Introduction
Orexin A and B, also known as hypocretins are
neuropeptides identified by two groups in 1998 (1,2).
They articulate their signaling cascades via two G-protein-coupled
receptors (GPCRs): orexin receptor 1 (OX1R) and orexin receptor 2
(OX2R) (2,3). Orexin A and B synthesize neurons
located in the lateral hypothalamus that project to other regions
of brain, including stem nuclei and thalamus, forebrain region and
spinal cord (4–6). Orexins and orexin receptors are
involved in many physiological processes, including food intake,
sleep-awake, reproductive behavior, and energy homeostasis
(7–10). OX1R has a wide tissue distribution
in the central nervous system and in peripheral tissues, including
adipose tissue, gut, pancreatic, adrenal gland and testis (11,12). AKT is able to phosphorylate a
large number of intracellular targets (13). Two evolutionarily conserved
downstream effectors of AKT, FoxO1 and mTORC1, were investigated in
the present study.
Forkhead box O1 (FoxO1) is a prominent member of the
forkhead box family and is involved in the regulation of
metabolism, cell proliferation, differentiation, cell cycle
progression, apoptosis and cell death in many cell types (14–17). The PI3K/AKT signaling pathway
changes FoxO1 transcriptional activities by regulating threonine,
serine and the phosphorylation of FoxO1 (14,18). Phosphorylated FoxO1 translocates
from the nucleus to cytosol and loses its transcriptional activity
in liver (16–19).
Mammalian target of rapamycin 1 (mTORC1) is another
downstream target of the PI3K/AKT signaling pathway (20). mTORC1 regulates cell growth and
proliferation largely through the increase in protein synthesis
(20,21). mTORC1 elevates mRNA translation by
phosphorylating and activating S6K1, and phosphorylating and
inhibiting the elF4E-binding protein, a repressor of mRNA
translation (20–22). AKT can activate mTORC1 through
direct phosphorylation of the tuberous sclerosis complex 2
(21,22).
Orexin A is known to play a key role in the PI3K/AKT
signaling pathway in many tyeps of cells (13,23–25), but there is no exact evidence
concerning orexin A acting on PI3K/AKT transduction in hepatocytes.
The aim of this study was to determine whether AKT is required for
cell proliferation and apoptosis by changing activities of the
downstream targets, FoxO1 and mTORC1. The role of FoxO1 and mTORC1
in cell proliferation and apoptosis in the involvement of PI3K/AKT
pathways regulated by orexin A in rat hepatocytes was examined. The
results provide insight into the mechanisms by which orexin A
contributes to cell cycle processes and metabolisms, thus having
important implications for clinical therapy.
Materials and methods
Animals
Thirty male Sprague-Dawley rats (3–4 weeks old,
weighing 200–250 g) were obtained from China Medical University and
bred in our laboratory. The temperature was maintained at 22±2°C
with a constant 12-h light-dark cycle (6:00 a.m. to 6:00 p.m.). The
rats received a normal diet (from commercial diet) of standard
laboratory chow (20% protein, 15% fat, 65% carbohydrate diet)
(China Medical University Laboratory Animal Center, Shenyang,
China). The animal experiments were approved by the Ethics
Committee of the First Affiliated Hospital of China Medical
University.
Reagents
Orexin A was obtained from Sigma (St. Louis, MO,
USA). RPMI-1640 medium and fetal bovine serum were purchased from
Gibco (Grand Island, NY, USA). The AKT inhibitor, LY294002, was
purchased from Selleck Chemicals (Houston, TX, USA). FoxO1
inhibitor AS1842856 and mTORC1 inhibitor everolimus were obtained
from Abcam (Cambridge, UK). OX1R-specific antagonist SB334867 was
obtained from Tocris Bioscience (Minneapolis, MN, USA). The
Cell-Death Detection ELISA kit and Cell Proliferation ELISA BrdU
colorimetric kit were purchased from Roche Diagnostics (Penzberg,
Germany). Total/phospho-AKT (s473) polyclonal antibody,
total/phospho-FoxO1 polyclonal antibody and total/phospho-mTORC1
polyclonal antibody, β-actin (c4): sc-47778 and OX1R antibody were
all obtained from Abcam.
Isolation and culturing of rat
hepatocytes
Rat livers were decapsulated and incubated with 0.5
mg/ml collagenase type IV (Invitrogen, Grand Island, NY, USA) for
30 min at 37°C in a shaker at 160 cycles/min. The cell suspension
was collected by centrifugation at 800 rpm for 10 min. To obtain
purified hepatocytes, the crude cell suspension was centrifuged on
a Percoll gradient (20, 40, 60 and 90% Percoll in PBS solution;
Sigma) and subsequently centrifuged at 800 rpm for 20 min at 4°C.
Fractions containing hepatocytes were collected and centrifuged in
a continuous, self-generating density gradient starting with 60%
Percoll at 2,000 rpm for 30 min at 4°C. The purified hepatocytes
were cultured with RPMI-1640 medium (Invitrogen) supplemented with
10% bovine serum albumin (HyClone, Beijing, China), 100 IU/ml
penicillin and 100 μg/ml streptomycin (Xianfeng, Shanghai, China)
and cultured (106 cells/ml per dish) at 37°C in a
humidified incubator with 5% CO2 for 24 h.
Cell proliferation assays
Cells (2×103 cells/well) were seeded in
96-well plates and cultured for 24 h. To synchronize cell cycles,
the cells were serum-deprived for 24 h and then treated with test
agents for an additional 24 h. BrdU incorporation into DNA was
measured by the cell proliferation ELISA BrdU colorimetric kit
(Roche Diagnostics). The cells were incubated with BrdU fresh
medium at 37°C and 5% CO2 for 12 h and fixed with 200 μl
of fixative/denaturing solution for 30 min at room temperature.
Peroxidase-conjugated BrdUrd antibody was added to each well and
incubated for 1 h. After washing thoroughly, the bound
peroxidase-conjugated BrdUrd antibody was quantified with
peroxidase substrate tetramethylbenzidine. BrdUrd absorbance was
measured at 440 nm using an ELISA plate reader (PeproTech China,
Suzhou, China). A control without cells was used to measure the
background absorbance of the medium and was subtracted from the
results.
Annexin V/PI assays for apoptosis
For Annexin V/PI assays, cells were stained with
Annexin V-FITC and PI, and evaluated for apoptosis by flow
cytometry according to the manufacturer’s protocol (BD Biosciences
Pharmingen, San Diego, CA, USA). Cells were treated with different
concentrations of orexin A in the absence of serum for 48 h.
Briefly, cells (1×105)were washed twice with PBS, and
stained with 5 μl of Annexin V-FITC and 10 μl of PI in 500 μl
binding buffer for 15 min at room temperature in the dark.
Quantification of apoptosis was determined by counting the number
of cells stained by FITC-labeled Annexin V. Cell apoptosis was
detected using the Annexin V/PI apoptosis detection kit by FACS
analysis. Early apoptotic cells were identified as PI-negative and
FITC Annexin V-positive, while late apoptotic or dead cells were
considered FITC Annexin V- and PI-positive.
Total RNA isolation and reverse
transcription (RT)-PCR
Total-RNA was extracted from hepatocytes using
TRIzol reagent (Invitrogen). The expression of OX1R mRNA in
hepatocytes was detected by RT-PCR using TaqMan reagents (Takara
Bio, Otsu, Japan). The specific primers used were: OX1R forward,
5′-TGC GGC CAA CCC TAT CAT CTA-3′; and reverse, 5′-ACC GGC TCT GCA
AGG ACA A-3′. As an internal control for reverse transcription and
reaction efficiency, amplification of glyceraldehyde-3-phosphate
dehydrogenase (GAPDH) mRNA was carried out concomitantly for each
sample. The primers used were: GAPDH forward, 5′-GGC ACA GTC AAG
GCT GAG AAT G-3′; and reverse, 5′-ATG GTG GTG AAG ACG CCA GTA-3′.
The PCR reactions were carried out under the following conditions:
95°C for 30 sec, then 40 cycles of 95°C for 5 sec, 60°C for 30 sec,
and 95°C for 15 sec. Primers and TaqMan probes specific to OX1R and
GAPDH were designed using Primer Premier 5.0 software (Premier
Biosoft International, Palo Alto, CA, USA).
Protein preparations and western blot
analysis
Cell lysates were incubated on ice for 30 min and
centrifuged at 12,000 × g for 10 min at 4°C. The supernatants were
collected and mixed with 5× loading buffer, then denatured by
boiling for 10 min. Lysate protein samples were separated by
SDS-PAGE and transferred to polyvinylidene fluoride (PVDF)
membranes at 70 V for 1.5 h in a transfer buffer containing 20 mM
Tris, 150 mM glycine and 20% methanol. The membranes were incubated
in non-fat dry milk for 120 min at room temperature, and then
washed three times with TBST for 30 min. The PVDF membranes were
incubated in TBST with primary antibodies: phospho/total-OX1R at a
1:250 dilution, phospho/total-AKT at a 1:1,000 dilution,
phospho/total-FoxO1 at a 1:1,000 dilution and phospho/total-mTORC1
at a 1:1,000 dilution overnight at 4°C, respectively. The membranes
were washed and incubated with horseradish peroxidase-conjugated
anti-species secondary antibody for 1.5 h at room temperature, then
washed three times with TBST for 30 min. The proteins were
visualized by ECL and densities were measured using Quantity-One
software.
Statistical analysis
Data was shown as means ± SEM. One-way analysis of
variance (ANOVA) was used for multiple group comparisons. Unpaired
t-tests were used for two-group comparisons. Correlation analysis
was carried out using Pearson’s correlation analysis. P<0.05 was
considered statistically significant (n=6). Statistical analysis
was performed using the SPSS 15.0 software package (SPSS Inc.,
Chicago, IL, USA).
Results
Effects of orexin A on OX1R in mRNA
expression and protein activation in hepatocytes
Rat hepatocytes were were treated with orexin A at
concentrations of 0, 10−10, 10−8,
10−6 M for 30 min respectively. RT-PCR analysis with the
use of specific paired primers was used to demonstrate the OX1R
mRNA expression in hepatocytes. Orexin A (10−10,
10−8 and 10−6 M) induced a significant
increase of OX1R mRNA levels in a dose-dependent manner. A
10−6 M orexin A-induced OX1R mRNA expression in the
presence of OX1R antagonist SB334867 (10−5 M, 30 min)
showed no significant differences compared with the 10−6
M orexin A treatment alone (Fig.
1A). The level of OX1R phosphorylation was determined by
western blot analysis. We observed that exogenous orexin A
upregulated OX1R in the protein level in a dose-dependent manner.
Maximum stimulation was identified at 10−6 M orexin A
treatment and minimum at 10−10 M treatment (Fig. 1B). OX1R phosphorylation was
reduced in the presence of 10−5 M SB334867, an
OX1R-specific antagonist (Fig.
1B).
Effects of orexin A on AKT
phosphorylation in hepatocytes
To determine the correlation of OX1R-induced AKT
phosphorylation in hepatocytes, the cells were treated with orexin
A at different concentrations (0, 10−10, 10−8
and 10−6 M) for 30 min. The total AKT and phospho-AKT
levels were determined by western blot analysis. As compared to
basal levels, orexin A induced maximum AKT phosphorylation at the
concentration of 10−6 M and minimum at 10−10
M (Fig. 2). Total AKT remained
unchanged (Fig. 2). OX1R
antagonist (SB334867, 10−5 M, 30 min) and AKT inhibitor
(LY294002, 25 μmol/l, 24 h) were then used to detect the
intracellular mechanisms of the orexin-induced signaling pathway.
AKT phosphorylation was abolished in cells treated with the two
blockers. Thus, orexin A induced AKT phosphorylation through an
OX1R-dependent signaling pathway in hepatocytes (Fig. 2).
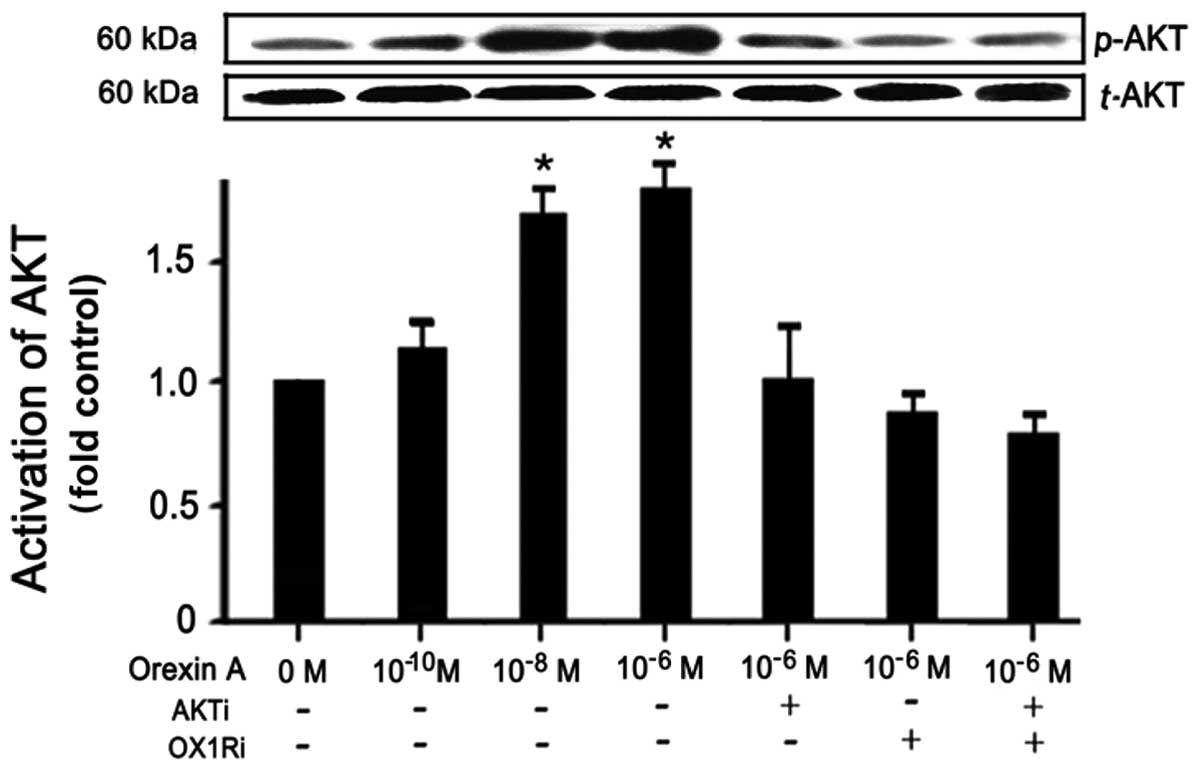 | Figure 2Effects of orexin A on AKT
phosphorylation in hepatocytes. To determine the correlation of
orexin receptor 1 (OX1R)-induced AKT phosphorylation in
hepatocytes, the cells were treated with orexin A at different
concentrations (0, 10−10, 10−8 and
10−6 M) for 30 min. Total-AKT and phospho-AKT levels
were determined by western blot analysis. Then OX1R antagonist
(OX1Ri, SB334867, 10−5 M, 30 min) and AKT inhibitor
(AKTi, LY294002, 25 μmol/l, 24 h) were used to detect the
intracellular mechanisms of the orexin-induced signaling pathway.
The results are expressed by means ± SEM. P<0.05 was considered
to be statistically significant, n=6. |
Effects of orexin A on FoxO1
phosphorylation in hepatocytes
To determine the effects of orexin A on FoxO1
phosphorylation in hepatocytes, the cells were treated with orexin
A at different concentrations (0, 10−10, 10−8
and 10−6 M) for 30 min. Total FoxO1 and phospho-FoxO1
levels were determined by western blot analysis. Orexin A induced
maximum FoxO1 phosphorylation at the concentration of
10−6 M and minimum at 10−10 M, while total
FoxO1 remained unchanged (Fig.
3A). OX1R antagonist (SB334867, 10−5 M) was used to
pre-incubate hepatocytes for 30 min in the presence of orexin A
(10−6 M, 30 min). FoxO1 phosphorylation was abolished in
the cells treated with SB334867. Thus, orexin A induced FoxO1
phosphorylation through OX1R-mediated signaling cascades.
Hepatocytes were treated with orexin A (10−6 M) in the
presence of PI3K/AKT inhibitor LY294002 pretreatment (25 μmol/l, 24
h before) and FoxO1 phosphorylation was measured by western
blotting. In the presence of LY294002, the phosphorylated activity
of PI3K/AKT was blocked, thus FoxO1 phosphorylation was
significantly reduced compared with cells incubated with orexin A
(10−6 M) alone. Additionally, hepatocytes were
pretreated with a combination of SB334867 and LY294002
simultaneously, then orexin A (10−6 M) was treated for
30 min. Results of the western blot analysis revealed that either
SB334867 or LY294002 significantly suppressed orexin A-induced
FoxO1 phosphorylation, and that there is no differences between the
two inhibitory effects. The blockage of either OX1R or PI3K/AKT
suppressed FoxO1 phosphorylation. However, with the simultaneous
combination of SB334867 and LY294002, FoxO1 phosphorylation was
significantly suppressed, compared with treated orexin A
(10−6 M) alone. It suggested an excitatory effect of
orexin A on FoxO1 phosphorylation through the OX1R-mediated
PI3K/AKT signaling pathway (Fig.
3B).
Effects of orexin-A on mTORC1
phosphorylation in hepatocytes
Hepatocytes were incubated and stimulated with
various concentrations (10−10 to 10−6 M) of
orexin A for 30 min. Alternatively, the cells were treated with
10−6 M orexin A combined with the AKT antagonist
LY294002 (25 μmol/l) or OX1R antagonist SB334867 (10−6
M). Western blot analysis was used to detect phospho-mTORC1
(p-mTORC1) and total mTORC1 proteins. The effect of 10−6
and 10−8 M orexin A reached statistical significance
compared to the control (p<0.05) (Fig. 4A). p-mTORC1 was then measured to
determine whether it was affected by the OX1R antagonist and AKT
inhibitor. This effect disappeared in the presence of AKT
antagonist LY294002 (25 μmol/l) or OX1R antagonist (SB334867,
10−6 M), as well as their simultaneous combination
(Fig. 4B). No statistical
difference was identified for the two blockers alone or in
combination (Fig. 4B).
Effects of orexin A on cell proliferation
and apoptosis in hepatocytes
To confirm the effects of orexin A-mediated
proliferation and apoptosis in hepatocytes, we stimulated
hepatocytes with orexin A (10−10 to 10−6 M)
for 24 h. BrdU analysis was used to determine cell proliferation.
Orexin A upregulated cell proliferation in a dose-dependent manner
(Fig. 5A). This
proliferation-stimulating effect was blocked by the OX1R antagonist
SB334867 (10−6 M), AKT inhibitor LY294002
(10−6 M) or mTORC1 inhibitor everolimus (10−5
M) (Fig. 5A). A significant
decrease in the presence of all three blockers was observed
although no significant difference was identified between the
individual blockers. However, in the presence of FoxO1 inhibitor,
AS1842856, cell proliferation was significantly increased compared
with the orexin A treatment alone. This is in contrast with other
inhibitors (Fig. 5A). Orexin A
treatment (10−10 to 10−6 M) resulted in a
decrease in the apoptotic index as measured by Annexin V/PI
analysis. Orexin A (10−10 to 10−6 M)
downregulated cell apoptosis in a dose-dependent manner and
protected hepatocytes from apoptosis (Fig. 5B). However, it failed to prevent
cells from apoptosis in the presence of OX1R antagonist, AKT
inhibitor or mTORC1 inhibitor (Fig.
5B). In the presence of FoxO1 inhibitor, AS1842856, the effect
of preventing cells from apoptosis was enhanced, which was in
contrast with other inhibitors (Fig.
5B).
 | Figure 5(A and B) Effects of orexin A on cell
proliferation and apoptosis in hepatocytes. To confirm the effects
of orexin A-mediated proliferation and apoptosis in hepatocytes, we
stimulated hepatocytes with orexin A (10−10 to
10−6 M) for 24 h. Orexin receptor 1 (OX1R) antagonist
SB334867 (OX1Ri, 10−5 M, 30 min), AKT inhibitor LY294002
(AKTi, 25 μmol/l, 24 h), FoxO1 inhibitor AS1842856 (FoxO1i,
10−6 M, 24 h) and mammalian target of rapamycin 1
(mTORC1) inhibitor everolimus (mTORC1i, 10−5 M, 18 h)
were then used to investigate the intracellular mechanism. BrdU and
Annexin V/PI analysis were used to determine cell proliferation and
apoptosis. The results are expressed as means ± SEM. P<0.05 was
considered statistically significant, n=6. |
Discussion
Orexin A is involved in the activation of PI3K/AKT
signaling pathways in many peripheral organs and cells (3,26).
Orexins elicit their biological effects via GPCRs, OX1R and OX2R,
which can signal through multiple G proteins (27,28). The RT-PCR and western blot
analysis showed the expression of OX1R in the mRNA and protein
levels in hepatocytes, which appears to be hypersensitive to
exogenous orexin A stimulation in a dose-dependent manner. The
results of this study suggest that orexin A exerted its biological
effects by the ligand-induced upregulation of OX1R. A higher
concentration of orexin A increases the expression of OX1R.
Findings of previous studies have demonstrated the expression of
OX1R in many cell types in human and rodents, suggesting that the
effects of orexin A are mediated through a direct interaction with
OX1R (29–31). The effects of orexin A may be
mediated through a specific interaction with the corresponding
GPCR. However, the physiological relevance of OX1R expression in
hepatocytes remains to be investigated.
PI3K/AKT is an important signaling pathway involved
in many cell processes and metabolisms (23,24). As shown in the present study,
orexin A upregulated AKT phosphorylation in a dose-dependent
manner, while SB334867, an OX1R-specific antagonist was able to
block this transduction. We suggest that orexin A activates AKT
through an OX1R-mediated pathway. Orexin A stimulates hepatocyte
proliferation and protects against apoptotic cell death via the
PI3K/AKT-dependent mechanism. Prolonged (24 h) incubation of
hepatocytes with orexin A enhanced cell proliferation and prevented
apoptotic cell death. These effects of orexin A were reversed by
blocking the PI3K/AKT pathway, indicating that orexin A stimulates
hepatocyte proliferation and protects against apoptotic cell death
via the PI3K/AKT pathway. The inhibition of AKT by LY294002 is well
established, however, the deactivation of AKT inhibited
proliferation and failed to prevent apoptosis. Although in this
study, activation of AKT by orexin A increased cell proliferation,
the manner in which AKT affects these processes and the identity of
critical downstream effectors remains largely unknown. To elucidate
the mechanism through which AKT affects cell cycle progression, we
focused on two evolutionarily conserved downstream effectors of
PI3K/AKT, FoxO1 and mTORC1 (32).
The two factors play key roles but have an opposite effect in
regulating cell proliferation and apoptosis (33–37). This is consistent with the results
obtained in the present study.
FoxO1 is a dominant regulator of hepatic gene
expression that is usually inactivated through the PI3K/AKT branch
of the exogenous stimulator-induced signaling system (32,38). Findings of previous studies have
shown that FoxO1 regulates cell proliferation through the
transcriptional activation of certain genes (32,38–40). Our data show that FoxO1 was
phosphorylated by orexin A and thus lost its transcriptional
activity in regulating genes associated with cell processes
(14,15,39,40). FoxO1 has been found to play a
critical role in the cell cycle processes of rats (41,42) as it suppresses cell proliferation
and promotes apoptotic cell death (41–44). FoxO1 suppresses cell proliferation
as it activates the expression of the eukaryotic initiation factor
4E-BP-1, which is a major potent proliferation suppressor (39,40). FoxO1 has been shown to directly
increase the expression of certain pro-apoptotic proteins such as
Bim and BAD (43,44). In previous studies, FoxO1 is
defined as a tumor suppressor and its activation has a therapeutic
advantage for cancer (45–47).
As another downstream target of PI3K/AKT signaling
pathway, mTORC1 promotes cell proliferation and prevents apoptosis
(21,32). It regulates cell proliferation
largely through an increase in protein synthesis (32,44). mTORC1 regulates proliferation in
eukaryotic cells (33–35,45) and inhibits the key proliferation
repressor 4E-BP-1 by phosphorylation, thereby increasing cell
proliferation (39,40). However, as an evolutionarily
conserved downstream target of AKT, FoxO1 is capable of suppressing
mTORC1 activity (21,32,42). FoxO1 is thought to have tumor
suppressive activity (46,47),
whereas mTORC1 is frequently activated in cancer cells (48).
Orexin A is identified as an endocrine effector that
activates mTORC1 but which inhibits FoxO1-dependent gene regulation
in a PI3K/AKT-dependent manner. As shown in results of the present
study, orexin A promotes hepatocyte proliferation and protects
against apoptosis. Orexin A deactivates FoxO1 and activates mTORC1
activity by phosphorylation, which co-affects
proliferation-promotion and apoptosis-prevention in hepatocytes.
Inhibition of FoxO1 with AS1842856 increases cell proliferation and
suppresses apoptosis. Inhibition of mTORC1 with everolimus
suppresses cell proliferation and increases apoptosis. In this
study, it was shown that orexin A enhances cell proliferation and
attenuates apoptosis. These effects are signaled by altering FoxO1
(deactivate) and mTORC1 (activate) activity. In this cell cycle
process, FoxO1 and mTORC1 play two opposing roles in the regulation
of cell proliferation and apoptosis.
In summary, our findings provide evidence that
orexin A increased cell proliferation and protected cells from
apoptosis, an effect that is signaled via PI3K/AKT cascades by
suppressing FoxO1 and enhancing mTORC1 activity by phosphorylation.
Orexin A may exert apoptotic function in a FoxO1 and
mTORC1-dependent manner. Therefore, understanding the cellular
mechanisms of action of orexin A in hepatocytes is crucial in
gaining insight into the therapeutic role of orexin A in the
regulation of a variety of cell cycle processes. For these reasons,
we predict that drug interference in conjunction with mTORC1 or
FoxO1 action may be used in clinical practice to improve human
health in the future. Orexin A has evolved to accelerate
proliferation and suppress apoptosis, while also increasing the
incidence of cancer, metabolic derangement and aging in patients.
Ongoing investigations should therefore focus on the relationship
between orexin A and its clinical implications.
Acknowledgements
We would like to thank the China Medical University
Affiliated Hospital Laboratory Center for kindly providing
equipment required. This study was supported by the National
Natural Science Foundation of China (grant nos. 30872724, 81071460,
and 81271996), the Natural Science Foundation of Liaoning Province
(grant no. 201202292).
References
|
1
|
De Lecea L, Kilduff TS, Peyron C, Gao X,
et al: The hypocretins: hypothalamus-specific peptides with
neuroexcitatory activity. Proc Natl Acad Sci USA. 95:322–327.
1998.PubMed/NCBI
|
|
2
|
Sakurai T, Amemiya A, Ishii M, Matsuzaki
I, Chemelli RM, Tanaka H, et al: Orexins and orexin receptors: a
family of hypothalamic neuropeptides and G protein-coupled
receptors that regulate feeding behavior. Cell. 92:573–585. 1998.
View Article : Google Scholar
|
|
3
|
Karteris E and Randeva HS: Orexin
receptors and G-protein coupling: evidence for another
‘promiscuous’ seven transmembrane domain receptor. J Pharmacol Sci.
93:126–128. 2003.
|
|
4
|
Date Y, Ueta Y, Yamashita H, Yamaguchi H,
Matsukura S, Kangawa K, Sakurai T, Yanagisawa M and Nakazato M:
Orexins, orexigenic hypothalamic peptides, interact with autonomic,
neuroendocrine and neuroregulatory systems. Proc Natl Acad Sci USA.
96:748–753. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nambu T, Sakurai T, Mizukami K, Hosoya Y,
Yanagisawa M and Goto K: Distribution of orexin neurons in the
adult rat brain. Brain Res. 827:243–260. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Peyron C, Tighe DK, van den Pol AN, de
Lecea L, Heller HC, Sutcliffe JG and Kilduff TS: Neurons containing
hypocretin (orexin) project to multiple neuronal systems. J
Neurosci. 18:9996–10015. 1998.PubMed/NCBI
|
|
7
|
Lin L, Faraco J, Li R, Kadotani H, Rogers
W, Lin X, Qiu X, de Jong PJ, Nishino S and Mignot E: The sleep
disorder canine narcolepsy is caused by a mutation in the
hypocretin (orexin) receptor 2 gene. Cell. 98:365–376. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yokobori E, Kojima K, Azuma M, et al:
Stimulatory effect of intracerebroventricular administration of
orexin A on food intake in the zebrafish. Peptides. 32:1357–1362.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Spinazzi R, Rucinski M, Neri G,
Malendowicz LK and Nussdorfer GG: Preproorexin and orexin receptors
are expressed in cortisol-secreting adrenocortical adenomas, and
orexins stimulate in vitro cortisol secretion and growth of tumor
cells. J Clin Endocrinol Metab. 90:3544–3549. 2005. View Article : Google Scholar
|
|
10
|
Ramanjaneya M, Conner AC, Chen J,
Stanfield PR and Randeva HS: Orexins stimulate steroidogenic acute
regulatory protein expression through multiple signaling pathways
in human adrenal H295R cells. Endocrinology. 149:4106–4115. 2008.
View Article : Google Scholar
|
|
11
|
Beiras-Fernández A, Gallego R, Blanco M,
García-Caballero T, Diéguez C and Beiras A: Merkel cells, a new
localization of prepro-orexin and orexin receptors. J Anat.
204:117–122. 2004.PubMed/NCBI
|
|
12
|
Jöhren O, Neidert SJ, Kummer M, Dendorfer
A and Dominiak P: Prepro-orexin and orexin receptor mRNAs are
differentially expressed in peripheral tissues of male and female
rats. Endocrinology. 142:3324–3331. 2001.PubMed/NCBI
|
|
13
|
Lawlor MA and Alessi DR: PKB/Akt: A key
mediator of cell proliferation, survival and insulin response? J
Cell Sci. 114:2903–2910. 2001.PubMed/NCBI
|
|
14
|
Gross DN, Wan M and Birnbaum MJ: The role
of FOXO in the regulation of metabolism. Curr Diab Rep. 9:208–214.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Matsumoto M, Pocai A, Rossetti L, Depinho
RA and Accili D: Impaired regulation of hepatic glucose production
in mice lacking the forkhead transcription factor Foxo1 in liver.
Cell Metab. 6:208–216. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Accili D and Arden KC: FoxOs at the
crossroads of cellular metabolism, differentiation, and
transformation. Cell. 117:421–426. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yuan Z, Lehtinen MK, Merlo P, Villén J,
Gygi S and Bonni A: Regulation of neuronal cell death by MST1-FOXO1
signaling. J Biol Chem. 284:11285–11292. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kajihara T, Jones M, Fusi L, Takano M,
Feroze-Zaidi F, Pirianov G, Mehmet H, Ishihara O, Higham JM, Lam EW
and Brosens JJ: Differential expression of FOXO1 and FOXO3a confers
resistance to oxidative cell death upon endometrial
decidualization. Mol Endocrinol. 20:2444–2455. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shen B, Chao L and Chao J: Pivotal role of
JNK-dependent FOXO1 activation in downregulation of kallistatin
expression by oxidative stress. Am J Physiol Heart Circ Physiol.
298:H1048–H1054. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Thomas GV: mTOR and cancer: Reason for
dancing at the crossroads? Curr Opin Genet Dev. 16:78–84. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen CC, Jeon SM, Bhaskar PT, Nogueira V,
Sundararajan D, Tonic I, Park Y and Hay N: FoxOs inhibit mTORC1 and
activate Akt by inducing the expression of Sestrin3 and Rictor. Dev
Cell. 18:592–604. 2010. View Article : Google Scholar
|
|
22
|
Zoncu R, Efeyan A and Sabatini DM: mTOR:
from growth signal integration to cancer, diabetes and ageing. Nat
Rev Mol Cell Biol. 12:21–35. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen ML, Xu PZ, Peng XD, Chen WS, Guzman
G, Yang X, Di Cristofano A, Pandolfi PP and Hay N: The deficiency
of Akt1 is sufficient to suppress tumor development in Ptenmice.
Genes Dev. 20:1569–1574. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hahn-Windgassen A, Nogueira V, Chen CC,
Skeen JE, Sonenbreg N and Hay N: Akt activates the mammalian target
of rapamycin by regulating cellular ATP level and AMPK activity. J
Biol Chem. 280:32081–32089. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Robey RB and Hay N: Mitochondrial
hexokinases: Guardians of the mitochondria. Cell Cycle. 4:654–658.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Holmqvist T, Johansson L, Ostman M, Ammoun
S, Akerman KE and Kukkonen JP: OX1 orexin receptors couple to
adenylyl cyclase regulation via multiple mechanisms. J Biol Chem.
280:6570–6579. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Malendowicz LK, Tortorella C and
Nussdorfer GG: Orexins stimulate corticosterone secretion of rat
adrenocortical cells, through the activation of the adenylate
cyclase-dependent signaling cascade. J Steroid Biochem Mol Biol.
70:185–188. 1999. View Article : Google Scholar
|
|
28
|
López M1, Señarís R, Gallego R,
García-Caballero T, Lago F, Seoane L, Casanueva F and Diéguez C:
Orexin receptors are expressed in the adrenal medulla of the rat.
Endocrinology. 140:5991–5994. 1999.PubMed/NCBI
|
|
29
|
Göncz E, Strowski MZ, Grötzinger C, Nowak
KW, Kaczmarek P, Sassek M, Mergler S, El-Zayat BF, Theodoropoulou
M, Stalla GK, Wiedenmann B and Plöckinger U: Orexin-A inhibits
glucagon secretion and gene expression through a Foxo1-dependent
pathway. Endocrinology. 149:1618–1626. 2008.PubMed/NCBI
|
|
30
|
Ramanjaneya M1, Conner AC, Chen J, Kumar
P, Brown JE, Jöhren O, Lehnert H, Stanfield PR and Randeva HS:
Orexin-stimulated MAP kinase cascades are activated through
multiple G-protein signaling pathways in human H295R adrenocortical
cells: diverse roles for orexins A and B. J Endocrinol.
202:249–261. 2009. View Article : Google Scholar
|
|
31
|
Zhu Y, Miwa Y, Yamanaka A, Yada T,
Shibahara M, Abe Y, Sakurai T and Goto K: Orexin receptor type-1
couples exclusively to pertussis toxin insensitive G-proteins,
while orexin receptor type-2 couples to both pertussis
toxin-sensitive and -insensitive G-proteins. J Pharmacol Sci.
92:259–266. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Skeen JE, Bhaskar PT, Chen CC, Chen WS,
Peng XD, Nogueira V, Hahn-Windgassen A, Kiyokawa H and Hay N: Akt
deficiency impairs normal cell proliferation and suppresses
oncogenesis in a p53-independent and mTORC1-dependent manner.
Cancer Cell. 10:269–280. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kapahi P, Chen D, Rogers AN, Katewa SD, Li
PW, Thomas EL and Kockel L: With TOR, less is more: a key role for
the conserved nutrient-sensing TOR pathway in aging. Cell Metab.
11:453–465. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang X and Proud CG: Nutrient control of
TORC1, a cell-cycle regulator. Trends Cell Biol. 19:260–267. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gera JF, Mellinghoff IK, Shi Y, Rettig MB,
Tran C, Hsu JH, Sawyers CL and Lichtenstein AK: AKT activity
determines sensitivity to mammalian target of rapamycin (mTOR)
inhibitors by regulating cyclin D1 and c-myc expression. J Biol
Chem. 279:2737–2746. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Buteau J and Accili D: Regulation of
pancreatic beta-cell function by the forkhead protein FoxO1.
Diabetes Obes Metab. 9:140–146. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Cheng Z and White MF: Targeting forkhead
box O1 from the concept to metabolic diseases: lessons from mouse
models. Antiox Redox Signal. 14:649–661. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Puthanveetil P, Wan A and Rodrigues B:
FoxO1 is crucial for sustaining cardiomyocyte metabolism and cell
survival. Cardiovasc Res. 97:393–403. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Jünger MA, Rintelen F, Stocker H,
Wasserman JD, Végh M, Radimerski T, Greenberg ME and Hafen E: The
Drosophila forkhead transcription factor FOXO mediates the
reduction in cell number associated with reduced insulin signaling.
J Biol. 2:202003.
|
|
40
|
Puig O, Marr MT, Ruhf ML and Tjian R:
Control of cell number by Drosophila FOXO: downstream and
feedback regulation of the insulin receptor pathway. Genes Dev.
17:2006–2020. 2003.
|
|
41
|
Zeng FY, Cui J, Liu L and Chen T:
PAX3-FKHR sensitizes human alveolar rhabdomyosarcoma cells to
camptothecin-mediated growth inhibition and apoptosis. Cancer Lett.
284:157–164. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Liu P, Kao TP and Huang H: CDK1 promotes
cell proliferation and survival via phosphorylation and inhibition
of FOXO1 transcription factor. Oncogene. 27:4733–4744. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Fendler A1, Jung M, Stephan C,
Erbersdobler A, Jung K and Yousef GM: The antiapoptotic function of
miR-96 in prostate cancer by inhibition of FOXO1. PLoS One.
8:e808072013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Harada H, Andersen JS, Mann M, Terada N
and Korsmeyer SJ: p70S6 kinase signals cell survival as well as
growth, inactivating the pro-apoptotic molecule BAD. Proc Natl Acad
Sci USA. 98:9666–9670. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Budanov AV and Karin M: p53 target genes
sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling.
Cell. 134:451–460. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Hay N: Interplay between FOXO, TOR, and
Akt. Biochim Biophys Acta. 1813:1965–1970. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Dansen TB and Burgering BM: Unravelling
the tumor- suppressive functions of FOXO proteins. Trends Cell
Biol. 18:421–429. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Bhaskar PT and Hay N: The two TORCs and
Akt. Dev Cell. 12:487–502. 2007. View Article : Google Scholar : PubMed/NCBI
|

















