Introduction
Acute promyelocytic leukemia (APL) is genetically
characterized by the PML/RARα fusion gene that forms as a
consequence of the 15;17 translocation (1). PML-RARα expression is required for
the development of APL. However, the role of fusion protein
PML-RARα as a whole is not unchangeable. For example, some
researchers have found that the phosphorylation of promyelocytic
leukemia (PML) by mitogen-activated protein kinase plays a key role
in PML-dependent apoptosis in response to
As2O3 exposure (2). PML can activate transcription by
protecting homeodomain-interacting protein kinase 2 (HIPK2) and
p300 from SCFFbx3-mediated degradation, and casein kinase 2 (CK2)
regulates the PML protein levels by promoting its
ubiquitin-mediated degradation which depends on direct
phosphorylation at Ser517 (3,4).
Meanwhile, PML is required for fas and caspase-dependent apoptosis
and is involved in non-caspase-dependent apoptosis. PML plays an
important role in cell growth by regulating the expression of
several cyclin proteins (5).
Notably, neutrophil elastase (NE) is important for PML-RARα
activity in early myeloid cells, and PML-RARα was found to be
rapidly and predictably cleaved by NE in an early myeloid cell line
(U937 cells) generating 69-and 53-kDa products (6,7).
PML-RARα cleavage products play an important role in the
development of APL. Thus, to investigate the interaction among
these cleavage products is critical to elucidate the molecular
pathogenesis of APL. WT-PML consists of three domains: PML-C (a
coiled-coil domain close to carboxyl), PML-B (a B-BOX domain close
to amino) and NLS (a nuclear localization signal), which is lost in
the pathogenesis of APL instead of binding to RARα, and is named
PML (NLS−).
The yeast two-hybrid assay has demonstrated the
interaction between PML-C and GINS2. The plasmids of PML
(NLS−) bait-protein and PML-B exhibit transcription
factor activity and toxic effects to yeast cells (8). Thus, the yeast two-hybrid cannot be
used to examine whether there is an interaction between PML
(NLS−) and GINS2. Thus, co-immunoprecipitation was
employed to directly identify the interaction between PML
(NLS−) and GINS2, which compensates for the deficient of
the yeast two-hybrid assay.
GINS complex subunit 2 (GINS2) is a member of the
tetrameric complex termed GINS, composed of GINS1, GINS2, GINS3 and
GINS4, which most likely serves as the replicative helicase,
unwinding duplex DNA ahead of the moving replication fork (9–11).
The GINS complex has been shown to be associated with DNA
replication in humans (12–16) and DNA damage (17–19). Several recent reports suggest a
role of GINS in cancer cells. For example, GINS has been suggested
to be related to cell division, more precisely in chromosome
segregation (20,21). However, the manners in which GINS
functions in leukemia cells is not yet clear. In the present study,
GINS2 expression was knocked down to observe changes in apoptosis
and cell division and the signaling pathway mediating these
changes.
Materials and methods
Reagents
Lipofectamine™ 2000 (Invitrogen); antibodies against
Bcl-2 and Bax (Santa Cruz Biotechnology, Inc., Santa Cruz, CA,
USA); antibodies against cyclin B1, cyclin D1 and cyclin A, and
anti-p-p38, anti-p-ERK and anti-p-JNK antibodies (Cell Signaling
Technology, Inc.); antibodies against cyclin B1 (Epitomics), GINS2
and PML (both from Abcam), and SB203580 (Beyotime) were used in the
present study.
Cell culture
Human embryonic kidney (HEK) 293 cells, chronic
myelocytic leukemia cells (K562) and acute promyelocytic leukemia
cells (NB4) were purchased from ATCC (Rockville, MD, USA). HEK293
cells were grown in Dulbecco’s modified Eagle’s medium (DMEM)
containing 10% fetal bovine serum (FBS) (both from HyClone). K562
and NB4 cells were grown in RPMI-1640 (Gibco) containing 10% FBS
(HyClone), 10 μg/ml penicillin and streptomycin (Sigma) and
2 mM L-glutamine (Sigma). Cells in passages 3–20 in logarithmic
growth phase were collected for the experiments.
Yeast two-hybrid screening
The screening was performed with the interaction
mating assay, a variation of the classic yeast TH screening. To
obtain the bait plasmid, PML-C was amplified from genomic DNA and
cloned into PGBKT7 (our laboratory). The AH109 yeasts with stable
expression of BD-PML-C and the Y187 yeasts transformed with human
leukemia cell cDNA library (our laboratory) were added to 2X
YPDA/Kan liquid medium for hybridization for 20 h. One drop of
solution was collected for observation under a phase contrast
microscope. Once zygotes were present, hybridization was carried
out for another 4 h, and the whole medium was added to
SD/-Trp/-Leu/-His/-Ade (QDO) nutrient deficiency medium. Screening
by nutrient deficiency and galactosidase was conducted thrice, and
the blue colonies were collected as positive colonies which were
used for extraction of plasmids of the AD library. The plasmids of
the AD library and bait plasmids pGBKT7-PML-C were used to
co-transfect AH109 yeasts followed by inverse incubation on a plate
pre-coated with SD/-Trp/-Leu/-His/-Ade/X-α-gal at 30°C for 7–10
days. The bacterial growth and presence of blueness were observed.
The positive colonies were collected for sequencing and analysis of
biological information.
Plasmids and constructs
Fusion protein eukaryotic expression vectors
pCMV-HA-PML-C/pCMV-HA-PML and pCMV-Myc-GINS2 were constructed and
then co-transfected into HEK293 cells. The plasmids of 5 groups of
GINS2 short hairpin RNA (shRNA) were constructed and contained a
reporter gene such as green fluorescence protein (GFP) and the
neomycin-resistant gene, respectively. These vectors were designed
for the cloning and expression of shRNA in mammalian cells under
the control of the human U6 promoter.
Reverse transcription-polymerase chain
reaction and western blot assay
For quantitative reverse transcription-polymerase
chain reaction (qRT-PCR), the total cellular RNA was isolated using
TRIzol reagent (Invitrogen). Total RNA was reverse-transcribed
using the PrimeScript RT reagent kit (Takara Bio, Inc.). The cDNA
was diluted 1:10, and SYBR Premix Ex Taq™ analysis was performed
with a Bio-Rad CFX Manager, using primers for GINS2 (Invitrogen):
forward, 5′-AATGCCCAGC CCTTACTA-3′ and reverse,
5′-GGATTTCGTCTGCCTTCG-3′. Following qRT-PCR, the shRNA possessing
the highest interfering efficacy was used for further experiments.
The relative expression of messenger RNA (mRNA) was determined
using β-actin as an endogenous control. Primers for β-actin
(Invitrogen) were; forward, 5′-ACGAGACCACCTTCAACTCC ATC-3′ and
reverse, 5′-TAGAAGCATTTGCGGTGGACGA-3′. Each experiment was
performed twice with triplicate samples, and data were averaged for
further analysis.
For the western blot assay, aliquots of total cell
lysates were separated on sodium dodecyl sulfate polyacrylamide
gels and were then transferred onto PVDF membranes (Beyotime). The
membranes were blocked in 5% skim milk and then probed with the
corresponding antibody overnight at 4°C (GINS2, 1:1,000; Abcam) and
the antibody against β-actin (1:4,000; Cell Signaling Technology,
Inc.). The secondary antibody was goat-anti-mouse IgG (1:3,000;
Santa Cruz Biotechnology, Inc.). Visualization was carried out with
the ECL kit.
Immunofluorescence staining
Immunofluorescence staining for cyclin B1 was
performed using standard procedures. Cells on coverslips were fixed
in 4% paraformaldehyde for 20 min and permeabilized with 0.5%
Triton X-100 in PBS for 15 min, and the background was excluded by
blocking with 5% bovine serum albumin (BSA) in PBS for 1 h before
incubation with the primary and secondary antibodies. Primary
antibody rabbit against cyclin B1 (1:50; rabbit anti-cyclin B1;
Epitomics) was diluted in 0.5% Triton X-100, and incubation with
the primary antibody was performed for 90 min at 37°C. The
secondary antibody was TRITC-conjugated goat anti-rabbit IgG
(1:100; Santa Cruz Biotechnology, Inc.) and diluted in 0.5% Triton
X-100. Incubation with the secondary antibody was performed for 60
min at room temperature. Cells were then stained with DAPI for 3
min. Rinsing with PBS was carried out at each step.
For immunofluorescence staining of HA-PML
(NLS−) and Myc-GINS2, HEK293 cells were cultured on
coverslip in a 24-well plate and stained using the same procedures
as described above. Primary antibodies against HA-PML-C (1:100,
rabbit anti-HA polyclone antibody) and Myc-GINS2 (1:100, mouse
anti-Myc monoclonal antibody) (both from Clontech) were diluted in
0.5% Triton X-100, and incubation with the primary antibody was
performed for 90 min at 37°C. The secondary antibody was
TRITC-conjugated goat anti-rabbit IgG and FITC-conjugated goat
anti-mouse IgG (1:100; Santa Cruz Biotechnology, Inc.). Cells were
then stained with DAPI for 3 min. Rinsing was carried out with PBS
at each step.
Protein extraction
Cells were lysed in RIPA containing 1% PMSF on ice
for 30 min and shaking was conducted once every 5 min. After
centrifugation at 12,000 × g for 30 min at 4°C, the supernatant was
collected.
Transfection, immunoprecipitation (IP)
and western blot assay
To evaluate the interaction between PML-C and GINS2
by co-immunoprecipitation, pCMV-HA-PML-C and pCMV-Myc-GINS2 were
co-transfected with Lipofectamine™ 2000 into HEK 293 cells.
pCMV-HA-PML and pCMV-Myc-GINS2 were independently transfected into
HEK 293 cells as the control groups. Protein extracts, aliquots
(500 μl) of the platelet suspension (109
cells/ml) were immunoprecipitated by incubation with 2 μg/ml
rabbit anti-HA antibody (Clontech) overnight at 4°C and 40
μl of protein A-agarose. Immunoprecipitates were resolved in
12% SDS-PAGE. Separated proteins were electrophoretically
transferred onto PVDF membranes for subsequent incubation with
mouse anti-myc antibody (1:1,000) overnight at 4°C. The primary
antibody was detected by incubation with horseradish
peroxide-conjugated goat anti-mouse IgG antibody (1:3,000) for 1 h
and exposed to ECL solution. Consistently, the interaction between
PML (NLS−) and GINS2 was also detected by
co-immunoprecipitation using the same protocol.
Four groups of plasmids of GINS2 shRNA were
respectively named 1, 2, 3, 4 and then transfected independently
into K562 and NB4 cells. siRNA sequences are as follows: siGINS2:
1, forward, 5′-CACCGCTGGCGATTAACCTGAA
ACATTCAAGAGATGTTTCAGGTTAATCGCCAGCTTT TTT-3′ and reverse,
5′-GATCCAAAAAAGCTGGCGATTAA CCTGAAACATCTCTTGAATGTTTCAGGTTAATCGCC
AGC-3′; 2, forward, 5′-CACCGGATCATGAACGAAAGGA
AGATTCAAGAGATCTTCCTTTCGTTCATGATCCTTT TTT-3′ and reverse,
5′-GATCCAAAAAAGGATCATGAAC GAAAGGAAGATCTCTTGAATCTTCCTTTCGTTCATG
ATCC-3′; 3, forward, 5′-CACCGCCCTTACTACATGGAAC
TTATTCAAGAGATAAGTTCCATGTAGTAAGGGCTTTT TTG-3′ and reverse,
5′-GATCCAAAAAAGCCCTTACTAC ATGGAACTTATCTCTTGAATAAGTTCCATGTAGTAA
GGGC-3′; 4, forward, 5′-CACCGAGCGCTCAACCACTAG
TACAATTCAAGAGATTGTACATGTGGTTGAGCG CTTTTTTTG-3′ and reverse,
5′-GATCCAAAAAAAGCGC TCAACCACATGTACAATCTCTTGAATTGTACATGTGG
TTGAGCGCTC-3′; negative control, forward, 5′-CACCGTT
CTCCGAACGTGTCACGTCAAGAGATTACGTGACACG TTCGGAGAATTTTTTG-3′ and
reverse, 5′-GATCCAAAA AATTCTCCGAACGTGTCACGTAATCTCTTGACGTGA
CACGTTCGGAGAA-3′.
Cells with low transfection efficiency were treated
with G418 until most of the non-transfected cells were removed to
prepare a population of cells stably expressing the siRNA. The
GINS2 siRNA is essential for survival, and thus the cells
transfected with the plasmid target which effectively reduced the
expression of the target gene may die. Thus, a less stringent
antibiotic selection was performed for 21 days. The surviving cells
were maintained and assessed for target gene expression. The
interfering efficiency was detected by immunoblot assay and
qRT-PCR.
Western blot assay was employed to measure the
protein expression of GINS2, cyclin B1, cyclin D1, cyclin A, p-p38,
t-p38, p-ERK, t-ERK, p-JNK and β-actin according to the above
mentioned procedures.
Cell cycle distribution by flow
cytometry
To investigate the effect of GINS2 siRNA on cell
cycle distribution, PI staining assay was performed. K562 cells
(8×105 cells/ml) were seeded in 6-well plates and
transfected with the plasmid. After screening, K562 cells were
collected by centrifugation, followed by washing, fixation and PI
staining. The cell cycle distribution was examined by flow
cytometry (Becton-Dickinson). Data were analyzed using the Modfit
program (Becton-Dickinson).
Detection of apoptosis by Annexin V/PI
staining
K562 cells were transfected with GINS2 siRNA. Cells
were harvested, washed and resuspended in PBS. Apoptotic cells were
identified by staining with Annexin V and PI, using the Annexin
V/PI Apoptosis Detection kit (KeyGene) according to the
manufacturer’s instructions. Flow cytometry was performed
immediately after supravital staining. The percentages of early
apoptotic and late apoptotic/necrotic cells were determined,
respectively, as the percentage of Annexin
V+/PI−or Annexin V+/PI+
cells. Data acquisition and analysis were performed using a
Becton-Dickinson flow cytometer with CellQuest software.
Statistical analysis
Data are presented as the means ± SD, and Student’s
t-test was used for comparisons among the different groups. A value
of P<0.05 was considered to indicate a statistically significant
result.
Results
Yeast two-hybrid assay and
co-immunoprecipitation for evaluating the interaction between PML-C
and GINS2
Our group had applied yeast two-hybrid assay to
primarily indicate the interaction between PML-C and GINS2. To
further confirm this interaction, the plasmid of the bait protein
PML-C and GINS2 protein were co-transformed into AH109 yeasts which
were inversely grown in SD/-Trp/-Leu/-His/-Ade (QDO) at 30°C for
7–10 days. The blue bacteria were continuously observed. Sequencing
showed that transformation was successful. These positive plasmids
and blank pGBKT7 were then used to co-transform AH109 yeasts as a
control, which were also inversely grown in SD/-Trp/-Leu/-His/-Ade
(QDO) at 30°C for 7–10 days; while the blue bacteria were absent
(Fig. 1A). Whether PML-C
interacts with GINS2 in mammalian cells is still unclear. We then
constructed eukaryotic expression vectors, pCMVHA-PML-C and
pCMV-Myc-GINS2, both of which were transfected into HEK293 cells.
The HEK293 cells which were transfected with one of the vectors
served as a negative control. Protein bands were present in cells
undergoing co-transfection but absent in cells transfected with one
of the vectors. The expression of the target protein was also found
in the cell lysate. These findings together with those of the
co-immunoprecipitation and western blot assay demonstrated the
interaction between PML-C and GINS2 (Fig. 1B).
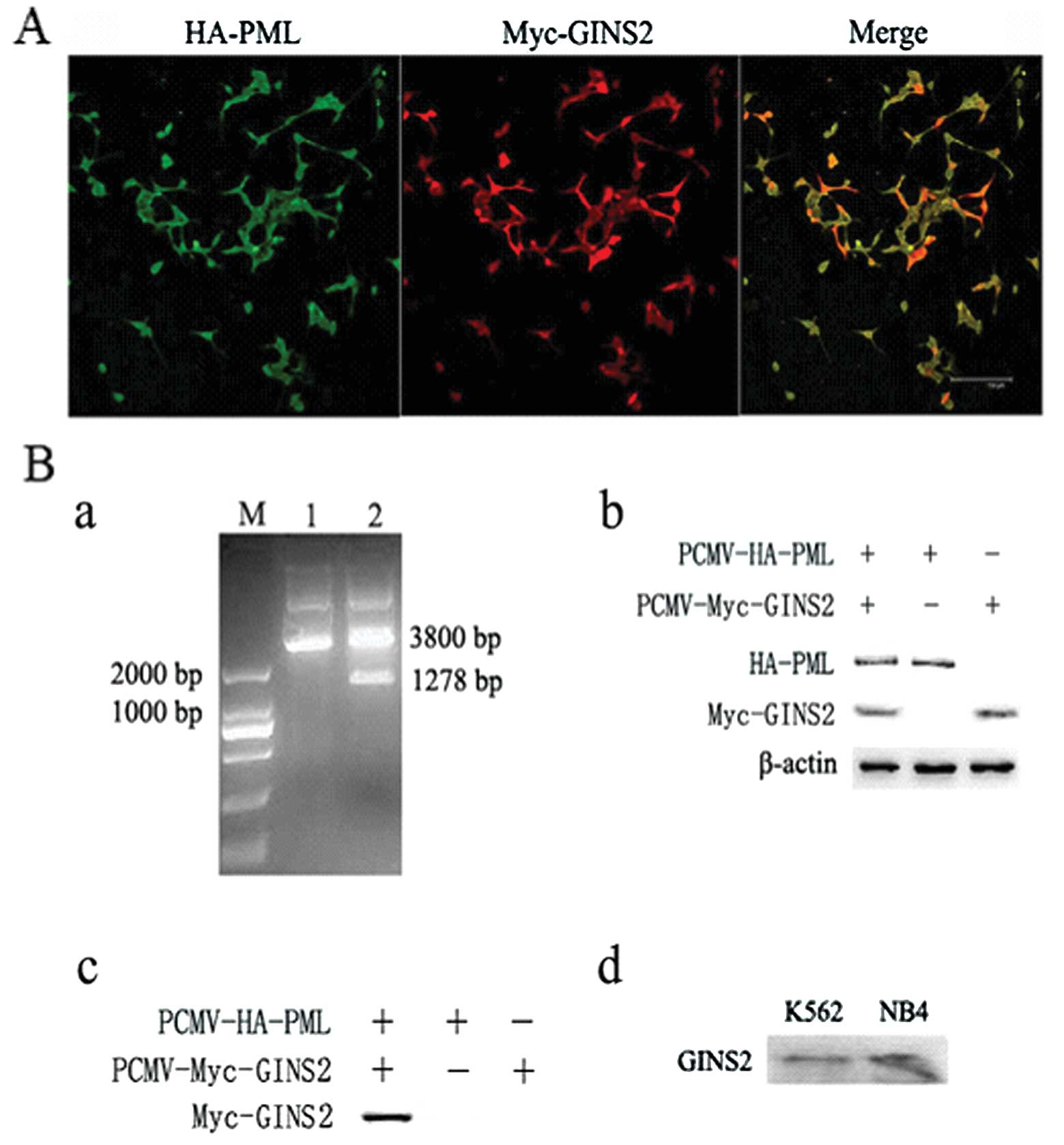
Immunofluorescence and
co-immunoprecipitation evaluating the interaction between PML
(NLS−) and GINS2
The plasmids of PML (NLS−) and PML-B were
toxic to AH109 yeasts and underwent self-activation, and thus these
plasmids were not used in the yeast two-hybrid screening. In the
present study, we aimed to directly investigate the interaction
between PML (NLS−) and GINS2 by immunofluorescence
staining and co-immunoprecipitation. The HEK293 cells undergoing
co-transfection were then subjected to immunofluorescence staining
with the rabbit anti-HA polyclonal and mouse anti-myc monoclonal
antibodies. Results showed that HA-PML (NLS−) and
Myc-GINS2 were expressed in both the nucleus and cytoplasm
(Fig. 2A). The vectors
pCMV-HA-PML (NLS−) and pCMV-Myc-GINS2 were used to
co-transfect HEK293 cells, and cells transfected with one of the
vectors served as a negative control. Results from the
co-immunoprecipitation indicated an interaction between PML
(NLS−) and GINS2 (Fig.
2B), which provides evidence for the interaction between PML
(NLS−) and GINS2, which may serve as a supplement to the
results from the yeast two-hybrid screening. This method also
breaks through the limitations of yeast two-hybrid screening and
provides a basis for the investigation of the role of GINS2 in
leukemia cells. In order to validate the endogenous interaction
between PML (NLS−) and GINS2 in leukemia cells, the PML
(NLS−) protein was immunoprecipitated by the anti-PML
polyclonal antibody, and GINS2 protein was measured by western
blotting with the anti-GINS2 polyclonal antibody from the
immunoprecipitared complex. Results from co-immunoprecipitation
indicated an interaction between PML and GINS2 (Fig. 2B).
Validation of GINS2 interference effect
in K562 and NB4 cells
K562 and NB4 cells were independently transfected
with different GINS2 plasmids. Forty-eight hours after
transfection, a few green fluorescent proteins were found under
inverted fluorescence microscope. G418 was then used to select
positive clones. Three weeks after G418 screening, polyclonal cell
lines were collected and the expression level of green fluorescent
GINS2 was ∼70% in cells which underwent transfection and 90% in the
NC group (only one interference group and NC were presented herein)
(Fig. 3A). Four weeks after
screening, the expression level of fluorescence protein was
markedly reduced, but the number of apoptotic cells increased.
These findings were consistent with the finding that GINS2
silencing may increase the number of dead polyploid cells and
inhibit cell growth and activity. However, the cell growth remained
unchanged in the NC group. Thus, the polyclonal cell lines at 3
weeks were collected for further study. In addition, fluorescent
quantitative PCR was employed to detect the GINS2 expression, and
the results demonstrated the reduction of interference efficacy.
The interference efficacy in K562 cells was 29, 30, 38 and 5%, and
that in NB4 cells was 50, 17, 9 and 28% (Fig. 3B). Western blot assay was used to
measure the GINS2 protein expression and results were consistent
with those of PCR (Fig. 3B). The
K562 and NB4 cells with the best interference efficacy were
employed for subsequent experiments.
Changes in cell cycle distribution and
expression of cell cycle-related proteins in K562 and NB4 cells
after GINS2 knockdown
To confirm the role of GINS2 in leukemia, K562 and
NB4 cells were employed, and changes in both types of cells were
observed after GINS2 gene silencing. Flow cytometry was carried out
to detect cell cycle distribution. Results showed that the
proportion of both types of cells in G2 phase was markedly
increased (P<0.05). The proportion of K562 cells in G1 phase was
increased while that of NB4 cells in G1 phase was reduced but a
significant difference was absent (Fig. 4A). To further explore the effect
of GINS2 silencing on the cell cycle, western blot assay was
employed to detect the protein expressions of cyclin B1, cyclin D1
and cyclin A. Results showed that the protein expression of the
three proteins was markedly reduced when compared with the control
and the NC group (Fig. 4B). Of
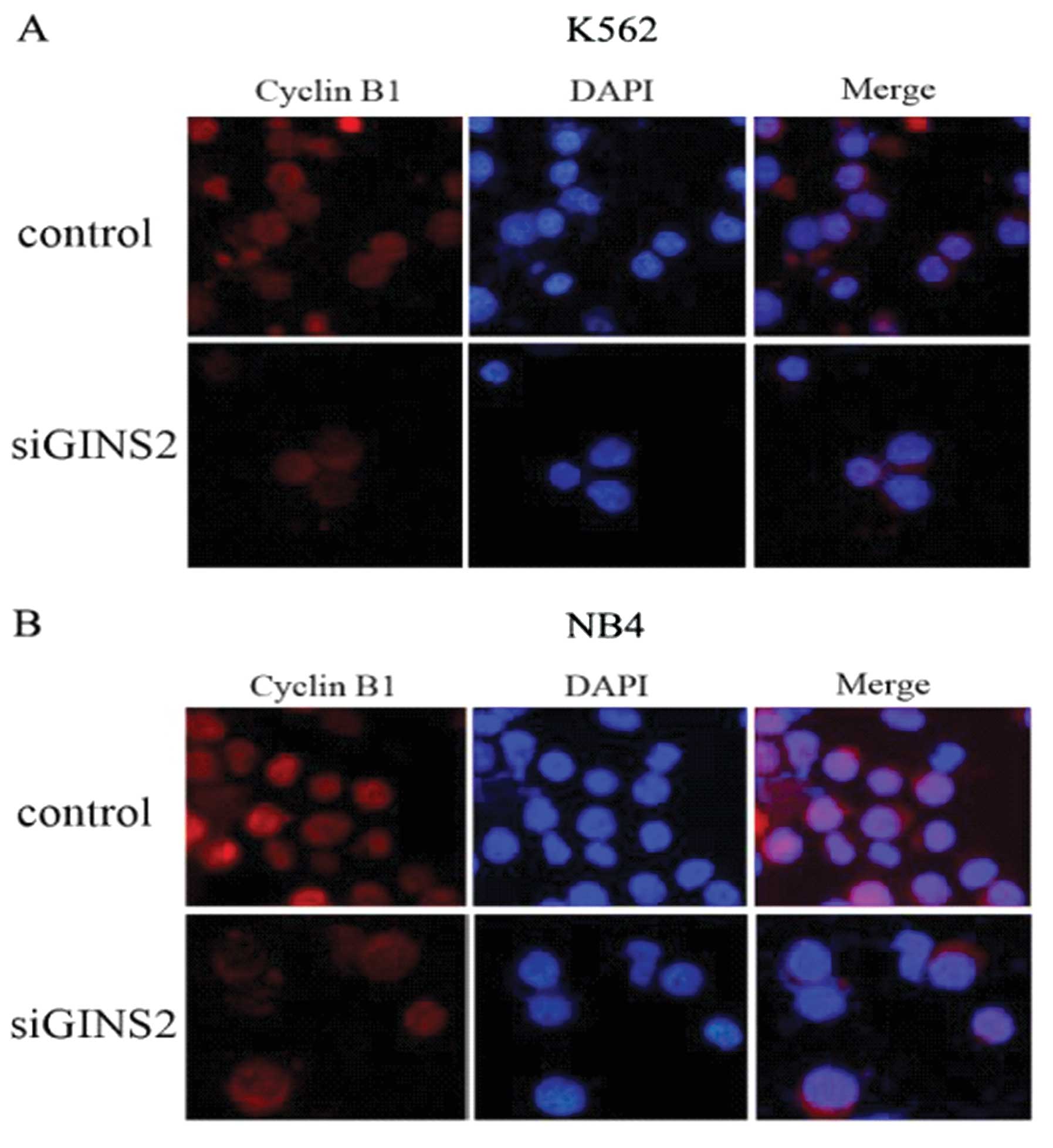
note, cyclin B1 aggregates in the cytoplasm and is a regulator of
G2 phase. Thus, immunofluorescence staining was performed to locate
cyclin B1 in the K562 and NB4 cells using rabbit anti-cyclin B1
monoclonal antibody. Results showed that cyclin B1 exhibited high
expression in the cytoplasm and nuclei in the control group but
high expression was only found in the cytoplasm following GINS2
silencing (Fig. 5). These
findings were consistent with arrest in the G2 phase in both types
of cells. These results demonstrated that GINS2 gene silencing
inhibits the transport of cyclin B1 from the cytoplasm into the
nucleus resulting in aggregation of cyclin B1 in the cytoplasm.
This may be the cause of cell cycle arrest in the G2 phase.
Detection of apoptotic cells and
proteins
The dysregulation of the cell cycle has been
reported to correlate with apoptosis induction. Based on the G2
peak in the cell cycle, apoptosis was analyzed by Annexin V/PI
staining. We further characterized the GINS2 siRNA-induced
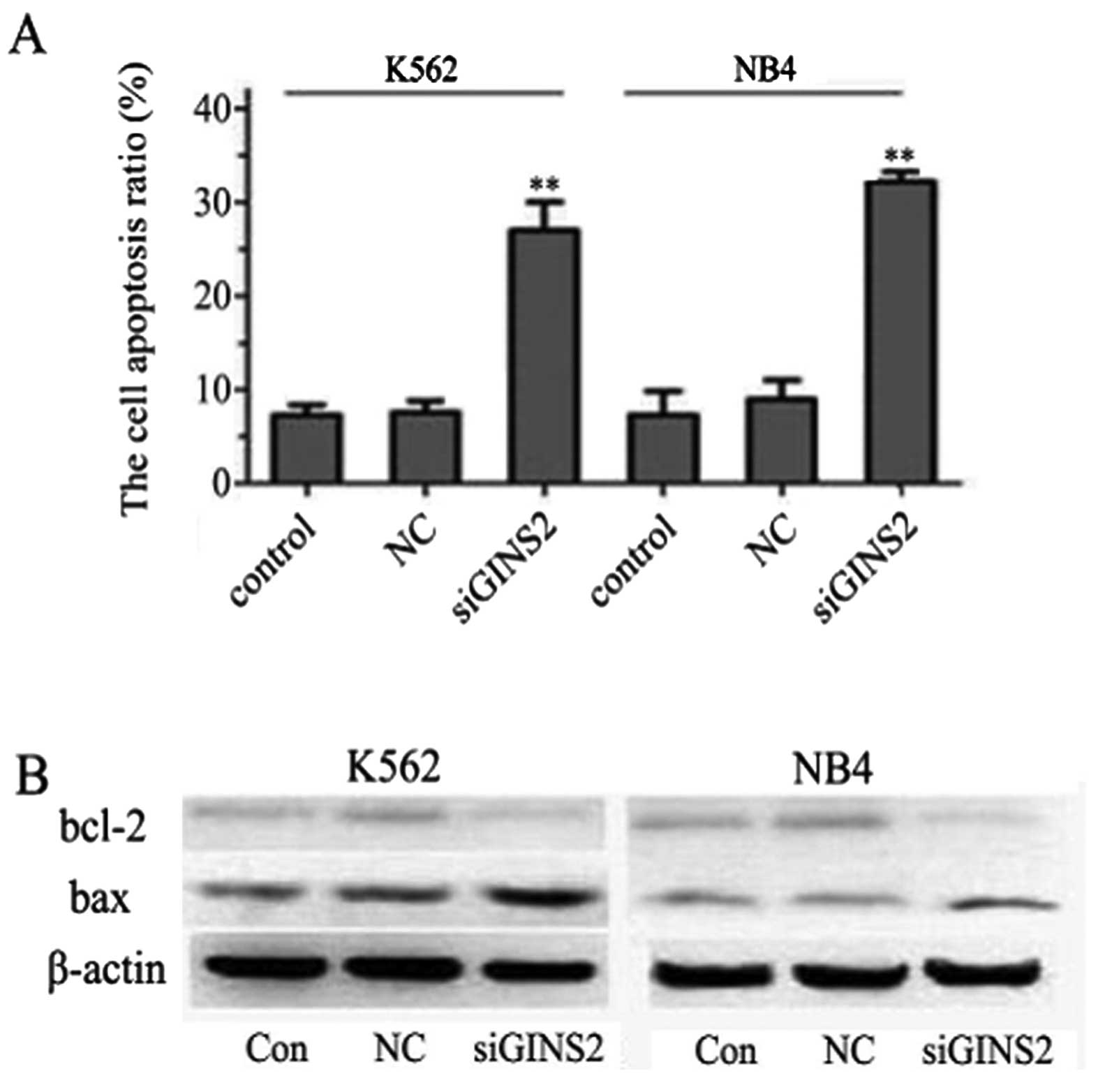
apoptosis by Annexin V/PI staining. The proportions of apoptotic
cells in the GINS2 siRNA groups were 26.99 and 32.54% in the K562
and NB4 cells, respectively, and the proportions in the NC group
were 7.85 and 9.36% in K562 and NB4 cells, respectively; that in
the control group was 6.53 and 7.07% in K562 and NB4 cells,
respectively. A statistically significant difference was noted
between the GINS2 siRNA groups and the control group (P<0.001)
but no marked difference was noted between the NC group and the
control group (Fig. 6A). In
attempt to further clarify the mechanisms underlying the inhibitory
effect of GINS2 siRNA on leukemia cells, the levels of Bcl-2 family
members were detected by western blot assay. A significant decrease
in Bcl-2 was noted while an increase in Bax was noted in the K562
cells. Similar findings were observed in NB4 cells (Fig. 6B). These results suggest that
GINS2 siRNA induces apoptosis of K562 and NB4 cells by upregulating
Bax expression and downregulating Bcl-2 expression.
p38MAPK signaling transduction
pathway
Some reports suggest that the MAPK signaling pathway
mediates cell cycle arrest. To study the relationship between the
MAPK signaling pathway and cell cycle arrest, the activation of MAP
kinases was detected by western blot assay using a specific
anti-phospho-antibody. The K562 cells expressing GINS2 siRNA had
increased expression of phosphorylated p38, and a slight increase
in phosphorylated ERKl/2 expression, while phosphorylated JNK
expression was not affected. Similarly, the NB4 cells expressing
GINS2 siRNA had increased expression of phosphorylated p38, while
the expression of phosphorylated ERKl/2 and phosphorylated JNK
remained unchanged (Fig. 7A).
To further investigate whether p38MAP kinase
activation is associated with GINS2 siRNA-induced G2 phase arrest,
specific p38MAPK inhibitor, SB203580 (10 nM, 1 h), was used to
inhibit p38. Western blot assay indicated that SB203580 attenuated
the decrease in cyclin B1 expression in the GINS2 siRNA groups, but
had no significant effect on cyclin B1 expression in the control
group (Fig. 7B). Moreover,
SB203580 had no evident influence on cell morphology and growth.
These results suggest that GINS2 knockdown influences the cell
cycle through activation of p38MAPK.
Discussion
Extensive research has been conducted aiming to
discover novel and effective strategies for the treatment of
leukemia. At present, the therapeutic strategies for leukemia
mainly include traditional chemotherapy and bone marrow
transplantation. However, chemotherapy has poor specificity and its
side effects often compromise the therapeutic efficacy of
chemotherapy. In addition, donors are insufficient, which
significantly limits the wide application of bone marrow
transplantation. Thus, increasing attention has been given to find
new chemopreventive and key role targets and develop novel methods
for molecular-targeted therapy. In the present study, our results
demonstrated an interaction between PML-C and GINS2 by the yeast
two-hybrid assay and co-immunoprecipitation, and
co-immunoprecipitation was employed to identify the interaction
between PML (NLS−) and GINS2. It is well known that the
yeast two-hybrid assay requires bait-protein without transcription
factor activity and toxic effects to yeast cells, yet the plasmid
of PML (NLS−) bait protein itself has transcription
factor activity and toxic effects to yeast cells (8). Consequently, through the interaction
between PML-C and GINS2 by the yeast two-hybrid, we further
investigated the interaction between PML (NLS−) and
GINS2 by co-immunoprecipitation. This suggests that GINS2 may be a
therapeutic target for leukemia treatment.
Several previous reports have confirmed the roles of
GINS. For example, GINS components were found to be over-expressed
in aggressive melanoma (22) and
upregulation of GINS1 promoted the growth of breast cancer cells
(23). It has been reported that
DNA replication-associated proteins have diverse functions in
different cells; however, the role of its components in mammalian
cells is not yet clear. In the present study, GINS2 knockdown was
found to result in growth inhibition and induction of apoptosis in
NB4 and K562 cells by suppressing G2 phase progression, indicating
that GINS2 may affect, in addition to DNA replication initiation
essential for S-phase progression (in the GINS complex), cell
division and probably chromosome segregation in human leukemic
cells. Consistent with other studies, our results demonstrated that
GINS2 exerts obvious effects on cell survival. Cell cycle analysis
revealed an increased proportion of cells in the G2 phase following
GINS2 silencing. To further clarify the effect of GINS2 knockdown
on the cell cycle, the expression of cyclin A, cyclin D1 and cyclin
B1 was measured. The results showed that the expression of these
proteins was significantly decreased. Moreover, it is well
acknowledged that the complex of cdc2 and cyclin B1
[M-phase-promoting factor (MPF)] is a key regulator of the G2/M
cell cycle transition (24,25). Since cyclin Bl is known to
localize in the cytoplasm in G2 phase and to be transported into
the nucleus during the M phase (26), we examined the subcellular
localization of cyclin B1 after GINS2 knockdown. In control cells,
cyclin Bl was expressed in both the cytoplasm and nucleus. However,
cyclin B1 accumulated in the cytoplasm after GINS2 knockdown. These
findings suggest that the nuclear transport of cyclin B1 is
inhibited by GINS2 knockdown, which further leads to cell cycle
arrest in the G2 phase.
Some studies have suggested that the MAPK signaling
pathway mediates cell cycle arrest. For example, ERK kinase is
required for G2/M phase arrest induced by DNA damage, and p38 is
associated with the G2/M checkpoint (27–30). Therefore, the activation of MAP
kinase was detected by western blot assay. The expression of
phosphorylated p38 was increased after GINS2 knockdown in both K562
and NB4 cells, while that of phosphorylated ERK and phosphorylated
JNK remained unchanged in the K562 and NB4 cells.
Taken together, our results demonstrated an
interaction between PML and GINS2 by co-immunoprecipitation which
is a breakthrough and widens the scope of application of the yeast
two-hybrid assay. In addition, GINS2 siRNA inhibits the cell
survival and apoptosis by activating p38MAP kinase and altering the
expression of Bax/Bcl-2 in K562 and NB4 cells. Our findings suggest
that GINS2 siRNA can exert potent inhibition on leukemia cell
growth by inducing apoptosis. Thus, GINS2 siRNA may be developed as
a promising chemotherapeutic method for the treatment of
leukemia.
Abbreviations:
|
GINS
|
Japanese Go-Ichi-Ni-San meaning
5-1-2-3, for the four-related subunits of the complex Sld5, Psf1,
Psf2 and Psf3;
|
|
PML
|
promyelocytic leukemia;
|
|
RARα
|
retinoic acid receptor α;
|
|
APL
|
acute myelocytic leukemia;
|
|
HIPK2
|
homeodomain-interacting protein kinase
2;
|
|
CK2
|
casein kinase 2;
|
|
NE
|
neutrophil elastase;
|
|
K562
|
chronic myelocytic leukemia K562
cells;
|
|
NB4
|
acute promyelocytic leukemia NB4
cells;
|
|
MAPK
|
mitogen-activated protein kinases;
|
|
HEK293
|
human embryonic kidney 293 cells;
|
|
GFP
|
green fluorescence protein;
|
|
FBS
|
fetal bovine serum;
|
|
BSA
|
bovine serum albumin;
|
|
NC
|
negative control;
|
|
shRNA
|
short haipin RNA
|
Acknowledgements
This study was supported by the
National Nature Science Foundation of China (NSFC, 81171658) and
the Chongqing Natural Science Foundation of China (CSTC,
2011BA5037).
References
|
1
|
de Thé H, Lavau C, Marchio A, Chomienne C,
Degos L and Dejean A: The PML-RAR alpha fusion mRNA generated by
the t(15;17) translocation in acute promyelocytic leukemia encodes
a functionally altered RAR. Cell. 66:675–684. 1991.PubMed/NCBI
|
|
2
|
Hayakawa F and Privalsky ML:
Phosphorylation of PML by mitogen-activated protein kinases plays a
key role in arsenic trioxide-mediated apoptosis. Cancer Cell.
5:389–401. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shima Y, Shima T, Chiba T, Irimura T,
Pandolfi PP and Kitabayashi I: PML activates transcription by
protecting HIPK2 and p300 from SCFFbx3-mediated degradation. Mol
Cell Biol. 28:7126–7138. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Scaglioni PP, Yung TM, Cai LF, et al: A
CK2-dependent mechanism for degradation of the PML tumor
suppressor. Cell. 126:269–283. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
So CW, Dong S, So CK, et al: The impact of
differential binding of wild-type RARalpha, PML-, PLZF- and
NPM-RARalpha fusion proteins towards transcriptional co-activator,
RIP-140, on retinoic acid responses in acute promyelocytic
leukemia. Leukemia. 14:77–83. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lane AA and Ley TJ: Neutrophil elastase is
important for PML-retinoic acid receptor alpha activities in early
myeloid cells. Mol Cell Biol. 25:23–33. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lane AA and Ley TJ: Neutrophil elastase
cleaves PML-RARα and is important for the development of acute
promyelocytic leukemia in mice. Cell. 115:305–318. 2003.PubMed/NCBI
|
|
8
|
Zhu D, Wang C, Liu B, Zhong L, Wang C and
Wu Y: Screening and identification of target proteins interacting
with structural domain of PML-C by yeast two-hybrid system
techniques. Yi Xue Fen Zi Sheng Wu Xue Za Zhi. 7:242–246. 2010.(In
Chinese).
|
|
9
|
Walther A, Houlston R and Tomlinson I:
Association between chromosomal instability and prognosis in
colorectal cancer: a meta-analysis. Gut. 57:941–950. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
MacNeill SA: Structure and function of the
GINS complex, a key component of the eukaryotic replisome. Biochem
J. 425:489–500. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Takayama Y, Kamimura Y, Okawa M, Muramatsu
S, Sugino A and Araki H: GINS, a novel multiprotein complex
required for chromosomal DNA replication in budding yeast. Genes
Dev. 17:1153–1165. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kanemaki M, Sanchez-Diaz A, Gambus A and
Labib K: Functional proteomic identification of DNA replication
proteins by induced proteolysis in vivo. Nature. 423:720–724. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Moyer SE, Lewis PW and Botchan MR:
Isolation of the Cdc45/Mcm2–7/GINS (CMG) complex, a candidate for
the eukaryotic DNA replication fork helicase. Proc Natl Acad Sci
USA. 103:10236–10241. 2006.
|
|
14
|
Pacek M, Tutter AV, Kubota Y, Takisawa H
and Walter JC: Localization of MCM2–7, Cdc45, and GINS to the site
of DNA unwinding during eukaryotic DNA replication. Mol Cell.
21:581–587. 2006.
|
|
15
|
Chang YP, Wang G, Bermudez V, Hurwitz J
and Chen XS: Crystal structure of the GINS complex and functional
insights into its role in DNA replication. Proc Natl Acad Sci USA.
104:12685–12690. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
De Falco M, Ferrari E, De Felice M,
Hübscher U and Pisani FM: The human GINS complex binds to and
specifically stimulates human DNA polymerase alpha-primase. EMBO
Rep. 8:99–103. 2007.PubMed/NCBI
|
|
17
|
Boskovic J, Coloma J, Aparicio T, et al:
Molecular architecture of the human GINS complex. EMBO Rep.
8:678–684. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Barkley LR, Song IY, Zou Y and Vaziri C:
Reduced expression of GINS complex members induces hallmarks of
pre-malignancy in primary untransformed human cells. Cell Cycle.
8:1577–1588. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Matsuoka S, Ballif BA, Smogorzewska A, et
al: ATM and ATR substrate analysis reveals extensive protein
networks responsive to DNA damage. Science. 316:1160–1166. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hayashi R, Arauchi T, Tategu M, Goto Y and
Yoshida K: A combined computational and experimental study on the
structure-regulation relationships of putative mammalian DNA
replication initiator GINS. Genomics Proteomics Bioinformatics.
4:156–164. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rantala JK, Edgren H, Lehtinen L, et al:
Integrative functional genomics analysis of sustained polyploidy
phenotypes in breast cancer cells identifies an oncogenic profile
for GINS2. Neoplasia. 12:877–888. 2010.
|
|
22
|
Ryu B, Kim DS, Deluca AM and Alani RM:
Comprehensive expression profiling of tumor cell lines identifies
molecular signatures of melanoma progression. PLoS One. 2:e5942007.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nakahara I, Miyamoto M, Shibata T, et al:
Up-regulation of PSF1 promotes the growth of breast cancer cells.
Genes Cells. 15:1015–1024. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Singh SV, Herman-Antosiewicz A, Singh AV,
et al: Sulforaphane-induced G2/M phase cell cycle arrest involves
checkpoint kinase 2-mediated phosphorylation of cell division cycle
25C. J Biol Chem. 279:25813–25822. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Morgan DO: Principles of CDK regulation.
Nature. 374:131–134. 1995. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Toyoshima F, Moriguchi T, Wada A, Fukuda M
and Nishida E: Nuclear export of cyclin B1 and its possible role in
the DNA damage-induced G2 checkpoint. EMBO J. 17:2728–2735. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tang D, Wu D, Hirao A, et al: ERK
activation mediates cell cycle arrest and apoptosis after DNA
damage independently of p53. J Biol Chem. 277:12710–12717. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Goulet AC, Chigbrow M, Frisk P and Nelson
MA: Selenomethionine induces sustained ERK phosphofylation leading
to cell-cycle arrest in human colon cancer cells. Carcinogenesis.
26:109–117. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bulavin DV, Amundson SA and Fornace AJ:
p38 and Chk l kinases: different conductors for the
G(2)/M checkpoint symphony. Curr Opin Genet Dev.
12:92–97. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bulavin DV, Higashimoto Y, Popoff IJ, et
al: Initiation of G2/M checkpoint after ultraviolet radiation
requires p38 kinase. Nature. 411:102–107. 2001. View Article : Google Scholar : PubMed/NCBI
|