Introduction
Rheumatoid arthritis (RA) is a chronic autoimmune
disease characterized by chronic inflammation, mainly of the
synovial joints, which affects numerous organs and systems, and may
lead to cartilage destruction and deformation, resulting in chronic
pain, severe disability and increased mortality rates (1). Although the causes of RA are not yet
fully understood, laboratory and clinical evidence suggests that
pro-inflammatory cytokines, particularly interleukin (IL)-17, play
an important role in the pathogenesis of this disease (2–5).
IL-17A is a T cell-derived pro-inflammatory cytokine produced in
the rheumatoid synovium. Some RA synovial T cells producing IL-17
can activate mesenchymal cells, leading to an increased
pro-inflammatory pattern sensitive to regulation by Th2-type
cytokines. IL-17, particularly when combined with tumor necrosis
factor (TNF)-α, may contribute to the progression of RA, notably
through their combined effect on synoviocyte aggressiveness
(6,7). In patients with RA, high local
levels of IL-17 are detected in both the synovium and synovial
fluid (8,9). IL-17 induces the production of
pro-inflammatory cytokines (IL-1β, TNF-α and IL-6) (10,11) and CXC chemokines, which recruit
neutrophils into the joints (12,13), stimulate angiogenesis (14,15), and are implicated in joint
degradation (16). The relevance
of IL-17 for RA has been further highlighted by in vitro
studies performed in the presence of TNF-α and IL-1β, two key
pathogenic pro-inflammatory cytokines overexpressed in the joints
of patients with RA (17,18). IL-17 synergistically acts with
these two cytokines to induce inflammatory mediators in
synoviocytes, human osteoblasts, myoblasts and chondrocytes
(19–21). Furthermore, a clinical study
demonstrated that synovial IL-17 overexpression is associated with
damage to the joints (22).
Although IL-17A shows affinity to the IL-17 receptor
A (IL-17RA), binding assays have suggested that it can bind
additional receptors. To date, four additional receptors have been
identified in the IL-17R family based on sequence homology to
IL-17R (IL-17Rh1, IL-17RC, IL-17RD and IL-17RE) (23). Functional IL-17R is a heteromeric
complex of IL-17RA and IL-17RC proteins (24). A number of different components of
the complex exist, including IL-17RB (25), which binds preferentially to
IL-17B and IL-17E (26,27). IL-17RA and IL-17RC have been found
to be overexpressed in RA peripheral whole blood cells and their
expression has been detected in the synovium of patients with RA
(28). Their levels are increased
in serum and synovial fluid in patients with RA and with other
autoimmune and inflammatory conditions.
In inflammatory arthritis, IL-17 plays a key role in
the propagation of joint inflammation, cartilage destruction and
bone erosion (29). In recent
years, the use of anti-IL-17 antibodies in RA therapy has been
investigated by an increasing number of researchers. Lubberts et
al (30) reported that
treatment with a neutralizing anti-murine IL-17 antibody after the
onset of collagen-induced arthritis (CIA) reduced joint
inflammation, cartilage destruction and bone erosion. Genovese
et al (31) reported that
a humanized anti-IL-17 monoclonal antibody is an efficient
anti-rheumatic agent, in a clinical trial in patients with RA.
Nardelli et al (32)
demonstrated that the treatment of interferon (IFN)-γ-deficient
mice vaccinated and challenged with Borrelia burgdorferi
with a murine anti-IL-17 antibody exerted therapeutic effects
against severe destructive arthritis. Kotake et al (9) demonstrated that the levels of IL-17
were significantly higher in the synovial fluid of patients with RA
compared with patients with osteoarthritis and that a murine
anti-IL-17 antibody significantly inhibited osteoclast formation.
Although numerous studies have suggested that anti-IL-17 antibodies
alleviate the symptoms of RA, a considerable number of improvements
in this type of therapy are required with regards to safety and
immunomodulatory effects. In this study, we investigated whether
the recombinant chimeric anti-IL-17 full-length monoclonal antibody
(CMa17Aab) exerts therapeutic effects by neutralizing IL-17. DNA
encoding the murine anti-human IL-17 was linked to DNA encoding the
Fc portion of a human IgG1 molecule, and the combined DNA was then
expressed in a mammalian cell line. CMa17Aab may be used to reduce
anti-xenogeneic immunoglobulin response and anti-idiotypic response
by employing alternative portions of the murine Fc fragment.
Differences in the constant region of the heavy chain of CMa17Aab
determine the immunoglobulin isotypes, which can alter the effects
of the antibody in vivo, such as complement-dependent
cytotoxicity (CDC), antibody-dependent cell-mediated cytotoxicity
(ADCC) and immunomodulatory effects. Our results demonstrate that
CMa17Aab acts as a competitive inhibitor of IL-17 and prevents the
binding of IL-17 to the cell surface receptor, IL-17R, thereby
reducing the biological activity of IL-17. No adverse effects were
detected following the intravenous administration of CMa17Aab in
DBA/1J mice. There were trends toward a reduction in disease
activity in a safety and dose-finding study of CMa17Aab
administered for four weeks to a small number of mice with
refractory RA. Based on these findings, we conclude that CMa17Aab
may be an ideal therapeutic agent for clinical application in
patients with active, refractory RA.
Materials and methods
Cell lines and reagents
CHO-K1SV and HeLa cells were purchased from the
American Type Culture Collection (ATCC; Rockville, MD, USA). RA
synoviocytes were kindly provided by Harbin Medical University. The
CHO-K1SV cell line was cultured in Iscove’s modified Dulbecco’s
medium (IMDM) supplemented with 10% fetal bovine serum (FBS) (both
from Sigma-Aldrich, St. Louis, MO, USA) and 2 mM L-glutamine. HeLa
cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM;
Sigma-Aldrich) supplemented with 10% FBS. RA synoviocytes were
cultured in DMEM (Invitrogen Life Technologies, Carlsbad, CA, USA)
supplemented with 10% fetal calf serum (FCS), 2 mM L-glutamine, 100
U/ml penicillin and 100 U/ml streptomycin, at 37°C in a humidified
5% CO2 incubator. Restriction enzymes and the DNA
ligation and purification systems were purchased from New England
Biolabs (Hitchin, UK).
Construction of expression vectors and
stable expression of CMa17Aab in CHO-K1SV cells
The construction of the neutralizing mouse
anti-human IL-17 monoclonal antibody was confirmed by
fluorescence-activated cell sorting (FACS). The single chain
variable fragments (ScFvs) of the mouse anti-human IL-17 monoclonal
antibody were cloned and inserted into the Pklight vector (termed
Pklight-anti-IL-17 vector). The constant domain heavy chain (CH)-Fc
and light chain (CL) fragments were subcloned into the
Pklight-anti-IL-17 vector, subsequently termed Pklight-CMa17Aab.
Pklight-CMa17Aab and IREX-enhanced green fluorescent protein (EGFP)
were subcloned into the Peedual12.4 vector (termed
Peedual12.4-CMa17Aab vector) which contained the glutamine
synthetase (GS) gene. Peedual12.4-CMa17Aab was transfected into the
CHO-K1SV cells using Lipofectamine 2000. Thus, CMa17Aab and EGFP
were both stably expressed in the CHO-K1SV cells. The stable and
high expression of CMa17Aab in the CHO-K1SV cells was confirmed
using a flow cytometer (BD FACSAria™ Cell Sorter; BD Biosciences,
Franklin Lakes, NJ, USA).
Purification of CMa17Aab from the
supernatant of CHO-K1SV cells
The supernatant of serum-free CHO-K1SV cells was
harvested. CMa17Aab was purified using protein-A affinity and
size-exclusion chromatography selecting for purity of the monomer
>90%.
Sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE)
The purified CMa17Aab fragment was resolved by 12%
SDS-PAGE under reducing conditions with β-mercaptoethanol
(β-ME).
ELISA
In the cross-linking assay, CMa17Aab was examined
for its capacity to simultaneously bind two target antigens: hIL-17
and mIL-17. Purified CMa17Aab was determined by ELISA. CMa17Aab or
anti-IL-17 antibody (ScFv) (0.2–1.0 mg/ml) were added into a
96-well microplate and incubated at 4°C for 12 h. hIL-17 or mIL-17
(2 μg/ml) were then added to the wells and incubated for 30 min at
37°C, with CMa17Aab replaced by BSA in the controls. After five
washes with PBST [20 mmol/l phosphatase-buffered saline (PBS),
0.05% Tween-20], 100 μl mouse HRP-conjugated anti-hIL-17 monoclonal
antibody (eBioscience, Inc., San Diego, CA, USA) was added followed
by incubation for 1 h. Following five washes with PBST, the
DAB/H2O2 system was used for detection. The
results were recorded at 450 nm on an ELISA plate reader.
ADCC and CDC assays
Peripheral blood mononuclear cells (PBMCs) were
prepared by centrifugation in a Ficoll-Hypaque density gradient.
ADCC and CDC of CMa17Aab were measured by lactate dehydrogenase
(LDH) assay (Merck KGaA, Darmstadt, Germany), which measures the
activity of LDH released from the cytosol of damaged cells. HeLa
cells stably expressing transmembrane IL-17A were incubated with
various concentrations of CMa17Aab for 1 h in assay medium (DMEM +
5% FBS) in a 5% CO2 incubator at 37°C, followed by the
addition of either human PBMCs as effector cells (effector to
target ratio, 20:1 for the ADCC assay) or human complement serum
(Quidel Corp., San Diego, CA, USA) (1.25% vol/vol for the CDC
assay). Following an additional incubation at 37°C for 16 h for the
ADCC assay and 5 h for the CDC assay, 100 μl of supernatant from
each well were transferred to a clean flat-bottom 96-well plate.
LDH substrate (100 μl) was added to each well followed by
incubation for 30 min at room temperature in the dark. The
absorbance of the samples was measured at 490 nm on an ELISA plate
reader.
RNA extraction and real-time RT-PCR
Total RNA (1 μg) was reverse transcribed using the
ThermoScript RT-PCR System (Invitrogen Life Technologies). The
concentration of RNA was quantified by spectrophotometry at 260 nm
(SmartSpec™ 3000; Bio-Rad, Hercules, CA, USA). Briefly, total RNA
was denatured by incubation for 5 min at 70°C with 4 μM oligo(dT)
primers and then reverse transcribed using 0.5 mM dNTP, 40 U/μl
RNaseOUT, 0.01 M DTT and 10 U/μl of the ThermoScript Reverse
Transcriptase enzyme (final concentrations). Reverse transcription
was performed by incubation at 42°C for 180 min. The obtained cDNA
was diluted 1/10 with distilled water and 10 μl of the dilution
were used for amplification. Specific primer sets for IL-6, IL-8,
matrix metalloproteinase (MMP)-3, IL-17, IL-1β, TNF-α, receptor
activator for nuclear factor-κB ligand (RANKL) and IFN-γ were
conserved in our laboratory. Primer sets for IL-17RA (GenBank
accession no. NM_014339) were synthesized by Invitrogen as follows:
IL-17RA forward, 5′-AGACACTCCAGAACCAATTCC-3′ and reverse,
5′-TCTTAGAGTTGCTCTCCACCA-3′. PCR was performed using the
LightCycler FastStart DNA SYBR-Green I kit (Roche Molecular
Biochemicals, Indianapolis, IN, USA) following the manufacturer’s
instructions on the parameters: 45 amplification cycles,
denaturation at 96°C, primer annealing at 68°C with touchdown at
58°C, and amplicon extension at 72°C.
Animal experiments
To establish the mouse model of CIA, 8–10-week-old
DBA/1J mice were purchased from the Shanghai SLAC Laboratory Animal
Co., Ltd. (Shanghai, China). The mice were kept in a
temperature-controlled environment in a 12-h light/dark cycle. The
induction of type II CIA was achieved as previously described
(33) by the subcutaneous
injection of 2, 4 or 8 mg collagen (ModiQuest Research, Oss, The
Netherlands) per mouse (n=9 in each group) (34). Clinical arthritis scores were
evaluated using a scale of 0–2 for each paw for a total score of 8.
Paws were assigned a clinical core based on the following scoring
method (ModiQuest Research): 0, normal; 0.25, one or two swollen
toes; 0.5, three to four swollen toes; 0.75, slightly swollen
footpad or ankle; 1, swollen footpad or ankle; 1.25, one or two
swollen toes and swollen footpad or ankle; 2.0, swollen toes,
footpad and ankle. Treatment commenced on day 21 after the initial
immunization. Mice were administered 10 mg/kg CMa17Aab by
subcutaneous injection for nine consecutive days and were
sacrificed on day 70.
This study was carried out in strict accordance with
the recommendations in the Guide for the Care and Use of Laboratory
Animals of the National Institutes of Health. The protocol was
approved by the Chinese Association For Laboratory Animal Sciences
(CALAS), Animal Health Products, Committee on the Ethics of Animal
Experiments Defence Research and Development, China and Animal
Experiments of the University of Northeast Agricultural (approval
number: SCXK-2012-0002). All surgical procedures and euthanasia
were performed under sodium pentobarbital anesthesia, and all
efforts were made to minimize suffering.
Statistical analysis
Statistical significance was determined with a
two-tailed Student’s t-test using Excel software. One-way ANOVA,
the Mann-Whitney U test, Kaplan-Meier and log-rank statistical
analyses were performed using MedCalc software (MedCalc, Ostend,
Belgium).
Results
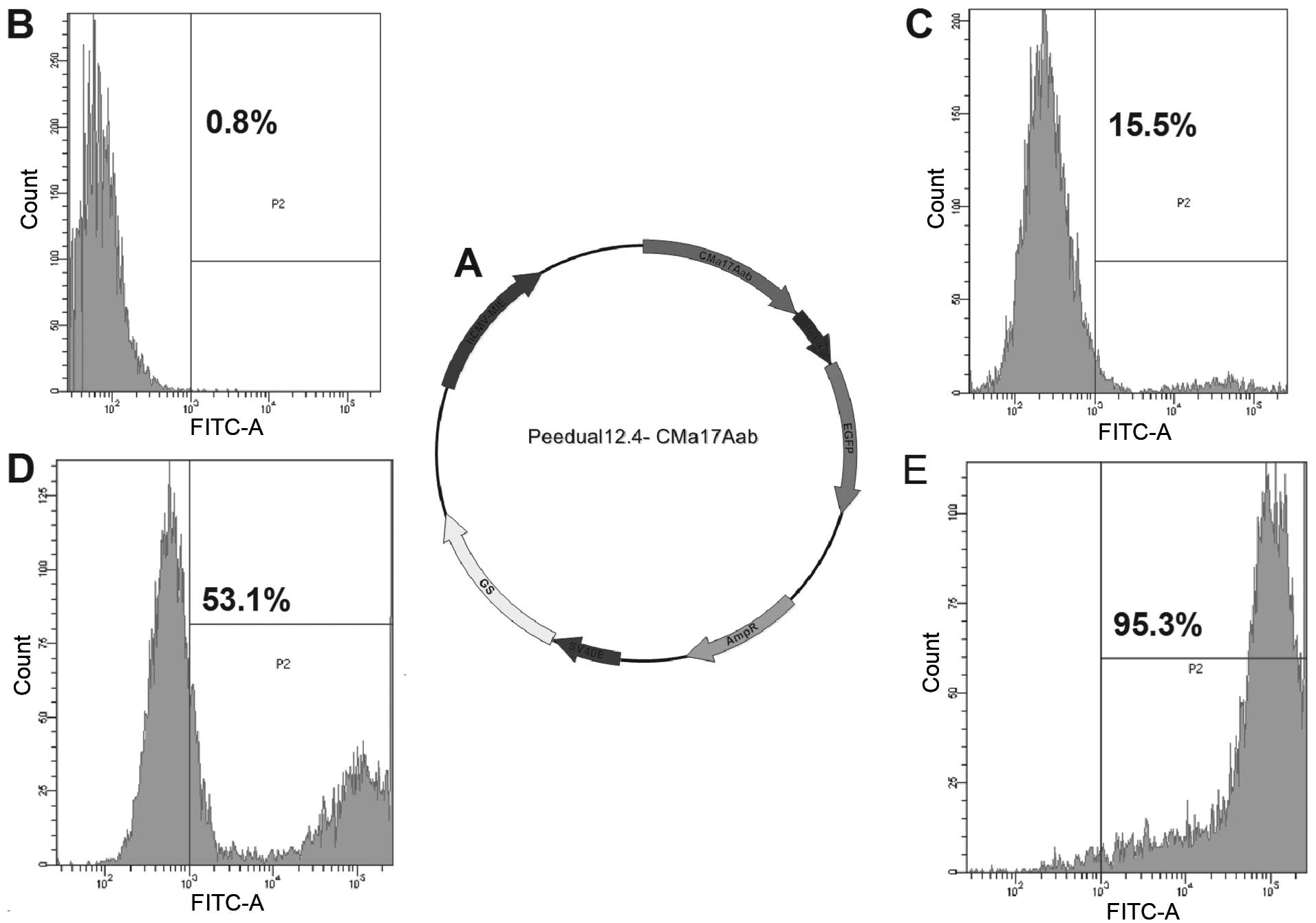
Construction of vector and stable
expression of the recombinant anti-human IL-17A high-affinity
antibody in CHO-K1SV cells
The recombinant vector Peedual12.4, expressing
CMa17Aab was constructed as described in Materials and methods
(Fig. 1A). The resulting vector
containing EGFP and GS genes, was termed Peedual-12.4-CMa17Aab. The
expression of CMa17Aab in the CHO-K1SV cells was determined by
screening with EGFP using FACS, a simple, rapid, sensitive and
reliable method. The expression of CMa17Aab in the CHO-K1SV cells
was also determined by screening with GS, using the methionine
sulfoximine (MSX) system. A screening system using EGFP and GS has
the advantage of emitting fluorescence without the addition of any
substrates. The system was excitated by a 488-nm laser and the
emission was detected through a 530/30 band-pass filter. For each
sample, 10,000 or 20,000 populations of cells were analyzed, and
cells with a higher fluorescence intensity as compared with the
background-defined threshold (from cells not transfected with the
Peedual-12.4-CMa17Aab vector) were calculated. The selected cells
were plated into 96-well culture plates, and subcloned one month
later. The results revealed that a polyclone with a stable and high
expression of CMa17Aab was successfully obtained.
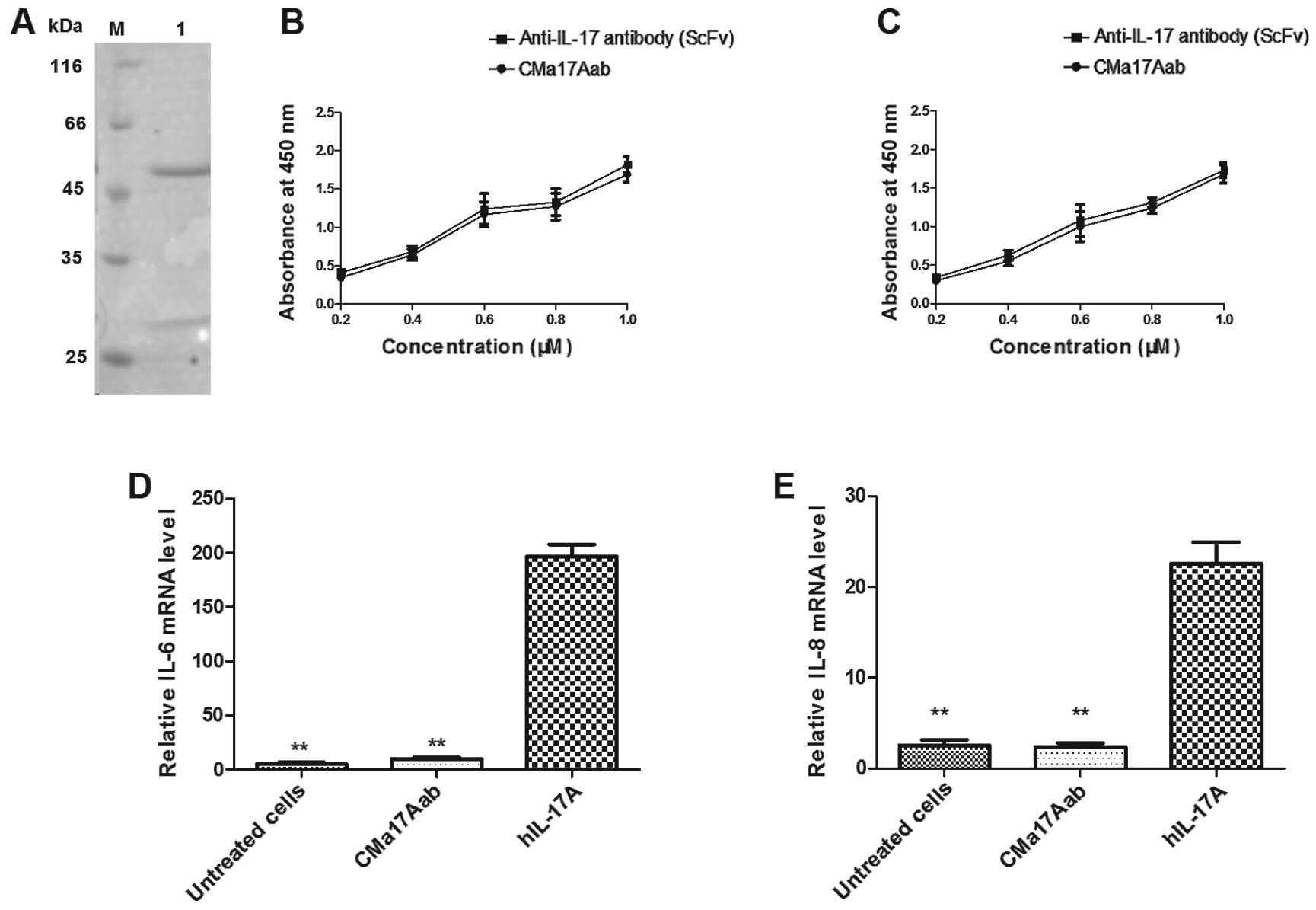
CMa17Aab blocks the gene expression of
IL-6 and IL-8 stimulated by IL-17A
The supernatant of the CHO-K1SV serum-free cells was
harvested after three days of culture and the CMa17Aab antibody was
purified using protein-A affinity and size-exclusion
chromatography. Purified CMa17Aab was analyzed on a 12% SDS-PAGE
gel (Fig. 2A), which confirmed
purification. DNA and endotoxins were removed from purified
CMa17Aab for the subsequent animal experiments. To evaluate the
binding specifity of purified CMa17Aab, a series of ELISAs were
performed. Binding to IL-17 was assessed using an anti-IL-17 scFv
antibody as the positive control. As shown in Fig. 2B, the results revealed the binding
of CMa17Aab to mIL-17 and hIL-17. We also evaluated the ability of
CMa17Aab to block the binding of hIL-17A with IL-17RA. hIL-17A (at
a final concentration of 100 ng/ml) was added to the HeLa cells,
the cells were cultured at 37°C for 12 h, and the gene expression
of IL-6 and IL-8 was measured by real-time RT-PCR. The
hIL-17A-stimulated cells showed an increase in the gene expression
of IL-6 (CMa17Aab-treated cells and untreated cells vs.
IL-17-treated cells, P=0.0036 and P=0.0024, respectively) and IL-8
(CMa17Aab-treated cells and untreated cells vs. IL-17-treated
cells, P=0.0075 and P=0.0078, respectively) (Fig. 2D and E). Student’s paired
two-tailed t-tests revealed that these differences were
statistically significant (P<0.05, P<0.01 vs. IL-17-treated
cells).
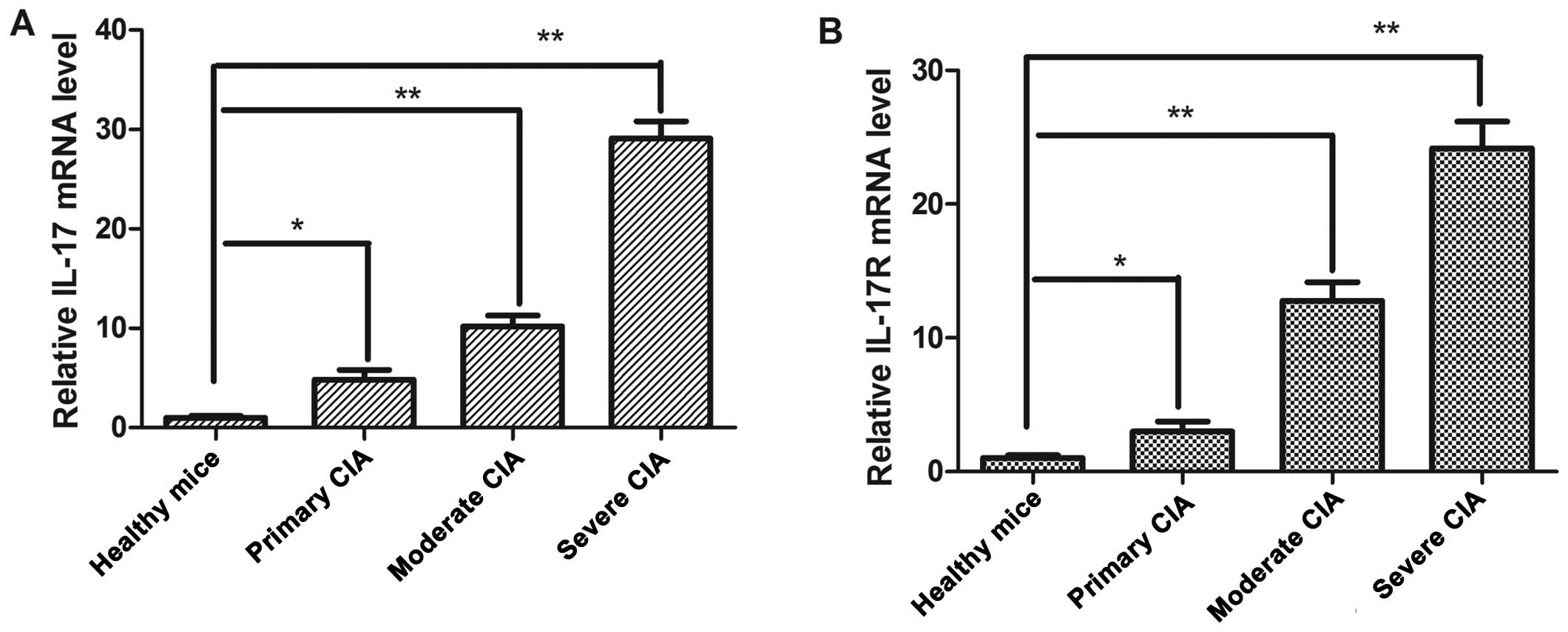
IL-17 detection in peripheral whole blood
and IL-17R overexpression in synovial tissue from mice with
CIA
To determine whether IL-17 and IL-17R are
differentially expressed in mice with CIA as compared with healthy
mice, we examined IL-17 expression at the mRNA level in whole blood
samples by real-time RT-PCR (Fig.
3A). A significant upregulation of IL-17 expression in
peripheral whole blood was observed in the mice with CIA (n=9) as
compared with the healthy mice (n=3). This difference mainly
reflects the overexpression of IL-17 in mice with severe CIA
(healthy mice vs. mice with primary, moderate and severe CIA: 4.9-,
10.22- and 29.09-fold, respectively; P<0.05, P<0.01 and
P<0.01, respectively). These results revealed that IL-17 mRNA
expression was higher in the mice with primary, moderate and severe
CIA mice as compared with the healthy mice.
As it has been previously suggested that IL-17 acts
as a heterodimer to transduce IL-17R signals (35), we examined the expression of
IL-17R in synovial tissue (n=9). We demonstrated a correlation
between the mRNA expression of IL-17R and the development of
illness in the synovial tissue of mice with CIA (primary, moderate
and severe CIA: P<0.05, P<0.01 and P<0.01, respectively).
Moreover, it should be noted that the IL-17R mRNA levels in mice
with CIA were higher than those in the healthy mice (Fig. 3B).
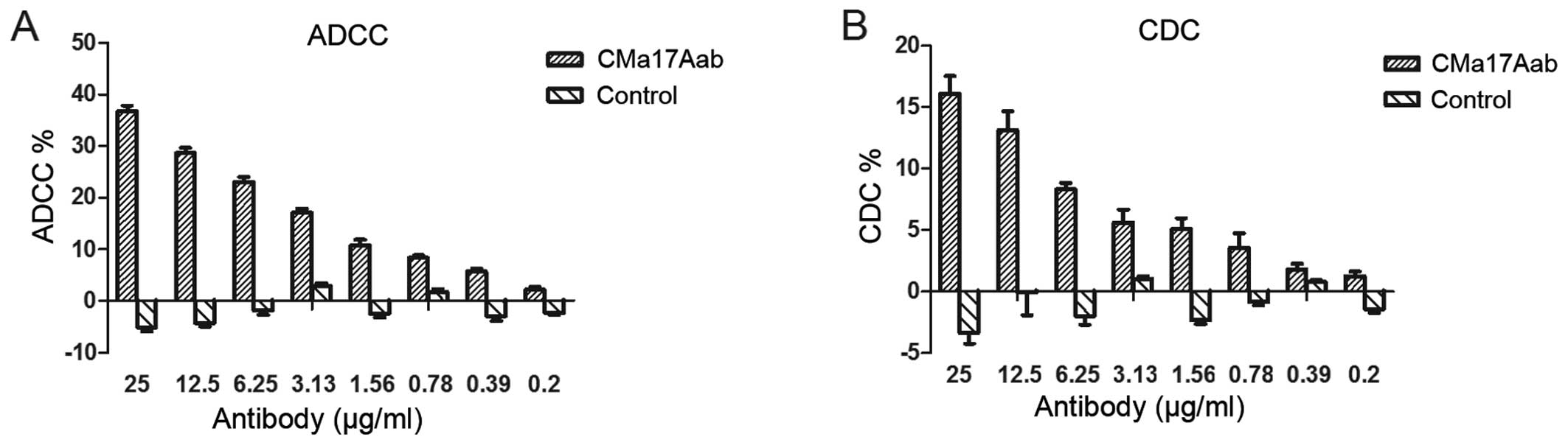
Ability of CMa17Aab to mediate ADCC and
CDC
Previous studies have reported that the binding of
antibodies to the free form of IL-17 is important in the treatment
of RA, since it can lead to the suppression of cell
surface-expressed IL-17A through ADCC or CDC (30,36,37); this may be one of the reasons
responsible for the differential effects observed during the
clinical treatment of diseases with anti-IL-17 antibodies. The cell
surface expression of IL-17RA on macrophages and monocytes plays a
critical role in granulomatous diseases such as Crohn’s disease and
Wegener’s granulomatosis, and the cells can be directly killed
through ADCC or CDC. When CMa17Aab binds to IL-17A following
IL-17-antagonist binding to cells expressing the transmembrane form
of IL-17RA, these cells are targeted by natural killer cells,
triggering systemic complement activation. The presence of the Fc
region of human IgG1 in CMa17Aab may induce cell lysis in
IL-17RA-producing cells. The ability of CMa17Aab to mediate ADCC
and CDC in cells expressing the transmembrane form of IL-17R was
examined in this study. In the ADCC assay, >20% of the
IL-17RA-bearing HeLa target cells were lysed by CMa17Aab at 6.25
mg/ml at a 20:1 effector to target ratio (Fig. 4A). In the CDC assay, CMa17Aab
induced the lysis of transmembrane IL-17RA cells in the presence of
human complement serum (Fig. 4B).
These data indicate that CMa17Aab mediates ADCC and CDC upon
binding to transmembrane IL-17RA expressed on the cell surface and
therefore, there is considerable potential to develop CMa17Aab into
a more effective IL-17A-neutralizing antibody, similar to other
therapeutic antibodies already in use with the ability to induce
ADCC and CDC.
In vivo inhibition of murine CIA with
CMa17Aab and reduction of the humoral immune response against type
II collagen
Heterologous type II collagen is widely used as an
immunogen for the development of the model of CIA. Antibodies to
type II collagen are elevated in mice with CIA. In this study, to
assess the therapeutic effects of CMa17Aab on the development of
RA, the mouse model of CIA was adopted, in which the disease was
induced by the systemic administration of a cocktail of monoclonal
antibodies that target various regions of type II collagen, which
is one of the major constituents of articular cartilage matrix
proteins, followed by lipopolysaccharides (38). The mice were subcutaneously
injected with 10 mg/kg CMa17Aab or the same volume of dextrose
(DEX) or sodium chloride (NaCl) (untreated group) for nine
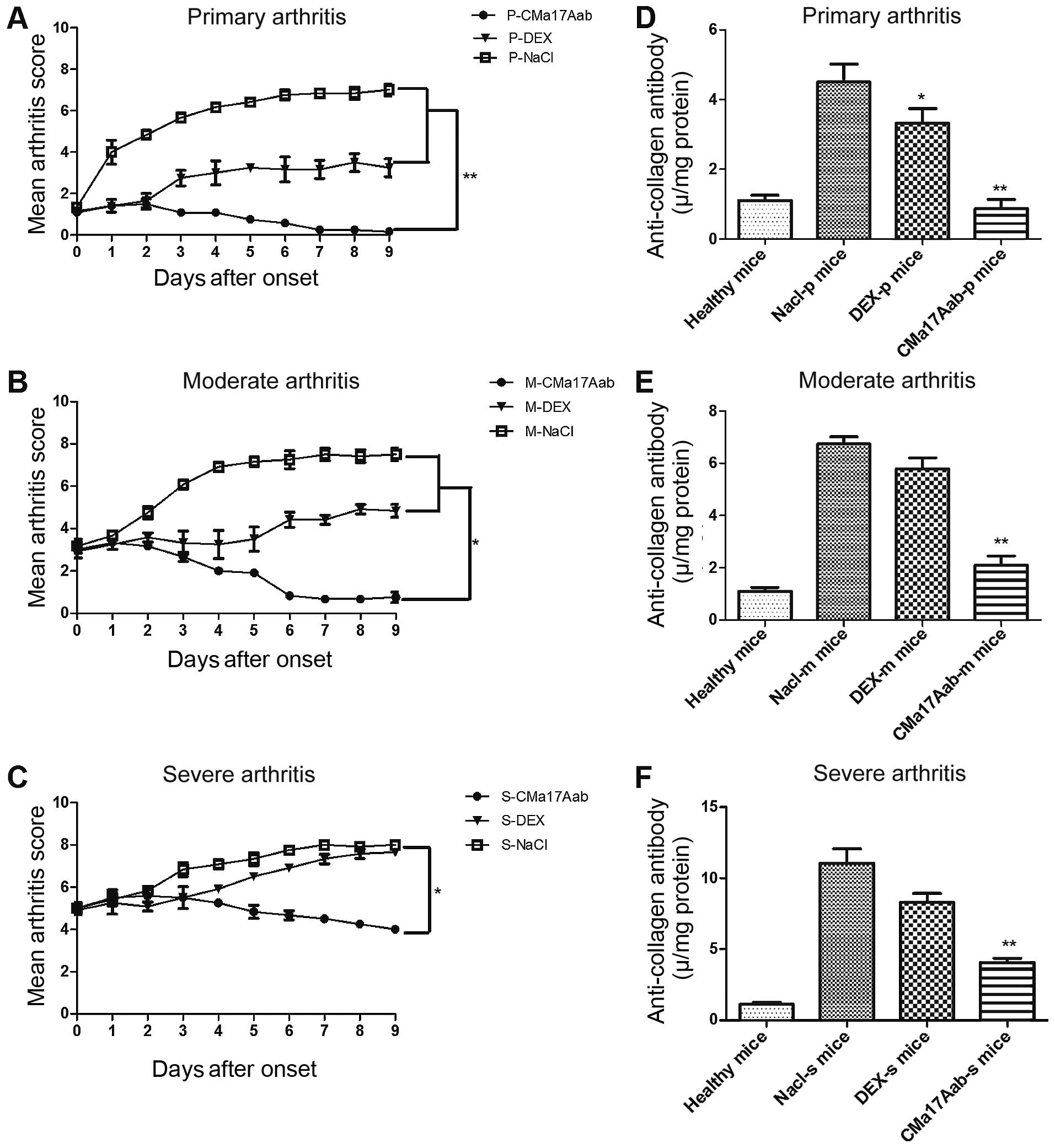
consecutive days. As shown in Fig.
5A–C, the CMa17Aab-treated mice showed a significantly reduced
progression to severe disease symptoms compared with the
saline-treated mice (P<0.05, P<0.01).
Since the humoral immune response against type II
collagen plays a pivotal role in the development of arthritis, we
examined the potentially beneficial effects of CMa17Aab on the
humoral anti-collagen response, by quantifying the anti-type II
collagen IgG level in serum. As shown in Fig. 5D–F, a significant decrease in the
serum level of anti-type II collagen IgG was observed in the
CMa17Aab-treated mice, compared with the controls (P<0.01).
Real-time RT-PCR analysis of RA-related
cytokine expression and histopathological analyses of RA synovial
grafts in mouse model of CIA
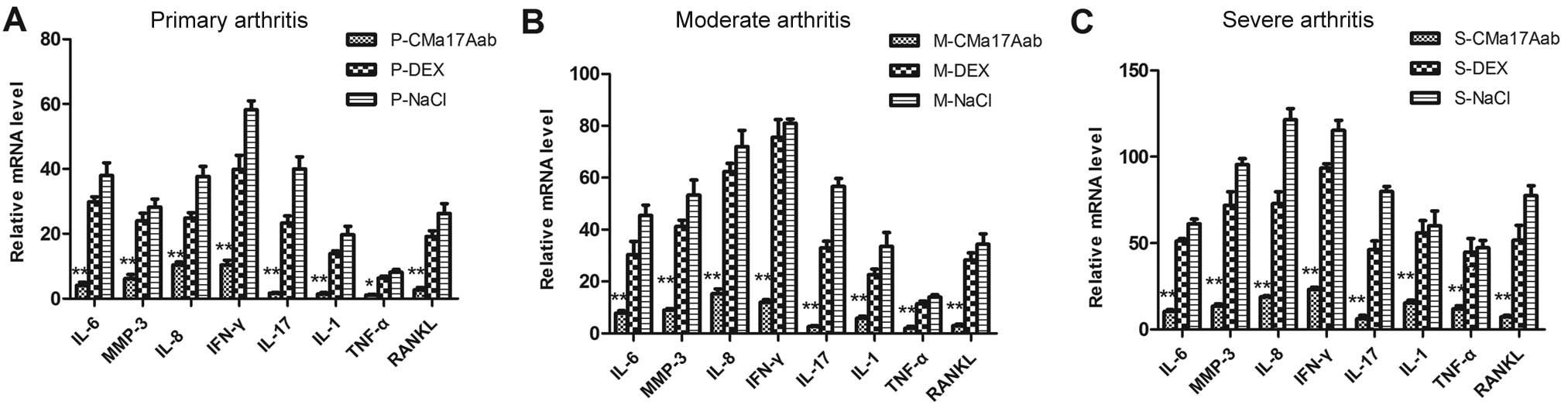
The regulatory effects of CMa17Aab on RA
synoviocytes were examined by real-time RT-PCR for eight major
genes involved in the pathogenesis of RA: IL-6, IL-8, MMP-3, IL-17,
IL-1β, TNF-α, RANKL and IFN-γ. The upregulation of IL-6, MMP-3,
IL-8 and IFN-γ by collagen antibody stimulation has been previously
demonstrated (39). To evaluate
the therapeutic effects of CMa17Aab in mice with varying degrees of
CIA, we analyzed the expression of the target genes following
independent treatments of mice with varying degrees of CIA with
CMa17Aab by RT-PCR (Fig. 5A–C).
Our results demonstrated that CIA alone induced an increase in
IL-6, IL-8, IFN-α and MMP-3 mRNA expression. Following treatment
with CMa17Aab, IFN-α, IL-6, MMP-3 and IL-8 mRNA expression
decreased in the mice with primary, moderate and severe CIA
compared with the controls (CMa17Aab vs. DEX and NaCl, P<0.05
and P<0.01, respectively). These results indicate that CMa17Aab
downregulates the expression of IL-6, IL-8, MMP-3, IL-17, IL-1β,
TNF-α, RANKL and IFN-γ.
In order to observe the changes occurring in RA
synovial tissue following treatment with CMa17Aab, X-rays of the
ankle joints of the experimental mice were acquired and the ankle
joints were then removed and fixed in formalin and decalcified with
formic acid. Ten micrometer-thick paraffin-embedded sections were
stained with hematoxylin and eosin and the histological changes
were observed. As shown in Fig.
6C–E, the untreated mice with CIA showed marked synovial
hyperplasia, inflammatory cell influx and destruction of the
cartilage and bone. As shown in Fig.
6F–J, there were fewer infiltrating cells in the joints of the
CMa17Aab-treated mice compared with the joints from the control
mice (treated with DEX) with CIA. This was also accompanied by a
decrease in joint synovial proliferation. These changes were
statistically significant (P<0.01). The pathological symptoms of
the CMa17Aab- and DEX-treated CIA mice were attenuated (data not
shown).
Discussion
IL-17A is a 17 kDa protein that is secreted
predominantly by human memory T cells α and β TCR+
CD4−CD8− thymocytes, mouse peripheral Th17
cells and some innate immune cells (40–42). IL-17A is spontaneously produced in
the RA synovium, and is highly expressed in the synovial fluid of
pateints and mice with RA. The synergistic action of IL-17 with
other pro-inflammatory cytokines, such as IL-1β, TNF-α and IFN-γ
provokes local inflammation and amplification of inflammatory
responses in RA. IL-17 exerts its effects by binding to IL-17R to
stimulate inflammatory cells, which is a central event in mediating
cell migration in the pathogenesis of RA (43). In our previous study, we
demonstrated that the anti-IL-17 antibody (ScFv) reduced joint
inflammation, bone damage and cartilage destruction in RA (44). In addition, treatment with siRNA
of the IL-17A receptor I in rat adjuvant arthritis led to a
significant suppression of joint inflammation and bone erosion
(28). In this study, we
constructed a chimeric anti-human IL-17A full-length monoclonal
antibody targeting IL-17A, so as to inhibit the pathway generating
pathogenic effector TH17 and regulatory T cells (45). In addition, we detected the
expression of IL-17 and IL-17RA in the synovium of mice with CIA.
Our results demonstrated that IL-17 and IL-17RA were highly
expressed in the mice with CIA compared with the healthy mice.
RA is one of the most common human autoimmune
diseases, characterized by a chronic inflammatory reaction in the
synovium of joints (46). RA is
characterized by symmetrical joint swelling. According to the
extent of damage to the articular cartilage, RA is divided into
primary, moderate and severe RA (47,48). In the primary state of RA, the
damage to the articular cartilage is apparent as periarticular soft
tissue swelling and joint osteoporosis. In moderate RA, articular
cartilage space becomes narrow, indicating extensive cartilage
destruction. In severe RA, articular cartilage appears eroded. In
this study, based on the clinical arthritis scores of CIA, we
examined the therapeutic effects of CMa17Aab in mice with primary,
moderate and severe RA. The results confirmed that CMa17Aab is
effective in the treatment of RA (compared with the DEX-treated and
NaCl-treated mice). However, CMa17Aab was not as effective in the
treatment of mice with severe RA compared with mice with primary
and moderate RA.
There are a number of known molecular mechanisms
that could explain the synergism of IL-17A with other cytokines,
such as IL-6, IL-8, MMP-3, IL-17, IL-1β, TNF-α and IFN-γ (49). In this study, we examined the
in vivo expression of the cytokines, IL-6, MMP-3, IL-8 and
IFN-γ, following treatment with CMa17Aab. Our results suggested
that these cytokines were highly expressed in the control group
(untreated group) and downregulated in the CMa17Aab-treated group.
Furthermore, IL-17A plays an important role in human destructive
arthritis (50). A previous study
demonstrated that in tissue-specific autoimmunity, the IL-17A level
is a key determinant of resistance or susceptibility (51). According to our results, the
expression of cytokines, such as IL-6, IL-17A, IFN-γ and TNF-α, was
upregulated in mice with CIA. However, the expression of these
cytokines was relatively lower in the mice with primary RA,
compared with the mice with severe RA. As expected, treatment with
CMa17Aab resulted in significant decreases in the levels of
pro-inflammatory cytokines in the mice with CIA.
Type II collagen is considered the major constituent
of articular cartilage in the joings of patients with RA (52). A previous study demonstrated that
the immune response to type II collagen may play a role in the
damage induced to the articular cartilage of joints (52). The mechanisms underlying
CII-induced RA have been clarified (53,54), with anti-type II collagen antibody
secretion from B cells being associated with the development of
CIA. Therefore, the serum levels of anti-type II collagen-specific
antibodies are markedly high in of patients with RA (55,56). In this study, we examined the
effects of CMa17Aab on the abnormal immune response in mice with
CIA. The level of serum IgG in the untreated mice with CIA was
slightly higher compared with the healthy mice. In addition,
following treatment with CMa17Aab, we analyzed the pathological
sections of joint issue from the different treatment groups. As
expected, the clinical arthritis scores (Fig. 5) indicated that the untreated mice
with CIA had marked synovial hyperplasia and inflammatory cell
influx, as well as cartilage and bone destruction. In the untreated
mice with severe RA, the extent of damage to the articular
cartilage was more pronounced.
In conclusion, our study demonstrates that treatment
with CMa17Aab exerts beneficial effects by alleviating joint
inflammation, cartilage destruction and bone damage in mice with
varying degrees of CIA (primary, moderate and severe). Our study
provides evidence that targeting pro-inflammatory cytokines
simultaneously can be used as a novel therapeutic approach for
patients with RA.
Acknowledgements
We are thankful for the support of the
industrialization fund of the Educational Bureau of Heilongjiang
Province (1252CGZH29).
References
|
1
|
van den Berg WB and Miossec P: IL-17 as a
future therapeutic target for rheumatoid arthritis. Nat Rev
Rheumatol. 5:549–553. 2009.PubMed/NCBI
|
|
2
|
Onishi RM and Gaffen SL: Interleukin-17
and its target genes: mechanisms of interleukin-17 function in
disease. Immunology. 129:311–321. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Leipe J, Grunke M, Dechant C, et al: Role
of Th17 cells in human autoimmune arthritis. Arthritis Rheum.
62:2876–2885. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Maione F, Paschalidis N, Mascolo N, Dufton
N, Perretti M and D’Acquisto F: Interleukin 17 sustains rather than
induces inflammation. Biochem Pharmacol. 77:878–887. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen H, Wang W, Xie H, et al: A pathogenic
role of IL- 17 at the early stage of corneal allograft rejection.
Transpl Immunol. 21:155–161. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hot A, Zrioual S, Lenief V and Miossec P:
IL-17 and tumour necrosis factor α combination induces a
HIF-1α-dependent invasive phenotype in synoviocytes. Ann Rheum Dis.
71:1393–1401. 2012.
|
|
7
|
Korn T, Bettelli E, Oukka M and Kuchroo
VK: IL-17 and Th17 Cells. Annu Rev Immunol. 27:485–517. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chabaud M, Durand JM, Buchs N, et al:
Human interleukin-17: A T cell-derived proinflammatory cytokine
produced by the rheumatoid synovium. Arthritis Rheum. 42:963–970.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kotake S, Udagawa N, Takahashi N, et al:
IL-17 in synovial fluids from patients with rheumatoid arthritis is
a potent stimulator of osteoclastogenesis. J Clin Invest.
103:1345–1352. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chabaud M, Fossiez F, Taupin JL and
Miossec P: Enhancing effect of IL-17 on IL-1-induced IL-6 and
leukemia inhibitory factor production by rheumatoid arthritis
synoviocytes and its regulation by Th2 cytokines. J Immunol.
161:409–414. 1998.PubMed/NCBI
|
|
11
|
Jovanovic DV, Di Battista JA,
Martel-Pelletier J, et al: IL-17 stimulates the production and
expression of proinflammatory cytokines, IL-beta and TNF-alpha, by
human macrophages. J Immunol. 160:3513–3521. 1998.PubMed/NCBI
|
|
12
|
Katz Y, Nadiv O and Beer Y: Interleukin-17
enhances tumor necrosis factor alpha-induced synthesis of
interleukins 1,6, and 8 in skin and synovial fibroblasts: a
possible role as a ‘fine-tuning cytokine’ in inflammation
processes. Arthritis Rheum. 44:2176–2184. 2001.PubMed/NCBI
|
|
13
|
Kehlen A, Thiele K, Riemann D and Langner
J: Expression, modulation and signalling of IL-17 receptor in
fibroblast-like synoviocytes of patients with rheumatoid arthritis.
Clin Exp Immunol. 127:539–546. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Numasaki M, Lotze MT and Sasaki H:
Interleukin-17 augments tumor necrosis factor-alpha-induced
elaboration of proangiogenic factors from fibroblasts. Immunol
Lett. 93:39–43. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Takahashi H, Numasaki M, Lotze MT and
Sasaki H: Interleukin-17 enhances bFGF-, HGF- and VEGF-induced
growth of vascular endothelial cells. Immunol Lett. 98:189–193.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Miossec P: Interleukin-17 in rheumatoid
arthritis: if T cells were to contribute to inflammation and
destruction through synergy. Arthritis Rheum. 48:594–601. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Granet C, Maslinski W and Miossec P:
Increased AP-1 and NF-kappaB activation and recruitment with the
combination of the proinflammatory cytokines IL-1beta, tumor
necrosis factor alpha and IL-17 in rheumatoid synoviocytes.
Arthritis Res Ther. 6:R190–R198. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chabaud M and Miossec P: The combination
of tumor necrosis factor alpha blockade with interleukin-1 and
interleukin-17 blockade is more effective for controlling synovial
inflammation and bone resorption in an ex vivo model. Arthritis
Rheum. 44:1293–1303. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chabaud M, Page G and Miossec P: Enhancing
effect of IL-1, IL-17, and TNF-alpha on macrophage inflammatory
protein-3alpha production in rheumatoid arthritis: regulation by
soluble receptors and Th2 cytokines. J Immunol. 167:6015–6020.
2001. View Article : Google Scholar
|
|
20
|
Granet C and Miossec P: Combination of the
pro-inflammatory cytokines IL-1, TNF-alpha and IL-17 leads to
enhanced expression and additional recruitment of AP-1 family
members, Egr-1 and NF-kappaB in osteoblast-like cells. Cytokine.
26:169–177. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chevrel G, Page G, Granet C,
Streichenberger N, Varennes A and Miossec P: Interleukin-17
increases the effects of IL-1 beta on muscle cells: arguments for
the role of T cells in the pathogenesis of myositis. J
Neuroimmunol. 137:125–133. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kirkham BW, Lassere MN, Edmonds JP, et al:
Synovial membrane cytokine expression is predictive of joint damage
progression in rheumatoid arthritis: a two-year prospective study
(the DAMAGE study cohort). Arthritis Rheum. 54:1122–1131.
2006.PubMed/NCBI
|
|
23
|
Yao Z, Spriggs MK, Derry JM, et al:
Molecular characterization of the human interleukin (IL)-17
receptor. Cytokine. 9:794–800. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tachihara A, Jin E, Matsuoka T, et al:
Critical roles of capillary endothelial cells for alveolar
remodeling in nonspecific and usual interstitial pneumonias. J
Nippon Med Sch. 73:203–213. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tian E, Sawyer JR, Largaespada DA, Jenkins
NA, Copeland NG and Shaughnessy JD Jr: Evi27 encodes a novel
membrane protein with homology to the IL17 receptor. Oncogene.
19:2098–2109. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shi Y, Ullrich SJ, Zhang J, et al: A novel
cytokine receptor-ligand pair. Identification, molecular
characterization, and in vivo immunomodulatory activity. J Biol
Chem. 275:19167–19176. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lee J, Ho WH, Maruoka M, et al: IL-17E, a
novel proinflammatory ligand for the IL-17 receptor homolog
IL-17Rh1. J Biol Chem. 276:1660–1664. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zrioual S, Toh ML, Tournadre A, et al:
IL-17RA and IL-17RC receptors are essential for IL-17A-induced
ELR+ CXC chemokine expression in synoviocytes and are
overexpressed in rheumatoid blood. J Immunol. 180:655–663. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Miljkovic D, Cvetkovic I, Vuckovic O,
Stosic-Grujicic S, Mostarica Stojkovic M and Trajkovic V: The role
of interleukin-17 in inducible nitric oxide synthase-mediated
nitric oxide production in endothelial cells. Cell Mol Life Sci.
60:518–525. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lubberts E, Koenders MI, Oppers-Walgreen
B, et al: Treatment with a neutralizing anti-murine interleukin-17
antibody after the onset of collagen-induced arthritis reduces
joint inflammation, cartilage destruction, and bone erosion.
Arthritis Rheum. 50:650–659. 2004. View Article : Google Scholar
|
|
31
|
Genovese MC, Van den Bosch F, Roberson SA,
et al: LY2439821, a humanized anti-interleukin-17 monoclonal
antibody, in the treatment of patients with rheumatoid arthritis: A
phase I randomized, double-blind, placebo-controlled,
proof-of-concept study. Arthritis Rheum. 62:929–939. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Nardelli DT, Burchill MA, England DM,
Torrealba J, Callister SM and Schell RF: Association of
CD4+ CD25+ T cells with prevention of severe
destructive arthritis in Borrelia burgdorferi-vaccinated and
challenged gamma interferon-deficient mice treated with
anti-interleukin-17 antibody. Clin Diagn Lab Immunol. 11:1075–1084.
2004.
|
|
33
|
Nakajima H, Takamori H, Hiyama Y and
Tsukada W: The effect of treatment with interferon-gamma on type II
collagen-induced arthritis. Clin Exp Immunol. 81:441–445. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Stuart JM, Townes AS and Kang AH: Type II
collagen-induced arthritis. Ann NY Acad Sci. 460:355–362. 1985.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Toy D, Kugler D, Wolfson M, et al: Cutting
edge: interleukin 17 signals through a heteromeric receptor
complex. J Immunol. 177:36–39. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Fan Y, Weifeng W, Yuluan Y, Qing K, Yu P
and Yanlan H: Treatment with a neutralizing anti-murine
interleukin-17 antibody after the onset of coxsackievirus
b3-induced viral myocarditis reduces myocardium inflammation. Virol
J. 8:172011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ziolkowska M, Koc A, Luszczykiewicz G,
Ksiezopolska-Pietrzak K, Klimczak E, Chwalinska-Sadowska H and
Maslinski W: High levels of IL-17 in rheumatoid arthritis patients:
IL-15 triggers in vitro IL-17 production via cyclosporin
A-sensitive mechanism. J Immunol. 164:2832–2838. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bas DB, Su J, Sandor K, et al: Collagen
antibody-induced arthritis evokes persistent pain with spinal glial
involvement and transient prostaglandin dependency. Arthritis
Rheum. 64:3886–3896. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Nandakumar KS and Holmdahl R: Collagen
antibody induced arthritis. Methods Mol Med. 136:215–223. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yao Z, Painter SL, Fanslow WC, et al:
Human IL-17: a novel cytokine derived from T cells. J Immunol.
155:5483–5486. 1995.PubMed/NCBI
|
|
41
|
Miyamoto M, Prause O, Sjostrand M, Laan M,
Lötvall J and Lindén A: Endogenous IL-17 as a mediator of
neutrophil recruitment caused by endotoxin exposure in mouse
airways. J Immunol. 170:4665–4672. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kennedy J, Rossi DL, Zurawski SM, et al:
Mouse IL-17: a cytokine preferentially expressed by alpha beta TCR
+ CD4-CD8-T cells. J Interferon Cytokine Res. 16:611–617. 1996.
|
|
43
|
Zhang R, Qian J, Guo J, Yuan YF and Xue K:
Suppression of experimental autoimmune uveoretinitis by Anti-IL-17
antibody. Curr Eye Res. 34:297–303. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhang Y, Ren G, Guo M, et al: Synergistic
effects of interleukin-1beta and interleukin-17A antibodies on
collagen-induced arthritis mouse model. Int Immunopharmacol.
15:199–205. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Miossec P: IL-17 and Th17 cells in human
inflammatory diseases. Microbes Infect. 11:625–630. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Bazzani C, Filippini M, Caporali R, et al:
Anti-TNFalpha therapy in a cohort of rheumatoid arthritis patients:
clinical outcomes. Autoimmun Rev. 8:260–265. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Gandjbakhch F, Conaghan PG, Ejbjerg B, et
al: Synovitis and osteitis are very frequent in rheumatoid
arthritis clinical remission: results from an MRI study of 294
patients in clinical remission or low disease activity state. J
Rheumatol. 38:2039–2044. 2011. View Article : Google Scholar
|
|
48
|
Emery P, McInnes IB, van Vollenhoven R and
Kraan MC: Clinical identification and treatment of a rapidly
progressing disease state in patients with rheumatoid arthritis.
Rheumatology (Oxford). 47:392–398. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Hornung N, Ellingsen T, Attermann J,
Stengaard-Pedersen K and Poulsen JH: Patients with rheumatoid
arthritis treated with methotrexate (MTX): concentrations of
steady-state erythrocyte MTX correlate to plasma concentrations and
clinical efficacy. J Rheumatol. 35:1709–1715. 2008.
|
|
50
|
Liu Y, Mei J, Gonzales L, et al: IL-17A
and TNF-alpha exert synergistic effects on expression of CXCL5 by
alveolar type II cells in vivo and in vitro. J Immunol.
186:3197–3205. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Leung S, Liu X, Fang L, Chen X, Guo T and
Zhang J: The cytokine milieu in the interplay of pathogenic
Th1/Th17 cells and regulatory T cells in autoimmune disease. Cell
Mol Immunol. 7:182–189. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Myers LK, Rosloniec EF, Cremer MA and Kang
AH: Collagen-induced arthritis, an animal model of autoimmunity.
Life Sci. 61:1861–1878. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Griffiths MM, Nabozny GH, Hanson J, et al:
Collagen-induced arthritis and TCRs in SWR and B10. Q mice
expressing an Ek alpha transgene. J Immunol. 153:2758–2768.
1994.PubMed/NCBI
|
|
54
|
Durie FH, Fava RA and Noelle RJ:
Collagen-induced arthritis as a model of rheumatoid arthritis. Clin
Immunol Immunopathol. 73:11–18. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Nabozny GH, Bull MJ, Hanson J, Griffiths
MM, Luthra HS and David CS: Collagen-induced arthritis in T cell
receptor V beta congenic B10. Q mice J Exp Med. 180:517–524. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Kadowaki KM, Matsuno H, Tsuji H and Tunru
I: CD4+ T cells from collagen-induced arthritic mice are
essential to transfer arthritis into severe combined
immunodeficient mice. Clin Exp Immunol. 97:212–218. 1994.
|