robust
phosphorylation and activation of AMPK. In addition, COH-SR4
decreased the phosphorylation of p70 kDa ribosomal protein S6
kinase (S6K) and eukaryotic initiation factor 4E (eIF4E) binding
protein (4E-BP), two key downstream effectors of mTOR that regulate
protein synthesis (22–25). Together, these results
demonstrated that the activation of AMPK by COH-SR4 and its
subsequent inhibition of mTORC1 may be involved in the cell cycle
arrest and inhibition of adipogenesis in 3T3-L1 cells.
We next examined whether COH-SR4 treatment modulates
mTORC2. mTORC2 phosphorylates Akt on S473 (28). Therefore, to determine whether
mTORC2 is also inhibited by COH-SR4 under similar conditions,
differentiating 3T3-L1 cells were treated with COH-SR4, and the
phosphorylation of Akt was determined. COH-SR4 treatment did not
alter Akt phosphorylation (Fig.
4C). These results provide evidence that COH-SR4 only inhibits
mTORC1 in differentiating adipocytes.
Knockdown of AMPK catalytic subunits by
siRNA suppresses the inhibitory effects of COH-SR4 on 3T3-L1
adipogenesis
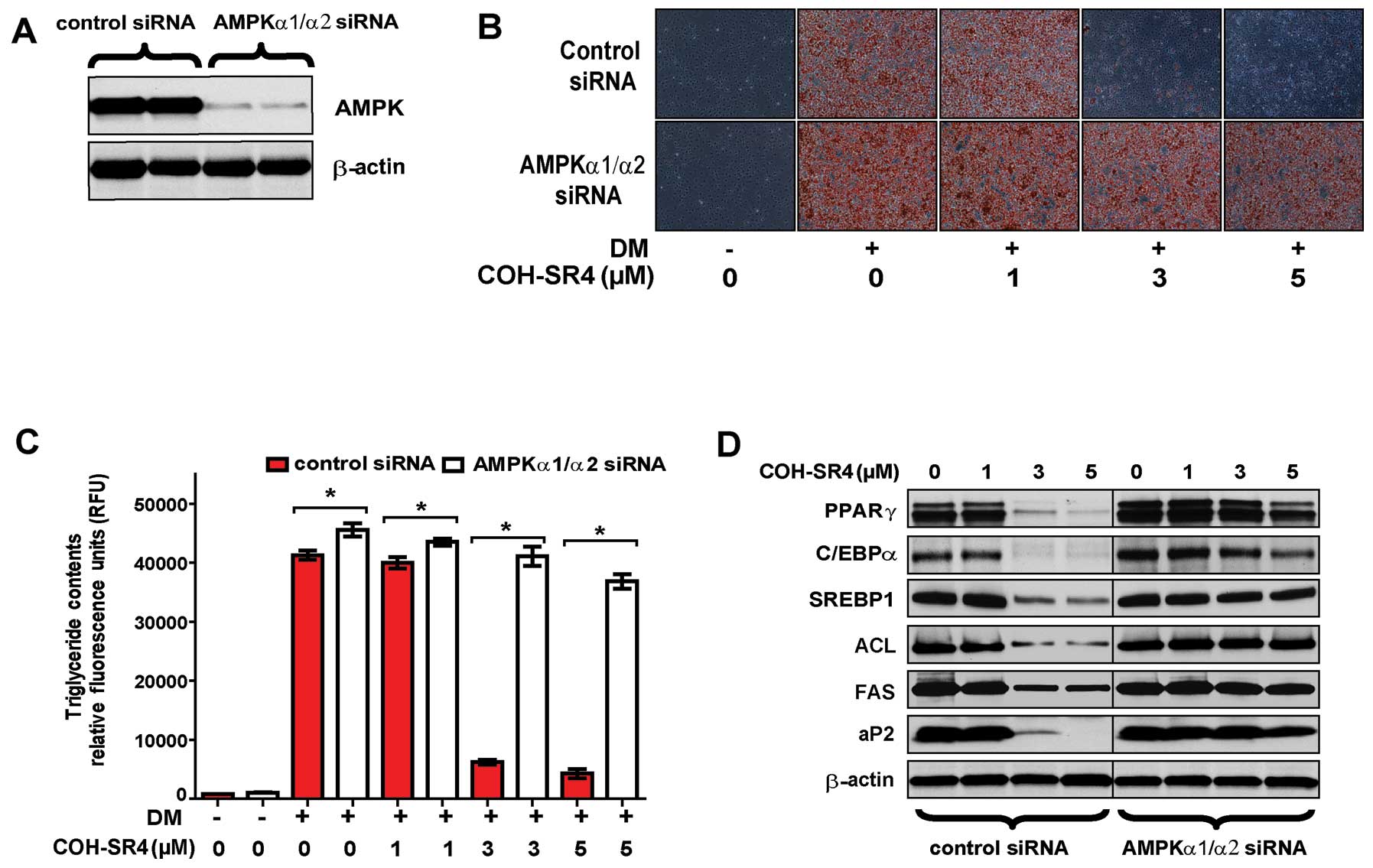
To investigate whether COH-SR4-mediated AMPK
activation is mainly responsible for inhibition of adipocyte
differentiation, we employed siRNA interference that reduced the
protein levels of AMPK catalytic subunits (α1 and α2) to <20% of
the control after 24 h (Fig. 5A).
As expected, AMPK knockdown prevented the ability of COH-SR4 to
inhibit adipocyte development and lipid accumulation after 7 days
as shown by Oil Red O staining and AdipoRed assay of intracellular
triglycerides (Fig. 5B and C).
Knockdown of AMPKα1/α2 also suppressed the effects of COH-SR4 on
the expression of key adipogenic transcription factors and their
target lipogenic genes. As shown by western blotting results, the
increased protein levels of PPARγ, SREBP1, C/EBPα, aP2, FAS and ACL
during adipocyte differentiation were all minimally affected by
COH-SR4 treatment, even at the highest concentration tested (5 μM)
(Fig. 5D).
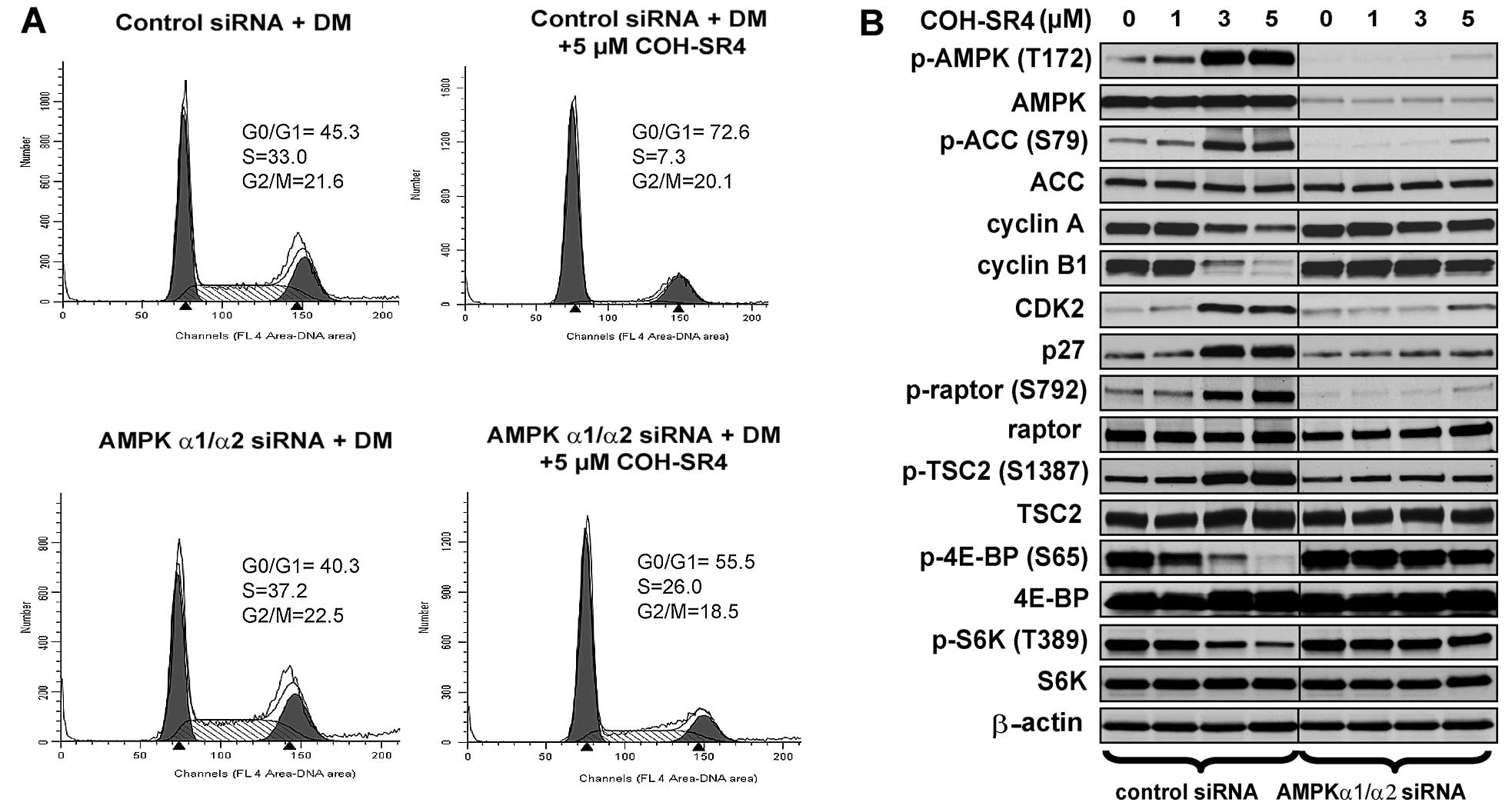
Although the above findings clearly showed the
involvement of AMPK activation in 3T3-L1 differentiation, we raised
the question whether COH-SR4 induces cell cycle arrest and the
inhibition of mTORC1 requires AMPK. Silencing of AMPKα1/α2 also
prevented COH-SR4 to induce cell cycle arrest. In control
siRNA-transfected cells, treatment of 5 μM COH-SR4 arrested ~70% of
differentiating cells at the G1 phase (Fig. 6A) which was almost similar to
non-transfected cells (Fig. 4A,
lower panel). In AMPKα1/α2 siRNA-transfected cells, COH-SR4 only
arrested ~55% of the cells at this stage and increased the number
of S phase cells from 7 to 26% (Fig.
6A). Further support was provided by western blot analyses.
Pre-treatment of AMPKα1/α2 siRNA and COH-SR4 in differentiating
3T3-L1 cells had no or little noticeable effects on the protein
levels of cyclin A, cyclin B1 and CDK2, while the amount of p27
protein remain unchanged (Fig.
6B). Additionally, AMPKα1/α2 knockdown abolished
COH-SR4-mediated mTORC1 inhibition as the compound did not induce
phosphorylation of both TSC2 and raptor, and failed to block the
mTORC1-dependent phosphorylation of both S6K and 4EBP (Fig. 6B). Thus, these results further
confirm that COH-SR4’s action on adipocyte differentiation is
mainly due to its ability to indirectly activate the AMPK/mTORC1
signaling pathways and influence cell cycle progression and cell
proliferation.
Discussion
AMPK, an energy-sensing enzyme involved in the
regulation of carbohydrate and fat metabolism, has been suggested
to play a role in the pathogenic development of obesity, type 2
diabetes and cancer, thus, making it an attractive drug target for
the treatment of these metabolic diseases (12,29). Our laboratory recently identified
several novel small molecules with potential anticancer activities,
including the lead compound COH-SR4, which exhibited strong
anti-proliferative effects against leukemia and other types of
human cancers in vitro and in vivo (19,20, unpublished data). Since an increase
in de novo lipid synthesis is a hallmark of proliferating
cancer cells (30), we
investigated in the present study the potential of this compound to
inhibit adipocyte differentiation and the underlying molecular
mechanisms. Accordingly, we hypothesized that AMPK-mediated
activation may be responsible for its anticancer and
anti-adipogenic effects. The following new findings are reported in
this study: i) COH-SR4 represents a new class of compounds that
indirectly activates AMPK via increased AMP:ATP ratio; ii) COH-SR4
prevents adipocyte differentiation by inducing cell cycle arrest
and preventing MCE concurrent with AMPK activation during the
proliferation phase; iii) COH-SR4 inhibits the expression of key
adipogenesis-related transcription factors and enzymes important in
fatty acid and cholesterol synthesis; iv) COH-SR4 stimulates AMPK
phosphorylation and inhibits mTORC1 activity as evidenced by
increased phosphorylation of raptor and TSC2, and reduced
phosphorylation of S6K and 4EBP; and v) knockdown of AMPKα1/α2
subunits suppresses the inhibitory effects of COH-SR4 on
adipogenesis, including cell cycle arrest, expression of
adipogenesis-related transcription factors and proteins, as well as
mTORC1 inhibition. Taken together, these results imply that COH-SR4
inhibits adipocyte differentiation primarily by its effects on
AMPK/mTORC1 signaling and associated downstream pathways related to
cell growth, proliferation, and protein and lipid synthesis.
AMPK acts as a metabolic master switch regulating
glucose and lipid metabolism (10,11). At the cellular level, AMPK is
activated by metabolic stressors that deplete ATP and activate AMP
levels. Many of the well-known pharmacological drugs (biguanide
derivatives, thiazolinediones, statins) as well as
phytochemicals/nutraceuticals (berberine, resveratrol, genistein
and capsaicin) with a wide variety of structures, are known to
indirectly activate AMPK by inhibiting mitochondrial ATP production
and altering the AMP:ATP ratios (29,31). In this study, we showed that
COH-SR4 did not activate recombinant AMPK in vitro,
suggesting that COH-SR4 did not bind nor directly activate AMPK.
Instead, COH-SR4 treatment resulted in increased intracellular
AMP:ATP ratio, which suggests a potential effect on the
mitochondria. A recent study showed that chemicals that can cause
depolarization of mitochondrial membrane potential (Δψm) can also
activate AMPK (32).
Interestingly, we previously found that COH-SR4 decreased Δψm in
HL-60 leukemia cells (19). We
are currently investigating the effects of COH-SR4 on the
mitochondria to identify the exact molecular target of the
compound.
Adipocyte differentiation is regulated by a
transcriptional cascade that coordinates changes in the expression
of specific adipocyte genes. Previous studies using agents to
activate AMPK have linked this energy-sensing enzyme to the
inhibition of adipogenesis. Both direct AMPK activators such as
AICAR (15,16) and A-76692 (17), as well as indirect activators such
as resveratrol (33) and its
analogs (18), and berberine
(34) have been demonstrated to
inhibit the expression of key lipogenic transcription factors and
their target genes such as C/EBPα, PPARγ, SREPB1, FAS, aP2, ACC and
other proteins involved in fatty acid and cholesterol synthesis. In
this study, we also observed a dose-dependent suppression of the
protein levels of these various transcription factors and
adipogenic genes by COH-SR4 treatment, consequently leading to the
prevention of the adipocyte phenotype as demonstrated by Oil Red O
and AdipoRed stainings (IC50 of ~2 μM). Further analysis
of the inhibitory effects of COH-SR4 showed that the compound was
more effective in blocking adipocyte differentiation during the
proliferation phase (days 0–2), which was strongly associated with
the early activation of AMPK during this period. Indeed, in 3T3-L1
cells where AMPKα1/α2 were knocked down and then treated with
COH-SR4 during the proliferation phase, the compound failed to
inhibit the expression of adipogenic transcription factors and
proteins and suppress lipid droplet formation and triglyceride
accumulation, confirming that activation of AMPK was primarily
responsible for inhibition of differentiation. It has been
speculated that this early activation of AMPK could be sensed by
adipocytes as a signal of energy depletion, thus leading to
inhibition of ATP-consuming pathways, such as lipogenesis (18).
Cell cycle analyses and cell proliferation assays
supported the early effects of COH-SR4 treatment on adipocyte
differentiation. AMPK activation by COH-SR4 during the
proliferation phase induced cell cycle arrest and significantly
suppressed preadipocyte MCE, the latter an important prerequisite
for adipocyte differentiation. In particular, COH-SR4 blocked the
cell cycle at the G1/S transition. Similarly, an AMPK activator,
AICAR, has been recently reported to induce G1/S cell cycle arrest
and inhibit proliferation in cancer cells (35). We also observed cell cycle arrest
in human leukemia melanoma and lung and breast cancer cells
(19,20, unpublished data). Overall, these
data indicate that COH-SR4 induction of AMPK in both adipocytes and
cancer cells results in cell cycle arrest, and thus, imply a common
molecular target.
How AMPK modulates cell cycle progression, cell
growth and proliferation in adipocytes is not totally clear, but
accumulating evidence points to the AMPK/mTORC1 axis (23–25,36,37). Cell growth and proliferation are
energetically demanding, and AMPK acts as an ‘energy checkpoint’
that permits growth and proliferation only when energy reserves are
sufficient (10,11,31). One of the key targets of AMPK is
mTORC1, the master orchestrator of cell growth and proliferation.
At the cellular level, mTORC1 promotes cellular anabolic processes,
including ribosome biogenesis, protein and lipid synthesis, cell
growth and cell cycle progression, which drive cell proliferation.
Under energetic stress conditions, AMPK phosphorylates TSC2, and
increases the activity of the TSC1-TSC2 complex to inhibit mTOR
(27). In addition, AMPK also
phosphorylates the mTORC1 component raptor leading to
14-3-3-binding and allosteric inhibition of mTORC1 (26). As shown in this study, COH-SR4
treatment dose-dependently increased the phosphorylation of both
TSC2 and raptor, which correlated well with cell cycle arrest and
prevention of MCE. Silencing of AMPKα1/α2 subunits prevented the
ability of COH-SR4 to induce phosphorylation in both proteins,
concomitant with the absence of cell cycle arrest and inhibition of
lipid accumulation. Thus, COH-SR4 inhibits mTORC1 function
indirectly via its AMPK activation effects. Consistent with our
studies, the knockdown of raptor inhibited adipogenesis in 3T3-L1
cells (38), while loss of TSC2
enhanced 3T3-L1 differentiation (39). Moreover, we also observed
increased phosphorylation of both TSC2 and raptor in
growth-arrested human lung cancer cells treated with COH-SR4
(unpublished data) further confirming the importance of mTORC1
signaling in cell cycle progression and proliferation, and again
indicating a common downstream AMPK target by COH-SR4 in both
cancer cells and adipocytes.
The mechanisms underlying mTORC1-mediated inhibition
of cell growth and proliferation in adipocytes remain incompletely
defined but likely involve reduced protein synthesis and induction
of the translation of mRNA coding for key components of the
adipogenic process. Activation of mTORC1 positively stimulates mRNA
translation via its downstream substrates S6K and 4E-BPs/eIF4E
(22,25). Phosphorylation of 4E-BP by mTORC1
results in its dissociation from eIF4E, promoting assembly of the
eIF4F complex and allowing eIF4E to initiate cap-dependent
translation, while mTORC1-mediated phosphorylation of S6K1 promotes
mRNA translation by phosphorylating and activating eIF4B; in turn,
eIF4B enhances the activity of eIF4A, an RNA helicase that unwinds
the structured 5′ untranslated regions (5′ UTRs) of many mRNAs
(23,24). 4E-BPs are crucial elements of the
mTORC1 pathway that regulate cell number and proliferation by
selectively inhibiting the translation of messenger RNAs that
encode proliferation-promoting proteins and proteins involved in
cell cycle progression (40). The
activation of eIF4E, via inhibition of 4EBP activity, enhances cell
proliferation by modulating the cell cycle through regulation of
the expression of G1/S proteins including cyclins A, B, D1 and E by
promoting the export of specific mRNAs from the nucleus to the
cytoplasm (41). Because eIF4E is
thought to increase the translation of C/EBPs (42), which are key components required
for the establishment of the adipogenic cascade, and that both
C/EBPβ and C/EBPδ are known to drive the expression of C/EBPα and
PPARγ to trigger the activation of a feed-forward loop in which
these two transcription factors reciprocally induce their
expression (43), we speculate
that AMPK activation by COH-SR4 during the early proliferation
phase of differentiation leads to inhibition of mTORC1-dependent
translation of proteins and transcription factors associated with
the adipogenic cascade. Thus, during adipocyte differentiation
where the intracellular energy demands are higher as a consequence
of increased protein biosynthetic rates that drive cell growth and
MCE, the ability of COH-SR4 to inhibit the high mTORC1 activity
during this phase via AMPK activation would negatively affect the
expression and activation of PPARγ and C/EBPα. In support of this
view, mTOR inhibition by rapamycin has been observed to induce G1/S
cell cycle arrest, inhibit MCE, downregulate PPARγ, SREBP1 and
C/EBPα protein expression, as well as prevent lipid accumulation in
differentiating 3T3 cells (44–47). Additionally, our present study
showed that COH-SR4 treatment inhibited both S6K and 4E-BP by
decreasing their phosphorylation during adipocyte differentiation.
Consequently, knockdown of AMPKα1/α2 prevented dephosphorylation of
both mTORC1 effectors, with noticeable absence of cell cycle arrest
and inhibition of protein expression of key transcription factors
and their target lipogenic genes. These findings are consistent
with several studies where AMPK activators were shown to block S6K
and 4E-BP phosphorylation (48,49), and the failure of AMPK agonist
AICAR to inhibit mTORC activity in AMPKα1/α2 double knockout mice
embryonic fibroblasts (50).
Nonetheless, to better understand whether inhibition of adipocyte
differentiation by COH-SR4 is directly mediated through AMPK-mTORC1
signaling and cell cycle arrest, future investigations assessing
the effects of the compound on raptor and/or TCS2-deficient, as
well as p27-knockdown 3T3 cells are warranted.
In summary, we demonstrated that COH-SR4 suppressed
adipogenesis in 3T3-L1 cells through indirect activation of AMPK
and downstream modulation of the mTORC1 signaling pathway, which
blocked important regulators involved in protein synthesis, cell
cycle progression, and expression of key transcription factors and
their target adipogenic genes involved in lipid synthesis. In
addition to exhibiting potent anticancer properties, COH-SR4 is a
potential therapeutic candidate for the treatment and prevention of
obesity and related metabolic disorders. We are currently assessing
the pharmacological effects of COH-SR4 in diet-induced obese (DIO)
mice as well as type 2 diabetic db/db mice.
Acknowledgements
The authors are grateful to Mr. and Mrs. Isaac
Moradi for their yearly financial support at the City of Hope. The
authors are also thankful to Mariko Lee (Microscope Core
Laboratory, COH) and Lucy Brown (Analytical Cytometry Core
Facility, COH) for the technical assistance in the fluorescence
microscopic and flow cytometric analyses, respectively. We also
acknowledge the help of Dr Tim Synold and Lisa Powell (Analytical
Pharmacology Laboratory) in the nucleotide measurements.
References
|
1
|
Grundy SM: Obesity, metabolic syndrome,
and cardiovascular disease. J Clin Endocrinol Metab. 89:2595–2600.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Khandekar MJ, Cohen P and Spiegelman BM:
Molecular mechanisms of cancer development in obesity. Nat Rev
Cancer. 11:886–895. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jo J, Gavrilova O, Pack S, Jou W, Mullen
S, Sumner AE, Cushman SW and Periwal V: Hypertrophy and/or
hyperplasia: dynamics of adipose tissue growth. PLoS Comput Biol.
5:e10003242009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pilch PF and Bergenhem N: Pharmacological
targeting of adipocytes/fat metabolism for treatment of obesity and
diabetes. Mol Pharmacol. 70:779–785. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rosen ED and MacDougald OA: Adipocyte
differentiation from the inside out. Nat Rev Mol Cell Biol.
7:885–896. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lowe CE, O’Rahilly S and Rochford JJ:
Adipogenesis at a glance. J Cell Sci. 124:2681–2686. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tang QQ, Otto TC and Lane MD: Mitotic
clonal expansion: a synchronous process required for adipogenesis.
Proc Natl Acad Sci USA. 100:44–49. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fajas L: Adipogenesis: a cross-talk
between cell proliferation and cell differentiation. Ann Med.
35:79–85. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Farmer SR: Transcriptional control of
adipocyte formation. Cell Metab. 4:263–273. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hardie DG: AMP-activated protein kinase:
an energy sensor that regulates all aspects of cell function. Genes
Dev. 25:1895–1908. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Carling D, Mayer FV, Sanders MJ and
Gamblin SJ: AMP-activated protein kinase: nature’s energy sensor.
Nat Chem Biol. 7:512–518. 2011.
|
|
12
|
Steinberg GR and Kemp BE: AMPK in health
and disease. Physiol Rev. 89:1025–1078. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Villena JA, Viollet B, Andreelli F, Kahn
A, Vaulont S and Sul HS: Induced adiposity and adipocyte
hypertrophy in mice lacking the AMP-activated protein kinase-alpha2
subunit. Diabetes. 53:2242–2249. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang W, Zhang X, Wang H, et al:
AMP-activated protein kinase α1 protects against diet-induced
insulin resistance and obesity. Diabetes. 61:3114–3125. 2012.
|
|
15
|
Habinowski SA and Witters LA: The effects
of AICAR on adipocyte differentiation of 3T3-L1 cells. Biochem
Biophys Res Commun. 286:852–856. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Giri S, Rattan R, Haq E, Khan M, Yasmin R,
Won JS, Key L, Singh AK and Singh I: AICAR inhibits adipocyte
differentiation in 3T3L1 and restores metabolic alterations in
diet-induced obesity mice model. Nutr Metab. 3:312006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhou Y, Wang D, Zhu Q, Gao X, Yang S, Xu A
and Wu D: Inhibitory effects of A-769662, a novel activator of
AMP-activated protein kinase, on 3T3-L1 adipogenesis. Biol Pharm
Bull. 32:993–998. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Vingtdeux V, Chandakkar P, Zhao H, Davies
P and Marambaud P: Small-molecule activators of AMP-activated
protein kinase (AMPK), RSVA314 and RSVA405, inhibit adipogenesis.
Mol Med. 17:1022–1030. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Figarola JL, Weng Y, Lincoln C, Horne D
and Rahbar S: Novel dichlorophenyl urea compounds inhibit
proliferation of human leukemia HL-60 cells by inducing cell cycle
arrest, differentiation and apoptosis. Invest New Drugs.
30:1413–1425. 2012. View Article : Google Scholar
|
|
20
|
Singhal SS, Figarola J, Singhal J, et al:
1,3-Bis(3,5-dichlorophenyl) urea compound ‘COH-SR4′ inhibits
proliferation and activates apoptosis in melanoma. Biochem
Pharmacol. 84:1419–1427. 2012.
|
|
21
|
Vermeulen K, Van Bockstaele DR and
Berneman ZN: The cell cycle: a review of regulation, deregulation
and therapeutic targets in cancer. Cell Prolif. 36:131–149. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fingar DC, Richardson CJ, Tee AR, Cheatham
L, Tsou C and Blenis J: mTOR controls cell cycle progression
through its cell growth effectors S6K1 and 4E-BP1/eukaryotic
translation initiation factor 4E. Mol Cell Biol. 24:200–216. 2004.
View Article : Google Scholar
|
|
23
|
Laplante M and Sabatini DM: mTOR signaling
in growth control and disease. Cell. 149:274–293. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zoncu R, Efeyan A and Sabatini DM: mTOR:
from growth signal integration to cancer, diabetes and ageing. Nat
Rev Mol Cell Biol. 12:21–35. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ma XM and Blenis J: Molecular mechanisms
of mTOR-mediated translational control. Nat Rev Mol Cell Biol.
10:307–318. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gwinn DM, Shackelford DB, Egan DF,
Mihaylova MM, Mery A, Vasquez DS, Turk BE and Shaw RJ: AMPK
phosphorylation of raptor mediates a metabolic checkpoint. Mol
Cell. 30:214–226. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Inoki K, Zhu T and Guan KL: TSC2 mediates
cellular energy response to control cell growth and survival. Cell.
115:577–590. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sarbassov DD, Guertin DA, Ali SM and
Sabatini DM: Phosphorylation and regulation of Akt/PKB by the
rictor-mTOR complex. Science. 307:1098–1101. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fogarty S and Hardie DG: Development of
protein kinase activators: AMPK as a target in metabolic disorders
and cancer. Biochim Biophys Acta. 1804:581–591. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Menendez JA and Lupu R: Fatty acid
synthase and the lipogenic phenotype in cancer pathogenesis. Nat
Rev Cancer. 7:763–777. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hardie DG: Sensing of energy and nutrients
by AMP-activated protein kinase. Am J Clin Nutr. 93:891S–896S.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Qiu BY, Turner N, Li YY, et al:
High-throughput assay for modulators of mitochondrial membrane
potential identifies a novel compound with beneficial effects on
db/db mice. Diabetes. 59:256–265. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chen S, Li Z, Li W, Shan Z and Zhu W:
Resveratrol inhibits cell differentiation in 3T3-L1 adipocytes via
activation of AMPK. Can J Physiol Pharmacol. 89:793–799.
2011.PubMed/NCBI
|
|
34
|
Lee YS, Kim WS, Kim KH, et al: Berberine,
a natural plant product, activates AMP-activated protein kinase
with beneficial metabolic effects in diabetic and insulin-resistant
states. Diabetes. 55:2256–2264. 2006. View Article : Google Scholar
|
|
35
|
Rattan R, Giri S, Singh AK and Singh I:
5-Aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside inhibits
cancer cell proliferation in vitro and in vivo via AMP-activated
protein kinase. J Biol Chem. 280:39582–39593. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Inoki K, Kim J and Guan KL: AMPK and mTOR
in cellular energy homeostasis and drug targets. Annu Rev Pharmacol
Toxicol. 52:381–400. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Laplante M and Sabatini DM: An emerging
role of mTOR in lipid biosynthesis. Curr Biol. 19:R1046–R1052.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Polak P, Cybulski N, Feige JN, Auwerx J,
Rüegg MA and Hall MN: Adipose-specific knockout of raptor results
in lean mice with enhanced mitochondrial respiration. Cell Metab.
8:399–410. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang HH, Huang J, Düvel K, et al: Insulin
stimulates adipogenesis through the Akt-TSC2-mTORC1 pathway. PLoS
One. 4:e61892009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Dowling RJ, Topisirovic I, Alain T, et al:
mTORC1-mediated cell proliferation, but not cell growth, controlled
by the 4E-BPs. Science. 328:1172–1176. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Culjkovic B, Topisirovic I, Skrabanek L,
Ruiz-Gutierrez M and Borden KL: eIF4E is a central node of an RNA
regulon that governs cellular proliferation. J Cell Biol.
175:415–426. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Calkhoven CF, Muller C and Leutz A:
Translational control of C/EBPalpha and C/EBPbeta isoform
expression. Genes Dev. 14:1920–1932. 2000.PubMed/NCBI
|
|
43
|
Rosen ED, Hsu C-H, Wang X, Sakai S,
Freeman MW, Gonzalez FJ and Spiegelman BM: C/EBPalpha induces
adipogenesis through PPARgamma: a unified pathway. Genes Dev.
16:22–26. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yeh WC, Bierer BE and McKnight SL:
Rapamycin inhibits clonal expansion and adipogenic differentiation
of 3T3-L1 cells. Proc Natl Acad Sci USA. 92:11086–11090. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hashemolhosseini S, Nagamine Y, Morley SJ,
Desrivières S, Mercep L and Ferrari S: Rapamycin inhibition of the
G1 to S transition is mediated by effects on cyclin D1 mRNA and
protein stability. J Biol Chem. 273:14424–14429. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Cho HJ, Park J, Lee HW, Lee YS and Kim JB:
Regulation of adipocyte differentiation and insulin action with
rapamycin. Biochem Biophys Res Commun. 321:942–948. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
El-Chaar D, Gagnon A and Sorisky A:
Inhibition of insulin signaling and adipogenesis by rapamycin:
effect on phosphorylation of p70 S6 kinase vs eIF4E-BP1. Int J Obes
Relat Metab Disord. 28:191–198. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kimura N, Tokunaga C, Dalal S, et al: A
possible linkage between AMP-activated protein kinase (AMPK) and
mammalian target of rapamycin (mTOR) signalling pathway. Genes
Cells. 8:65–79. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Chiang PC, Lin SC, Pan SL, Kuo CH, et al:
Antroquinonol displays anticancer potential against human
hepatocellular carcinoma cells: a crucial role of AMPK and mTOR
pathways. Biochem Pharmacol. 79:162–171. 2010. View Article : Google Scholar
|
|
50
|
Kalender A, Selvaraj A, Kim SY, et al:
Metformin, independent of AMPK, inhibits mTORC1 in a rag
GTPase-dependent manner. Cell Metab. 11:390–401. 2010. View Article : Google Scholar : PubMed/NCBI
|