Introduction
Dexmedetomidine, which is known as a potent
α2-adrenoceptor agonist, induces analgesia, anxiolysis,
sedation and sympatholysis (1).
The neuroprotective effects of dexmedetomidine have been shown in
transient cerebral ischemia models (1,2).
Dexmedetomidine directly affects neurons and exerts protective
effects through the attenuation of catecholamine and glutamate
levels or the prevention of neuronal apoptosis (1–3).
Astrocytes regulate neurons by releasing neurotransmitters and
other extracellular signaling molecules and exert neuroprotective
effects through the uptake of synaptic glutamate and the production
of glutathione (4).
Dexmedetomidine also affects astrocytes (1,2).
Dexmedetomidine protects neurons by increasing glutamate uptake by
astrocytes in hippocampal slices exposed to hypoxia (5), promoting the release of glial cell
line-derived neurotrophic factor (GDNF) from rat cultured
astrocytes following oxygen-glucose deprivation (6), and upregulating the release of
hypoxia-inducible factor (HIF)-1α and vascular endothelial growth
factor (VEGF) from rat C6 glioma cells following oxygen-glucose
deprivation (7). However, the
effects of dexmedetomidine and the exact mechanisms involving
astrocytes have not yet been fully elucidated.
In the central nervous system (CNS) of healthy
individuals, interleukin (IL)-1β, a pro-inflammatory cytokine, is
expressed at low levels (8). IL-1
plays an important role in some physiological functions, including
sleep and synaptic plasticity (8). The levels of IL-1β are increased in
cerebrospinal fluid in patients following traumatic brain injury,
stroke and neurodegenerative diseases (8,9).
The main source of brain IL-1β is microglia following acute insults
(8,9). Astrocytes also produce IL-1β at a
later stage than microglia (8,9).
IL-1β induces the production of other cytokines, such as tumor
necrosis factor (TNF)-α and IL-6, from microglia and astrocytes
(8). Cytokines have been
implicated not only in neuroinflammation, but also in astrogliosis,
brain ischemia and chronic diseases of the CNS (8,10).
Dexmedetomidine suppresses systemic immune function similar to
other sedative medications (11).
Dexmedetomidine decreases transient global cerebral
ischemia-induced neuronal apoptosis and plasma TNF-α levels
(12), suppresses plasma IL-6
levels in septic rats (13),
decreases serum IL-6 levels compared with propofol administration
in post-operative patients (14)
and decreases serum TNF-α, IL-1β or IL-6 levels compared with
midazolam administration in patients with sepsis (15). C6 cells, a rat glioma cell line,
have frequently been used for the investigation of the mechanisms
of IL-6 production or release (16–18). We previously reported that
midazolam, a sedative agent used in intensive care units, inhibits
the IL-1β-induced IL-6 release from C6 cells (17). However, the effects of
dexmedetomidine on cytokine synthesis in brain cells have not yet
been fully elucidated. In the present study, we investigated the
effects and mechanism of action of dexmedetomidine on the
IL-1β-induced release of IL-6 from glial cells.
Materials and methods
Materials
Dexmedetomidine was kindly provided by Orion Pharma
(Turku, Finland). 8-Bromo-adenosine-3′,5′-cyclic monophosphate
(8-bromo-cAMP), forskolin or
12-O-tetradecanoylphorbol-13-acetate (TPA) were purchased
from Sigma Chemical Co. (St. Louis, MO, USA). 8-Bromo-guanosine
3′,5′-cyclic monophosphate (8-bromo-cGMP) was purchased from
Calbiochem-Novabiochem Co. (La Jolla, CA, USA). Yohimbine
hydrochloride (yohimbine) was purchased from Wako Pure Chemical
Industries (Osaka, Japan). IL-6 enzyme-linked immunosorbent assay
(ELISA) kit and IL-1β were obtained from R&D Systems
(Minneapolis, MN, USA). The cAMP ELISA kit was purchased from Enzo
Life Sciences Inc. (Farmingdale, NY, USA).
α2A-adrenoceptor antibody was purchased from Santa Cruz
Biotechnology (Santa Cruz, CA, USA). Phospho-specific p38
mitogen-activated protein (MAP) kinase, p38 MAP kinase,
phospho-specific stress-activated protein kinase/c-Jun N-terminal
kinase (SAPK/JNK), SAPK/JNK, phospho-c-Jun, c-Jun, phospho-specific
IκB, IκB, phospho-nuclear factor (NF)-κB (serine 536) or NF-κB
antibodies were purchased from Cell Signaling Technology (Beverly,
MA, USA). An enhanced chemiluminescence western blotting detection
system was obtained from GE Healthcare Ltd. (Buckinghamshire, UK).
Other materials and chemicals were obtained from commercial
sources. Forskolin, TPA and yohimbine were dissolved in dimethyl
sulfoxide (DMSO), and others were dissolved in assay buffer (5 mM
HEPES, pH7.4, 150 mM NaCl, 5 mM KCl, 5.5 mM glucose, 0.8 mM
MgSO4, 1 mM CaCl2, containing 0.01% albumin)
as the vehicle. The maximum concentration of DMSO was 0.1%, which
did not affect the assay for IL-6 or cAMP.
Cell culture
Rat C6 glioma cells, obtained from the American Type
Culture Collection (ATCC; Rockville, MD, USA), were seeded into
35-mm (5×104 cells/dish) or 90-mm (2×105
cells/dish) diameter dishes and maintained in Dulbecco’s modified
Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS) at
37°C in a humidified atmosphere of 5% CO2/95% air. The
medium was exchanged for serum-free DMEM after 6 days. The cells
were then used for the experiments after 24 h. The cells were
pre-treated with dexmedetomidine, forskolin, 8-brom-cAMP,
8-bromo-cGMP, TPA or yohimbine for 60 min where indicated.
Assay for IL-6
Cultured cells (35-mm diameter dishes) were
stimulated with 10 ng/ml IL-1β in serum-free DMEM for 36 h. The
conditioned medium was collected at the end of the incubation
period and the IL-6 concentration was measured using an ELISA kit.
The absorbance of each sample at 450 and 540 nm, respectively, was
measured with a Multiscan JX ELISA reader (Thermo Labsystems,
Helsinki, Finland). Absorbance was corrected with reference to a
standard curve.
Assay for cAMP
Cultured cells (35-mm diameter dishes) were
pre-incubated with 0.5 mM 3-isobutyl-1-methylxanthine, a cyclic
nucleotide phosphodiesterase inhibitor, for 20 min for the
inhibition of cAMP decomposition, and then stimulated with 10 ng/ml
IL-1β or 10 μM forskolin for 20 min. The reaction was terminated by
aspiration of the assay buffer. Intracellular cAMP was extracted
with 0.1 M HCl. The extracted cAMP was measured using an ELISA kit.
The absorbance of each sample at 405 nm was measured using a
Multiscan JX ELISA reader. Absorbance was corrected with reference
to a standard curve.
Real-time reverse transcription
(RT)-polymerase chain reaction (PCR)
Cultured cells (35-mm diameter dishes) were
stimulated with 10 ng/ml IL-1β for 6 h. Total RNA was isolated and
transcribed into cDNA using TRIzol reagent and the Omniscript
Reverse Transcriptase kit (QIAGEN, Hilden, Germany). Real-time
RT-PCR was performed using a LightCycler system (Roche Diagnostics,
Basel, Switzerland) in capillaries and FastStart DNA Master
SYBR-Green I provided with the kit. Sense and antisense primers for
mouse IL-6 mRNA or rat GAPDH mRNA were purchased from Takara Bio
Inc. (Tokyo, Japan) (primer set ID: MA039013 or RA015380,
respectively). The amplified products were determined by melting
curve analysis and agarose electrophoresis. The IL-6 mRNA levels
were normalized to those of GAPDH mRNA.
Western blot analysis
Cultured cells (90-mm diameter dishes) were
stimulated with 10 ng/ml IL-1β in serum-free DMEM for the indicated
periods of time. The cells were washed twice with
phosphate-buffered saline and then lysed and sonicated in a lysis
buffer containing 62.5 mM Tris-HCl (pH 6.8), 2% sodium dodecyl
sulfate (SDS), 50 mM dithiothreitol and 10% glycerol. The sample
was used for the analysis by western blotting as previously
described (17).
SDS-polyacrylamide gel electrophoresis (PAGE) was performed
according to the method of Laemmli (19) in 10% polyacrylamide gels. Western
blot analysis was performed using antibodies against
α2A-adrenoceptor, phospho-specific p38 MAP kinase, p38
MAP kinase, phospho-specific SAPK/JNK, SAPK/JNK, phospho-c-Jun,
c-Jun, phospho-specific IκB, IκB, phospho-NF-κB or NF-κB with
peroxidase-labeled antibodies raised in goat against rabbit IgG
being used as secondary antibodies. The peroxidase activity on
polyvinylidene difluoride membranes was visualized on X-ray film by
means of an enhanced chemiluminescence western blotting detection
system.
Statistical analysis
The data were analyzed by ANOVA followed by
Bonferroni’s method for multiple comparisons between pairs. A value
of P<0.05 was considered to indicate a statistically significant
difference. All data are presented as the means ± SD of triplicate
determinations. Each experiment was repeated 3 times with similar
results.
Results
Effects of dexmedetomidine on the
IL-1β-induced IL-6 release and IL-6 mRNA expression in C6
cells
It has been reported that there are
α2A-adrenoceptors, but not α2B- and
α2C-adrenoceptors, in C6 glioma cells (20). We confirmed the expression of
α2A-adrenoceptors in these cells (Fig. 1). Subsequently, we investigated
effects of dexmedetomidine on the IL-1β-induced release of IL-6. It
has been reported that IL-1β induces IL-6 mRNA expression and IL-6
release from C6 glioma cells (16). Dexmedetomidine, which on its own
had little effect on the IL-6 levels, significantly suppressed the
IL-1β-induced IL-6 release. The inhibitory effects of
dexmedtomidine were concentration-dependent between 1 and 100 μM
(Fig. 2A). Dexmedetomidine (100
μM) suppressed the effects of IL-1β by approximately 40%. The
viability of the cells treated with 100 μM dexmedetomidine and 10
ng/ml IL-1β for 36 h was >97% compared to the cells without
treatment, as shown by trypan blue staining (data not shown).
Moreover, dexmedetomidine (30 μM) markedly suppressed the
IL-1β-induced IL-6 mRNA expression, and caused approximately a 50%
inhibition of the effect of IL-1β (Fig. 2B).
Effects of 8-bromo-cAMP on the
IL-1β-induced IL-6 release and IL-6 mRNA expression in C6
cells
Dexmedetomidine inhibits the adenylyl cyclase
activity through the pertussis toxin-sensitive GTP-binding protein
(Gi/o) coupling with its receptors,
α2-adrenoceptors (1,21).
Adenylyl cyclase produces cAMP from adenosine triphosphate (ATP)
(1,21). We investigated whether the
activation of the adenylyl cyclase-cAMP pathway is involved in the
IL-1β-induced release of IL-6 from C6 cells. IL-1β on its own did
not affect the accumulation of cAMP in the C6 cells (Table I). It has been reported that
forskolin, a direct activator of adenylyl cyclase, or dibutyryl
cAMP, a permeable analogue of cAMP, significantly enhances the
IL-1β-induced release of IL-6 from C6 cells (18). We found that 8-bromo-cAMP, another
plasma membrane-permeable cAMP analogue, which alone slightly
increased the release of IL-6, significantly enhanced the
IL-1β-induced release of IL-6 (Fig.
3A). The amplifying effects of 8-bromo cAMP were
concentration-dependent between 0.1 and 2 mM (Fig. 3A). 8-Bromo cAMP (1 mM) markedly
enhanced the IL-1β-induced IL-6 mRNA expression (Fig. 3B). In addition,
α2-adrenoceptor agonists have been shown to decrease
cerebellar cGMP levels (18). We
investigated whether cGMP affects the IL-1β-induced IL-6 release
from C6 cells. However, 8-bromo-cGMP failed to affect the
IL-1β-induced release of IL-6 at up to 2 mM (Fig. 3A).
 | Table IEffects of dexmedetomidine on cAMP
accumulation in C6 cells following treatment with IL-1β. |
Table I
Effects of dexmedetomidine on cAMP
accumulation in C6 cells following treatment with IL-1β.
| Dexmedetomidine (50
μM) | IL-1β (10
ng/ml) | cAMP (pmol/ml) |
|---|
| − | − | 8.7±0.8 |
| − | + | 8.0±1.4 |
| + | − | 6.1±3.4 |
| + | + | 9.8±0.3 |
Effect of dexmedetomidine on the
forskolin-induced cAMP accumulation in C6 cells
We then investigated the association between
dexmedetomidine and the adenylyl cyclase-cAMP pathway in the
IL-1β-stimulated IL-6 synthesis in C6 cells. Dexmedetomidine (50
μM) had little effect on cAMP accumulation regardless of the
presence of IL-1β (Table I).
Additionally, we confirmed that forskolin increased cAMP
accumulation in the C6 cells (Table
II). However, dexmedetomidine (50 μM) did not affect the
forskolin-induced cAMP accumulation (Table II). Therefore, it seems likely
that the suppression of the IL-1β-stimulated IL-6 release by
dexmedetomidine is not due to the suppression of the adenylyl
cyclase-cAMP pathway.
| Table IIEffects of dexmedetomidine on the
forskolin-induced cAMP accumulation in C6 cells. |
Table II
Effects of dexmedetomidine on the
forskolin-induced cAMP accumulation in C6 cells.
| Dexmedetomidine (50
μM) | Forskolin (10
μM) | cAMP (pmol/ml) |
|---|
| − | − | 13.4±2.9 |
| − | + | 814.9±50.4a |
| + | − | 16.8±5.6 |
| + | + | 787.7±22.3 |
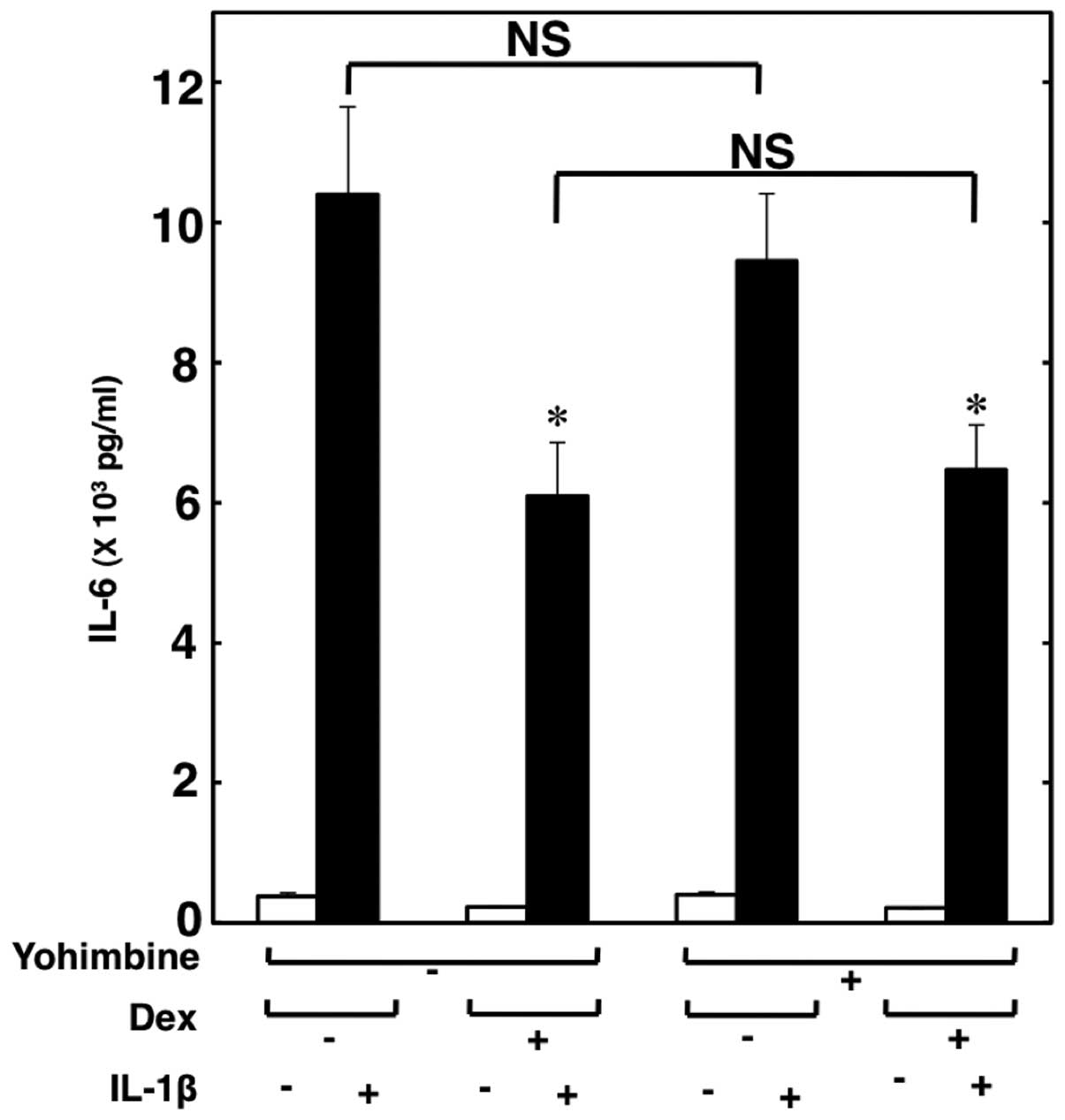
Effect of yohimbine on the suppression of
IL-1β-induced IL-6 release from C6 cells by dexmedetomidine
Subsequently, we investigated whether the
suppressive effects of dexmedetomidine are mediated through
α2-adrenoceptors. Yohimbine (10 μM), an
α2-adrenoceptor antagonist, did not reverse the
suppressive effects of dexmedetomidine on the IL-1β-induced IL-6
release (Fig. 4). Therefore, it
seems unlikely that dexmedetomidine suppresses the IL-1β-induced
release of IL-6 through α2-adrenoceptors.
Effect of TPA on the IL-1β-induced IL-6
release from C6 cells
It has been reported that α2-adrenoceptor
agonists also bind to imidazoline receptors, which activate
phospholipase C and protein kinase C (1,2,22).
Finally, we investigated the association between the activation of
protein kinase C and the IL-1β-induced release of IL-6 from C6
cells. TPA, a specific protein kinase C activator (23), significantly enhanced the release
of IL-6 and markedly enhanced the IL-1β-induced release of IL-6
(Fig. 5).
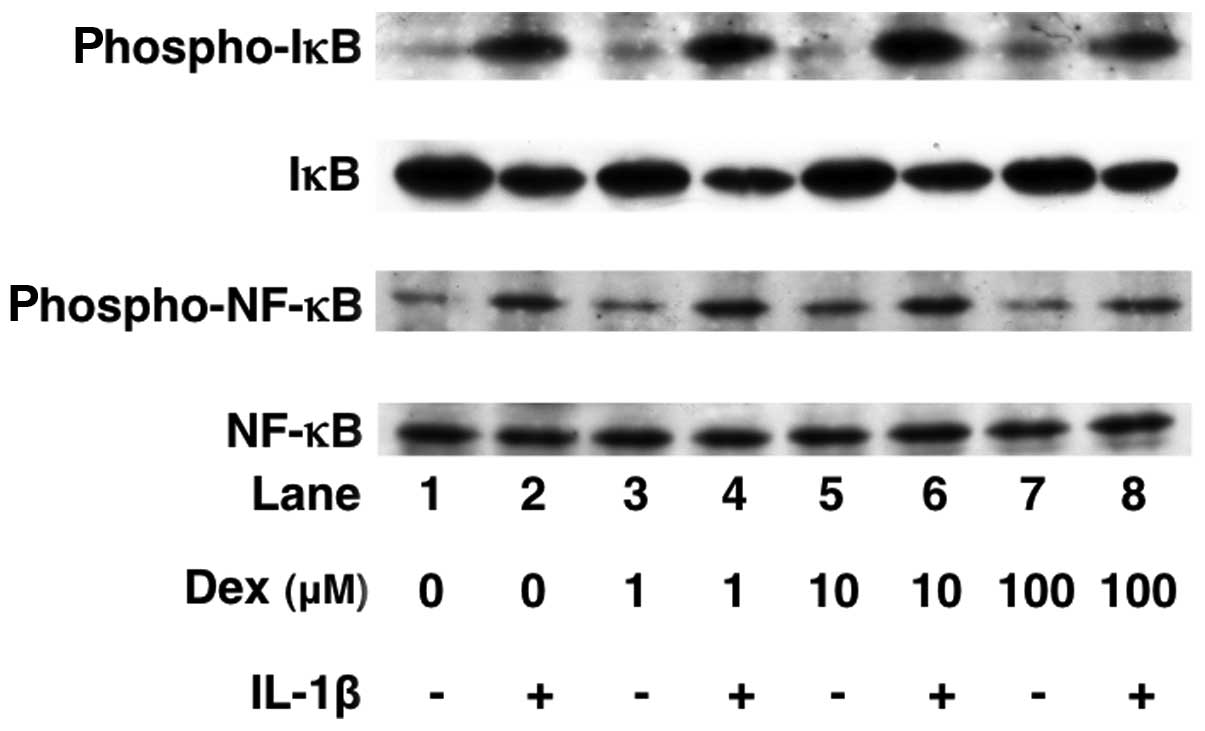
Effects of dexmedetomidine on the
IL-1β-induced phosphorylation of p38 MAP kinase, SAPK/JNK, c-Jun,
IκB and NF-κB or the levels of IκB in C6 cells
We have previously reported that IL-1β stimulates
IL-6 synthesis through p38 MAP kinase, SAPK/JNK or the IκB/NF-κB
pathway in C6 cells (17). In
order to elucidate the mechanisms through which dexmedetomidine
alters IL-1β intracellular signaling, we examined the effects of
dexmedetomidine on the IL-1β-induced phosphorylation of p38 MAP
kinase, SAPK/JNK, c-Jun, IκB and NF-κB or the IκB levels.
Dexmedetomidine (100 μM) did not affect the IL-1β-induced
phosphorylation of p38 MAP kinase, SAPK/JNK, c-Jun, IκB or NF-κB
(Figs. 6 and 7). In addition, dexmedetomidine failed
to affect the levels of IκB (Fig.
7).
Discussion
In this study, we first confirmed the expression of
α2A-adrenoceptors in C6 cells. Thereafter, we showed
that dexmedetomidine significantly suppressed the IL-1β-induced
IL-6 synthesis in C6 glioma cells. Dexmedetomidine, a specific
agonist of α2-adrenoceptor, binds to
α2-adrenoceptors, [all 3 subtype receptors
(α2A/D, α2B and α2C)], which are
coupled to pertussis toxin-sensitive Gi/o protein
(2,21). Pertussis toxin-sensitive
Gi/o protein suppresses adenylyl cyclase activity, which
produces cAMP from ATP (2). In
the present study, IL-1β by itself did not increase cAMP
accumulation in C6 cells. It has been reported that the activation
of adenylyl cyclase-cAMP pathway enhances the IL-1β-induced IL-6
release from C6 cells (18). We
found that 8-bromo-cAMP enhanced the IL-1β-induced IL-6 mRNA levels
and release in these cells, whereas 8-bromo-cGMP did not affect the
IL-6 release. Based on these findings, it is possible that the
adenylyl cyclase-cAMP pathway, but not the guanylyl cyclase-cGMP
system, enhances the IL-1β-induced IL-6 release and its mRNA
expression in C6 cells. Therefore, we investigated whether the
adenylyl cyclase-cAMP system is involved in suppressive effect of
dexmedetomidine on IL-6 synthesis in C6 cells. We showed that
dexmedetomidine had little effect on the cAMP levels in the
presence or absence of IL-1β. Forskolin is known as a direct
activator of adenylyl cyclase (24) and increases cAMP accumulation in
C6 cells (25). It is well known
that α2-adrenoceptor agonists suppress adenylyl cyclase
activity (1,2,21).
However, we found that dexmedetomidine failed to affect the
accumulation of cAMP in the cells treated with IL-1β or forskolin.
Yohimbine, an α2-adrenoceptor antagonist, did not
reverse the suppressive effects of dexmedetomidne. It seems
unlikely that dexmedetomidine suppresses the IL-1β-induced release
of IL-6 through α2-adrenoceptors. In addition to
α2-adrenoceptors, dexmedetomidine contains an imidazol
ring, which can bind imidazoline receptors (1,2).
Imidazoline receptors are coupled to the activation of
phospholipase C and protein kinase C (22). However, we found that TPA, a
direct activator of protein kinase C (23), markedly enhanced the IL-1β-induced
relesae of IL-6 and that bisindolylmaleimide I, a protein kinase C
inhibitor, did not reverse the suppression of IL-6 release by
dexmedetomidine in C6 cells (data not shown). Based on our
findings, it seems unlikely that dexmedetomidine suppresses the
IL-1β-induced IL-6 release through α2-adrenoceptors,
imidazoline receptors or the cGMP pathway in C6 glioma cells. We
previously reported that IL-1β stimulates IL-6 synthesis through
p38 MAP kinase signaling, SAPK/JNK signaling and IκB/NF-κB
signaling in C6 cells (17).
SAPK/JNK activates the transcription factor, c-Jun, by
phosphorylation (26). The
heterodimer complex of c-Jun and c-Fos is known as activator
protein-1 (AP-1) (26). On the
other hand, IκB is phosphorylated and degradated, and subsequently,
NF-κB is freed from IκB and translocates to the nucleus (26). Transcription factors, such as AP-1
and NF-κB, bind to DNA-regulatory sequences to modulate the rate of
gene transcription (26). In the
present study, dexmedetomidine did not affect the IL-1β-induced
phosphorylation of p38 MAP kinase, SAPK/JNK, IκB, NF-κB and c-Jun
or the IκB level. Based on our findings, it seems unlikely that
dexmedetomidine suppresses the IL-1β-induced release of IL-6 at the
point between IL-1β receptors and transcription factors in C6
cells. Further studies are required to clarify the exact mechanisms
underlying the inhibitory effects of dexmedetomidine on IL-6
synthesis.
In a previous study, the blood peak concentration of
dexmedetomidine in human subjects after the initiation of 3–6
μg/kg/h intravenous infusion was approximately 4–6 ng/ml (27). The neuroprotective effects of
dexmedetomidine against transient focal cerebral ischemia have also
been observed at 4 ng/ml (0.02 μM) in rabbits (1,3).
In the present study, the suppressive effects of dexmedetomidine on
IL-6 expression were observed at the dose of 3 μM, which is higher
than the concentration used in clinical practice. However, in a
number of studies using cultured astocytes, dexmedetomidine has
exerted its effects at higher concentrations than those used in
clinical practice. For example, dexmedetomidine has been shown to
induce the biphasic increase in the accumulation of glutamine
(second peak is observed over 25 μM) in mouse astrocytes (28), the intracellular calcium
concentration (second peak is observed over 1 μM) in mouse
astrocytes (29), and to inhibit
oxygen-glucose deprivation-induced apoptosis at a dose of >1 μM
in C6 cells (7). Therefore, it is
possible that the differences observed in the effective
concentration between clinical and in vitro studies are due
to the differences in the experimental conditions between in
vivo and in vitro experiments.
Astrocytes are considered to be the main source of
IL-6 in the CNS (10). IL-6 is
involved in multiple physiological CNS functions, such as neuron
homeostasis, astrogliosis and neuronal differentiation (10). IL-6 has also been implicated in
both acute and chronic diseases of the CNS, including infection,
traumatic brain injury, ischemia, multiple sclerosis, Alzheimer’s
disease and Parkinson’s disease (10). IL-6 plays a key role in
neuroinflammation, which accompanies those disorders, as both a
pro-inflammatory cytokine and an anti-inflammatory cytokine
(10). As a pro-inflammatory
cytokine, IL-6 induces astrogliosis, which is characterized by
hypertrophy and the hyperplasia of astrocytes, and produces other
inflammatory mediators, such as prostaglandins, cytokines,
chemokines and acute phase proteins (10). On the other hand, as an
anti-inflammatory cytokine, IL-6 affects neuronal survival,
proliferation, differentiation and regeneration (10). It is possible that IL-6 may be a
valid therapeutic target for the treatment of disorders of the CNS
(10,30).
Some sedative agents used in critical care have
anti-inflammatory properties (11). α2-adrenoceptor agonists
interact with the immune system in a complex manner.
Dexmedetomidine also suppresses immune function, but improves
outcome, including mortality (11,14,15). It has been reported that
dexmedetomidine improves the early survival rate and suppresses
plasma IL-6 levels in septic rats and that patients administered
with dexmedetomodine show a decrease in serum IL-6 levels compared
with midazolam or propofol administration (13–15). There are some reports that
dexmedetomidine does not affect plasma IL-6 levels in patients
undergoing laparoscopic cholecystectomy or spine surgery (31,32). However, the effects of
dexmedetomidine on cytokine synthesis, including IL-6 in the brain
have not yet been fully elucidated. We previously reported that
midazolam, but not propofol, inhibits the IL-1β-induced IL-6
synthesis in C6 cells (17). The
suppressive effects of midazolam were greater than those of
dexmedetomidine, which were observed in the present study.
Midazolam, propofol or dexmedetomidine may have different effects
on the CNS in patients with elevated IL-6 levels.
Although astrocytes play important roles in the CNS
(4), the effects of
dexmedetomidine on astrocytes remain to be clarified (1). Astrocytes are known to protect
neurons though the uptake of glutamine and the conversion to
glutamate, both neurotoxicants (4). It has been reported that
dexmedetomidine increases the uptake of glutamine in rat
hippocampal slices or cultured mouse astrocytes (5,28).
Astrocytes protect neurons by the release of neurotrophic factors
against brain ischemia, brain injury and neurodegenerative diseases
(33). It has been shown that
dexmedetomidine stimulates GDNF, one of the neurotrophic factors,
released from rat primary cultured astrocytes through
α2A-adrenoceptor and the stimulation of GDNF by
dexmedetomidine decreases neuronal cell death induced by
oxygen-glucose deprivation (6).
In C6 cells, it has been reported that dexmedetomidine inhibits the
decrease in cell viability induced by HIF-1α and VEGF expression
under conditions of oxygen-glucose deprivation (7). Taken together with our results, it
is possible that astrocytes may be an important target of
dexmedetomidine. However, the effects of dexmedetomidine on the CNS
and the exact mechanisms involved are not yet fully understood.
Further investigations into the effects of dexmedetomidine on
astrocytes are required.
In conclusion, our findings strongly suggest that
dexmedetomidine inhibits the IL-1β-induced IL-6 synthesis
independently of the adenylyl cyclase-cAMP pathway through
α2-adrenoceptors in C6 glioma cells. It is possible that
dexmedetomidine may affect the immune system in the CNS by
regulating the production of IL-6.
Acknowledgements
We are very grateful to Yumiko Kurokawa for her
skillful technical assistance. The present study was supported in
part by a Grant-in-Aid for Scientific Research (23592248) from the
Ministry of Education, Science, Sports and Culture of Japan.
References
|
1
|
Ma D, Rajakumaraswamy N and Maze M:
α2-Adrenoceptor agonists: shedding light on
neuroprotection? Br Med Bull. 71:77–92. 2004.
|
|
2
|
Zhang Y and Kimelberg HK: Neuroprotection
by alpha 2 adrenergic agonists in cerebral ischemia. Curr
Neuropharmacol. 3:317–323. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sanders RD and Maze M:
α2-Adrenoceptor agonists. Curr Opin Invest. 8:25–33.
2007.
|
|
4
|
Benarroch EE: Neuron-astrocyte
interactions: partnership for normal function and disease in the
central nervous system. Mayo Clin Proc. 80:1326–1338. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Talke P and Bickler PE: Effects of
dexmedetomidine on hypoxia-evoked glutamate release and glutamate
receptor activity in hippocampal slices. Anesthesiology.
85:551–557. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yan M, Dai H, Ding T, Dai A, Zhang F, Yu
L, Chen G and Chen Z: Effects of dexmedetomodine on the release of
glial cell line-derived neurotrophic factor from rat astrocyte
cells. Neurochem Int. 58:549–557. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang F, Ding T, Yu L, Zhong Y, Dai H and
Yan M: Dexmedetomidine protects against oxygen-glucose
deprivation-induced injury through the I2 imidazoline
receptor-PI3K/AKT pathway in rat C6 glioma cells. J Pharm
Pharmacol. 64:120–127. 2012. View Article : Google Scholar
|
|
8
|
Allan SM, Tyrrell PJ and Rothwell NJ:
Interleukin-1 and neuronal injury. Nat Rev Immunnol. 5:629–640.
2005. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Simi A, Tsakiri N, Wang P and Rothwell NJ:
Interleukin-1 and inflammatory neurodegeneration. Biochem Soc
Trans. 35:1122–1126. 2007. View Article : Google Scholar
|
|
10
|
Spooren A, Kolmus K, Laureys G, Clinckers
R, De Keyser J, Haegeman G and Gerlo S: Interleukin-6, a mental
cytokine. Brain Res Rev. 67:157–183. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sanders RD, Hussell T and Maze M: Sedation
& immunomodulation. Anesthesiol Clin. 29:687–706. 2011.
View Article : Google Scholar
|
|
12
|
Eser O, Fidan H, Sahin O, Cosar M, Yaman
M, Mollaoglu H, Songur A and Buyukbas S: The influence of
dexmedtomidine on ischemic rat hippocampus. Brain Res.
1218:250–256. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Qiao H, Sanders RD, Ma D, Wu X and Maze M:
Sedation improves early outcome in severely septic Sprague Dawley
rats. Crit Care. 13:R1362009. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Venn RM, Bryant A, Hall GM and Grounds RM:
Effects of dexmedetomidine on adrenocortical function, and the
cardiovascular, endocrine and inflammatory responses in
post-operative patients needing sedation in the intensive care
unit. Br J Anaesth. 86:650–656. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Meniş D, Hekimoğlu S, Vatan İ, Yandım T,
Yüksel M and Süt N: Effects of midazolam and dexmedetomidine on
inflammatory responses and gastric intramucosal pH to sepsis, in
critically ill patients. Br J Anaesth. 98:550–552. 2007.PubMed/NCBI
|
|
16
|
Kawashima A, Harada T, Imada K, Yano T and
Mizuguchi K: Eicosapentaenoic acid inhibits interleukin-6
production in interleukin-1β-stimulated C6 glioma cells through
peroxisome proliferator-activated receptor-gamma. Prostaglandins
Leukot Essent Fatty Acids. 79:59–65. 2008.PubMed/NCBI
|
|
17
|
Tanabe K, Kozawa O and Iida H: Midazolam
suppresses interleukin-1β-induced interleukin-6 release from rat
glial cells. J Neuroinflammation. 8:682011.PubMed/NCBI
|
|
18
|
Zumwalt JW, Thunstrom BJ and Spangelo BL:
Interleukin-1β and catecholamines synergistically stimulate
interleukin-6 release from rat C6 glioma cells in vitro: a
potential role for lysophosphatidylcholine. Endocrinology.
140:888–896. 1999.
|
|
19
|
Laemmli UK: Cleavage of structural
proteins during the assembly of the head of bacteriophage T4.
Nature. 227:680–685. 1970. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Morioka N, Sugimoto T, Tokuhara M, Dohi T
and Nakata Y: Noradrenaline induces clock gene Per1 mRNA expression
in C6 glioma cells through β2-adrenergic receptor
coupled with protein kinase A-cAMP response element binding protein
(PKA-CREB) and Src-tyrosine kinase-glycogen synthase kinase-3β
(Src-GSK-3β). J Pharmacol Sci. 113:234–245. 2010.PubMed/NCBI
|
|
21
|
Aantaa R, Marjamäki A and Scheinin M:
Molecular pharmacology of α2-adrenoceptor subtypes. Ann Med.
27:439–449. 1995.
|
|
22
|
Edwards LP, Brown-Bryan TA, McLean L and
Ernsberger P: Pharmacological properties of the central
antihypertensive agent, moxonidine. Cardiovasc Ther. 30:199–208.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nishizuka Y: Studies and perspectives of
protein kinase C. Science. 233:305–312. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Simonds WF: G protein regulation of
adenylate cyclase. Trends Pharmacol Sci. 20:66–73. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gallagher HC, Bacon CL, Odumeru OA,
Gallagher KF, Fitzpatrick T and Regan CM: Valproate activates
phosphodiesterase-mediated cAMP degradation: relevance to C6 glioma
G1 phase progression. Neurotoxicol Teratol. 26:73–81. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Adcock IM and Caramori G: Cross-talk
between pro-inflammatory transcription factors and glucocorticoids.
Immunol Cell Biol. 79:376–384. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Iirola T, Ihmsen H, Laitio R, Kentala E,
Aantaa R, Kurvinen JP, Scheinin M, Schwilden H, Schüttler J and
Olkkola KT: Population pharmacokinetics of dexmedetomidine during
long-term sedation in intensive care patients. Br J Anaesth.
108:460–468. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Huang R and Hertz L: Receptor subtype and
dose dependence of dexmedetomidine-induced accumulation of
[14C]glutamine in astrocytes suggests glial involvement
in its hypnotic-sedative and anesthetic-sparing effects. Brain Res.
873:297–301. 2000.PubMed/NCBI
|
|
29
|
Chen Y, Zhao Z, Code WE and Hertz L: A
correlation between dexmedetomidine-induced biphasic increases in
free cytosolic calcium concentration and energy metabolism in
astrocytes. Anesth Analg. 91:353–357. 2000.
|
|
30
|
Lambertsen KL, Biber K and Finsen B:
Inflammatory cytokines in experimental and human stroke. J Cereb
Blood Flow Metab. 32:1677–1698. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bekker A, Haile M, Kline R, Didehvar S,
Babu R, Martiniuk F and Urban M: The effect of intraoperative
infusion of dexmedetomidine on the quality of recovery after major
surgery. J Neurosurg Anesthesiol. 25:16–24. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kang AH, Kim YS, Hong TH, Chae MS, Cho ML,
Her YM and Lee J: Effects of dexmedetomidine on inflammatory
responses in patients undergoing laparoscopic cholecystectomy. Acta
Anaesthesiol Scand. 57:480–487. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Saavedra A, Baltazar G and Duarte EP:
Driving GDNF expression: the green and the red traffic light. Prog
Neurobiol. 86:186–215. 2008. View Article : Google Scholar : PubMed/NCBI
|