Introduction
Diabetes mellitus (DM) is a ~3,000-year-old disease,
already dating back to the Egyptian era (1). In 2011 ~366 million individuals
worldwide suffered from diabetes and an estimated number of ~553
million persons will be affected by 2030 (2).
Since 1936, DM is classified into insulin-dependent
DM (IDDM) [nowadays known as type 1 DM (T1DM)] and
non-insulin-dependent DM (NIDDM) [currently referred to as type 2
DM (T2DM)] (3). The latter form,
which is characterised by a relative lack of insulin, is much more
common comprising ~90–95% of DM cases, whereas T1DM comprises the
residual ~5% of all DM cases (4).
The relative insulin deficiency in T2DM is mainly due to the
insulin resistance of target tissues, secretory defects and/or
failure of the receptors (5). A
combination of genetic predisposition and lifestyle contribute to
the prevalence of T2DM (6).
Common risk factors for T2DM include physical inactivity, smoking,
alcohol abuse and environmental toxins, such as bisphenol A. In
addition, hypertension, obesity and hyperglycaemia, 3 major
hallmarks of metabolic syndrome, have been associated with
increased plasma levels of angiotensin(Ang) II, a central component
of the renin-angiotensin system (RAS) (7–9).
RAS, in particular Ang II, has been implicated in
the onset and progression of T2DM. Systemic RAS is an endocrine
system mainly known for its role in the regulation of blood
pressure, fluid and electrolyte balance, as well as in volume
homeostasis through different active metabolites. From the
precursor of these active peptides, angiotensinogen, a decapeptide
(Ang I) is cleaved off by renin (10). A key enzyme of RAS,
angiotensin-converting enzyme (ACE) then converts Ang I into the
biological active octapeptide, Ang II, which preferentially binds
to its receptors, Ang II type 1 receptor (AT1R) or Ang II type 2
receptor (AT2R) (11).
AT1R-mediated signaling leads to vasoconstriction, increased
production of aldosterone and the secretion of vasopressin and in
addition, promotes responses, such as inflammation, proliferation,
fibrosis and atherosclerosis. On the contrary, AT2R signaling often
exerts opposite effects, such as vasodilation or growth inhibition
(12,13). With the discovery of the ACE
homologue, ACE2 in the year 2000, and the subsequent recognition of
its crucial role in the generation of the Ang peptide,
Ang-(1-7), an important alternative RAS axis has
been established (14,15). The effects of Ang-(1-7),
which can also be produced by prolyl endopeptidase (PEP) or neutral
endopeptidase 24.11 (NEP), are mediated by its putative receptor,
Mas (16,17). These effects are
anti-proliferative, anti-fibrotic, anti-thrombotic and
anti-arrhythmogenic; therefore, they generally oppose the effects
of the classical ACE/Ang II/AT1R axis (18). A second alternative RAS axis
consists of the aminopeptidases A (APA) and N (APN), which
successively convert Ang II into Ang III and then into Ang IV. In
addition to its function in Ang peptide processing, APN has been
implicated in the regulation of immune cell function (19–21). Ang IV binds to the Ang IV receptor
(AT4R) that has been identified as insulin-regulated aminopeptidase
(IRAP). Similar to APN, IRAP also serves multiple functions in
different organs. In adipocytes and muscle, IRAP is co-localised
with the insulin-responsive glucose transporter, GLUT4, and is
redistributed from the endosomes by GLUT4 specialised vesicles
(GSVs) to the cell surface in response to insulin (22,23). This translocation of IRAP to the
cell membrane has been shown to be impaired in patients with T2DM
(24). Further evidence linking
the APN/Ang IV/IRAP axis to glucose homeostasis includes decreased
basal and insulin-stimulated glucose uptake into muscle and fat in
IRAP-deficient mice (25).
Furthermore, Ang IV or its more stable analogue, Nle-Ang IV, have
been reported to stimulate insulin secretion in INS-1 cells, and to
reduce the increase in blood glucose during a glucose tolerance
test or to improve insulin signaling in diet-induced hyperglycaemic
mice (26,27). The different signaling pathways of
RAS are illustrated in Fig.
1.
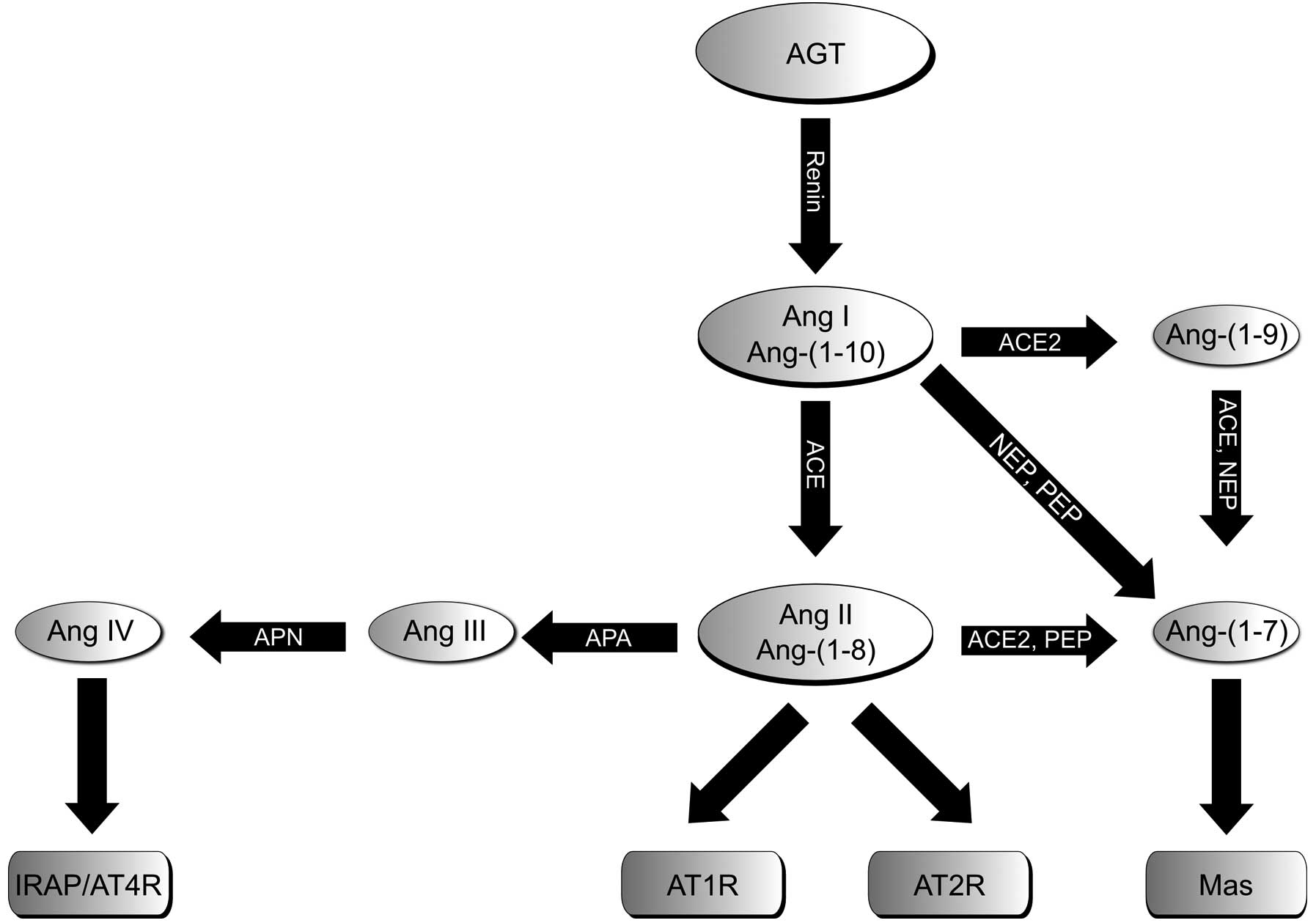 | Figure 1Schematic illustration of RAS
consisting of the classical Ang II/ACE/AT1R axis and AT2R, as well
as the two alternative axes, ACE2/Ang-(1-7)/Mas and APN/Ang IV/IRAP. RAS, renin
angiotensin system; AGT, angiotensinogen; Ang, angiotensin; AT1bR,
angiotensin II type 1b receptor; AT2R, angiotensin II type 2
receptor; ACE, angiotensin-converting enzyme; NEP, neutral
endopeptidase 24.11; PEP, prolyl endopeptidase; Mas, Mas receptor;
APA/APN, aminopeptidases A and N; IRAP, insulin-regulated
aminopeptidase. |
Local tissue-specific RAS acts independently from
circulatory RAS, but can interact with the latter in an endocrine
manner (28,29). This has been shown to exist in the
pancreas, including the islets of Langerhans (30–32). A close association between RAS
activation and diabetes has been confirmed by clinical trials
showing the beneficial effects of ACE inhibitors (ACEis)
and AT1R blockers (ARBs) on the incidence of DM, as well as on the
reduction of cardiovascular complications in patients with DM
[Heart Outcomes Prevention Evaluation (HOPE) and ONTARGET studies
among others] (33,34). Mechanistically, Ang II, by AT1R,
impairs the phosphorylation of insulin receptor substrate 1 (IRS-1)
by alternative phosphorylation on a serine, instead of a tyrosine
residue, thereby decreasing phosphatidylinositol 3-kinase (PI3K)
activity and enhancing mitogen-activated protein kinase (MAPK)
pathways (35). This diminishes
insulin secretion and enforces insulin resistance. Accordingly,
these detrimental effects of Ang II can be abolished by ARBs and/or
ACEis (35,36). Similar beneficial effects on
insulin signaling and hyperglycaemia resulting from the blockade of
the ACE/Ang II/AT1R axis have been demonstrated for Ang-(1-7)
and the concomitant activation of the alternative
ACE2/Ang-(1-7)/Mas axis (37). This view is supported by studies
demonstrating that Ang-(1-7)
prevents metabolic syndrome and improves insulin resistance
(38,39). In adipocytes, Ang-(1-7)/Mas has been shown to affect glucose
uptake and to suppress the production of reactive oxygen species
(ROS) (40).
In this study, using the BRIN-BD11 rat insulinoma
cell line, we examined the expression/activity of three RAS axes.
The effects of the increased concentration of glucose on insulin
production/secretion were assessed in parallel to glucose-dependent
alterations in the expression and activity of local pancreatic
islet RAS and β-cell function. The findings of the present study
suggest a shift from the classical ACE/Ang II/AT1R axis to the
Ang-(1-7)- and Ang IV-triggered alternative RAS
pathways.
Materials and methods
Cultivation and treatment of BRIN-BD11
cells
The BRIN-BD11 cells were cultured in Dulbecco’s
modified Eagle’s medium containing 5.5 mM glucose, 4 mM
L-glutamine, 10% (v/v) fetal calf serum, 100 U/ml penicillin and
100 μg/ml streptomycin at 37°C, 5% CO2 in a humidified
atmosphere for 18–24 h prior to the experiments (all reagents from
PAA Laboratories, Pasching, Austria). The cells were exposed to
various concentrations of glucose (5.5, 11, 15 and 25 mM) or to 40
mM potassium chloride (KCl) in combination with 15 mM glucose and
were incubated under the conditions described above for the periods
of time indicated in the figure legends. For insulin secretion and
expression analyses, in each case 2 million cells in 6 ml medium
were seeded into 60×15 mm cell culture petri dishes; for the
determination of enzyme activities, 0.5 million cells in 4 ml
medium were seeded per well of a 6-well plate.
RNA preparation and reverse transcription
quantitative PCR (RT-qPCR)
RNA was extracted using the innuPrep RNA Mini kit
(Analytik Jena, Jena, Germany) following the manufacturer’s
instructions and the concentration was measured with a
spectrophotometer NanoDrop 2000c (Thermo Fisher Scientific,
Wilmington, DE, USA). RNA (1 μg) was reverse-transcribed using a
Revert Aid™ First Strand cDNA Synthesis kit (Thermo Fisher
Scientific, Braunschweig, Germany) following the instruction manual
using oligo(dT) primers in a 30 μl reaction mixture. RT-qPCR was
performed in a CFX96 thermocycler (Bio-Rad, Munich, Germany). A
typical 20 μl reaction mixture consisted of 1X SensiMix™ SYBR
Hi-ROX Mastermix (Bioline, Luckenwalde, Germany), 250–1,000 nM
primer mix (sense and antisense) and 1 μl cDNA. Initial
denaturation at 95°C for 10 min was followed by 45 cycles at 95°C
for 10 sec, 57–65°C for 15 sec and 72°C for 30 sec. Melt curve
analysis of the amplificates was carried out at 65–95°C with ΔT =
0.5°C every 5 sec. Data obtained by RT-qPCR were evaluated using
the ΔΔ Cq-method included in the CFX96™/C1000 RT-qPCR detection
system evaluation-software (Bio-Rad). Ribosomal protein L13a
(Rpl13a) was used for normalization. The size and the purity of the
PCR products were determined by melt curve analysis and by
visualization on RedSafe™-stained agarose gels (iNtRON
Biotechnology, Seoul, Korea). Amplificates having a Cq value >39
were considered as not expressed. Primers were designed using the
Invitrogen OligoPerfect Designer and were obtained from Invitrogen
(Darmstadt, Germany). Primer sequences (sense and antisense), the
size of the amplificates in base pairs (bp) and the optimised
annealing temperatures were as follows: ACE,
5′-AGTGGGTGCTGCTCTTCCTA-3′ and 5′-ATGGG ACACTCCTCTGTTGG-3′, 188 bp,
57–65°C; ACE2, 5′-GTGGAGCACTGACTGGAGC-3′ and 5′-GACAGGA
GGCTCGTAAGGTG-3′, 403 bp, 59°C; APA, 5′-CCTCAC ATCCGGTGGTTGTC-3′
and 5′-TGGGTGACGTT CTGCTTTCC-3, 304 bp, 61°C; APN, 5′-CATCATAGCTCT
GTCGGTGG-3′ and 5′-AGCGGACAGTACTGGAACC-3′, 238 bp, 61°C; AT1aR,
5′-CAGCGTGAGCTTCAACCTC TAC-3′ and 5′-CAGCCAGATGATGATGCAGGTG-3′, 145
bp, 61°C; AT1bR, 5′-TGTTGACAAGCCTGCGTGTGAC-3′ and
5′-GACATTGTGGACACCGCTATGC-3′, 165 bp, 61°C; AT2R,
5′-CACACTACGGAGCTTCTGTTGG-3′ and 5′-TTGGATGCTCTGACCTGGATGG-3′, 165
bp, 61°C; insulin 1, 5′-GCCCAGGCTTTTGTCAAACAG-3′ and
5′-GCAGATGCTGGTGCAGCACTG-3′, 237 bp, 57°C; insulin 2,
5′-CAGCACCTTTGTGGTTCTCAC-3′ and 5′-CAGTGCCAAGGTCTGAAGGTC-3′, 165
bp, 57°C; IRAP, 5′-GCCTACATCCAAACCTAACCTC-3′ and 5′-GCAG
ATCTTGCTGCCAAAGG-3′, 367 bp, 57–65°C; Mas,
5′-CAGATGTCACCGCCCCAAGCA-3′ and 5′-GTGTTGCC ATTGCCCTCCTGA-3′, 534
bp, 62°C; NEP, 5′-CCAGACT GATTCGTCAGGAAC-3′ and 5′-CGGCTGAGGCTGC
TTACAAG-3′, 397 bp, 57–65°C; Rpl13a, 5′-CTGGTACTTCC ACCCGACCTC-3′
and 5′-GGATCCCTCCACCCTAT GACA-3′, 131 bp, 57–65°C.
Protein preparation and analyses
The cells were washed with ice-cold PBS and
collected by centrifugation (1,900 × g, 4°C, 5 min). Cells were
homogenised in lysis buffer, which contained 50 mM Tris-HCl (pH
7.5), 100 mM NaCl, 5 mM EDTA, 0.5% Triton X-100, 10% glycerol, 10
mM K2HPO4, 0.5% NP-40, 1 mM PMSF, 1 mM sodium
vanadate, 0.5% desoxycholate, 20 mM NaF, 20 mM glycerol-2-phosphate
(all from Sigma, Heidelberg, Germany) and a protease inhibitor
cocktail (Roche, Mannheim, Germany), kept on ice for 30 min, frozen
in liquid nitrogen, defrosted on ice and centrifuged at 16,000 × g
and 4°C for 30 min to separate the proteins from the cell debris.
Protein concentrations were determined using the Bradford method
(41). A total of 20 μg (40 μg
for Mas-detection) of protein in a final volume of 30 μl 1X Laemmli
buffer were separated by SDS-PAGE and transferred onto
nitrocellulose (NC; Whatman, Dassel, Germany) or
polyvinylidenefluoride (PVDF) membranes (Mas; Millipore, Bedford,
MA, USA). The membranes were incubated with primary antibodies
followed by incubation with horse radish peroxidase-conjugated
secondary antibodies. For detection, the SuperSignal®
West Dura Enhanced Chemiluminescence Substrate (Pierce, Rockford,
IL, USA) was used. Subsequently, protein amounts were normalised to
ribosomal protein, large, P0 (RPLP0) or actin signals. Protein
expression was quantified using ImageJ software (Wayne Rasband,
National Institute of Mental Health, Bethesda, MD, USA). The
antibodies used, antibody dilutions and dilution buffers for
western blotting were as follows: rabbit anti ACE2 (LS-B439;
Biozol, Eching, Germany) 1:10,000, mouse anti-NEP (CD10; ab951)
1:1,000, goat anti-APA (BP1; ab36122) 1:2,000, rabbit anti-APN
(CD13; ab108310; all from Abcam Cambridge, UK) 1:2,000, rabbit
anti-IRAP (#6918; NEB; Cell Signaling Technology, Frankfurt/Main,
Germany) 1:2,000, all in TBST/5% BSA/0.03% NaN3; rabbit
anti-Mas (LS-B2564; Biozol) 1:2,000 in PBS/5% skimmed milk/0.03%
NaN3; goat anti -actin (I-19, SC-1616; Santa Cruz
Biotechnology, Inc., Heidelberg, Germany) 1:500, rabbit anti-RPLP0
(Biozol) 1:2,000, both in TBST/5% BSA/0.03% NaN3.
HRP-coupled secondary antibodies (all purchased from Cell Signaling
Technology) were used in the following dilutions in 1X
Roti®-Block (Carl Roth, Karlsruhe, Germany): anti rabbit
IgG 1:10,000 (1:5,000 for Mas-detection), anti-mouse and anti-goat
IgG 1:10,000.
Determination of insulin secretion
Insulin concentrations in the supernatants of
BRIN-BD11 cells were determined by a sandwich enzyme-linked
immunosorbent assay (ELISA; Mercodia, Uppsala, Sweden) following
the recommendations of the manufacturer.
Enzymatic activities
The enzymatic activity of ACE, ACE2, APA, APN and
IRAP was determined using viable BRIN-BD11 cells. The cells were
scraped in PBS, centrifuged at 2,500 × g and resuspended in PBS.
The following substrates were used for the determination of
enzymatic activity: H-Ala-pNA-HCl for APN; H-Glu-pNA-2HCl (both
from Bachem, Weil am Rhein, Germany) for APA; H-Leu-pNA-HCl (Sigma,
Taufkirchen, Germany) plus RB3014 [specific inhibitor of APN,
kindly provided by B.P. Roques (Département de Pharmacochimie
Moléculaire et Structurale INSERM U266 - CNRS UMR 8600 UFR des
Sciences Pharmaceutiques et Biologiques, Paris, France) (42)] for IRAP; Mca-APK (Dnp;
Enzo/Biomol, Hamburg, Germany) for ACE2. All the assays were
carried out in 50 mM HEPES buffer containing 200 mM NaCl, 10 μM
ZnCl2 and 1% DMSO pH 6.8. The enzyme reactions were
performed in 1.5 ml tubes or in flat-bottom 96-well microtiter
plates (Greiner Bio-One, Frickenhausen, Germany) for the
measurement of the supernatants. Black plates were used for the
measurement of fluorescence (ACE2) and transparent plates for the
measurement of optical density. The total reaction volume amounting
to 200 μl consisted of 100 μl buffer, 50 μl cell suspension and 50
μl substrate. The reaction was started by the addition of the
substrate. For the determination of IRAP activity, a pre-incubation
with the inhibitor, RB3014, was performed for 30 min at 37°C prior
to the addition of the substrate. The optical density or
fluorescence measured without cells was used as the blank sample.
Following incubation at 37°C for 1 h in the dark and centrifugation
at 2,500 × g for 5 min, the optical density of the supernatants was
measured at 405 nm with 620 nm reference wavelength by a microtiter
plate reader Infinite M200 (Tecan, Crailsheim, Germany). The
fluorescence intensity in the ACE2 assay was determined using an
excitation wavelength of 320 nm and an emission wavelength of 405
nm using a microtiter plate reader (Infinite F200; Tecan). All
samples were determined in duplicates.
Statistical analysis
Statistical analyses were carried out using GraphPad
Prism 5 software (GraphPad, La Jolla, CA, USA). Data with n≥12 were
analysed using one-way ANOVA to compare the means of 3 or more
unmatched groups. If there was a statistically significant
difference, unpaired t-tests were applied to compare 2 sets of
measurements. Normally distributed data are illustrated as the
means + SEM.
In order to compare 3 or more groups of data (n≥4 to
<12), the non-parametric Kruskal-Wallis test was used. If there
was a significant difference, Mann-Whitney tests were applied to
compare 2 sets of measurements. Non-parametric data are illustrated
as boxplots with medians, quartiles and an interquartile range
(IQR) ± 1.5 × IQR. P-values <0.05 were considered to indicate
statistically significant differences.
Results
Effect of glucose on insulin production
and secretion
To verify that BRIN-BD11 cells can be used as a
suitable β-cell model, the short-time glucose-stimulated insulin
secretion (GSIS) was determined. Basal levels of insulin in the
cell culture supernatants (0.13 μg/l) were set to 100% (control).
Stimulation with 40 mM KCl and 15 mM glucose serving as the
positive control increased the amounts of insulin to ~247±65%
(P<0.01) of the control (Fig.
2A). In response to a 20-min exposure of the BRIN-BD11 cells to
elevated glucose concentrations, there was a dose-dependent
increase in insulin secretion (Fig.
2A). This increase was significant from the concentration of 11
mM glucose (172±31%, P<0.01) and reached 283±91% (P<0.01)
with 25 mM glucose (Fig. 2A).
Furthermore, the BRIN-BD11 cells, when exposed to elevated glucose
concentrations for 24 h, showed a dose-dependent increase in
insulin 1- and 2 mRNA levels (Fig. 2B
and C). Again, this effect was significant from the
concentration of 11 mM glucose, reaching 389% (Q1 = 352; Q3 = 412)
(P<0.01) for insulin 1 mRNA or 415% (203; 510) (P<0.01) for
insulin 2 mRNA (Fig. 2B and
C).
Basal RAS expression
After having confirmed that BRIN-BD11 rat insulinoma
cells exhibit an appropriate responsiveness to glucose, we then
determined the mRNA expression levels of components of the main RAS
axes. BRIN-BD11 cells express all essential components of RAS
investigated, including the constituents of the classical RAS axis,
ACE, AT1bR, AT2R (but not AT1aR), as well as those of the
alternative APN/Ang IV/IRAP and ACE2/Ang-(1-7)/Mas axes (Fig. 3).
Effect of glucose on RAS expression and
activity
We then determined whether the exposure of BRIN-BD11
cells to increasing concentrations of glucose alters the expression
and, if applicable, the enzymatic activity of RAS components. As
shown in Fig. 4A–C, the mRNA
expression of constituents of the classical RAS axis, ACE and
AT1bR, as well as those of AT2R was not altered after 24 h. By
contrast, glucose at higher concentrations induced a dose-dependent
increase in the mRNA expression of every component analysed within
the ACE2/Ang-(1-7)/Mas axis (Fig. 4D–F). More precisely, with the
concentration of 11 mM glucose a significant increase in ACE2 [245%
(202; 410), P<0.05], NEP [305% (188; 604), P<0.01] and Mas
[255% (164; 665), P<0.05] mRNA expression was observed. The mRNA
levels even further increased in response to higher concentrations
of glucose. In the case of NEP, glucose was additionally titrated
between 5.5 and 11 mM, as there was a marked increase in mRNA
expression between these 2 concentrations (Fig. 4E). As observed for the
ACE2/Ang-(1-7)/Mas axis, glucose also increased the
mRNA expression of APA, APN and IRAP, which are all constituents of
the APN/Ang IV/IRAP alternative RAS pathway (Fig. 4G–I). Whereas the mRNA levels of
APA [194% (177; 248), P<0.01] and IRAP [151% (143; 185),
P<0.01] were already elevated in response to 11 mM or higher
concentrations of glucose, APN mRNA levels [130% (116; 179),
P<0.01] were significantly upregulated from starting from the
concentration of 15 mM glucose.
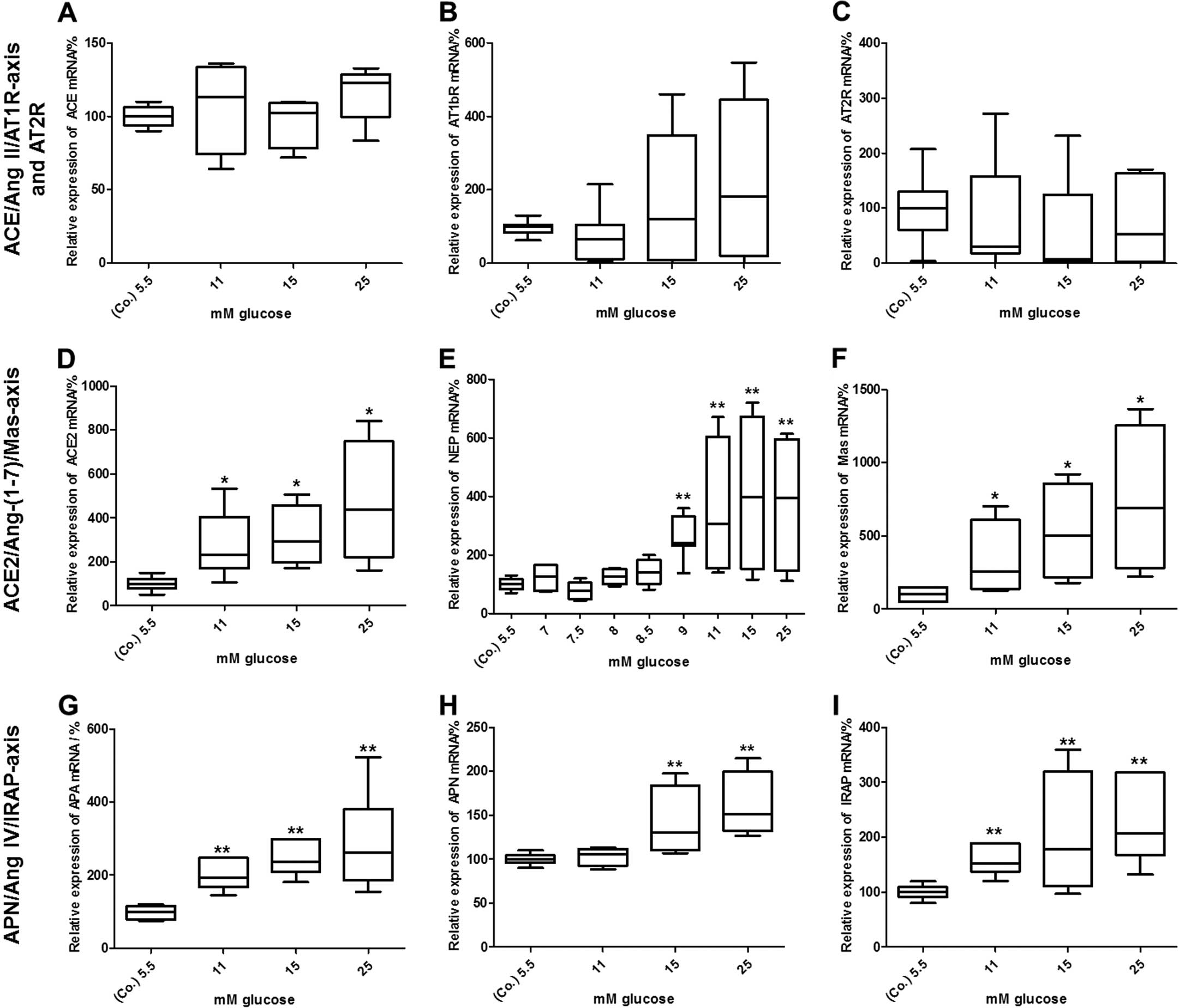 | Figure 4Effects of glucose on the mRNA
expression of renin-angiotensin system (RAS) components. Elevated
concentrations of glucose did not alter the mRNA levels of (A)
angiotensin-converting enzyme (ACE), (B) angiotensin II type 1b
receptor (AT1bR), or (C) angiotensin II type 2 receptor (AT2R) mRNA
levels in BRIN-BD11 cells after 24 h, whereas the levels of (D)
ACE2, (E) neutral endopeptidase 24.11 (NEP), (F) Mas, (G)
aminopeptidase A (APA), (H) aminopeptidase N (APN) and (I)
insulin-regulated aminopeptidase (IRAP) were dose-dependently
increased. mRNA levels were determined by quantitative PCR. Data
were evaluated and normalized to Rpl13a mRNA expression using the
ΔΔ Cq method. Data are presented as box plots with medians,
quartiles and an interquartile range (IQR) ± 1.5 × IQR of 5
independent experiments [Mann-Whitney *P<0.05,
**P<0.01, vs. control (Co.)]. |
Changes in the expression of constituents of the
alternative RAS axes observed at the mRNA level were subsequently
verified at the protein and enzymatic activity level. Immunoblot
analyses revealed a dose-dependent increase in the levels of ACE2,
NEP, Mas, APA, APN and IRAP protein in response to the exposure of
the BRIN-BD11 cells to 25 mM glucose for 24 h. As shown in Fig. 5, the levels of ACE2, NEP and Mas
increased to 172% [(166; 206), P<0.01], 120% [(118; 160),
P<0.05] or 623% [(482; 950), P<0.01] compared with the
control (Fig. 5A–C). Similarly,
the protein levels of APA (204%), APN [109% (100; 143), P<0.01]
and IRAP [130% (109; 153), P<0.05] were markedly increased by 25
mM glucose (Fig. 5D–F).
The results obtained by the analysis of mRNA and
protein expression clearly suggest an induction of the two
alternative RAS axes. To further support this view, the enzymatic
activity of relevant RAS proteases under conditions of low and high
glucose was measured. In full accordance with the observed changes
in mRNA and protein expression levels, there was a significant
increase in the enzymatic activities assigned to both the APN/Ang
IV/IRAP axis (APA, APN and IRAP) and the ACE2/Ang-(1-7)/Mas axis (ACE2). More precisely, the
enzymatic activities increased with the concentration of 25 mM
glucose as follows: ACE2, 141% [(119; 157), P<0.05]; APA, 124%
[(114; 127), P<0.01]; APN, 109% [(100; 143), P<0.05]; and
IRAP, 123% [(111; 131), P<0.01] (Fig. 6).
Discussion
Glucose and fatty acids represent the main
nutritional components. Their concentration in the blood differs
between food intake and fasting periods to a large extent.
Normally, the acute exposure of β-cells to elevated concentrations
of glucose following food intake leads to an adequate insulin
secretion. However, a sustained elevation of plasma levels of
glucose as in the case of patients suffering from impaired glucose
tolerance or DM may adversely affect islet function. Accumulating
evidence indicates that a systemic and/or local activation of
classical RAS is common to various diabetic risk factors, including
hypertension and obesity (43,44). Ang II, in particular, has been
reported to impair insulin signaling and islet function (45,46). Although first data suggest a
protective role of alternative RAS axes on β-cell function, this
needs to be further substantiated. Therefore, in this study, we
aimed i) to assess the basal expression levels and enzymatic
activity of the components of three major RAS axes in BRIN-BD11
insulinoma cells and ii) to evaluate possible alterations thereof
in response to the exposure of the cells to high glucose
concentrations. A crucial limitation when using immortalised β-cell
lines as a model system may arise from their lack of an adequate
responsiveness to glucose. The data from the present study
demonstrate an appropriate preservation of functionality in
BRIN-BD11 cells, showing a dose-dependent short-term insulin
secretion following exposure to various concentrations of glucose.
These findings are in full accordance with previous results from
the study by McClenaghan et al(47), who established this cell line and
confirmed that BRIN-BD11 cells are a suitable model for the study
of β-cell function.
In this study, we first demonstrated (on different
levels of expression) that the essential components of the three
main RAS axes are expressed in BRIN-BD11 cells under basal
conditions. In detail, we proved the following constituents of RAS
to be present in the BRIN-BD11 rat insulinoma cell line: ACE,
AT1bR, AT2R, ACE2, NEP, Mas, APA, APN and IRAP, but not AT1aR.
These results are in agreement with those of previous studies,
demonstrating the expression of (pro)renin (48,49), angiotensinogen (50–52), ACE (49–53) and ACE2 (49), AT1R (48,50,53) and AT2R (53,54), Mas (55) and IRAP (56,57) in several islet-specific cell
types, specifically in β-cells, in various species including
humans, rats, mice and canines (28). In this study, to our knowledge, we
demonstrate for the first time the expression of APA, APN and NEP
in the BRIN-BD11 pancreatic β-cell line, thus proving the presence
of local RAS on β-cells.
Despite the earlier detection of RAS components in
pancreatic islets, Lupi et al(58) were the first to study the effects
of glucose on RAS mRNA expression in islets of Langerhans. A second
study confirmed these findings, showing that in INS-1E rat
insulinoma cells, chronic hyperglycaemia induces AT1R gene and
protein expression, in parallel to enhanced ROS production and
impaired GSIS (59). As regards
other cell types, hyperglycaemia has been shown to increase
extracellular matrix (ECM) production through RAS activation in rat
pancreatic stellate cells (PSCs), which are involved in pancreatic
inflammation and fibrosis (60).
In these cells, 27.7 mM glucose induced an increase in ACE and
AT1aR mRNA expression after 72 h, whereas AT1bR and AT2R mRNA
levels remained unaltered (61).
Again, ACEis and ARBs prevented the increase in
proliferation and ECM production, emphasizing the pivotal role that
Ang II plays in the pathophysiology of pancreatic inflammation and
fibrosis, which are exacerbated by chronic hyperglycaemia (60). The increased mRNA expression and
enzymatic activity of NEP have been observed in human microvascular
endothelial cells (HMECs) in response to 40 mM glucose. ROS seems
to play a role in this process, since vitamins C and E attenuate
the effects of glucose (61,62). In rats fed a high sucrose diet for
30 days, Ang-(1-7)-levels, ACE2, AT1R and AT2R protein
expression have been shown to increase in adipose tissue, but
remain unaltered in the kidneys. By contrast, ACE activity is
decreased in adipose tissue, but increased in the kidneys (63).
In this study, we examined the effects of glucose on
the expression and/or enzymatic activities of the β-cell RAS
components in BRIN-BD11 cells. To the best of our knowledge, this
is the first comprehensive study of islet RAS i) considering both
the classical and the alternative axes of RAS and ii) analyzing RAS
at different expression and enzymatic activity levels in parallel.
Of note, under conditions of high glucose expression and activities
of the alternative RAS axes, ACE2/Ang-(1-7)/Mas and APN/IRAP levels were found to
increase after 24 h. In contrast to this, the classical ACE/Ang
II/AT1R axis and AT2R remained largely unaffected by glucose. Of
note, in isolated human islets, ACE and AT1R mRNA levels have been
shown to be upregulated by 22.2 mM glucose (58). The extent to which islet-specific
cell types other than β-cells contribute to the ACE/Ang II/AT1R
induction observed in whole islets remains to be elucidated. The
data presented in this study suggest that in β-cells, there is a
general shift from the classical to the alternative and supposedly
protective RAS axes under acute hyperglycaemic conditions. Whereas
22.2 mM glucose have been shown to decrease insulin secretion by
isolated human islets, possibly due to oxidative stress that was
concomitantly observed (58), in
the present study, glucose at concentrations of 25 mM enhanced
insulin expression and secretion in BRIN-BD11 cells. It can be
speculated that in human islets, the increased expression and
enzymatic activity of the classical Ang II/ACE/AT1R axis
contributes through increased ROS production to the compromised
insulin response under very high glucose conditions. This view is
supported by the well-established fact that Ang II upon binding to
the AT1R, induces the activation of NADPH oxidase and, thus,
enhances the production of ROS. Recent evidence indicates that this
mechanism works in islets or β-cells as well (64–66). Accordingly, as shown in a previous
study, impaired insulin secretion and elevated ROS production can
be abolished by ACEis (58). The authors concluded that
ACEis protect human islets from apparent glucotoxicity
(58). It is tempting to
speculate that BRIN-BD11 cells tolerate higher glucose
concentrations well, as they show no such induction of the Ang
II/ACE/AT1R axis. This hypothesis, however, needs to be verified in
future studies. Furthermore, this may be true for the short-term
exposure of β-cells to high glucose only, whereas sustained
hyperglycaemia provokes opposite effects. In support of this view,
chronically elevated glucose levels have been shown to increase Ang
II levels in several models, including diabetic rats, thereby
promoting Ang II/AT1R-mediated glucotoxicity and hampering islet
blood flow, insulin production and GSIS (58,60,67,68).
By contrast, the short-term exposure of BRIN-BD11
cells to high 25 mM glucose as performed in the present study did
not compromise cell viability (data not shown) or insulin
production/secretion. Our data clearly demonstrate a profound
induction of the alternative ACE2/Ang-(1-7)/Mas and APN/Ang IV/IRAP axes in the
BRIN-BD11 cells. The observed parallel increase in mRNA and protein
expression together with corresponding enzymatic activities
emphasise the relevance of this finding and point to a functional
role of these alternative RAS axes in maintaining β-cell viability
and function. As to how the activation of alternative RAS axes and,
in particular, the observed Ang-(1-7)-mediated activation of PI3K via Mas
aims to compensate Ang II/AT1R-mediated toxicity, remains to be
fully elucidated (69).
Acknowledgements
C.H. was supported by a fellowship of the Alfried
Krupp Wissenschaftskolleg Greifswald. The authors thank the student
apprentices, Ana Finzel-Pérez, Wiebke Malenke, Teresa Hellberg and
Daniel Galla for providing excellent experimental support.
References
|
1
|
Ahmed AM: History of diabetes mellitus.
Saudi Med J. 23:373–378. 2002.
|
|
2
|
International Diabetes Federation. The
Global Burden. IDR Diabetes Atlas. 5th edition. 2011, http://www.idf.org/diabetesatlas/5e/the-global-burden.
|
|
3
|
Himsworth HP: Diatebetes mellitus: its
differentiation into insulin-sensitive and insulin-insensitive
types. Lancet. 127–130. 1936.
|
|
4
|
Centers for Disease Control Prevention
(CDC). 2011 National Diabetes Fact Sheet Publications Division of
Diabetes Translation. U.S. Department of Health and Human Services,
Centers for Disease Control and Prevention; Atlanta, GA: 2011,
http://www.cdc.gov/diabetes/pubs/factsheet11.htm.
|
|
5
|
Reaven GM: Banting lecture 1988. Role of
insulin resistance in human disease. Diabetes. 37:1595–1607. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ripsin CM, Kang H and Urban RJ: Management
of blood glucose in type 2 diabetes mellitus. Am Fam Physician.
79:29–36. 2009.PubMed/NCBI
|
|
7
|
Hu FB, Manson JE, Stampfer MJ, Colditz G,
Liu S, Solomon CG and Willett WC: Diet, lifestyle, and the risk of
type 2 diabetes mellitus in women. N Engl J Med. 345:790–797. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Centers for Disease Control and Prevention
(CDC). Prevalence of Overweight and Obesity Among Adults with
Diagnosed Diabetes - United States, 1988–1994 and 1999–2002.
Morbidity and Mortality Weekly Report (MMWR). 53:pp. 1066–1068.
2004, http://www.cdc.gov/mmwr/preview/mmwrhtml/mm5345a2.htm.
|
|
9
|
Lang IA, Galloway TS, Scarlett A, Henley
WE, Depledge M, Wallace RB and Melzer D: Association of urinary
bisphenol A concentration with medical disorders and laboratory
abnormalities in adults. JAMA. 300:1303–1310. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tigerstedt R and Bergman PG: Niere und
Kreislauf. Skand Arch Physiol. 8:223–271. 1898. View Article : Google Scholar
|
|
11
|
Skeggs LT Jr, Kahn JR and Shumway NP: The
preparation and function of the hypertension-converting enzyme. J
Exp Med. 103:295–299. 1956. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Touyz RM and Schiffrin EL: Signal
transduction mechanisms mediating the physiological and
pathophysiological actions of angiotensin II in vascular smooth
muscle cells. Pharmacol Rev. 52:639–672. 2000.PubMed/NCBI
|
|
13
|
Zimmerman MC, Lazartigues E, Lang JA,
Sinnayah P, Ahmad IM, Spitz DR and Davisson RL: Superoxide mediates
the actions of angiotensin II in the central nervous system. Circ
Res. 91:1038–1045. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tipnis SR, Hooper NM, Hyde R, Karran E,
Christie G and Turner AJ: A human homolog of angiotensin-converting
enzyme. Cloning and functional expression as a
captopril-insensitive carboxypeptidase. J Biol Chem.
275:33238–33243. 2000. View Article : Google Scholar
|
|
15
|
Donoghue M, Hsieh F, Baronas E, Godbout K,
Gosselin M, Stagliano N, Donovan M, Woolf B, Robison K, Jeyaseelan
R, Breitbart RE and Acton S: A novel angiotensin-converting
enzyme-related carboxypeptidase (ACE2) converts angiotensin I to
angiotensin 1–9. Circ Res. 87:E1–E9. 2000.
|
|
16
|
Rice GI, Thomas DA, Grant PJ, Turner AJ
and Hooper NM: Evaluation of angiotensin-converting enzyme (ACE),
its homologue ACE2 and neprilysin in angiotensin peptide
metabolism. Biochem J. 383:45–51. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chappell MC, Modrall JG, Diz DI and
Ferrario CM: Novel aspects of the renal renin-angiotensin system:
angiotensin-(1-7), ACE2 and blood pressure regulation. Contrib
Nephrol. 143:77–89. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Santos RA and Ferreira AJ:
Angiotensin-(1-7) and the renin-angiotensin system. Curr Opin
Nephrol Hypertens. 16:122–128. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bank U, Tadje J, Täger M, Wolke C,
Bukowska A, Ittenson A, Reinhold D, Helmuth M, Ansorge S,
Shakespeare A, Vieth M, Malfertheiner P, Naumann M and Lendeckel U:
Inhibition of alanyl-aminopeptidase on
CD4+CD25+ regulatory T-cells enhances
expression of FoxP3 and TGF-β1 and ameliorates acute colitis in
mice. Int J Mol Med. 20:483–492. 2007.PubMed/NCBI
|
|
20
|
Wolke C, Tadje J, Bukowska A, Täger M,
Bank U, Ittenson A, Ansorge S and Lendeckel U: Assigning the
phenotype of a natural regulatory T-cell to the human T-cell line,
KARPAS-299. Int J Mol Med. 17:275–278. 2006.PubMed/NCBI
|
|
21
|
Lendeckel U, Arndt M, Frank K, Wex T and
Ansorge S: Role of alanyl aminopeptidase in growth and function of
human T cells (Review). Int J Mol Med. 4:17–27. 1999.PubMed/NCBI
|
|
22
|
Albiston AL, McDowall SG, Matsacos D, Sim
P, Clune E, Mustafa T, Lee J, Mendelsohn FA, Simpson RJ, Connolly
LM and Chai SY: Evidence that the angiotensin IV (AT(4)) receptor
is the enzyme insulin-regulated aminopeptidase. J Biol Chem.
276:48623–48626. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jordens I, Molle D, Xiong W, Keller SR and
McGraw TE: Insulin-regulated aminopeptidase is a key regulator of
GLUT4 trafficking by controlling the sorting of GLUT4 from
endosomes to specialized insulin-regulated vesicles. Mol Biol Cell.
21:2034–2044. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Takeuchi M, Itakura A, Okada M, Mizutani S
and Kikkawa F: Impaired insulin-regulated membrane aminopeptidase
translocation to the plasma membrane in adipocytes of Otsuka Long
Evans Tokushima Fatty rats. Nagoya J Med Sci. 68:155–163.
2006.PubMed/NCBI
|
|
25
|
Keller SR, Davis AC and Clairmont KB: Mice
deficient in the insulin-regulated membrane aminopeptidase show
substantial decreases in glucose transporter GLUT4 levels but
maintain normal glucose homeostasis. J Biol Chem. 277:17677–17686.
2002. View Article : Google Scholar
|
|
26
|
Siebelmann M, Wensing J and Verspohl EJ:
The impact of Ang II and IV on INS-1 cells and on blood glucose and
plasma insulin. J Recept Signal Transduct Res. 30:234–245. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wong YC, Sim MK and Lee KO:
Des-aspartate-angiotensin-I and angiotensin IV improve glucose
tolerance and insulin signalling in diet-induced hyperglycaemic
mice. Biochem Pharmacol. 82:1198–1208. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Paul M, Poyan Mehr A and Kreutz R:
Physiology of local renin-angiotensin systems. Physiol Rev.
86:747–803. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Leung PS: The physiology of a local
renin-angiotensin system in the pancreas. J Physiol. 580:31–37.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lavoie JL and Sigmund CD: Minireview:
overview of the renin-angiotensin system - an endocrine and
paracrine system. Endocrinology. 144:2179–2183. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ribeiro-Oliveira A Jr, Nogueira AI,
Pereira RM, Boas WW, Dos Santos RA and Simões e Silva AC: The
renin-angiotensin system and diabetes: an update. Vasc Health Risk
Manag. 4:787–803. 2008.
|
|
32
|
Ganten D, Marquez-Julio A, Granger P,
Hayduk K, Karsunky KP, Boucher R and Genest J: Renin in dog brain.
Am J Physiol. 221:1733–1737. 1971.PubMed/NCBI
|
|
33
|
Yusuf S, Sleight P, Pogue J, Bosch J,
Davies R and Dagenais G: Effects of an
angiotensin-converting-enzyme inhibitor, ramipril, on
cardiovascular events in high-risk patients. The Heart Outcomes
Prevention Evaluation Study Investigators. N Engl J Med.
342:145–153. 2000. View Article : Google Scholar
|
|
34
|
Jarvis S: Angiotensin receptor blockers in
clinical practice-implications of the ONTARGET study. J Int Med
Res. 40:10–17. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Folli F, Saad MJ, Velloso L, Hansen H,
Carandente O, Feener EP and Kahn CR: Crosstalk between insulin and
angiotensin II signalling systems. Exp Clin Endocrinol Diabetes.
107:133–139. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Nakashima H, Suzuki H, Ohtsu H, Chao JY,
Utsunomiya H, Frank GD and Eguchi S: Angiotensin II regulates
vascular and endothelial dysfunction: recent topics of angiotensin
II type-1 receptor signaling in the vasculature. Curr Vasc
Pharmacol. 4:67–78. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Passos-Silva DG, Verano-Braga T and Santos
RA: Angiotensin-(1-7): beyond the cardio-renal actions. Clin Sci
(Lond). 124:443–456. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Santos SH, Fernandes LR, Mario EG,
Ferreira AV, Pôrto LC, Alvarez-Leite JI, Botion LM, Bader M,
Alenina N and Santos RA: Mas deficiency in FVB/N mice produces
marked changes in lipid and glycemic metabolism. Diabetes.
57:340–347. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Giani JF, Mayer MA, Muñoz MC, Silberman
EA, Höcht C, Taira CA, Gironacci MM, Turyn D and Dominici FP:
Chronic infusion of angiotensin-(1-7) improves insulin resistance
and hypertension induced by a high-fructose diet in rats. Am J
Physiol Endocrinol Metab. 296:E262–E271. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Liu C, Lv XH, Li HX, Cao X, Zhang F, Wang
L, Yu M and Yang JK: Angiotensin-(1-7) suppresses oxidative stress
and improves glucose uptake via Mas receptor in adipocytes. Acta
Diabetol. 49:291–299. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bradford MM: A rapid and sensitive method
for the quantitation of microgram quantities of protein utilizing
the principle of protein-dye binding. Anal Biochem. 72:248–254.
1976. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chen H, Roques BP and Fournié-Zaluski MC:
Design of the first highly potent and selective aminopeptidase N
(EC 3.4.11.2) inhibitor. Bioorg Med Chem Lett. 9:1511–1516. 1999.
View Article : Google Scholar
|
|
43
|
Dzau VJ, Bernstein K, Celermajer D, Cohen
J, Dahlöf B, Deanfield J, Diez J, Drexler H, Ferrari R, van Gilst
W, Hansson L, Hornig B, Husain A, Johnston C, Lazar H, Lonn E,
Lüscher T, Mancini J, Mimran A, Pepine C, Rabelink T, Remme W,
Ruilope L, Ruzicka M, Schunkert H, Swedberg K, Unger T, Vaughan D
and Weber M; Working Group on Tissue Angiotensin-converting enzyme,
International Society of Cardiovascular Pharmacotherapy. The
relevance of tissue angiotensin-converting enzyme: manifestations
in mechanistic and endpoint data. Am J Cardiol. 88:1L–20L. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Goossens GH, Blaak EE and van Baak MA:
Possible involvement of the adipose tissue renin-angiotensin system
in the pathophysiology of obesity and obesity-related disorders.
Obes Rev. 4:43–55. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Prasannarong M, Santos FR and Henriksen
EJ: ANG-(1-7) reduces ANG II-induced insulin resistance by
enhancing Akt phosphorylation via a Mas receptor-dependent
mechanism in rat skeletal muscle. Biochem Biophys Res Commun.
426:369–373. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Tikellis C, Cooper ME and Thomas MC: Role
of the renin-angiotensin system in the endocrine pancreas:
implications for the development of diabetes. Int J Biochem Cell
Biol. 38:737–751. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
McClenaghan NH, Barnett CR, Ah-Sing E,
Abdel-Wahab YH, O’Harte FP, Yoon TW, Swanston-Flatt SK and Flatt
PR: Characterization of a novel glucose-responsive
insulin-secreting cell line, BRIN-BD11, produced by electrofusion.
Diabetes. 45:1132–1140. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Tahmasebi M, Puddefoot JR, Inwang ER and
Vinson GP: The tissue renin-angiotensin system in human pancreas. J
Endocrinol. 161:317–322. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Tikellis C, Wookey PJ, Candido R,
Andrikopoulos S, Thomas MC and Cooper ME: Improved islet morphology
after blockade of the renin-angiotensin system in the ZDF rat.
Diabetes. 53:989–997. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Lau T, Carlsson PO and Leung PS: Evidence
for a local angiotensin-generating system and dose-dependent
inhibition of glucose-stimulated insulin release by angiotensin II
in isolated pancreatic islets. Diabetologia. 47:240–248. 2004.
View Article : Google Scholar
|
|
51
|
Chu KY, Lau T, Carlsson PO and Leung PS:
Angiotensin II type 1 receptor blockade improves beta-cell function
and glucose tolerance in a mouse model of type 2 diabetes.
Diabetes. 55:367–374. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Regoli M, Bendayan M, Fonzi L, Sernia C
and Bertelli E: Angiotensinogen localization and secretion in the
rat pancreas. J Endocrinol. 179:81–89. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Chu KY, Cheng Q, Chen C, Au LS, Seto SW,
Tuo Y, Motin L, Kwan YW and Leung PS: Angiotensin II exerts
glucose-dependent effects on Kv currents in mouse pancreatic
beta-cells via angiotensin II type 2 receptors. Am J Physiol Cell
Physiol. 298:C313–C323. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Wong PF, Lee SS and Cheung WT:
Immunohistochemical colocalization of type II angiotensin receptors
with somatostatin in rat pancreas. Regul Pept. 117:195–205. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Bindom SM and Lazartigues E: The sweeter
side of ACE2: physiological evidence for a role in diabetes. Mol
Cell Endocrinol. 302:193–202. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Kobayashi H, Mitsui T, Nomura S, Ohno Y,
Kadomatsu K, Muramatsu T, Nagasaka T and Mizutani S: Expression of
glucose transporter 4 in the human pancreatic islet of Langerhans.
Biochem Biophys Res Commun. 314:1121–1125. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Kobayashi H, Nomura S, Mitsui T, Ito T,
Kuno N, Ohno Y, Kadomatsu K, Muramatsu T, Nagasaka T and Mizutani
S: Tissue distribution of placental leucine
aminopeptidase/oxytocinase during mouse pregnancy. J Histochem
Cytochem. 52:113–121. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Lupi R, Del Guerra S, Bugliani M, Boggi U,
Mosca F, Torri S, Del Prato S and Marchetti P: The direct effects
of the angiotensin-converting enzyme inhibitors, zofenoprilat and
enalaprilat, on isolated human pancreatic islets. Eur J Endocrinol.
154:355–361. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Leung KK and Leung PS: Effects of
hyperglycemia on angiotensin II receptor type 1 expression and
insulin secretion in an INS-1E pancreatic beta-cell line. JOP.
9:290–299. 2008.PubMed/NCBI
|
|
60
|
Ko SH, Hong OK, Kim JW, Ahn YB, Song KH,
Cha BY, Son HY, Kim MJ, Jeong IK and Yoon KH: High glucose
increases extracellular matrix production in pancreatic stellate
cells by activating the renin-angiotensin system. J Cell Biochem.
98:343–355. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Muangman P, Tamura RN and Gibran NS:
Antioxidants inhibit fatty acid and glucose-mediated induction of
neutral endopeptidase gene expression in human microvascular
endothelial cells. J Am Coll Surg. 200:208–215. 2005. View Article : Google Scholar
|
|
62
|
Muangman P, Spenny ML, Tamura RN and
Gibran NS: Fatty acids and glucose increase neutral endopeptidase
activity in human microvascular endothelial cells. Shock.
19:508–512. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Coelho MS, Lopes KL, Freitas Rde A, de
Oliveira-Sales EB, Bergasmaschi CT, Campos RR, Casarini DE, Carmona
AK, Araújo Mda S, Heimann JC and Dolnikoff MS: High sucrose intake
in rats is associated with increased ACE2 and angiotensin-(1-7)
levels in the adipose tissue. Regul Pept. 162:61–67. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Mohammed AM, Syeda K, Hadden T and Kowluru
A: Upregulation of phagocyte-like NADPH oxidase by cytokines in
pancreatic beta-cells: attenuation of oxidative and nitrosative
stress by 2-bromopalmitate. Biochem Pharmacol. 85:109–114. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Syed I, Kyathanahalli CN, Jayaram B,
Govind S, Rhodes CJ, Kowluru RA and Kowluru A: Increased
phagocyte-like NADPH oxidase and ROS generation in type 2 diabetic
ZDF rat and human islets: role of Rac1-JNK1/2 signaling pathway in
mitochondrial dysregulation in the diabetic islet. Diabetes.
60:2843–2852. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Michalska M, Wolf G, Walther R and
Newsholme P: Effects of pharmacological inhibition of NADPH oxidase
or iNOS on pro-inflammatory cytokine, palmitic acid or
H2O2-induced mouse islet or clonal pancreatic
β-cell dysfunction. Biosci Rep. 30:445–453. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Abdel-Rahman EM, Abadir PM and Siragy HM:
Regulation of renal 12(S)-hydroxyeicosatetraenoic acid in diabetes
by angiotensin AT1 and AT2 receptors. Am J Physiol Regul Integr
Comp Physiol. 295:R1473–R1478. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Carlsson PO, Berne C and Jansson L:
Angiotensin II and the endocrine pancreas: effects on islet blood
flow and insulin secretion in rats. Diabetologia. 41:127–133. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Giani JF, Gironacci MM, Muñoz MC, Peña C,
Turyn D and Dominici FP: Angiotensin-(1-7) stimulates the
phosphorylation of JAK2, IRS-1 and Akt in rat heart in vivo: role
of the AT1 and Mas receptors. Am J Physiol Heart Circ Physiol.
293:H1154–1163. 2007. View Article : Google Scholar : PubMed/NCBI
|


















