Introduction
Parkinson’s disease (PD) is a common
neurodegenerative disorder characterized by the selective and
progressive loss of dopaminergic neurons in the substantia nigra,
leading to a depletion of the dopamine neurotransmitter in the
striatum. Although the underlying mechanisms by which nigrostriatal
dopaminergic neuron degeneration are not completely understood,
accumulating data suggest that reactive oxygen species (ROS)
induced by oxidative stress play a crucial role in dopaminergic
cell death in PD (1,2). PC12 cells treated with
1-methyl-4-phenylpyridinium (MPP+) provide a reliable
in vitro model for insight into the pathogenesis of PD. PC12
cells are a rat pheochromocytoma cell line which has been shown to
produce dopamine (3), and
possesses a dopaminergic uptake system (4), indicating that these cells can be
used as a cell culture model for studying the dopaminergic neuron
degeneration. 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)
is a neurotoxin which selectively kills dopaminergic neurons and
causes irreversible parkinsonism-like symptoms in humans and
primates (5–8). MPTP is a lipophilic molecule and can
rapidly cross the blood-brain barrier. Once it crosses the barrier,
it is oxidized in the brain to its toxic metabolite MPP+
by type B monoamine oxidase (9),
and finally causes dopaminergic neuron death. The exact mechanism
for this toxic action of MPP+, although not fully
understood, appears to involve ROS generation, which initiates the
downstream apoptotic pathway. MPP+ is taken up by
dopaminergic neurons via DA transporter and accumulates in
mitochondria where it causes excessive ROS formation by inhibiting
the respiration complex I (10).
The interaction of ROS and mitochondria increases the permeability
in mitochondria transmembrane which results in cytochrome c
release, followed by caspase-3 activation, the crucial contributor
to the neuronal cell death in PD (11).
Currently, the main therapeutic approaches for
treating PD are limited to the replacement of dopamine in the brain
either by levodopa or by dopamine agonists. Although these drugs
can relieve the symptoms of PD, the degeneration of dopaminergic
neurons is not prevented and still slowly progresses. Promising new
concepts in the therapy of PD should prevent or reverse the
progressive dopaminergic neuron degeneration. Since
neurodegenerative diseases are mainly mediated by ROS generation,
attenuation of ROS levels may result in neuroprotection in these
disorders. LA, also known as 1,2-dithiolane-3-pentanoic acid or
thioctic acid, is a pleiotropic compound with potential
pharmacotherapeutic value against a range of pathophysiological
insults. Evidence has shown that LA has effective antioxidative
activities by scavenging ROS (12,13) and inhibits free radical formation
by chelating various metal types (14), indicating that it can exert
beneficial effects on various disorders correlated with oxidative
stress including PD. LA is also reported to have anti-inflammatory
properties and to increase intracellular glutathione (GSH)
formation in a range of cell types and tissues (15–17), which may be beneficial in
neurodegenerative conditions. Notably, LA serving as an effective
antioxidant has been in common clinical use for several diseases
that are associated with increased production of free radicals
(13,18–22), and has been administered in
moderate doses with no evidence of serious side-effects (23,24). These properties of LA may be
beneficial in PD, a neurodegenerative disease in which increasing
formation of ROS is involved. Therefore, in the present study, we
investigated whether LA attenuated MPP+-induced
oxidative damage in PC12 cells, which may provide potential
effective strategies for PD treatment.
Materials and methods
Drugs and chemicals
All reagents and chemicals were purchased from
Sigma-Aldrich (St. Louis, MO, USA) unless noted otherwise.
PC12 cell cultures
PC12 cells, obtained from the Cell Bank of the
Chinese Academy of Sciences (Shanghai, China), were maintained in
high glucose DMEM supplemented with 10% serum, 4.00 mM L-Glutamine,
100 U/ml of penicillin and 100 U/ml of streptomycin (Gibco, Grand
Island, NY, USA). The cultures were maintained in a humidified 5%
CO2 atmosphere at 37°C, and culture medium was changed
every 3–4 days. The cells were seeded at a density of 30,000
cells/cm2.
Cell viability assay
Cell viability was evaluated by using the modified
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay, which is a colormetric assay for measuring the activity of
mitochondrial dehydrogenases that convert MTT into blue formazan
crystals. Briefly, after plating at the density of 30,000
cells/cm2 on 96-well plates for 24 h, PC12 cells were
exposed to different concentrations of MPP+ (100-1,000
μM) for 24, 48 and 72 h. The wells were supplemented with
MTT solution (5 mg/ml) and incubated for 4 h, and the culture
medium was then removed and dimethyl sulfoxide was added to each
well to solubilize the formazan into a colored solution. The
absorbance of colored solution was measured at 570 nm using a
microplate reader (BioTek Epoch; BioTek Instruments, Inc., USA).
Results were expressed as the percentage of the absorbance of
vehicle-treated control culture wells. Following time- and
dose-response studies, the conditions of 500 μM
MPP+ for 72 h were chosen in all experiments.
To assess the neuroprotective effects of LA on
MPP+-induced toxicity in PC12 cells, the cells were
treated with different concentrations of LA (0,001–1,000
μM), 1 h prior to the addition of 500 μM
MPP+. Cell viability was assessed 72 h later by
measuring the absorbance of colored solution. Based on these
results, we used 0.01 μM LA in all subsequent
experiments.
Nuclear staining assay
Nuclear change induced by MPP+ was
evaluated using acridine orange/ethidium bromide staining (AO/EB).
Cells at the concentration of 30,000 cells/cm2 were
plated on 6-well plates and incubated in DMEM medium at 37°C. After
1 h of pretreatment with LA (0.01 μM), MPP+ (500
μM) was added for 72 h. The cells were washed three times
and resuspended in PBS. Then, the samples were stained with AO/EB
(final concentration 1 μg/ml) and at least 200 cells were
randomly observed under a fluorescence microscope (IX71; Olympus,
Japan). Viable cells with intact structures stained only with AO
showed bright green nuclear staining, the early apoptotic cells
were bright green and the later apoptotic cells were red-orange
with condensed chromatin. The number of apoptotic cells is
expressed as a percentage of total cells counted.
Flow cytometric analysis of
apoptosis
The membrane and nuclear events during apoptosis
were analyzed by flow cytometry using FITC-Annexin V and propidium
iodide (PI) staining. Following treatment, PC12 cells were
harvested and centrifuged for 10 min at room temperature at 1,000 ×
g. Cells were washed with PBS and resuspended in binding buffer,
and then 5 μl Annexin V-FITC (20 μg/ml) and 5
μl PI (50 μg/ml) were added. After incubating in the
dark for 15 min, the samples were analyzed by flow cytometry
(Becton-Dickinson). The assay was performed with a two-color
analysis of FITC-labeled Annexin V binding and the uptake of PI.
Living cells (Annexin V−/PI−, Q3), early apoptotic cells (Annexin
V+/PI−, Q4), late apoptotic cells (Annexin V+/PI+, Q2), and
necrotic cells (Annexin V−/PI+, Q1) were distinguished. Therefore,
the total apoptotic proportion included the percentage of cells
with fluorescence for Annexin V+/PI− and Annexin V+/PI+.
ROS activity assay
The production of ROS in the PC12 cells was measured
by DCFH-DA assay based on the ROS-dependent oxidation of DCFH-DA to
2′,7′-dichlorofluorescein (DCF). The cells were seeded at
3×105 cells/well in 6-well tissue culture plates. After
1 h of pretreatment with LA, MPP+ was added for 72 h.
The cells were then incubated in BSA-free DMEM with DCFH-DA at a
final concentration of 20 μM for 30 min at 37°C. DCFH-DA
crosses the cell membrane and is hydrolyzed by intracellular
esterases to non-fluorescent DCFH. It is then trapped in the cells
and becomes fluorescent following oxidation to DCF by ROS. The
intensity of fluorescence was recorded using a flow cytometer
(Becton-Dickinson). The excitation wavelength was 485 nm and the
reading was performed at 530 nm.
Measurement of mitochondrial
transmembrane potential
Mitochondrial transmembrane potential was measured
by flow cytometry with mitochondrial dye 3,3-dihexyloxacarbocyanine
iodide [DiOC6(3)]. DiOC6(3) is lipophilic
fluorescent stain that becomes highly fluorescent when incorporated
into membranes. The levels of mitochondria depolarization represent
membrane permeabilization. The cells at a concentration of 30,000
cells/cm2 were cultured in 24-well plates for 24 h,
followed by the treatment of 0.01 μM LA prior to the
addition of 500 μM MPP+ 1 h. After 72 h of
incubation, 1 ml of serum-free culture medium containing
DiOC6(3) was added to each well with the final
concentration of 1 μM and the cells were cultured in a
humidified incubator for 15 min. For the detection of mitochondrial
transmembrane potential, cells were collected and centrifuged at
1,000 × g for 5 min, and the cell pellets were resuspended in PBS
containing 0.5 mM EDTA. Thereafter, cells were analyzed by flow
cytometry using the FL1 flow cytometer detection channels.
Western blot analysis
Following treatment, PC12 cells were collected and
extracted in lysis buffer containing 7 M urea, 2 M thiourea, 4%
CHAPS 30 mM Tris, 1% protease inhibitor and 1% nuclease mix. The
protein concentration was determined with the 2-D Quant kit
(Amersham Biosciences, Uppsala, Sweden). Equal amounts of protein
were loaded onto a 12% SDS-polyacrylamide gel. Following
electrophoretic separation, the polyacrylamide gels were
transferred to PVDF transfer membrane (Amersham Biosciences), and
western blotted using rabbit anti-Bax, anti-Bcl-2. Horseradish
peroxidase (HRP)-conjugated anti-rabbit antibodies were used as the
secondary antibodies.
Evaluation of caspase-3 activity
Caspase-3 activity was determined using an
ApoAlert® caspase-3 assay kit according to the
manufacturer’s instructions. Following treatment, the cells were
collected and centrifuged at 400 × g for 10 min. The cell pellet
was lysed in 50 μl of lysis buffer. After incubating for 10
min on ice, the cells were centrifuged at 10,000 × g for 3 min at
4°C. The supernatant was collected and the reaction mixture
containing dithiothreitol and caspase-3 substrate
(N-Acetyl-Asp-Glu-Val-Asp-p-nitroanilide) was added. Samples were
incubated for an additional 1 h at 37°C. Absorbance of the
chromophore p-nitroanilide produced was measured at 405 nm. The
standard curves were obtained from the absorbance of p-nitroanilide
standard reagent diluted with cell lysis buffer (up to 20 nM). One
unit of the enzyme was defined as the activity producing 1 nmol of
p-nitroanilide.
Statistical analysis
Data are expressed as the means ± SEM. Statistical
analysis was performed by one-way analysis of variance, followed by
Dunnett’s multiple comparisons test. P<0.05 was considered to
indicate a statistically significant difference.
Results
The neurotoxicity of MPP+ in
PC12 cells
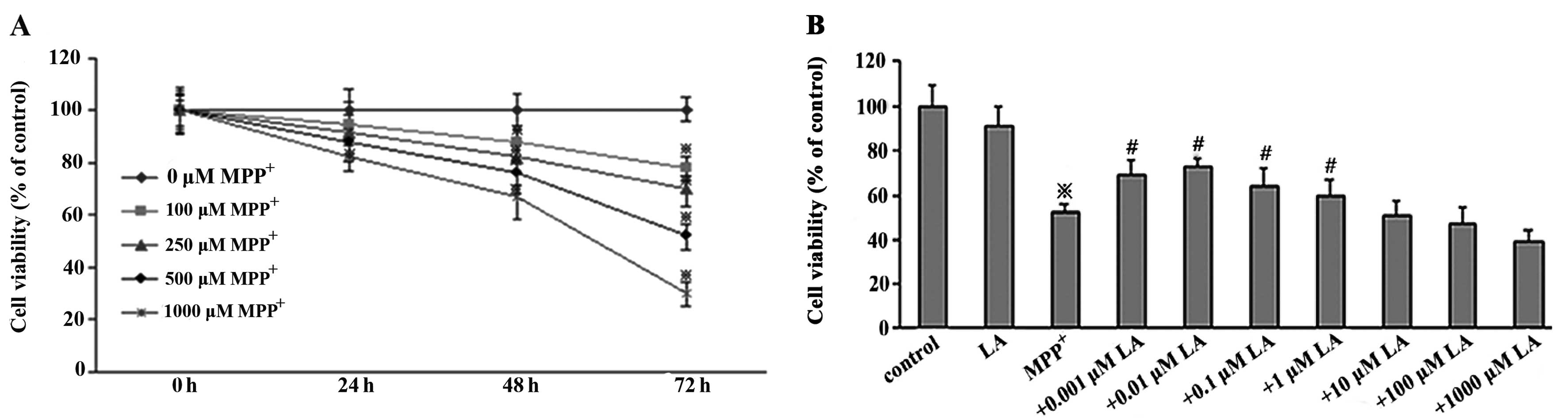
To determine the toxicity of MPP+ in PC12
cells, cell viability was assessed using MTT, a mitochondrial dye,
which can be converted into a blue formazan product by
mitochondrial dehydrogenases, thereby partially detecting the
levels of metabolically active cells. PC12 cells were treated with
different concentrations of MPP+ for different periods
of time. The measurements revealed that the treatment of the cells
for 72 h with 500 μM MPP+ caused ~48% cell
viability loss (Fig. 1A). The
higher concentrations of MPP+ resulted in significant
and irreversible neurotoxicity, which would prevent neuroprotection
measurement. Thus, the cells exposed to 500 μM
MPP+ for 72 h were selected as optimal conditions to
measure neuroprotection of LA against the toxicity of
MPP+.
LA reduces MPP+-induced cell
viability loss
The ability of LA to prevent the cytotoxicity of
MPP+ in PC12 cells was measured by the MTT assay. The
measurements revealed significant decrease in cell viability in
PC12 cells following exposure to 500 μM MPP+ for
72 h, but not in LA used alone. In the presence of 500 μM
MPP+, 0.01 μM LA showed a significant protective
effect on the cells (Fig.
1B).
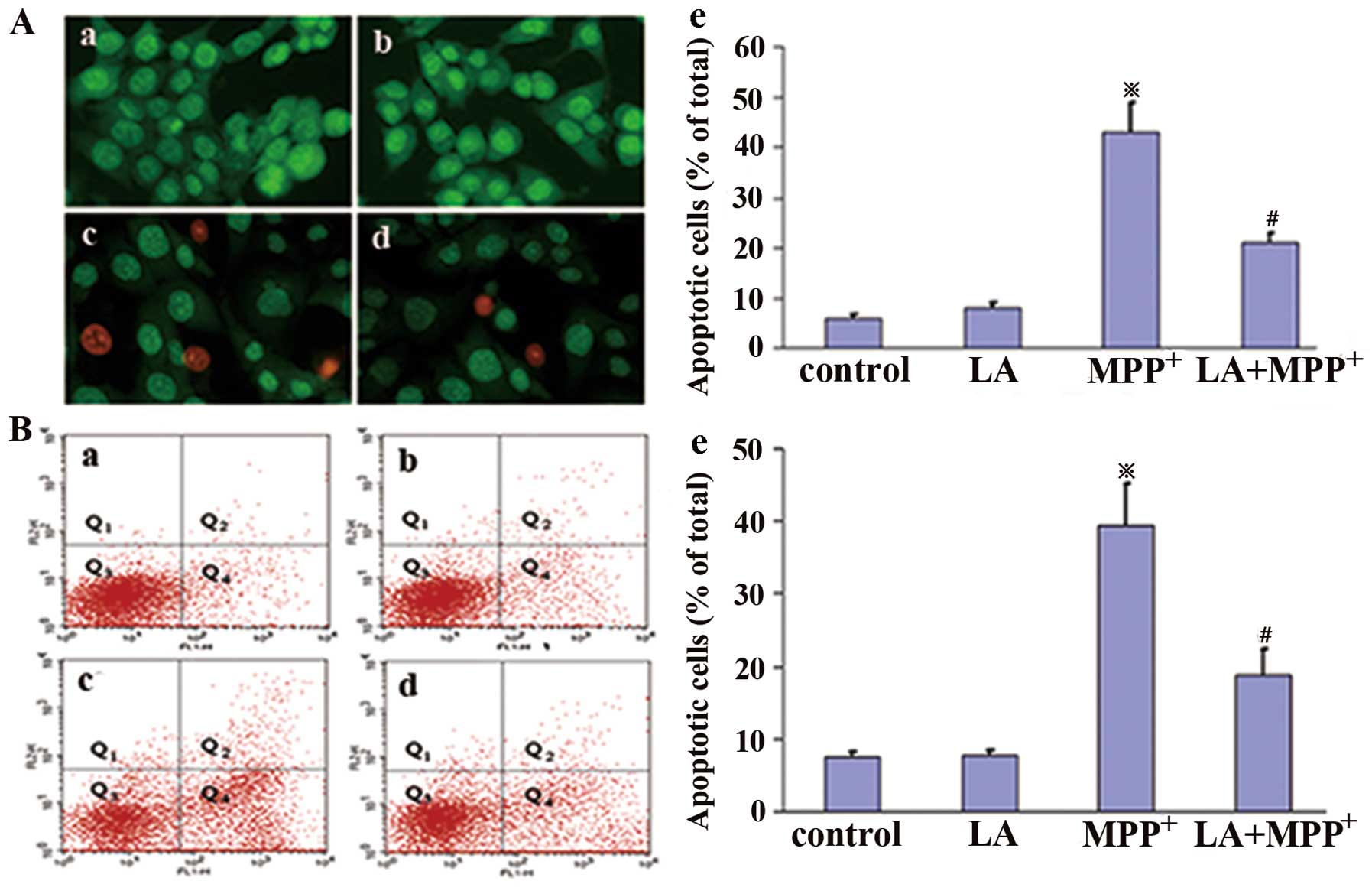
LA exerts anti-apoptotic effects against
the cytotoxicity of MPP+ in PC12 cells
To determine whether LA protects PC12 cells from
MPP+-induced apoptosis, we undertook AO/EB assay and
FITC-Annexin V and PI staining. Apoptosis is a major type of cell
death, characterized by a series of nuclear morphological changes.
These changes can be detected by AO/EB staining. The fluorescence
microscopic analysis results are shown in Fig. 2A, and three types of cells can be
recognized under fluorescence microscopy: live cells (green), early
apoptotic cells (bright green with condensed chromatin), and later
apoptotic cells (red-orange with condensed chromatin). Following
administration of 0.01 μM LA, the proportion of apoptotic
cells induced by MPP+ treatment was significantly
reduced, indicating that LA significantly prevents
MPP+-induced apoptosis in PC12 cells.
FITC-Annexin V and PI staining were analyzed by flow
cytometry, and the total apoptotic cells included early apoptotic
cells (Annexin V+/PI−, Q4) and late apoptotic cells (Annexin
V+/PI+, Q2). The results showed that LA administered alone did not
significantly induce changes in the number of apoptotic cells,
while the administration of MPP+ markedly increased
their number in comparison with control cells (Fig. 2B). This increase was strongly
prevented when LA was administered 1 h prior to MPP+,
confirming the anti-apoptotic activities of LA against the toxicity
of MPP+.
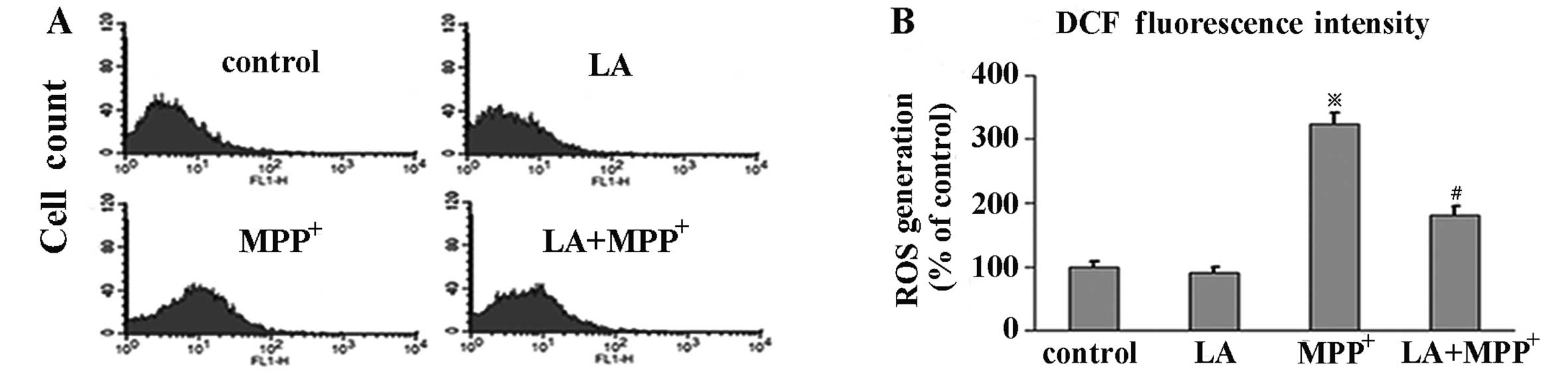
LA inhibits the formation of ROS
To investigate the effects of LA on ROS levels in
MPP+-induced PC12 cells, we evaluated the levels of ROS
by flow cytometry with DCFH-DA, a stable compound, which can easily
diffuse into cells and is converted into DCFH by intracellular
esterase. DCFH is then trapped within cells and oxidized to highly
fluorescent DCF by ROS, thereby the intensity of fluorescence
produced by DCF may reflect an intracellular oxidative state. Our
results showed that the administration of LA alone, compared with
the control group, did not elicit DCF fluorescence intensity
change. Exposure of PC12 cells to MPP+ alone induced a
significant increase of DCF fluorescence levels, which was
prevented by pre-treatment with LA, indicating a preventive role of
LA in ROS formation in PC12 cells elicited by MPP+
(Fig. 3).
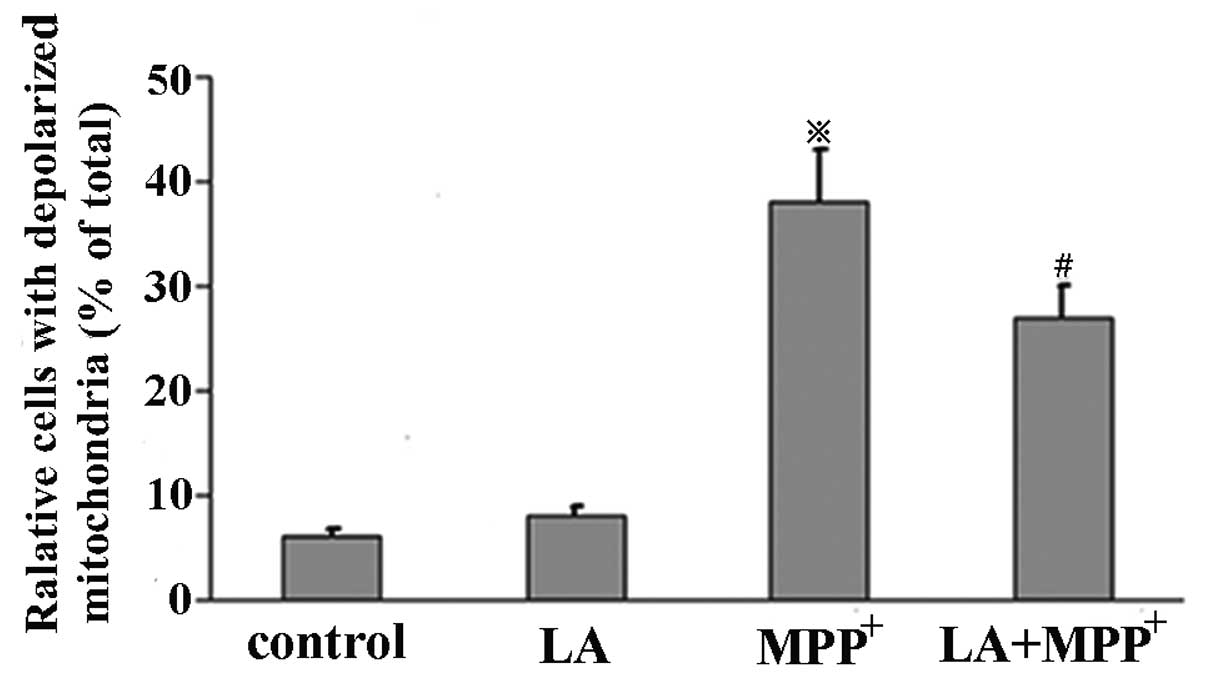
LA prevents MPP+-induced
mitochondrial transmembrane potential discharge
Mitochondrial membrane potential was assessed by
measuring the response to the mitochondrial dye
DiOC6(3), which is converted into highly green
fluorescence by the incorporation into mitochondrial membranes,
thereby allowing qualitative assessment of the mitochondrial
membrane potential. When MPP+ was administered alone, we
observed the increased percentage of cells with depolarized
mitochondrion in comparison with control cells. We also observed a
marked reduction in the number of cells with depolarized
mitochondrion when LA was added prior to MPP+ and LA
alone did not induce the change (Fig.
4). These results demonstrate that the mitochondrial membrane
permeability induced by MPP+ is restored by the
administration of LA.
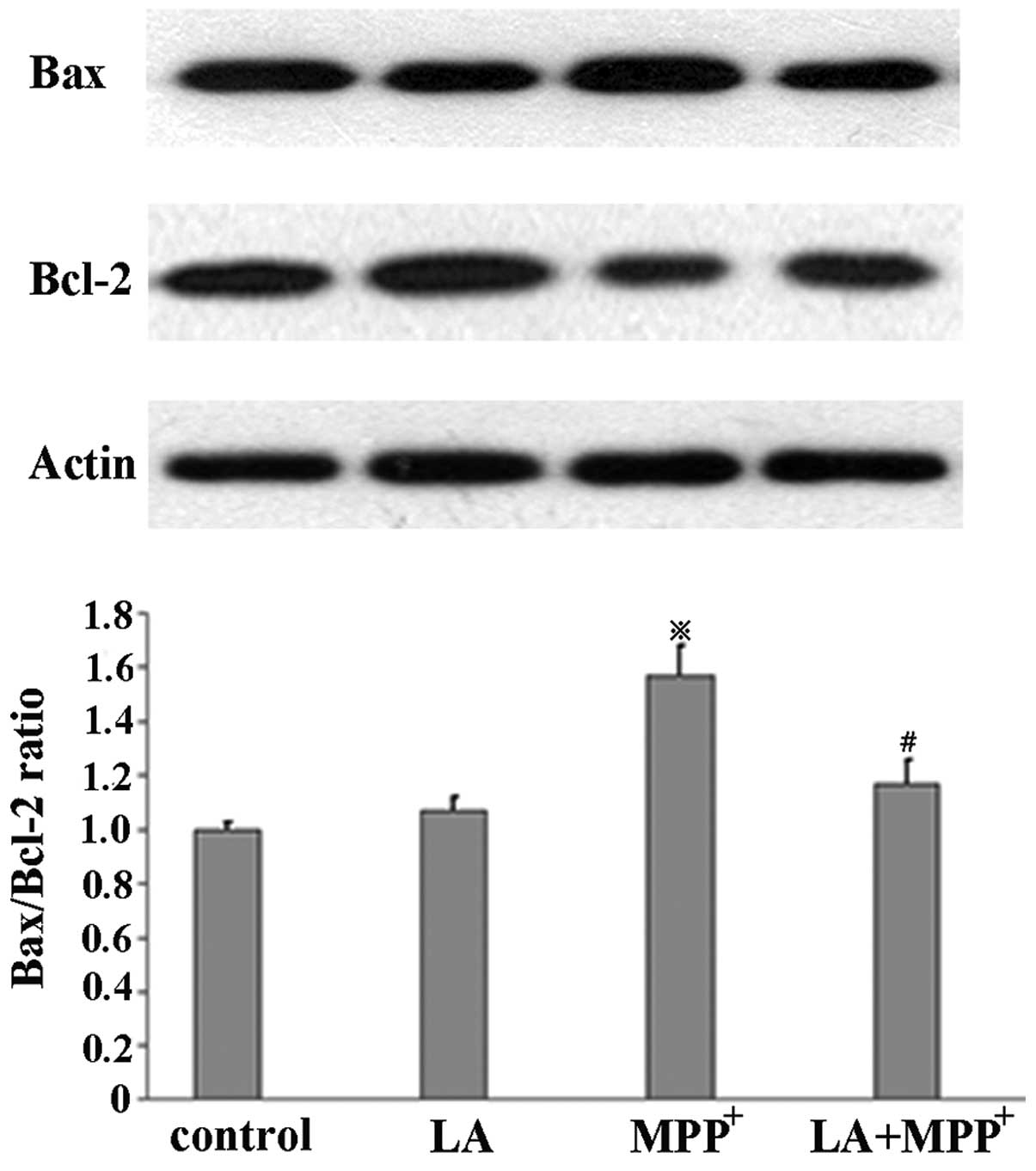
LA modulates Bax and Bcl-2 protein
expression
To examine the effects of LA on Bax and Bcl-2
protein levels induced by MPP+ in PC12 cells, western
blotting was performed in the cells untreated, or treated with 500
μM MPP+ alone, or treated with 500 μM
MPP+ in the presence of 0.01 μM LA. When
MPP+ was administered alone, we observed a significant
increase in the Bax/Bcl-2 ratio compared with control cells. We
also observed a marked reduction in the Bax/Bcl-2 ratio when LA was
added prior to MPP+ and LA alone did not induce the
change (Fig. 5).
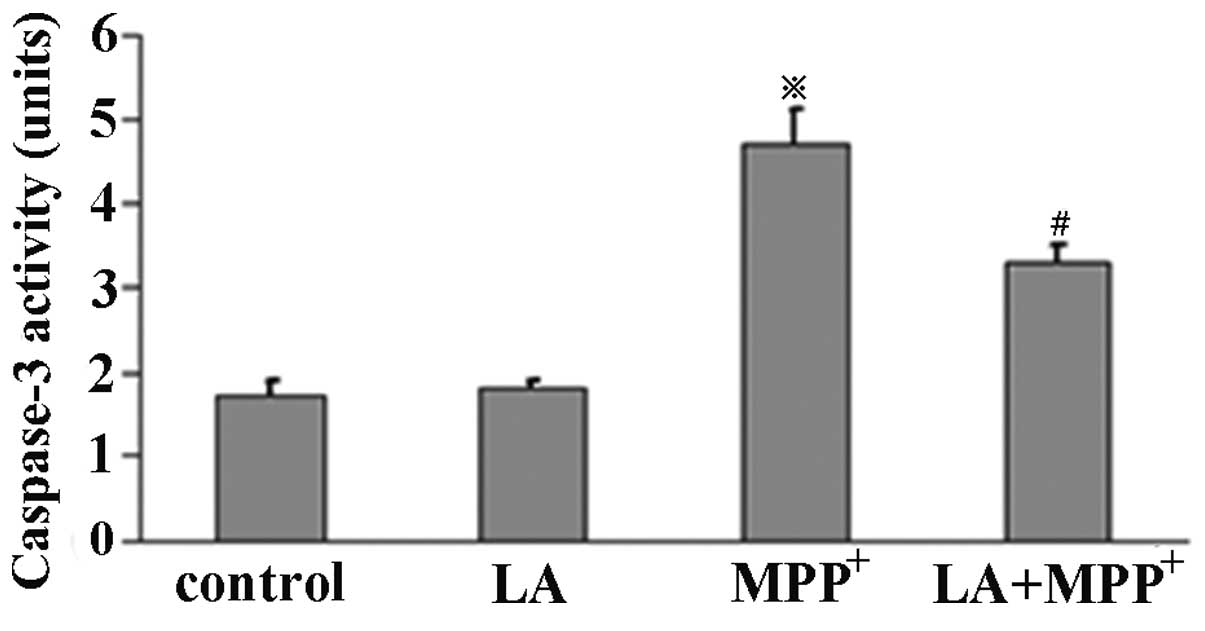
LA reduces caspase-3 activity
To investigate the effects of LA on caspase-3
activity, ELISA was performed in PC12 cells. The cells were
pretreated with LA for 1 h and then exposed to MPP+ or
not for 72 h. Caspase-3 activity was determined using an ApoAlert
caspase-3 assay kit. The results demonstrated that PC12 cells
treated with 500 μM MPP+ showed a significant
increase in caspase-3 activity, but not in LA used alone.
Pretreatment of LA significantly suppressed the
MPP+-induced caspase-3 activity (Fig. 6), suggesting a protective role of
LA against the MPP+ toxicity in PC12 cells.
Discussion
The present investigation indicates for the first
time that LA, a naturally occurring dithiol compound, can exert
protective effects against apoptotic cell death elicited by
MPP+ in PC12 cells, a reliable model for PD in which
dopaminergic neurons present a slow and progressive loss. LA
effectively inhibits MPP+-induced ROS formation,
mitochondrial potential discharge, caspase-3 activation, and
cytotoxic cell death in dopaminergic neurons.
Oxidative stress is a central event in a range of
pathological conditions. Mitochondria are generally considered to
be the primary source of ROS within most mammalian cells (25–28), and the generation of ROS is mainly
correlated with the inhibition of complex I (25,29–31). This production of ROS activates
the mitochondrial cell death pathway by oxidative damage to
mitochondria and/or by transferring redox signal from the organelle
to the nucleus. Such a pathway appears to underlie the pathological
processes of PD, in which the inhibition of the mitochondrial
complex I elicited by the neuron toxicant increases the formation
of ROS that cause the mitochondrial dysfunction (32,33), and finally cause cell death. MPTP,
a well known neurotoxin, has been widely used to study the
pathogenesis of PD. Its toxic metabolite MPP+ is thought
to selectively kill dopaminergic neurons and to elicit severe
parkinsonism-like symptoms in humans and primates (5–8).
ROS serving as a key mediator are responsible for
MPP+-induced oxidative damage in dopaminergic neuronal
cells. This contribution of ROS may be related to mitochondrial
membrane potential discharge, cytochrome c release and
caspase-3 activation, which together contribute to cell death. LA,
also known as 1,2-dithiolane-3-pentanoic acid, can be synthesized
enzymatically in the mitochondria from octanoic acid and absorbed
from dietary sources including animal and plant foods (34). LA functioning as an essential
cofactor for mitochondrial α-ketoacid dehydrogenases plays a
critical role in mitochondrial energy metabolism. Its protective
actions against oxidative stress and toxin insults have been
reported in vitro and in vivo studies (35–38). We investigated the anti-apoptotic
activity of LA in PC12 cells treated with MPP+, a
reliable cellular model of PD. These results showed that the number
of apoptotic cells significantly decreased when LA was administered
1 h prior to MPP+, and the activation of caspase-3 was
also prevented by the administration of LA. Moreover, in PC12 cells
treated by MPP+ we established that LA inhibited the
formation of ROS, an oxidative damage inducer and redox signal
transducer in a range of pathogeneses (27,39). Our studies demonstrated that when
MPP+ was administered, the fluorescence density of DCF
significantly increased, indicating high ROS levels. By contrast,
the treatment with LA prior to MPP+ administration
indicated low ROS levels. These results indicate that the
neuroprotective actions of LA may be mediated by the inhibition of
ROS formation, which reverses the neurodegenerative processes.
Previous evidence suggested that mitochondria play a key role in
neurodegenerative diseases, including Alzheimer’s disease (AD),
Huntington’s disease (HD), amyotrophic lateral sclerosis (ALS) and
Parkinson’s disease (PD) (40–43). Mitochondrial damage mediated by
ROS have been recognized as a central event in dopaminergic neuron
degeneration (44,45). Maintenance of mitochondrial
membrane potential is essential for living cells; its loss has
numerous consequences for the cells, including apoptosis (46). In agreement with these reports, in
the present study, mitochondrial transmembrane potential was
implicated in PD. We also observed a marked reduction in the number
of cells with depolarized mitochondrion when LA was administered,
supporting a neuroprotective role of LA in the neurodegenerative
diseases mediated by the mitochondrial cell death pathway. Similar
protection of LA by inhibiting ROS production and mitochondrial
membrane depolarization has previously been reported in other cell
types such as β cells (47).
Bax and Bcl-2 are key members of the Bcl-2 protein
family which plays a central role in the mitochondrial cell death
pathway. Bax increases the mitochondrial membrane permeabilization
by inducing the mitochondrial transmembrane pore formation
(48), leading to the
pro-apoptotic molecule release. By contrast, Bcl-2 preserves
mitochondrial membrane integrity, thereby inhibiting the release of
apoptogenic molecules such as cytochrome c (49). The ratio of pro-apoptotic Bax to
anti-apoptotic Bcl-2 has been reported to be strongly correlated
with apoptosis in various pathophysiological conditions (50). In this study, the administration
of MPP+ significantly increased the Bax/Bcl-2 ratio,
which was significantly prevented when LA was administered 1 h
prior to MPP+, further supporting the protective role of
LA against the toxicity of MPP+ in PC12 cells.
The protection of neuron cells from apoptosis has
been underlined in the therapeutic strategy in neurodegenerative
disorders. Studies in vitro and in vivo provide
considerable evidence for the neuroprotective activities of certain
agents, such as EPO or estrogens. However, the adverse effects
associated with these agents should be underlined in some
pathological conditions. Treatment with EPO, for example, increases
red blood cell count and platelet aggregation, the key contributors
to congestive heart failure and ischemic diseases, respectively. LA
serving as a necessary mitochondrial cofactor, is ubiquitous in
living organisms including neurons. Owing to its powerful
antioxidative activities, LA has been used in several disorders to
prevent oxidative stress (51,52). Preclinical and clinical data
demonstrate that LA is bioavailable and safe in moderate doses
(51). It was administered
intravenously in doses of 600 mg/day for three weeks with no
serious side-effects (23). Oral
doses of 1,800 mg LA (600 mg t.i.d.) for six months did not elicit
significant adverse effects (24). In addition to directly scavenging
ROS, LA is reported to increase intracellular glutathione (GSH)
that is an abundant natural thiol antioxidant and co-substrate for
detoxification enzymes in a range of cell types and tissues
(15–17), further supporting the
antioxidative activities of LA with potential clinical value
against oxidative damage. Evidence has also shown that exogenous LA
can easily cross the blood-brain barrier (53,54), suggesting it can exert
antioxidative effects in the neuronal system.
LA with strong antioxidative activities and clinical
safety record may be an efficient agent in protecting the
dopaminergic neurons against oxidative damage. However, the
majority of biological actions of LA have yet to be fully
elucidated, thus, significantly more research is required to
understand these actions and the underlying mechanisms in the
potential of LA to improve neurodegenerative disorders.
In conclusion, our results demonstrate the marked
protective activities of LA against MPP+-induced
apoptosis in PC12 cells. The exact mechanism for this action,
although incompletely understood, appears to relate to inhibiting
ROS formation and stabilizing mitochondrial membrane, thereby
preventing the downstream pro-apoptotic events including cytochrome
c release and caspase-3 activation. Further studies are
required to elucidate the precise cellular and molecular mechanisms
by which LA protects dopaminergic neurons from degeneration, which
may lead to potential advancement in the treatment of PD with a
neuroprotective strategy.
Abbreviations:
|
ROS
|
reactive oxygen species;
|
|
PD
|
Parkinson’s disease;
|
|
LA
|
α-lipoic acid;
|
|
MPP+
|
1-methyl-4-phenylpyridinium;
|
|
MPTP
|
1-methy-4-phenyl-1,2,3,6-tetrahydropyridine;
|
|
PI
|
propidium iodide;
|
|
DCF
|
2′7′-dichlorodihydrofluorescein
|
References
|
1.
|
Fleury C, Mignotte B and Vayssiere JL:
Mitochondrial reactive oxygen species in cell death signaling.
Biochimie. 84:131–141. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Jenner P: Oxidative stress in Parkinson’s
disease. Ann Neurol. 53(Suppl 3): S26–S38. 2003.
|
|
3.
|
Hatanaka H: Nerve growth factor-mediated
stimulation of tyrosine hydroxylase activity in a clonal rat
pheochromocytoma cell line. Brain Res. 222:225–233. 1981.
View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Rebois RV, Reynolds EE, Toll L and Howard
BD: Storage of dopamine and acetylcholine in granules of PC12, a
clonal pheochromocytoma cell line. Biochemistry. 19:1240–1248.
1980. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Langston JW and Irwin I: MPTP: current
concepts and controversies. Clin Neuropharmacol. 9:485–507. 1986.
View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Kopin IJ and Markey SP: MPTP toxicity:
implications for research in Parkinson’s disease. Annu Rev
Neurosci. 11:81–96. 1988.
|
|
7.
|
Heikkila RE, Sieber BA, Manzino L and
Sonsalla PK: Some features of the nigrostriatal dopaminergic
neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) in
the mouse. Mol Chem Neuropathol. 10:171–183. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Calon F, Lavertu N, Lemieux AM, et al:
Effect of MPTP-induced denervation on basal ganglia GABA(B)
receptors: correlation with dopamine concentrations and dopamine
transporter. Synapse. 40:225–234. 2001. View Article : Google Scholar
|
|
9.
|
Chiba K, Trevor A and Castagnoli N Jr:
Metabolism of the neurotoxic tertiary amine, MPTP, by brain
monoamine oxidase. Biochem Biophys Res Commun. 120:574–578. 1984.
View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Javitch JA, D’Amato RJ, Strittmatter SM
and Snyder SH: Parkinsonism-inducing neurotoxin,
N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine: uptake of the
metabolite N-methyl- 4-phenylpyridine by dopamine neurons explains
selective toxicity. Proc Natl Acad Sci USA. 82:2173–2177. 1985.
View Article : Google Scholar
|
|
11.
|
Hartley A, Stone JM, Heron C, Cooper JM
and Schapira AH: Complex I inhibitors induce dose-dependent
apoptosis in PC12 cells: relevance to Parkinson’s disease. J
Neurochem. 63:1987–1990. 1994.PubMed/NCBI
|
|
12.
|
Packer L, Witt EH and Tritschler HJ:
Alpha-lipoic acid as a biological antioxidant. Free Radic Biol Med.
19:227–250. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Packer L, Tritschler HJ and Wessel K:
Neuroprotection by the metabolic antioxidant alpha-lipoic acid.
Free Radic Biol Med. 22:359–378. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Bustamante J, Lodge JK, Marcocci L,
Tritschler HJ, Packer L and Rihn BH: Alpha-lipoic acid in liver
metabolism and disease. Free Radic Biol Med. 24:1023–1039. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Han D, Tritschler HJ and Packer L:
Alpha-lipoic acid increases intracellular glutathione in a human
T-lymphocyte Jurkat cell line. Biochem Biophys Res Commun.
207:258–264. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Bast A and Haenen GR: Interplay between
lipoic acid and glutathione in the protection against microsomal
lipid peroxidation. Biochim Biophys Acta. 963:558–561. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Busse E, Zimmer G, Schopohl B and
Kornhuber B: Influence of alpha-lipoic acid on intracellular
glutathione in vitro and in vivo. Arzneimittelforschung.
42:829–831. 1992.PubMed/NCBI
|
|
18.
|
Hagen TM, Ingersoll RT, Lykkesfeldt J, et
al: (R)-alpha-lipoic acid-supplemented old rats have improved
mitochondrial function, decreased oxidative damage, and increased
metabolic rate. FASEB J. 13:411–418. 1999.
|
|
19.
|
Packer L, Kraemer K and Rimbach G:
Molecular aspects of lipoic acid in the prevention of diabetes
complications. Nutrition. 17:888–895. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Yilmaz O, Ozkan Y, Yildirim M, Ozturk AI
and Ersan Y: Effects of alpha lipoic acid, ascorbic
acid-6-palmitate, and fish oil on the glutathione, malonaldehyde,
and fatty acids levels in erythrocytes of streptozotocin induced
diabetic male rats. J Cell Biochem. 86:530–539. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Wollin SD and Jones PJ: Alpha-lipoic acid
and cardiovascular disease. J Nutr. 133:3327–3330. 2003.PubMed/NCBI
|
|
22.
|
McNeilly AM, Davison GW, Murphy MH, et al:
Effect of α-lipoic acid and exercise training on cardiovascular
disease risk in obesity with impaired glucose tolerance. Lipids
Health Dis. 10:2172011.
|
|
23.
|
Ziegler D, Hanefeld M, Ruhnau KJ, et al:
Treatment of symptomatic diabetic peripheral neuropathy with the
anti-oxidant alpha-lipoic acid. A 3-week multicentre randomized
controlled trial (ALADIN Study). Diabetologia. 38:1425–1433. 1995.
View Article : Google Scholar
|
|
24.
|
Ziegler D, Hanefeld M, Ruhnau KJ, et al:
Treatment of symptomatic diabetic polyneuropathy with the
antioxidant alpha-lipoic acid: a 7-month multicenter randomized
controlled trial (ALADIN III Study). ALADIN III Study Group
Alpha-Lipoic Acid in Diabetic Neuropathy. Diabetes Care.
22:1296–1301. 1999. View Article : Google Scholar
|
|
25.
|
Andreyev AY, Kushnareva YE and Starkov AA:
Mitochondrial metabolism of reactive oxygen species. Biochemistry
(Mosc). 70:200–214. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Turrens JF: Mitochondrial formation of
reactive oxygen species. J Physiol. 552:335–344. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Balaban RS, Nemoto S and Finkel T:
Mitochondria, oxidants, and aging. Cell. 120:483–495. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Adam-Vizi V and Chinopoulos C:
Bioenergetics and the formation of mitochondrial reactive oxygen
species. Trends Pharmacol Sci. 27:639–645. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Brookes PS: Mitochondrial H(+) leak and
ROS generation: an odd couple. Free Radic Biol Med. 38:12–23.
2005.
|
|
30.
|
Kussmaul L and Hirst J: The mechanism of
superoxide production by NADH: ubiquinone oxidoreductase (complex
I) from bovine heart mitochondria. Proc Natl Acad Sci USA.
103:7607–7612. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Sipos I, Tretter L and Adam-Vizi V:
Quantitative relationship between inhibition of respiratory
complexes and formation of reactive oxygen species in isolated
nerve terminals. J Neurochem. 84:112–118. 2003. View Article : Google Scholar
|
|
32.
|
Dauer W and Przedborski S: Parkinson’s
disease: mechanisms and models. Neuron. 39:889–909. 2003.
|
|
33.
|
Przedborski S, Jackson-Lewis V, Djaldetti
R, et al: The parkinsonian toxin MPTP: action and mechanism. Restor
Neurol Neurosci. 16:135–142. 2000.PubMed/NCBI
|
|
34.
|
Kataoka H: Chromatographic analysis of
lipoic acid and related compounds. J Chromatogr B Biomed Sci Appl.
717:247–262. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Smith AR, Shenvi SV, Widlansky M, Suh JH
and Hagen TM: Lipoic acid as a potential therapy for chronic
diseases associated with oxidative stress. Curr Med Chem.
11:1135–1146. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Scott BC, Aruoma OI, Evans PJ, et al:
Lipoic and dihydrolipoic acids as antioxidants. A critical
evaluation. Free Radic Res. 20:119–133. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Liu J, Head E, Gharib AM, et al: Memory
loss in old rats is associated with brain mitochondrial decay and
RNA/DNA oxidation: partial reversal by feeding acetyl-L-carnitine
and/or R-alpha-lipoic acid. Proc Natl Acad Sci USA. 99:2356–2361.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Han D, Sen CK, Roy S, Kobayashi MS,
Tritschler HJ and Packer L: Protection against glutamate-induced
cytotoxicity in C6 glial cells by thiol antioxidants. Am J Physiol.
273:R1771–R1778. 1997.PubMed/NCBI
|
|
39.
|
Droge W: Free radicals in the
physiological control of cell function. Physiol Rev. 82:47–95.
2002.PubMed/NCBI
|
|
40.
|
Reddy PH: Mitochondrial medicine for aging
and neurodegenerative diseases. Neuromol Med. 10:291–315. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Beal MF: Mitochondria take center stage in
aging and neurodegeneration. Ann Neurol. 58:495–505. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Lin MT and Beal MF: Mitochondrial
dysfunction and oxidative stress in neurodegenerative diseases.
Nature. 443:787–795. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Reddy PH, Mao P and Manczak M:
Mitochondrial structural and functional dynamics in Huntington’s
disease. Brain Res Rev. 61:33–48. 2009.PubMed/NCBI
|
|
44.
|
Selvaraj S, Watt JA and Singh BB: TRPC1
inhibits apoptotic cell degeneration induced by dopaminergic
neurotoxin MPTP/MPP(+). Cell Calcium. 46:209–218. 2009.PubMed/NCBI
|
|
45.
|
Abou-Sleiman PM, Muqit MM and Wood NW:
Expanding insights of mitochondrial dysfunction in Parkinson’s
disease. Nat Rev Neurosci. 7:207–219. 2006.
|
|
46.
|
Shiba-Fukushima K, Imai Y, Yoshida S, et
al: PINK1-mediated phosphorylation of the Parkin ubiquitin-like
domain primes mitochondrial translocation of Parkin and regulates
mitophagy. Sci Rep. 2:10022012. View Article : Google Scholar : PubMed/NCBI
|
|
47.
|
Lee BW, Kwon SJ, Chae HY, et al:
Dose-related cytoprotective effect of alpha-lipoic acid on hydrogen
peroxide-induced oxidative stress to pancreatic beta cells. Free
Radic Res. 43:68–77. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
48.
|
Tsujimoto Y and Shimizu S: VDAC regulation
by the Bcl-2 family of proteins. Cell Death Differ. 7:1174–1181.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
49.
|
Gollapudi S, McCormick MJ and Gupta S:
Changes in mitochondrial membrane potential and mitochondrial mass
occur independent of the activation of caspase-8 and caspase-3
during CD95-mediated apoptosis in peripheral blood T cells. Int J
Oncol. 22:597–600. 2003.PubMed/NCBI
|
|
50.
|
Wu Y, Shang Y, Sun SG, Liu RG and Yang WQ:
Protective effect of erythropoietin against
1-methyl-4-phenylpyridinium-induced neurodegenaration in PC12
cells. Neurosci Bull. 23:156–164. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
51.
|
Shay KP, Moreau RF, Smith EJ, Smith AR and
Hagen TM: Alpha-lipoic acid as a dietary supplement: molecular
mechanisms and therapeutic potential. Biochim Biophys Acta.
1790:1149–1160. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
52.
|
Coleman MD, Eason RC and Bailey CJ: The
therapeutic use of lipoic acid in diabetes: a current perspective.
Environ Toxicol Pharmacol. 10:167–172. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
53.
|
Panigrahi M, Sadguna Y, Shivakumar BR, et
al: alpha-Lipoic acid protects against reperfusion injury following
cerebral ischemia in rats. Brain Res. 717:184–188. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
54.
|
Arivazhagan P, Shila S, Kumaran S and
Panneerselvam C: Effect of DL-alpha-lipoic acid on the status of
lipid peroxidation and antioxidant enzymes in various brain regions
of aged rats. Exp Gerontol. 37:803–811. 2002. View Article : Google Scholar
|