Introduction
Microglia are the resident immune cells in the
central nervous system (CNS) and function as the first and main
form of active immune defense in the CNS. Under normal conditions,
microglia play a major role in host defense and tissue repair in
the CNS (1,2). However, the prolonged activation of
microglia can cause chronic neuroinflammation due to the increased
production of neurotoxic and pro-inflammatory mediators, including
nitric oxide (NO), prostaglandin E2 (PGE2),
reactive oxygen species (ROS) and pro-inflammatory cytokines, such
as interleukin (IL)-1β, IL-6 and tumor necrosis factor-α (TNF-α)
(3–5), eventually leading to neuronal death.
This is a common characteristic found in the initiation and
progression of neurodegenerative diseases, including Alzheimer’s
disease (AD), Parkinson’s disease (PD), cerebral ischemia, multiple
sclerosis and trauma (6–8). Thus, regulating microglial
activation and the downregulation of pro-inflammatory molecules in
microglia may have the therapeutic potential to reduce neuronal
injury or death in the treatment of neurodegenerative diseases.
Flavonoids are a diverse group of plant natural
products synthesized from phenylpropanoid and acetate-derived
precursors. They are becoming an important source of novel agents
with pharmaceutical potential and have attracted a great deal of
attention over the years for their role in the prevention of
chronic diseases (9–12). Among them, purpurogallin
(2,3,4,5-tetrahydroxybenzo[7]annulen-6-one) is a
benzotropolone-containing natural product, which occurs in the nut
gall of Quercus spp. (13,14). Purpurogallin is synthesized by the
chemical oxidation of pyrogallol and has been shown to have
biological properties, such as hydroxyl radical-scavenging
properties (15) and to protect
erythrocytes against lysis induced by peroxyl radicals (16). Previous studies have indicated
that this compound inhibits prolyl endopeptidases (17), human immunodeficiency virus 1
integrase (18) and
hydroxyestradiol methylation by catechol-O-methyltransferase
(19). In addition purpurogallin
has also been known to have anticancer activities through the
inhibition of the DNA synthesis of tumor cells (20), tyrosine-specific protein kinases
(13) and the interaction between
Bcl-xL and BH3 peptides (20–22). Moreover, a number of studies have
reported the antioxidant and anti-inflammatory effects of
purpurogallin (15,23–27); however, the actual molecular
mechanisms involving signal transduction cascades underlying the
purpurogallin-induced anti-inflammatory effects have not yet been
elucidated.
In the present study, we investigated the the
anti-inflammatory effects of purpurogallin and the mechanisms by
which it inhibits inflammation and the production of inflammatory
mediators in lipopolysaccharide (LPS)-stimulated murine BV2
microglial cells. Our findings suggest that purpurogallin may be a
candidate for use in the treatment of various neurodegenerative
disorders.
Materials and methods
Cell culture and purpurogallin
treatment
BV2 murine microglial cells were obtained from
Professor I.W. Choi of Inje University College of Medicine (Busan,
Korea). The cells were cultured in Dulbecco’s modified Eagle’s
medium (DMEM; Gibco BRL, Gaithersburg, MD, USA) supplemented with
10% fetal bovine serum (FBS), 100 U/ml penicillin and 100 μg/ml
streptomycin and were maintained in a humidified incubator with 5%
CO2. Purpurogallin was obtained from Professor T.H. Kim
of Daegu Haany University (Gyeongsan, Korea) and dissolved in
dimethyl sulfoxide (DMSO) and dilutions were made in DMEM. The
final concentration of DMSO in the medium was <0.0005% (v/v)
which showed no effects on cell growth. In all the experiments, the
cells were pre-treated with the indicated concentrations of
purpurogallin for 1 h prior to the addition of LPS (Sigma-Aldrich,
St. Louis, MO, USA).
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay
Cell viability was measured based on the formation
of blue formazan metabolized from colorless MTT (Sigma-Aldrich) by
mitochondrial dehydrogenases, which are active only in live cells.
In brief, BV2 cells were plated into 24-well plates at a density of
2×105 cells/well for 24 h and then washed. The cells
incubated with various concentrations of purpurogallin for 1 h were
treated with or without 0.5 μg/ml LPS for 24 h and then incubated
in 0.5 mg/ml MTT solution. Three hours later, the supernatant was
removed and the formation of formazan was measured at 540 nm using
a microplate reader (Dynatech MR-7000; Dynatech Laboratories, El
Paso, TX, USA).
Measurement of NO production
Using the Griess reaction, nitrite was measured in
the culture supernatants as an indicator of NO production. Aliquots
of culture supernatants from each sample were mixed with an equal
volume of Griess reagent [1% sulfanilamide/0.1%
N-(1-naphthyl)-ethylenediamine dihydrochloride/2.5%
H3PO4]. NO concentration was determined by
measuring the absorbance at 540 nm using a microplate
spectrophotometer. Nitrite concentration was calculated with
reference to the standard curve of sodium nitrite generated by
known concentrations (28).
Measurement of PGE2
production
BV2 cells were incubated with purpurogallin in the
presence or absence of LPS (0.5 μg/ml) for 24 h. Following the
manufacturer’s instructions, a volume of 100 μl of culture
supernatant was collected for the determination of PGE2
concentration by ELISA (Cayman Chemical Co., Ann Arbor, MI,
USA).
Reverse transcriptase-polymerase chain
reaction (PCR)
Total RNA was prepared using an RNeasy kit (Qiagen,
La Jolla, CA, USA) and primed with random hexamers for the
synthesis of complementary DNA using AMV Reverse Transcriptase
(Amersham Corp., Arlington Heights, IL, USA) according to the
manufacturer’s instructions. PCR was performed using a Mastercycler
(Eppendorf, Hamburg, Germany). The PCR primers were as follows:
mouse iNOS (5′-ATG TCC GAA GCA AAC ATC AC-3′ and 5′-TAA TGT CCA GGA
AGT AGG TG-3′), COX-2 (5′-CAG CAA ATC CTT GCT GTT CC-3′ and 5′-TGG
GCA AAG AAT GCA AAC ATC-3′), IL-1β (5′-ATG GCA ACT GTT CCT GAA CTC
AAC T-3′ and 5′-TTT CCT TTC TTA GAT ATG GAC AGG AC-3′), and TNF-α
(5′-ATG AGC ACA GAA AGC ATG ATC-3′ and 5′-TAC AGG CTT GTC ACT CGA
ATT-3′). The following conditions were used for the PCR reactions:
1 × (94°C for 3 min); 35 × (94°C for 45 sec; 58°C for 45 sec; and
72°C for 1 min); and 1 × (72°C for 10 min). The resulting
amplification products were separated electrophoretically on a 1%
agarose gel and visualized by ethidium bromide (EtBr;
Sigma-Aldrich) staining. In a parallel experiment,
glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an
internal control.
Protein extraction and western blot
analysis
For the preparation of total proteins, the cells
were gently lysed for 30 min with lysis buffer (20 mM sucrose, 1 mM
EDTA, 20 μM Tris-Cl, pH 7.2, 1 mM DTT, 10 mM KCl, 1.5 mM
MgCl2, 5 μg/ml pepstatin A, 10 μg/ml leupeptin and 2
μg/ml aprotinin). Supernatants were collected and protein
concentrations were determined using the Bio-Rad Protein Assay kit
(Bio-Rad, Hercules, CA, USA). For western blot analysis, an equal
amount of protein was subjected to electrophoresis on
SDS-polyacrylamide gel and transferred onto a nitrocellulose
membrane (Schleicher & Schuell, Keene, NH, USA) by
electroblotting. The blots were probed with the desired antibodies
for 1 h, incubated with the diluted enzyme-linked secondary
antibodies and visualized by enhanced chemiluminescence (ECL)
western blotting detection reagents (SuperSignal; Thermo Fisher
Scientific, Rockford, IL, USA) according to the recommended
procedures. In a parallel experiment, cells were washed with
ice-cold PBS and scraped; cytoplasmic and nuclear proteins were
then extracted using NE-PER® Nuclear and Cytoplasmic
Extraction Reagents (Pierce Biotechnology, Rockford, IL, USA).
Actin and lamin B were used as the internal controls for the
cytosolic and nuclear fraction, respectively.
Enzyme immunosolvent assay (ELISA)
The levels of IL-1β and TNF-α were measured using
ELISA kits (R&D Systems, Minneapolis, MN, USA) according to the
manufacturer’s instructions. Briefly, BV2 cells (5×105
cells/ml) were plated in 24-well plates and pre-treated with the
indicated concentrations of purpurogallin for 1 h before treatment
with 0.5 μg/ml LPS for 24 h. A total of 100 μl of culture
supernatants were collected for the determination of the IL-1β and
TNF-α concentration by ELISA as previously described (29).
Statistical analyses
Data represent the means ± SD. Statistical
significance was determined using an analysis of variance, followed
by a Student’s t-test. A value of P<0.05 was considered to
indicate a statistically significant difference.
Results
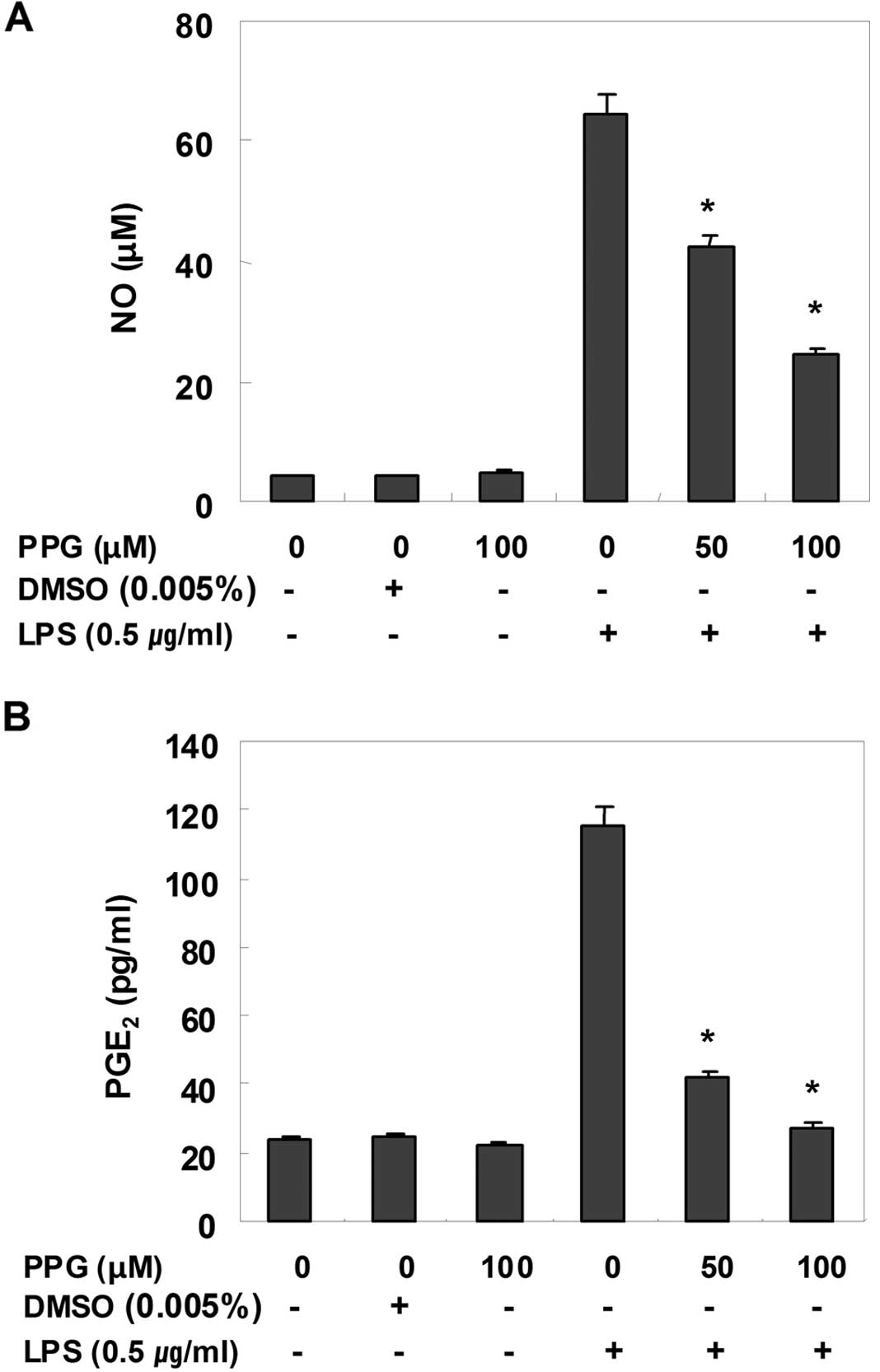
Purpurogallin inhibits NO and
PGE2 production in LPS-stimulated BV2 microglial
cells
To determine the inhibitory effects of purpurogallin
on LPS-induced NO production in BV2 microglial cells, NO levels in
the cell culture medium were measured by the Griess assay. As shown
in Fig. 1A, compared with the
control, treatment with LPS alone resulted in a marked induction of
NO production; however, treatment with purpurogallin resulted in a
significant inhibition of LPS-induced NO production in a
dose-dependent manner. PGE2, another important
inflammatory mediator, was also evaluated by ELISA. The effects of
purpurogallin on the production of PGE2 in
LPS-stimulated BV2 microglial cells were investigated. As shown in
Fig. 2B, the treatment of BV2
cells with LPS resulted in a marked increase in the release of
PGE2 when compared with the untreated control; however,
treatment with purpurogallin resulted in a dose-dependent decrease
in LPS-induced PGE2 production. These results suggest
that pre-treatment with purpurogallin significantly suppresses the
expression of LPS-mediated pro-inflammatory mediators, such as NO
and PGE2.
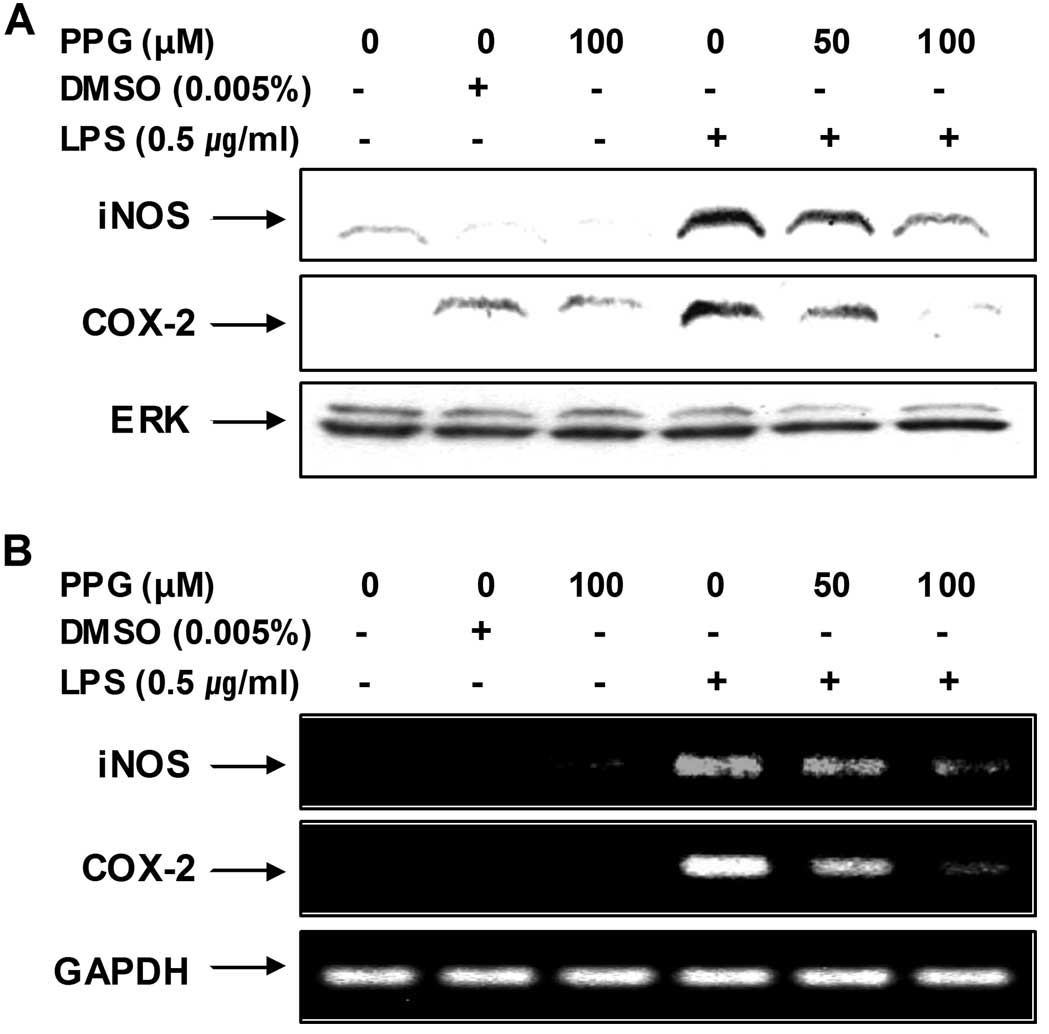
Purpurogallin decreases the expression of
LPS-induced inducible NO synthase (iNOS) and cyclooxygenase-2
(COX-2) mRNA and protein
In order to determine whether the inhibitory effects
of purpurogallin on NO and PGE2 production were
associated with the modulation of iNOS and COX-2 expression, we
examined the mRNA and protein levels of iNOS and COX-2 by RT-PCR
and western blot analysis, respectively. As shown in Fig. 2, iNOS and COX-2 mRNA levels were
observed 6 h following treatment with LPS, whereas the protein
levels of these enzymes were detected in the whole cell lysates 24
h after treatment with LPS. However, treatment with purpurogallin
resulted in a significant decrease in the mRNA and protein
induction of iNOS and COX-2 in the LPS-stimulated BV2 microglial
cells. These results indicate that the purpurogallin-induced
reduction in the expression of iNOS and COX-2 is responsible for
the inhibition of NO and PGE2 production.
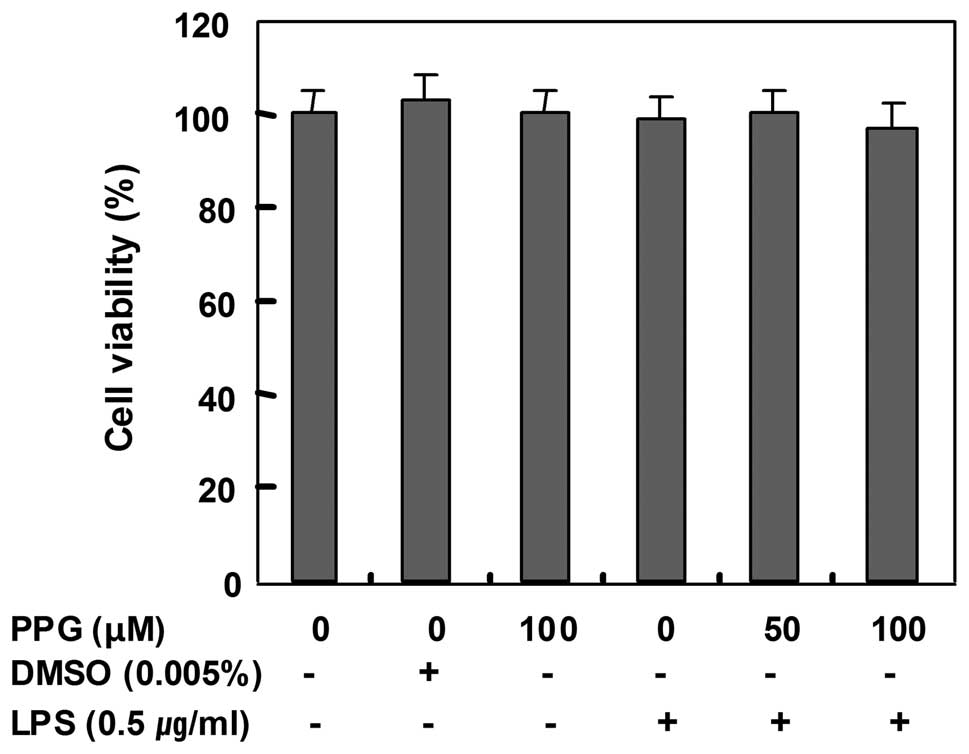
Effects of purpurogallin on cell
viability
In order to exclude any cytotoxic effects of
purpurogallin in BV2 microglial cells, we evaluated the viability
of BV2 cells incubated with or without 0.5 μg/ml LPS in the absence
or presence of purpurogallin using an MTT assay. The concentrations
(50 to 100 μM) used for the inhibition of NO and PGE2
production in the present study did not affect cell viability
(Fig. 3), confirming that the
inhibition of NO and PGE2 production in the
LPS-stimulated BV2 cells was not due to the cytotoxic effects of
purpurogallin.
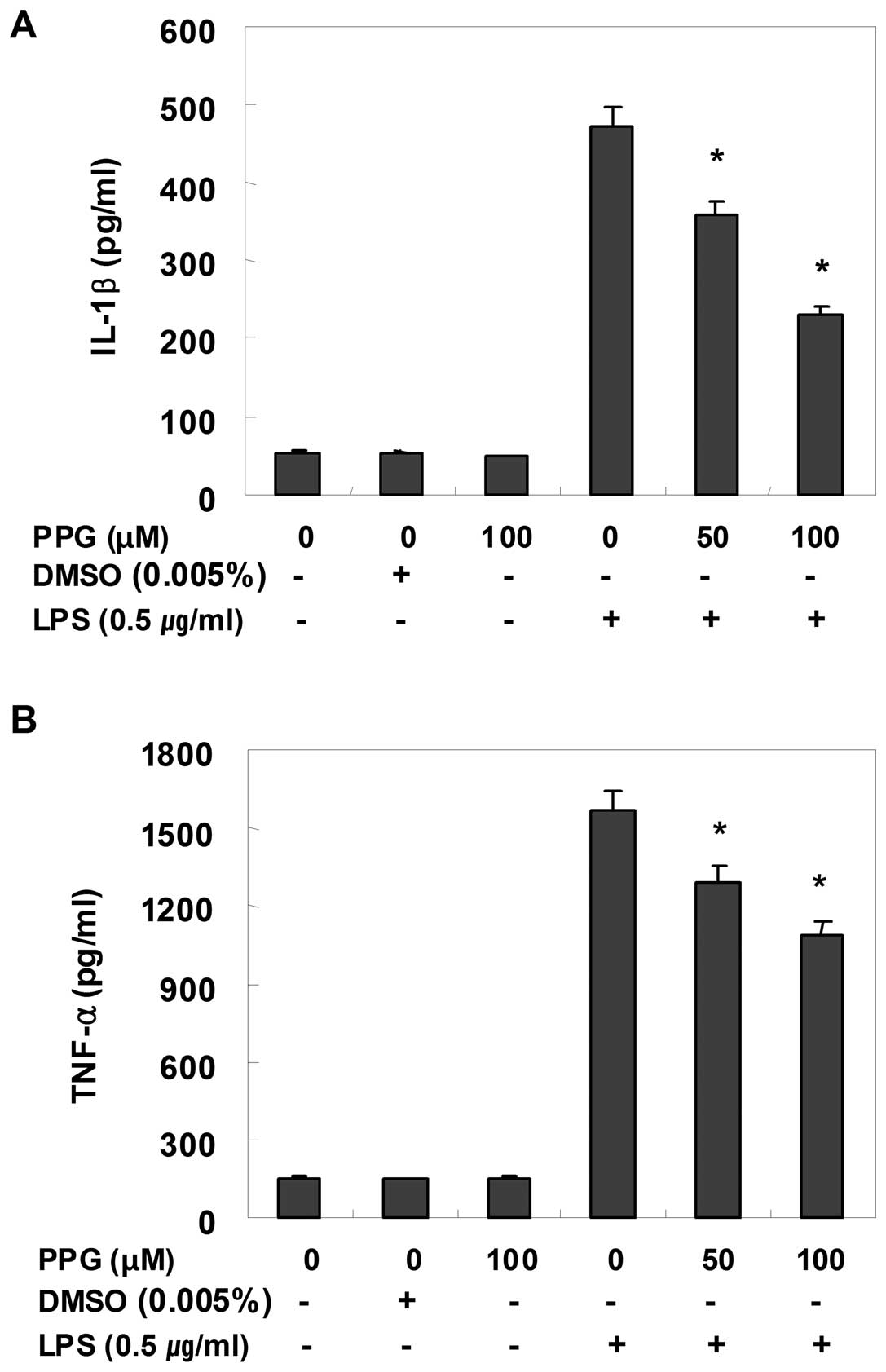
Purpurogallin suppresses the induction of
inflammatory cytokines in LPS-stimulated BV2 microglial cells
Using EIA and RT-PCR, we also analyzed the effects
of purpurogallin on pro-inflammatory cytokines, such as TNF-α and
IL-1β. For this study, BV2 cells were incubated with 50 and 100 μM
purpurogallin in the presence or absence of LPS for 24 h and then
cytokine levels in the culture medium were measured. As shown in
Fig. 4, TNF-α and IL-1β levels
were significantly increased in the culture medium of
LPS-stimulated BV2 microglial cells. However, pre-treatment with
purpurogallin resulted in a significant decrease in the levels of
these cytokines in a concentration-dependent manner. In a parallel
experiment, using RT-PCR, we studied the effects of purpurogallin
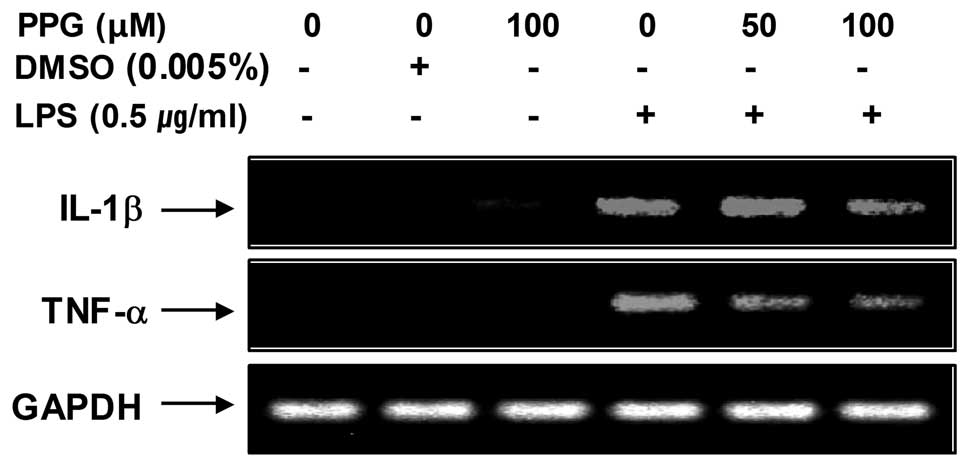
on LPS-induced IL-1β and TNF-α mRNA expression. As shown in
Fig. 5, IL-1β and TNF-α mRNA
transcription also decreased following treatment with
purpurogallin. These results suggest that purpurogallin is
effective in the suppression of pro-inflammatory cytokine
production through the alteration of the transcription levels of
IL-1β and TNF-α in activated microglia.
Purpurogallin blocks nuclear factor-κB
(NF-κB) activation in LPS-stimulated BV2 microglial cells
NF-κB is the most important transcription factor
involved in the regulation of pro-inflammatory mediators and
cytokines, including iNOS, COX-2, TNF-α and IL-1β in activated
microglial cells (30–32); therefore, we investigated whether
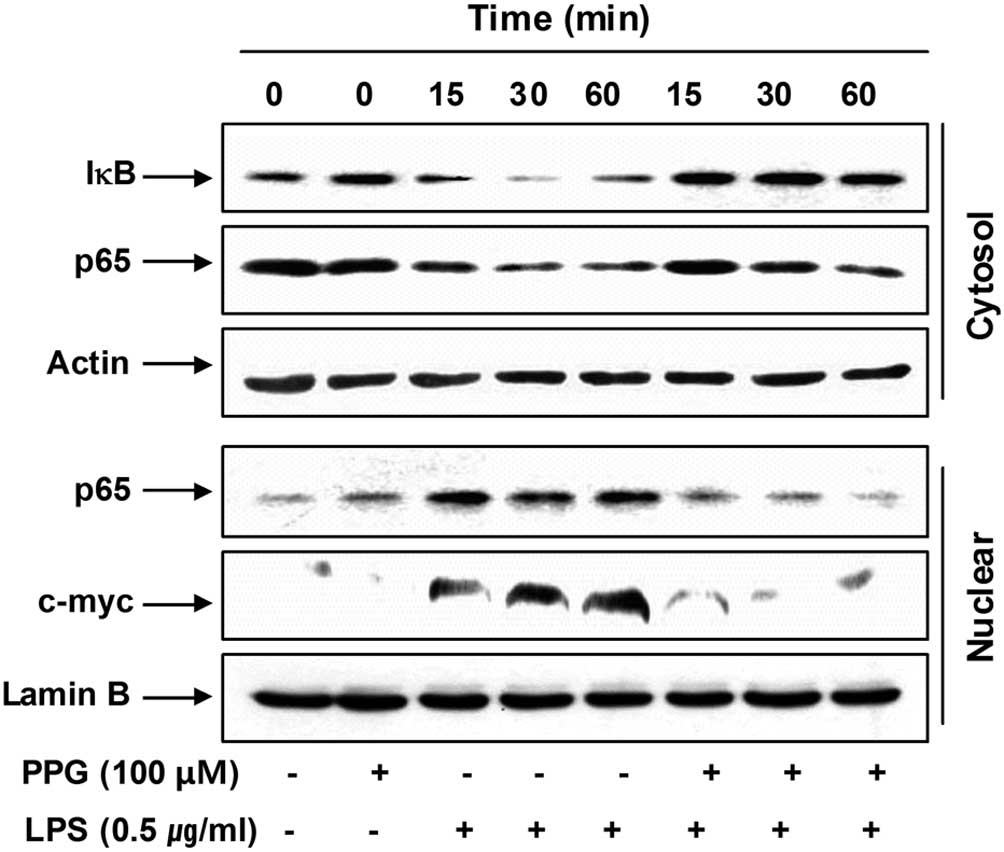
purpurogallin blocks the NF-κB signaling pathway. For this purpose,
cytosolic and nuclear levels of inhibitor of NF-κB (IκB) and NF-κB
p65 subunits were analyzed. As indicated in Fig. 6, LPS stimulation induced IκB
degradation in the cytosol and the translocation of the NF-κB p65
subunit into the nucleus of BV2 cells; however, treatment with
purpurogallin attenuated the degradation of IκB and the nuclear
levels of the NF-κB p65 subunit in LPS-stimulated BV2 cells.
As c-myc is one of the transcription factors
involved in inflammation and its transcription is regulated by
NF-κB (33), we then investigated
whether purpurogallin has an effect on LPS-induced c-myc
expression. As shown in Fig. 6,
although c-myc was expressed at very low levels, exposure to LPS
markedly induced the accumulation of c-myc proteins in the nucleus
of BV2 cells. However, the results demonstrated that, in the
presence of purpurogallin, the levels of c-myc expression were
significantly reduced to levels similar to those of the controls.
These results indicate that purpurogallin inhibits LPS-induced
c-myc expression mediated by NF-κB. Taken together, these findings
indicate that purpurogallin suppresses iNOS, COX-2, TNF-α and IL-1β
expression, at least in part, through an NF-κB-dependent
mechanism.
Purpurogallin reduces LPS-induced
phosphorylation of phosphatidylinositol 3-kinase (PI3K)/Akt and
mitogen-activated protein kinases (MAPKs) in LPS-stimulated BV2
microglial cells
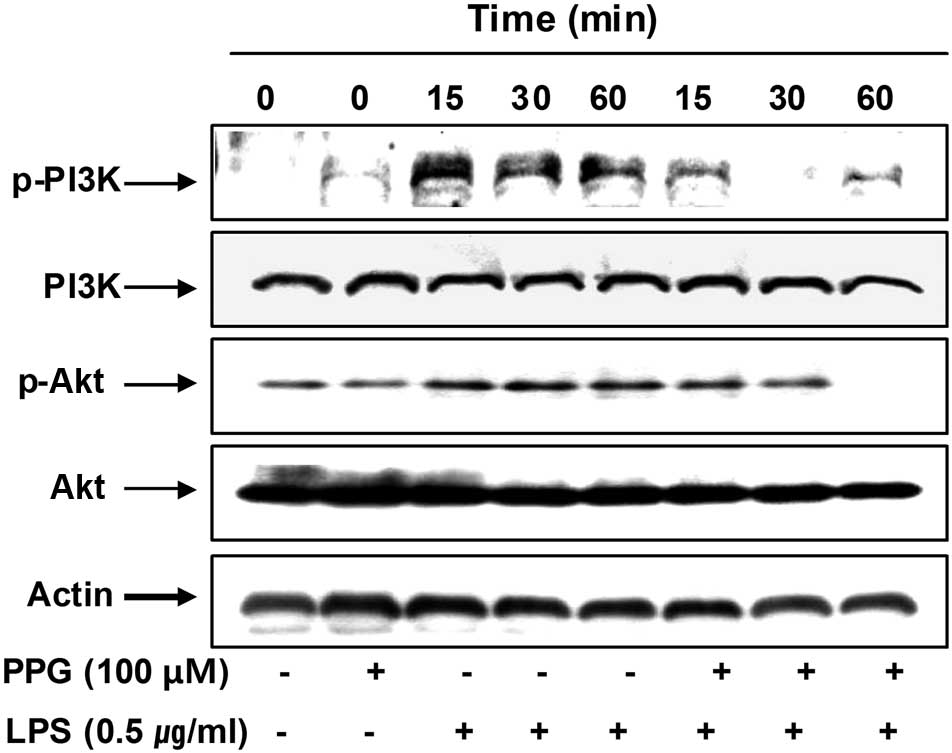
To investigate other intracellular mechanisms
responsible for the inhibitory effects of purpurogallin on
inflammatory mediators, we examined the effects of purpurogallin on
the PI3K/Akt and MAPK signaling pathways. As shown in Fig. 7, the phosphorylation of PI3K and
Akt was markedly increased within 15 min after LPS stimulation;
however, pre-treatment with purpurogallin resulted in significant
blockage of the LPS-induced PI3K and Akt phosphorylation,
indicating that the anti-inflammatory effects of purpurogallin are
associated with the inactivation of the PI3K/Akt signaling
pathway.
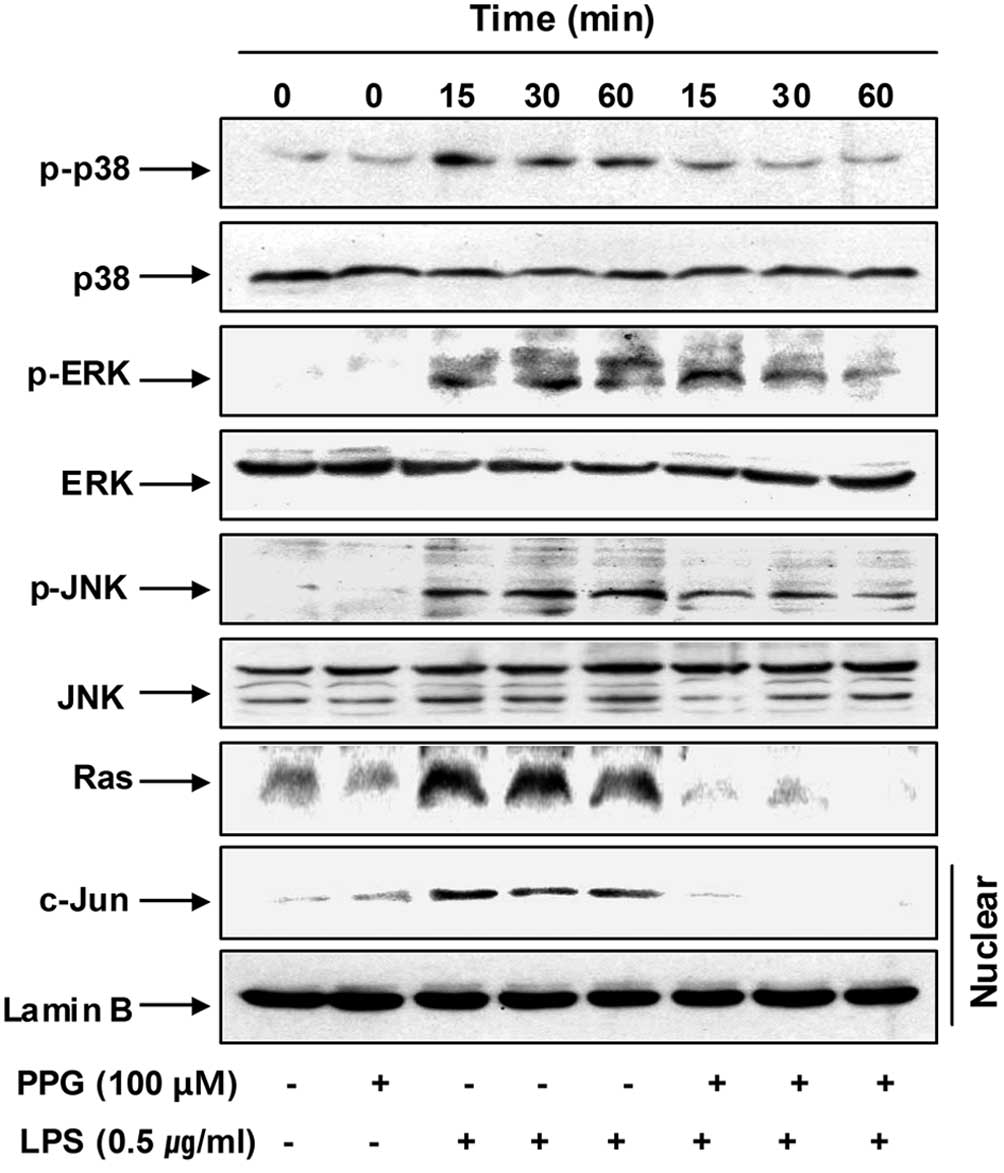
Furthermore, the stimulation of BV2 cells with LPS
induced the rapid activation of MAPKs, such as p38MAPK, ERK and
JNK, with the peak levels of each phospho-MAPK occurring 15 to 60
min after the addition of LPS. However, pre-treatment with
purpurogallin significantly inhibited the phosphorylation of MAPKs
in the LPS-stimulated BV2 microglial cells (Fig. 8). Moreover, the elevated nuclear
levels of c-Jun protein, a common target phosphorylated through the
stress-activated protein kinases, p38MAPK and JNK (34), were also markedly blocked by
purpurogallin.
To assess the mechanisms by which purpurogallin and
LPS co-treatment suppress ERK activation, we evaluated the effects
of LPS on the small molecular weight G protein, Ras, in BV2 cells,
given that Ras is a known upstream activator of ERK (35). As shown in Fig. 8, LPS potently stimulated Ras
expression as early as 15 min after treatment, which was an effect
that persistd for at least 60 min. Of note, upon co-stimulation
with LPS and purpurogallin, we observed a marked reduction in
LPS-mediated Ras expression. These data suggest that purpurogallin
blocks the phosphorylation of ERK through the downregulation of
Ras.
Discussion
In the present study, we demonstrate that
purpurogallin inhibits inflammation in BV2 microglial cells.
Purpurogallin significantly reduced the LPS-induced production of
pro-inflammatory mediators, such as NO and PGE2, as well
as the production of cytokines, including TNF-α and IL-1β.
Furthermore, the anti-inflammatory properties of purpurogallin were
mediated by the downregulation of NF-κB activity, and the
inactivation of the Akt and MAPK signaling pathways.
Among pro-inflammatory mediators released by
activated microglia, NO and PGE2 are the main cytotoxic
mediators participating in the innate response in mammals (36–38). Several lines of evidence have
shown that the activation of microglia induced by injury to the CNS
or infection is associated with neurodegenerative disorders and the
release of NO and PGE2 and with the subsequent release
of pro-inflammatory cytokines and chemokines (3–5). A
number of studies have shown that the expression of COX-2 and iNOS,
key enzymes for NO and PGE2, are upregulated in
activated glial cells. In addition, pro-inflammatory cytokines
activate the transcription of COX-2 and iNOS genes, and
anti-inflammatory drugs may also effectively reduce NO and
PGE2 production (3–5,39).
Thus, downregulators of these inflammatory molecules have been
considered as potential candidates of anti-inflammatory agents to
alleviate the progression of neurodegenerative diseases caused by
the activation of microglia. The results of this study demonstrated
that purpurogallin significantly inhibited NO and PGE2
production by downregulating iNOS and COX-2 mRNA and protein
expression, indicating that the effects of purpurogallin occur at
the transcriptional level. Notably, the inhibitory effects of
purpurogallin on the LPS-induced production of NO and
PGE2 in BV2 microglial cells were not due to the
cytotoxicity of this compound, as assessed by an MTT assay.
Several studies have demonstrated that
pro-inflammatory cytokines, such as IL-1β and TNF-α, are initiators
of the inflammatory response and mediators of the development of
chronic inflammatory diseases (40,41). Therefore, the consequent
overproduction of pro-inflammatory cytokines can be considered to
be a histopathological hallmark of various neurological diseases
and controlling microglial activation and neuroinflammatory
processes may prove to be a therapeutic benefit in the treatment of
depression. In the present study, our results revealed that
purpurogallin significantly inhibited the production of IL-1β and
TNF-α by suppressing their mRNA expression in LPS-treated BV2
microglial cells. Our data indicate that the inhibitory effects of
purpurogallin on the production of these pro-inflammatory cytokines
occur at the transcriptional level.
Various intracellular signaling molecules are
involved in the modulation of inflammatory responses. Among them,
the transcription factor, NF-κB, is a primary regulator of genes
that are involved in the production of pro-inflammatory cytokines
and enzymes involved in the inflammatory process (30–32). In addition, the involvement of the
PI3K/Akt pathway in the expression of inflammatory mediators in
microglia through the activation of NF-κB has been shown (42,43). As a result of its key role in
several pathological conditions, NF-κB is a major drug target in a
variety of diseases, and the blockade of NF-κB transcriptional
activity in microglia is known to suppress the expression of iNOS,
COX-2 and pro-inflammatory cytokines (44–46). Therefore, many putative
anti-inflammatory therapies seek to block NF-κB activity. Our
results demonstrated that purpurogallin markedly inhibited the
LPS-induced IκB-α degradation and NF-κB translocation, which was
associated with the inhibition of c-myc accumulation in the
nucleus. Furthermore, purpurogallin significantly inhibited
PI3K/Akt activation in LPS-stimulated BV2 microglial cells. These
data indicate that purpurogallin inhibits LPS-induced NF-κB
activation through the inactivation of the PI3K/Akt signaling
pathway, and that the inhibitory effects of purpurogallin on
LPS-induced NF-κB and PI3K/Akt activation may be associated with
its anti-inflammatory mechanism(s).
MAPKs are also important signaling molecules
involved in the production of pro-inflammatory mediators and
cytokines and the modulation of NF-κB in microglia (47–49). MAPKs have previously been
implicated in the signaling pathways relevant to LPS-induced
inflammation and LPS is also known to activate a series of MAPKs,
such as ERK, p38MAPK and JNK in microglial cells (50). Therefore, in this study, we
conducted experiments to determine whether purpurogallin tightly
regulates the activation of 3 MAPKs to induce anti-inflammatory
effects in LPS-stimulated BV2 microglial cells. LPS enhanced the
activation of MAPKs, whereas purpurogallin decreased the
LPS-induced activation of MAPKs, which was accompanied by
alterations in the nuclear levels of c-Jun, a common target of
p38MAPK and JNK (34) and Ras, an
upstream activator of ERK (35).
These results suggest that the purpurogallin-mediated inhibition of
pro-inflammatory mediators and cytokines is associated with the
downregulation of the MAPK signaling pathways.
In conclusion, the results presented in this study
demonstrate that purpurogallin inhibits LPS-induced NO and
PGE2 production by suppressing iNOS and COX-2 mRNA and
protein expression in BV2 microglial cells. Purpurogallin also
inhibits the production of pro-inflammatory cytokines (TNF-α and
IL-1β) by suppressing their transcriptional activity. The
inhibitory effects of purpurogallin were mediated by the prevention
of NF-κB activation and by the inhibition of IκB-degradation, which
was accompanied by the blocking of the PI3K/Akt and MAPKs pathways.
Our findings suggest that purpurogallin may provide an effective
treatment strategy for a number of inflammatory and
neurodegenerative disorders.
Acknowledgements
The present study was supported by the R&D
program of MKE/KEIT (10040391, Development of Functional Food
Materials and Device for Prevention of Aging-associated Muscle
Function Decrease), Korea.
References
|
1
|
Mosley RL, Benner EJ, Kadiu I, Thomas M,
Boska MD, Hasan K, Laurie C and Gendelman HE: Neuroinflammation,
oxidative stress and the pathogenesis of Parkinson’s disease. Clin
Neurosci Res. 6:261–281. 2006.
|
|
2
|
Reynolds A, Laurie C, Mosley RL and
Gendelman HE: Oxidative stress and the pathogenesis of
neurodegenerative disorders. Int Rev Neurobiol. 82:297–325. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Perry VH, Nicoll JA and Holmes C:
Microglia in neurodegenerative disease. Nat Rev Neurol. 6:193–201.
2010. View Article : Google Scholar
|
|
4
|
Rivest S: Molecular insights on the
cerebral innate immune system. Brain Behav Immun. 17:13–19. 2003.
View Article : Google Scholar
|
|
5
|
Das S and Basu A: Inflammation: a new
candidate in modulating adult neurogenesis. J Neurosci Res.
86:1199–1208. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Stadelmann C: Multiple sclerosis as a
neurodegenerative disease: pathology, mechanisms and therapeutic
implications. Curr Opin Neurol. 24:224–229. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lull ME and Block ML: Microglial
activation and chronic neurodegeneration. Neurotherapeutics.
7:354–365. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Long-Smith CM, Sullivan AM and Nolan YM:
The influence of microglia on the pathogenesis of Parkinson’s
disease. Prog Neurobiol. 89:277–287. 2009.
|
|
9
|
Dixon RA and Steele CL: Flavonoids and
isoflavonoids - a gold mine for metabolic engineering. Trends Plant
Sci. 4:394–400. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Le Marchand L, Murphy SP, Hankin JH,
Wilkens LR and Kolonel LN: Intake of flavonoids and lung cancer. J
Natl Cancer Inst. 92:154–160. 2000.PubMed/NCBI
|
|
11
|
Szejtli J and Szente L: Elimination of
bitter, disgusting tastes of drugs and foods by cyclodextrins. Eur
J Pharm Biopharm. 61:115–125. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen D, Chen MS, Cui QC, Yang H and Dou
QP: Structure-proteasome-inhibitory activity relationships of
dietary flavonoids in human cancer cells. Front Biosci.
12:1935–1945. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Abou-Karam M and Shier WT: Inhibition of
oncogene product enzyme activity as an approach to cancer
chemoprevention. Tyrosine-specific protein kinase inhibition by
purpurogallin from Quercus sp nutgall. Phytother Res.
13:337–340. 1999. View Article : Google Scholar
|
|
14
|
Wu TW, Zeng LH, Wu J, Fung KP, Weisel RD,
Hempel A and Camerman N: Molecular structure and antioxidant
specificity of purpurogallin in three types of human cardiovascular
cells. Biochem Pharmacol. 52:1073–1080. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Prasad K and Laxdal VA: Evaluation of
hydroxyl radical-scavenging property of purpurogallin using high
pressure liquid chromatography. Mol Cell Biochem. 135:153–158.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sugiyama H, Fung KP and Wu TW:
Purpurogallin as an antioxidant protector of human erythrocytes
against lysis by peroxyl radicals. Life Sci. 53:PL39–PL43. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Inamori Y, Muro C, Sajima E, Katagiri M,
Okamoto Y, Tanaka H, Sakagami Y and Tsujibo H: Biological activity
of purpurogallin. Biosci Biotechnol Biochem. 61:890–892. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Farnet CM, Wang B, Hansen M, Lipford JR,
Zalkow L, Robinson WE Jr, Siegel J and Bushman F: Human
immunodeficiency virus type 1 cDNA integration: new aromatic
hydroxylated inhibitors and studies of the inhibition mechanism.
Antimicrob Agents Chemother. 42:2245–2253. 1998.PubMed/NCBI
|
|
19
|
Lambert JD, Chen D, Wang CY, Ai N, Sang S,
Ho CT, Welsh WJ and Yang CS: Benzotropolone inhibitors of estradiol
methylation: kinetics and in silico modeling studies. Bioorg Med
Chem. 13:2501–2507. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fung KP, Wu TW and Lui CP: Purpurogallin
inhibits DNA synthesis of murine fibrosarcoma L-929 and human U-87
MG glioblastoma cells in vitro. Chemotherapy. 42:199–205. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kitada S, Leone M, Sareth S, Zhai D, Reed
JC and Pellecchia M: Discovery, characterization, and
structure-activity relationships studies of proapoptotic
polyphenols targeting B-cell lymphocyte/leukemia-2 proteins. J Med
Chem. 46:4259–4264. 2003. View Article : Google Scholar
|
|
22
|
Tang G, Yang CY, Nikolovska-Coleska Z, Guo
J, Qiu S, Wang R, Gao W, Wang G, Stuckey J, Krajewski K, Jiang S,
Roller PP and Wang S: Pyrogallol-based molecules as potent
inhibitors of the antiapoptotic Bcl-2 proteins. J Med Chem.
50:1723–1726. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Doulias PT, Nousis L, Zhu BZ, Frei B and
Galaris D: Protection by tropolones against
H2O2-induced DNA damage and apoptosis in
cultured Jurkat cells. Free Radic Res. 39:125–135. 2005.PubMed/NCBI
|
|
24
|
Méchin V, Baumberger S, Pollet B and
Lapierre C: Peroxidase activity can dictate the in vitro lignin
dehydrogenative polymer structure. Phytochemistry. 68:571–579.
2007.PubMed/NCBI
|
|
25
|
Moniruzzaman M, Kamiya N and Goto M:
Biocatalysis in water-in-ionic liquid microemulsions: a case study
with horseradish peroxidase. Langmuir. 25:977–982. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cheong MH, Lee SR, Yoo HS, Jeong JW, Kim
GY, Kim WJ, Jung IC and Choi YH: Anti-inflammatory effects of
Polygala tenuifolia root through inhibition of NF-κB
activation in lipopolysaccharide-induced BV2 microglial cells. J
Ethnopharmacol. 137:1402–1408. 2011.PubMed/NCBI
|
|
27
|
Kim TH, Ku SK, Lee IC and Bae JS:
Anti-inflammatory functions of purpurogallin in LPS-activated human
endothelial cells. BMB Rep. 45:200–205. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lee YA, Choi HM, Lee SH, Yang HI, Yoo MC,
Hong SJ and Kim KS: Synergy between adiponectin and interleukin-1β
on the expression of interleukin-6, interleukin-8, and
cyclooxygenase-2 in fibroblast-like synoviocytes. Exp Mol Med.
44:440–447. 2012.PubMed/NCBI
|
|
29
|
Lee SH, Kim DW, Eom SA, Jun SY, Park M,
Kim DS, Kwon HJ, Kwon HY, Han KH, Park J, Hwang HS, Eum WS and Choi
SY: Suppression of 12-O-tetradecanoylphorbol-13-acetate
(TPA)-induced skin inflammation in mice by transduced Tat-Annexin
protein. BMB Rep. 45:354–359. 2012.
|
|
30
|
Atreya I, Atreya R and Neurath MF:
NF-kappaB in inflammatory bowel disease. J Intern Med. 263:591–596.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wright JG and Christman JW: The role of
nuclear factor kappa B in the pathogenesis of pulmonary diseases:
implications for therapy. Am J Respir Med. 2:211–219. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sebban H and Courtois G: NF-kappaB and
inflammation in genetic disease. Biochem Pharmacol. 72:1153–1160.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Duyao MP, Buckler AJ and Sonenshein GE:
Interaction of an NF-kappa B-like factor with a site upstream of
the c-myc promoter. Proc Natl Acad Sci USA. 87:4727–4731. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Turjanski AG, Vaqué JP and Gutkind JS: MAP
kinases and the control of nuclear events. Oncogene. 26:3240–3253.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Montagut C and Settleman J: Targeting the
RAF-MEK-ERK pathway in cancer therapy. Cancer Lett. 283:125–134.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Garden GA and Möller T: Microglia biology
in health and disease. J Neuroimmune Pharmacol. 1:127–137. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sugama S, Takenouchi T, Fujita M, Conti B
and Hashimoto M: Differential microglial activation between acute
stress and lipopolysaccharide treatment. J Neuroimmunol. 207:24–31.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Schwartz M and Shechter R: Systemic
inflammatory cells fight off neurodegenerative disease. Nat Rev
Neurol. 6:405–410. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Gebicke-Haerter PJ: Microglia in
neurodegeneration: molecular aspects. Micros Res Tech. 54:47–58.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Inoue K: The function of microglia through
purinergic receptors: neuropathic pain and cytokine release.
Pharmacol Ther. 109:210–226. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Merson TD, Binder MD and Kilpatrick TJ:
Role of cytokines as mediators and regulators of microglial
activity in inflammatory demyelination of the CNS. Neuromolecular
Med. 12:99–132. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Madrid LV, Wang CY, Guttridge DC,
Schottelius AJ, Baldwin AS Jr and Mayo MW: Akt suppresses apoptosis
by stimulating the transactivation potential of the RelA/p65
subunit of NF-kappaB. Mol Cell Biol. 20:1626–1638. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lee JY, Jhun BS, Oh YT, Lee JH, Choe W,
Baik HH, Ha J, Yoon KS, Kim SS and Kang I: Activation of adenosine
A3 receptor suppresses lipopolysaccharide-induced TNF-alpha
production through inhibition of PI 3-kinase/Akt and NF-kappaB
activation in murine BV2 microglial cells. Neurosci Lett. 396:1–6.
2006. View Article : Google Scholar
|
|
44
|
Shin WS, Szuba A and Rockson SG: The role
of chemokines in human cardiovascular pathology: enhanced
biological insights. Atherosclerosis. 160:91–102. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Nam NH: Naturally occurring NF-kappaB
inhibitors. Mini Rev Med Chem. 6:945–951. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Luqman S and Pezzuto JM: NFkappaB: a
promising target for natural products in cancer chemoprevention.
Phytother Res. 24:949–963. 2010.PubMed/NCBI
|
|
47
|
Kaminska B: MAPK signalling pathways as
molecular targets for anti-inflammatory therapy - from molecular
mechanisms to therapeutic benefits. Biochim Biophys Acta.
1754:253–262. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kaminska B, Gozdz A, Zawadzka M,
Ellert-Miklaszewska A and Lipko M: MAPK signal transduction
underlying brain inflammation and gliosis as therapeutic target.
Anat Rec (Hoboken). 292:1902–1913. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Wei J and Feng J: Signaling pathways
associated with inflammatory bowel disease. Recent Pat Inflamm
Allergy Drug Discov. 4:105–117. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Pyo H, Jou I, Jung S, Hong S and Joe EH:
Mitogen-activated protein kinases activated by lipopolysaccharide
and beta-amyloid in cultured rat microglia. Neuroreport. 9:871–874.
1998. View Article : Google Scholar : PubMed/NCBI
|