Introduction
Hepatocellular carcinoma (HCC) is a malignant tumor
with a high prevalence in Asia and Africa (1). HCC is the third most common cause of
cancer-related mortalities (500,000 mortalities per year) and the
fifth most common cancer worldwide (2). Treatments for HCC include
percutaneous ethanol injection therapy, transcatheter arterial
chemoembolization, liver transplantation and surgical intervention.
Of these treatments, surgical intervention is the most effective
for improving the 5-year survival rate of patients (3). One major obstacle for the optimum
treatment of HCC is the high expression of anti-apoptotic genes,
which contributes to an extremely poor prognosis (4,5).
Therefore, novel or additional therapeutic strategies for HCC are
required to circumvent this challenge.
Catechins constitute ~40% of the dry weight of green
tea, and epigallocatechin-3-gallate (EGCG) is the most common
catechin (6–8). EGCG has great potential as an
inexpensive, bioavailable chemotherapeutic agent for cancer
prevention and complementary treatment (9). The anticancer role of EGCG has been
studied epidemiologically in in vitro and in vivo
systems, and in clinical trials (10–12). In vitro studies have
demonstrated that EGCG inhibits cancer by inducing apoptosis
(11–13). Several studies have also reported
its role in cancer prevention via the induction of apoptosis,
alteration of protein expression and inhibition of intracellular
communication pathways (14–17). Apoptosis is driven by two major
pathways: The cell death receptor-mediated extrinsic pathway and
the mitochondria-mediated intrinsic pathway. However, the precise
mechanism by which EGCG exerts its anticancer effects remains
unknown.
There is a lack of studies describing the effect of
EGCG on HCCs, compared to other cancers (18–20). To the best of our knowledge, no
comparative studies of the effects of EGCG on HCCLM6 (a human HCC
metastatic cell line) and HL-7702 (a non-cancerous, normal human
liver cell line) cells have been reported. The present study
investigated the potential effect of EGCG on these cell lines and
the molecular mechanism of EGCG induced apoptosis of HCCLM6
cells.
Materials and methods
Antibodies and reagents
Dulbecco’s modified Eagle’s medium (DMEM) and fetal
bovine serum (FBS) were purchased from Gibco (Grand Island, NY,
USA). EGCG (≥98% purity),
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
and 4′,6-diamidino-2-phenylindole (DAPI) were provided by Sigma
(St. Louis, MO, USA). The fluorescein isothiocyanate
(FITC)-conjugated anti-Annexin V and propidium iodide (PI) Dye
Apoptosis Detection kit, the JC-1 Mitochondrial Membrane Potential
Detection kit, the Cell Cycle Detection kit and the BCA Protein
Assay kit were purchased from Nanjing KeyGen Biotech., Co., Ltd.
(Nanjing, China). The SuperSignal® West Pico Trial kit
was purchased from Thermo Scientific (Rockford, IL, USA). Mouse
anti-human β-actin (sc-1496), mouse anti-human caspase-3
(sc-65497), mouse anti-human Bax (sc-7480), rabbit anti-human
B-cell lymphoma 2 (Bcl-2) (sc-783) and horseradish peroxidase
(HrP)-conjugated anti-rabbit or anti-mouse secondary antibodies
were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA,
USA). Mouse anti-human p53 (AP062), rabbit anti-human nuclear
factor-κB (NF-κB) (AN365), mouse anti-human caspase-9 (AC062),
mouse anti-human cytochrome c (AC909), rabbit anti-human
cyclooxygenase (COX) IV (AC610) were purchased from Beyotime
Institute of Biotechnology (Beijing, China).
Cells and cell culture
HCCLM6 cells (ATCC, Rockefeller, MD, USA) and
HL-7702 liver cells (Shenke Biological Technology, Co., Ltd.,
Shanghai, China) were grown in DMEM supplemented with 10% FBS, 0.1%
benzyl penicillin and streptomycin. The cells were maintained at
37°C in a humidified incubator containing 5% CO2. The
cells were treated with varying concentrations of EGCG dissolved in
serum-free medium.
Cell viability assay
HCCLM6 and HL-7702 cells (6×103
cells/well) were incubated in 96-well plates for 24 h. This was
followed by 24 h incubation of HCCLM6 cells with 0, 5, 10, 20, 30,
40, 60, 80, and 100 μg/ml EGCG and incubation of HL-7702 cells with
0, 5, 10, 20, 40, 60, 80, 100, 140, 180, 220, and 260 μg/ml EGCG.
At the end of the treatment, the culture supernatant was replaced
with fresh complete medium containing 0.5 mg/ml MTT, and the cells
were incubated at 37ºC for 4 h. The medium was discarded and
replaced by dimethyl sulfoxide, and the cells were subjected to 10
min of gentle agitation. Absorbance was subsequently measured at
490 nm. The assay was repeated three times. The growth inhibition
rates of the cells were calculated according to the formula:
Inhibition rate (%) = (1 - mean absorbance of treated group/mean
absorbance of untreated group) × 100%.
Characterization of nuclear
morphology
A nuclear DAPI dihydrochloride-staining assay was
conducted to detect apoptosis. HCCLM6 and HL-7702 cells
(1×105 cells/ml) were seeded onto 1-cm plates and
incubated at 37°C overnight. The cells were treated with 0, 10 and
30 μg/ml EGCG and incubated for an additional 24 h. The cells were
washed once with phosphate-buffered saline (PBS), fixed with 4%
paraformaldehyde for 15 min, washed with PBS and air-dried. The
cells were stained with DAPI at room temperature for 20 min, washed
with PBS and covered with Anti-Fade Fluoromount-G (Beyotime,
Shanghai, China). Stained cells were observed and images captured
at magnification, ×400, with a fluorescent microscope (Leica, ELS,
Wetzlar, Germany) equipped with a Nikon camera (Nikon, Tokyo,
Japan).
In vitro apoptosis assay
HCCLM6 and HL-7702 cells (3×105
cells/well) were seeded into 6-well plates. At 80% confluency, the
cells were treated with 0, 10, and 30 μg/ml EGCG and incubated for
24 h. The cells were harvested, washed with PBS and stained with
the FITC-conjugated anti-Annexin V and PI Dye Apoptosis Detection
kit according to the manufacturer’s protocol. The apoptotic stage
of the cells was analyzed using a flow cytometer (BD Biosciences,
Franklin Lakes, NJ, USA).
Cell cycle analysis
HCCLM6 and HL-7702 cells were seeded into 6-well
plates (6×105 cells/well). At 80% confluency, the cells
were treated with EGCG (0, 10 and 30 μg/ml) and incubated for 24 h.
The cells were harvested, washed with PBS and fixed in a 70%
methanol solution at 4°C for 24 h. Following fixation, the cells
were washed with PBS and stained using the Cell Cycle Detection kit
according to the manufacturer’s instructions. The cell cycle
distribution was analyzed using a flow cytometer (BD
Biosciences).
Mitochondrial membrane potential (MMP)
determination
HCCLM6 and HL-7702 cells were seeded into 6-well
plates (6×105 cells/well). At 80% confluency, the cells
were treated with EGCG (0, 10 and 30 μg/ml) and incubated for 24 h.
Cells were harvested, washed with PBS and stained with the JC-1
Mitochondrial Membrane Potential Detection kit according to the
manufacturer’s instructions. The MMP was analyzed using a flow
cytometer (BD Biosciences).
Western blot analysis
HCCLM6 cells (1×107) were seeded into a
T-25 culture flask and incubated until 80% confluency was achieved.
The cells were treated with 0, 10 and 30 μg/ml EGCG and incubated
for 24 h. Total cell protein was extracted from the treated cells
and quantified with the BCA Protein Assay kit. Bovine serum albumin
was used as the standard. Protein (80 μg) was separated on a 12%
SDS-PAGE gel and transferred to nitrocellulose membranes with a
semi-dry transfer unit (Jim-X Biotechnology, Co., Ltd., Dalian,
China). The membranes were treated with a 5% skimmed dry
milk-Tris-buffered saline with Tween 20 (TBST) solution at 37°C for
2 h. Antibodies against p53, NF-κB, caspase-3, caspase-9, Bcl-2,
Bax, cytochrome c, COX IV and β-actin in 5% skimmed dry
milk-TBST solution were employed to probe their respective
proteins. Incubation was performed at 4°C overnight. Membranes were
washed with TBST (3×10 min washes) and incubated with
HRP-conjugated secondary antibodies (1:1,000) of the appropriate
species (mouse or rabbit) at room temperature for 2 h. Following
thorough washing of membranes, immunoreactive bands were visualized
using the SuperSignal® West Pico Trial kit and detected
with the ECL Gel Documentation and Analysis System (Bio-Rad,
Hercules, CA, USA).
Statistical analysis
The data from all the experiments were expressed as
the mean ± standard deviation. Statistical analyses were performed
with SPSS 13.0 software (SPSS Inc., Chicago, IL, USA). P<0.05
was considered to indicate a statistically significant
difference.
Results
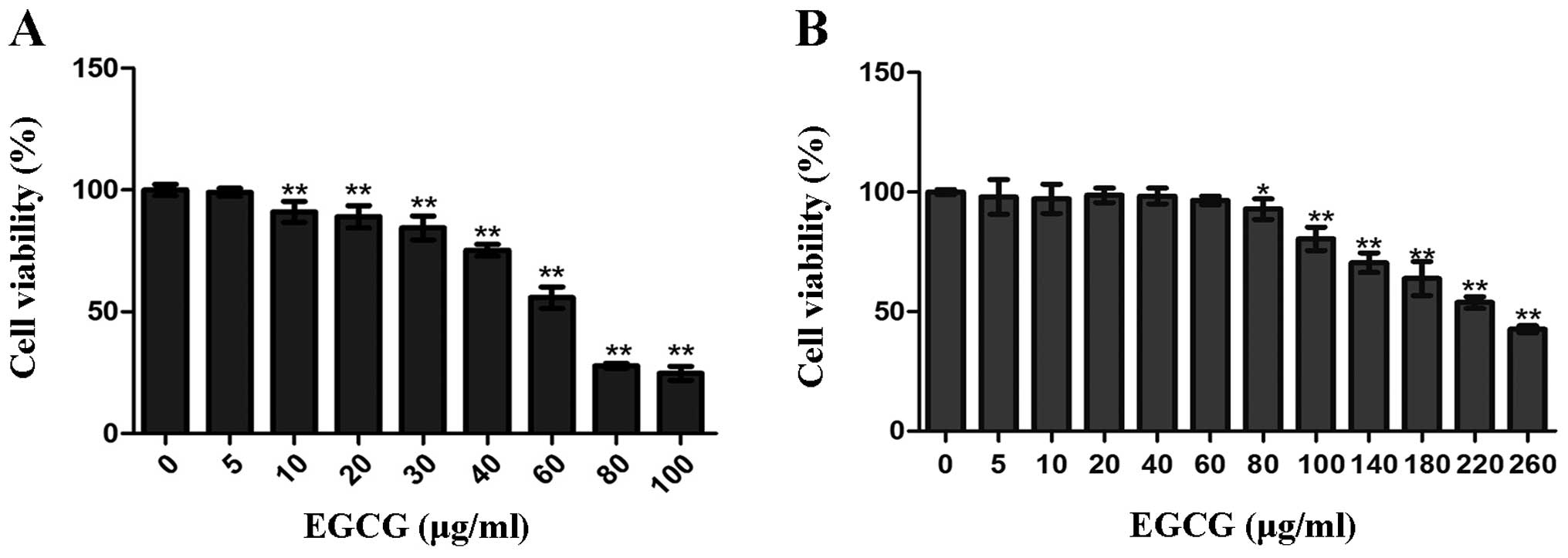
Effects of EGCG on cell viability
To study the growth inhibitory effect of EGCG,
HCCLM6 cells and HL-7702 liver cells were treated with 5–100 μg/ml
and 5–260 μg/ml EGCG, respectively (Fig. 1). EGCG significantly inhibited the
growth of HCCLM6 cells in a dose-dependent manner at concentrations
between 10 and 100 μg/ml (P<0.01; Fig. 1A). The IC50 for HCCLM6
cells was 62 μg/ml. By contrast, a much higher minimum dose of EGCG
(80 μg/ml) was required to significantly inhibit the growth of
HL-7702 liver cells (P<0.01; Fig.
1B), and the IC50 was 227 μg/ml.
Cell death assessment
To assess the type of cell death induced by EGCG,
the nuclei of EGCG-treated (0, 10 or 30 μg/ml) and untreated cells
were stained with DAPI. The nuclei of EGCG-treated non-viable
HCCLM6 cells were predominantly condensed with nuclear
morphological changes and DNA fragmentation (Fig. 2A-b and c) compared to untreated
cells (Fig. 2A-a). This indicates
that these cells underwent apoptotic-like cell death. Notably,
HL-7702 cells that received a similar treatment were round in shape
and had stable nucleus morphology with intact DNA integrity
(Fig. 2B-a to c). This indicates
that the HL-7702 cells did not undergo apoptotic-like cell
death.
Effect of EGCG on cell apoptotic
rate
To confirm the type of cell death induced by EGCG in
HCCLM6 cells and to further evaluate the effects of EGCG on HL-7702
cells, a double-staining assay was conducted with Annexin V-FITC
and PI. Annexin V-positive and PI-negative (Fig. 3, lower right quadrant) cell
staining indicated early apoptosis. The cells that were strongly
positive for Annexin V and PI (upper right quadrant) underwent late
apoptosis/necrosis (23). EGCG
treatment significantly increased the percentage of apoptotic
HCCLM6 cells in a dose-dependent manner compared with untreated
cells (P<0.05; Fig. 3A and B).
HCCLM6 cells exposed to 10 μg/ml EGCG exhibited significant early
apoptosis (P<0.01; Fig. 3B).
Significant late apoptosis (P<0.05; Fig. 3B) occurred only at 30 μg/ml EGCG.
By contrast, neither 10 nor 30 μg/ml EGCG caused significant early
or late apoptosis in HL-7702 cells compared to untreated cells
(Fig. 3C and D).
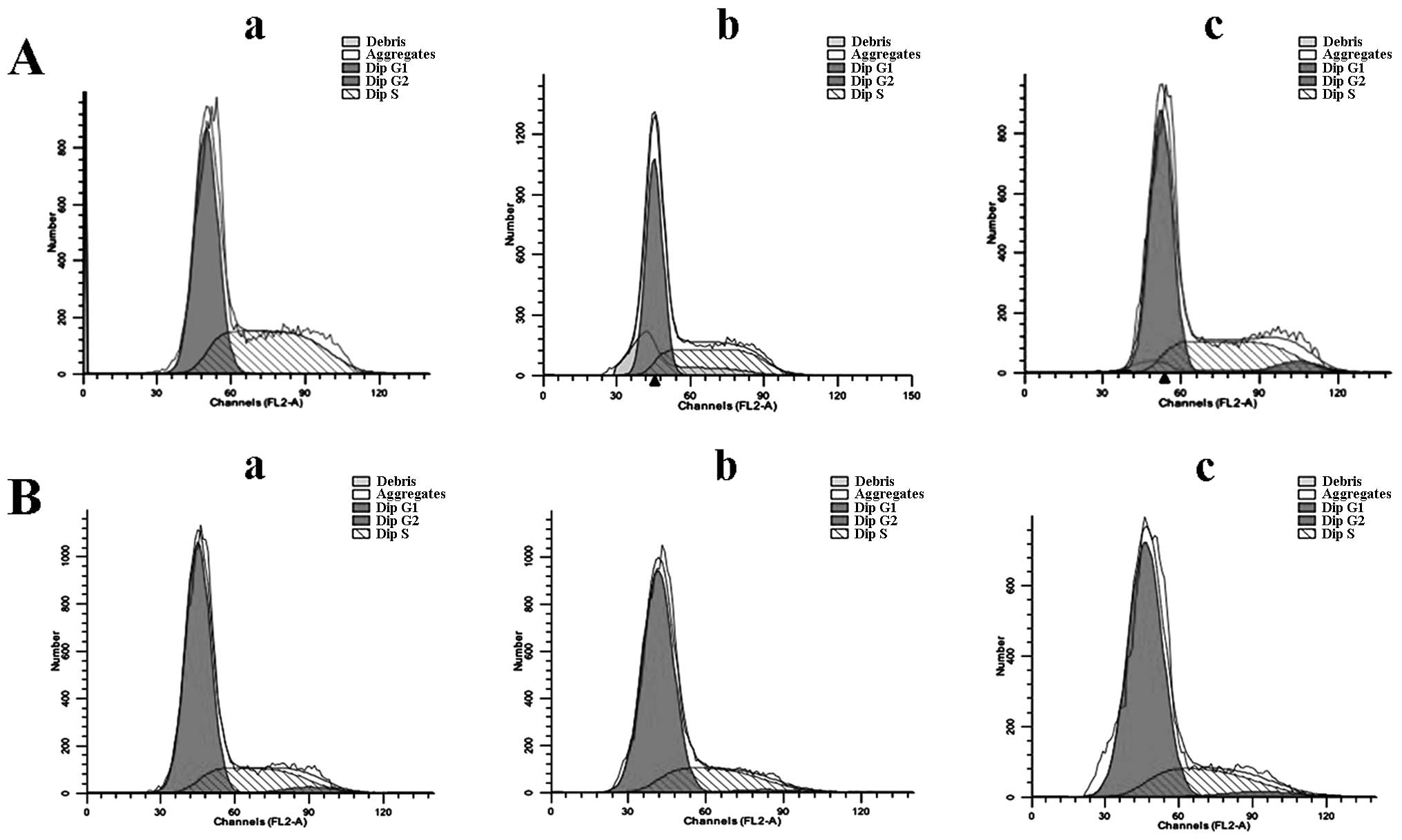
Effect of EGCG on the cell cycle
HCCLM6 cells treated with 30 μg/ml EGCG exhibited
significant G0/G1 phase arrest compared to
untreated cells (Fig. 4 and
Table I). HL-7702 cells, however,
did not exhibit phase arrest upon EGCG treatment (Fig. 4B and Table I). This indicates that EGCG is
able to selectively induce cell cycle phase arrest in cancerous
HCCLM6 cells but spares non-cancerous cells under similar treatment
conditions.
 | Table ISummary of the cell cycle phase
distribution for epigallocatechin-3-gallate (EGCG)-treated and
untreated HCCLM6 and HL-7702 cells. |
Table I
Summary of the cell cycle phase
distribution for epigallocatechin-3-gallate (EGCG)-treated and
untreated HCCLM6 and HL-7702 cells.
| EGCG, μg/ml | Phase
distribution |
|---|
|
|---|
|
G0/G1 |
G2/M | S |
|---|
| HCCLM6 cells |
| 0 | 55.58±3.34 | 0.0067±0.0058 | 44.41±3.34 |
| 10 | 60.50±0.38 | 1.66±1.22 | 37.84±0.92 |
| 30 | 63.15±4.92a | 6.41±1.25b | 30.11±6.63b |
| HL-7702 cells |
| 0 | 73.20±2.58 | 2.66±1.12 | 24.15±1.46 |
| 10 | 72.90±3.81 | 3.01±2.45 | 23.13±0.00 |
| 30 | 73.80±0.71 | 3.72±0.71 | 21.47±1.41 |
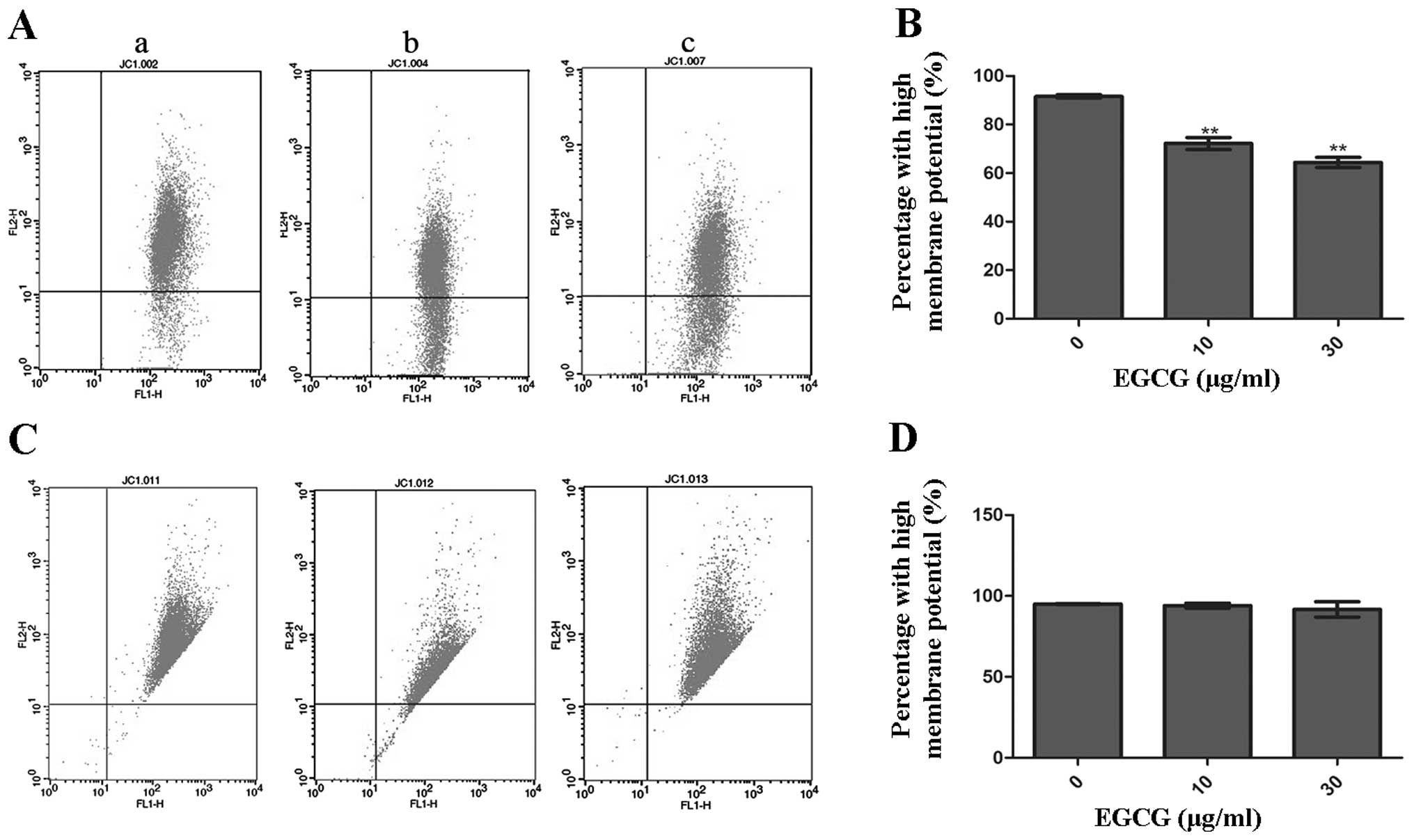
Measurement of ΔΨm in HCCLM6 and HL-7702
cells
MMP was measured using the mitochondria-specific
lipophilic cationic fluorescent dye, JC-1. As a monomer, JC-1 is
capable of selectively entering mitochondria. Under normal
conditions, JC-1 aggregates within mitochondria and emits red
fluorescence. However, when the MMP collapses during apoptosis,
JC-1 emits green fluorescence. The dissipation of ΔΨm was measured
as an increase in green fluorescence.
Fig. 5A and C show
the analysis of MMP in HCCLM6 and HL-7702 cells that were incubated
with or without EGCG. Following 10 and 30 μg/ml EGCG treatment,
HCCLM6 cells showed a significantly lower ratio of JC-1 aggregation
in mitochondria compared to untreated cells (Fig. 5B). However, there was no
significant change in the ΔΨm of HL-7702 cells, as indicated by the
fairly constant JC-1 aggregation in the mitochondria of
EGCG-treated and untreated cells (Fig. 5D).
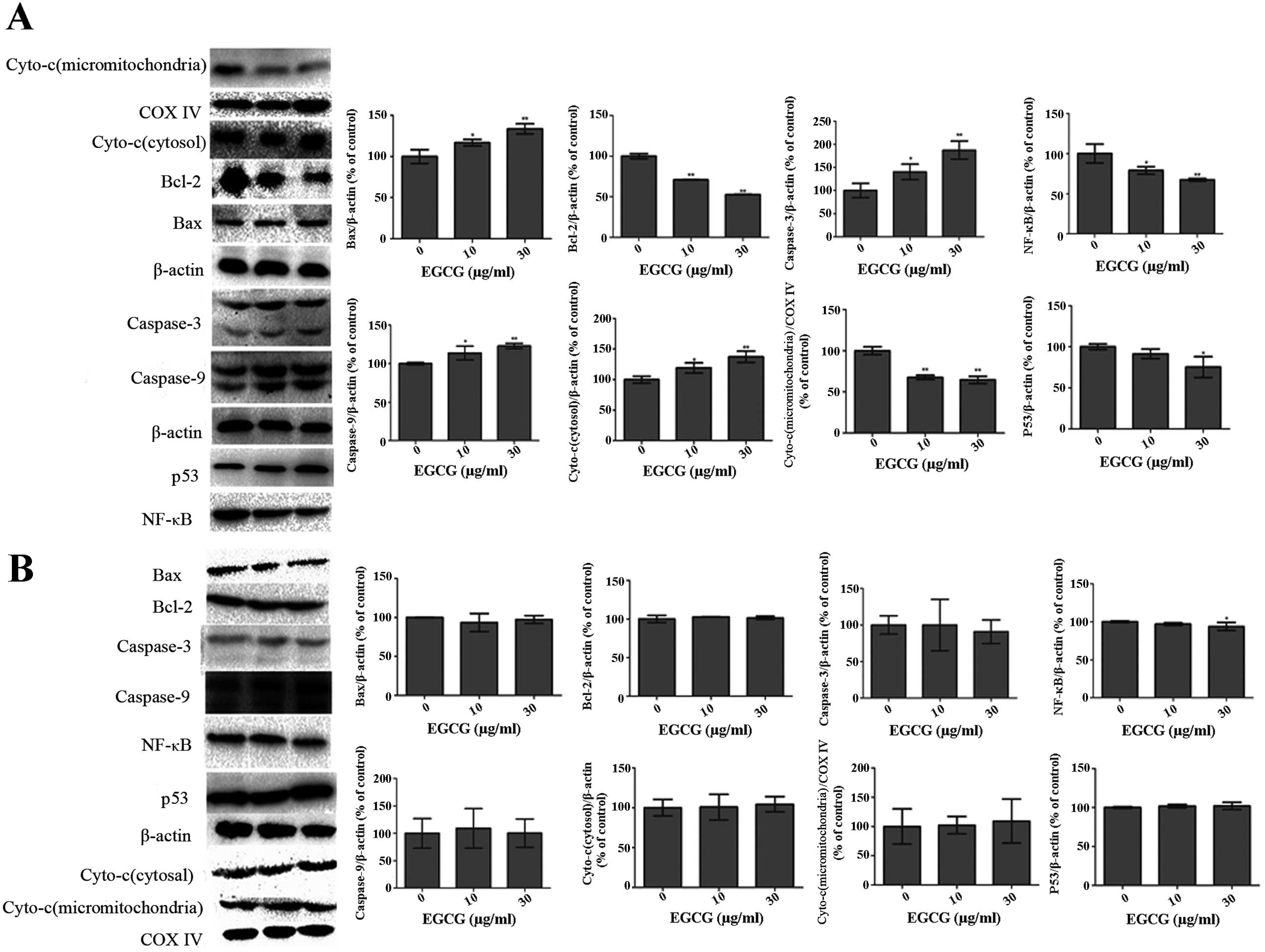
Effect of EGCG on apoptotic proteins
Western blot analysis showed significantly lower
expression of Bcl-2 and NF-κB (Fig.
6A; P<0.01) in EGCG-treated HCCLM6 cells. Conversely, the
expressions of Bax, p53, caspase-3 and caspase-9 were significantly
increased (P<0.05) in a dose-dependent manner (Fig. 6A). EGCG treatment significantly
reduced the expression of cytochrome c in mitochondria but
increased its expression in the cytosol of HCCLM6 cells (Fig. 6A). As shown in Fig. 6B, EGCG treatment did not
significantly affect the expression of any of these proteins in
HL-7702 cells, even at high EGCG concentrations.
Discussion
Apoptosis, a physiological death mechanism that
preserves homeostasis, is the most common form of eukaryotic cell
death (21). There are two major
apoptotic pathways: The receptor-mediated (extrinsic) pathway and
the mitochondrial-mediated (intrinsic) pathway (22). In the present study, the
anticancer activity of EGCG was investigated. EGCG-induced cell
death was observed to be apoptotic-like and characterized by
cellular shrinkage, DNA fragmentation and the reduction of MMP
(Figs. 1, 2 and 5). EGCG induced apoptosis of malignant
HCCLM6 cells without inducing a similar effect on non-cancerous
HL-7702 cells that received similar treatment (Figs. 3 and 4).
Apoptotic stimuli resulted in the loss of MMP and
the release of cytochrome c from the mitochondrial inner
membrane space into the cytosol (23–26). The released cytochrome c, a
potent caspase-activating protein, initiated the caspase-dependent
apoptotic cascade. EGCG treatment caused a significant increase in
the cytochrome c levels in the cytosol and a concomitant
reduction in the levels in the mitochondria of the HCCLM6 cells
(Fig. 6A). The significantly
increased expression of caspase-3 and -9 (Fig. 6) may be the direct result of the
increased cytosolic cytochrome c levels induced by EGCG in
HCCLM6 cells but not in HL-7702 cells (Fig. 6A and B). Thus, apoptosis is
increased in HCCLM6 cells.
Bax is present in its inactive form in the cytosol
or loosely attached to intracellular membranes as a monomer. In
apoptotic cells, activated Bax is translocated and integrated into
mitochondrial membranes (27,28). This event causes membrane
permeabilization and release of cytochrome c from the
mitochondria. Cytochrome c is subsequently complexed with
apoptotic protease activating factor-1 and procaspase-9 to form the
apoptosome. Cytochrome c recruits procaspase-3, which is
cleaved and activated by caspase-9 to induce apoptosis. The ratio
of Bax/Bcl-2 is critical for cell survival such that an increase in
Bax levels could shift the ratio in favor of apoptosis. EGCG
induced an increase in Bax expression but a reduction in Bcl-2 (a
major anti-apoptotic protein) expression in HCCLM6 cells. Thus, a
higher Bax/Bcl-2 ratio favored the apoptosis of these cells
compared to non-cancerous HL-7702 cells (Fig. 6A and B).
EGCG was found to effectively decrease NF-κB
translational activity in human HCCLM6 cells (Fig. 6A). The NF-κB protein stimulates
cell survival and promotes cell proliferation, and increased NF-κB
activity is associated with numerous cancers, including HCC. A
previous study also indicates that NF-κB activates the expression
of the anti-apoptotic protein Bcl-2 (29).
The increase in p53 expression in EGCG-treated
HCCLM6 cells (Fig. 6A) was
associated with increased cytosolic cytochrome c and Bax
expression. The p53 tumor suppressor gene is critically involved in
cell cycle regulation, DNA repair and programmed cell death. This
gene stimulates the release of cytochrome c by
transcriptionally increasing cytosolic Bax levels in certain human
cell lines (29,30). Ahmed et al (31) reported in 2000 that EGCG induces
G0/G1 phase cell cycle arrest, which is an
irreversible process that ultimately leads to apoptotic death of
human carcinoma cells. Cell cycle perturbations are an important
component of the cellular response to DNA damage (32,33). In addition, p53 plays an important
role in controlling the G1/S checkpoint. Some cells
undergo cell cycle arrest in the G1 phase in response to
DNA damage (34). The present
study results indicate that EGCG-induced
G0/G1 cell cycle phase arrest of HCCLM6 cells
occurs via increased expression of p53, which eventually results in
apoptotic cell death.
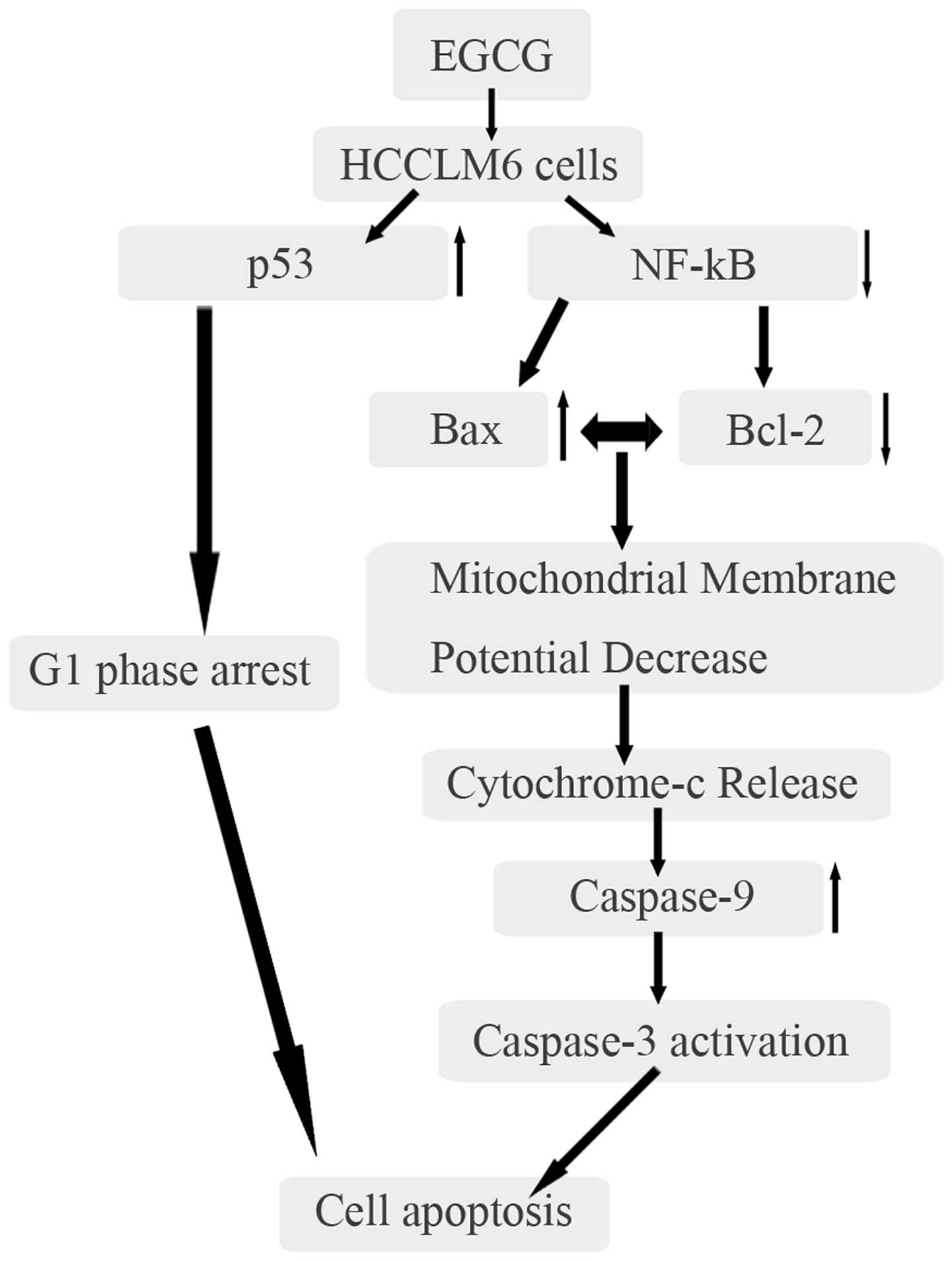
The data indicated that EGCG treatment results in
upregulation of Bax via p53 and downregulation of Bcl-2 via NF-κB.
The overexpression of Bax and low expression of Bcl-2 eventually
led to mitochondrial membrane disintegration and the release of
cytochrome c. During apoptosis, cytochrome c release
caused the activation of caspase-9 through the formation of the
multimeric apoptosome complex. Caspase-9 activated caspase-3, which
resulted in apoptosis (Fig. 7).
Additionally, EGCG caused G0/G1 phase arrest
of the HCCLM6 cell cycle via increased expression of p53.
In conclusion, EGCG affects HCCLM6 cells by inducing
apoptosis, predominantly through the mitochondria-mediated
apoptosis pathway. Although the concentrations of EGCG used in the
present study are higher than those achievable by drinking tea or
through EGCG oral administration (35,36), higher plasma and/or organ-specific
EGCG bioavailability is achievable through intravenous
administration (37). The present
study indicates that EGCG is an anticancer agent in the treatment
of HCC. EGCG selectively induced apoptosis in cancerous cells with
a minimal effect on non-cancerous cells. However, the present study
only approximately assessed the role of mitochondrial dysfunction
in EGCG-induced apoptosis. Therefore, further studies are required
to investigate other pathways that may induce apoptosis in
EGCG-treated cells, the effect of EGCG on diseased and healthy
tissues in vivo and the relevant mechanisms for maintaining
cell viability of non-tumor cells.
References
|
1
|
El-Serag HB and Mason AC: Rising incidence
of hepatocellular carcinoma in the United States. N Engl J Med.
340:745–750. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Parkin DM, Bray F, Ferlay J and Pisani P:
Estimating the world cancer burden: Globocan 2000. Int J Cancer.
94:153–156. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
lkai I, Yamaoka Y, Yamamoto Y, Ozaki N,
Sakai Y, Satoh S, Shinkura N and Yamamoto M: Surgical intervention
for patients with stage IV-A hepatocellular carcinoma without lymph
node metastasis: proposal as a standard therapy. Ann Surg.
227:433–439. 1998. View Article : Google Scholar
|
|
4
|
Lee JS, Chu IS, Heo J, Calvisi DF, Sun Z,
Roskams T, Durnez A, Demetris AJ and Thorgeirsson SS:
Classification and prediction of survival in hepatocellular
carcinoma by gene expression profiling. Hepatology. 40:667–676.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Song HY, Liu YK, Feng JT, Cui JF, Dai Z,
Zhang LJ, Feng JX, Shen HL and Tang ZY: Proteomic analysis on
metastasis-associated proteins of human hepatocellular carcinoma
tissues. J Cancer Res Clin Oncol. 132:92–98. 2006. View Article : Google Scholar
|
|
6
|
Yang CS and Wang ZY: Tea and cancer. J
Natl Cancer Inst. 85:1038–1049. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Balentine DA, Wiseman SA and Bouwens LC:
The chemistry of tea flavonoids. Crit Rev Food Sci Nutr.
37:693–704. 1997. View Article : Google Scholar
|
|
8
|
Mukhtar H and Ahmad N: Green tea in
chemoprevention of cancer. Toxicol Sci. 52(Suppl 2): 111–117. 1999.
View Article : Google Scholar
|
|
9
|
Singh BN, Shankar S and Srivastava RK:
Green tea catechin, epigallocatechin-3-gallate (EGCG): mechanisms,
perspectives and clinical applications. Biochem Pharmacol.
82:1807–1821. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Thawonsuwan J, Kiron V, Satoh S, Panigrahi
A and Verlhac V: Epigallocatechin-3-gallate (EGCG) affects the
antioxidant and immune defense of the rainbow trout, Oncorhynchus
mykiss. Fish Physiol Biochem. 36:687–697. 2010. View Article : Google Scholar
|
|
11
|
Mukhtar H and Ahmad N: Tea polyphenols:
prevention of cancer and optimizing health. Am J Clin Nutr.
71(Suppl 6): 1698S–1702S. 2000.PubMed/NCBI
|
|
12
|
Thangapazham RL, Singh AK, Sharma A,
Warren J, Gaddipati J and Maheshwari RK: Green tea polyphenols and
its constituent epigallocatechin gallate inhibits proliferation of
human breast cancer cells in vitro and in vivo. Cancer Lett.
245:232–241. 2007. View Article : Google Scholar
|
|
13
|
Stuart EC, Scandlyn MJ and Rosengren RJ:
Role of epigallocatechin gallate (EGCG) in the treatment of breast
and prostate cancer. Life Sci. 79:2329–2336. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ahmad N, Feyes DK, Nieminen AL, Agarwal R
and Mukhtar H: Green tea constituent epigallocatechin-3-gallate and
induction of apoptosis and cell cycle arrest in human carcinoma
cells. J Natl Cancer Inst. 89:1881–1886. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Masuda M, Suzui M and Weinstein IB:
Effects of epigallocatechin-3-gallate on growth, epidermal growth
factor receptor signaling pathways, gene expression, and
chemosensitivity in human head and neck squamous cell carcinoma
cell lines. Clin Cancer Res. 7:4220–4229. 2001.PubMed/NCBI
|
|
16
|
Nihal M, Ahmad N, Mukhtar H and Wood GS:
Anti-proliferative and proapoptotic effects of
(−)-epigallocatechin-3-gallate on human melanoma: possible
implications for the chemoprevention of melanoma. Int J Cancer.
114:513–521. 2005. View Article : Google Scholar
|
|
17
|
Gupta S, Hussain T and Mukhtar H:
Molecular pathway for (−)-epigallocatechin-3-gallate-induced cell
cycle arrest and apoptosis of human prostate carcinoma cells. Arch
Biochem Biophys. 410:177–185. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wei DZ, Yang JY, Liu JW and Tong WY:
Inhibition of liver cancer cell proliferation and migration by a
combination of (−)-epigallocatechin-3-gallate and ascorbic acid. J
Chemother. 15:591–595. 2003. View Article : Google Scholar
|
|
19
|
Uesato S, Kitagawa Y, Kamishimoto M,
Kumagai A, Hori H and Nagasawa H: Inhibition of green tea catechins
against the growth of cancerous human colon and hepatic epithelial
cells. Cancer Lett. 170:41–44. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nishikawa T, Nakajima T, Moriguchi M, Jo
M, Sekoguchi S, Ishii M, et al: A green tea polyphenol,
epigalocatechin-3-gallate, induces apoptosis of human
hepatocellular carcinoma, possibly through inhibition of Bcl-2
family proteins. J Hepatol. 44:1074–1082. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wyllie AH, Kerr JF and Currie AR: Cell
death: the significance of apoptosis. Int Rev Cytol. 68:251–306.
1980. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Soldatenkov VA and Smulson M: Poly
(ADP-ribose) polymerase in DNA damage-response pathway:
implications for radiation oncology. Int J Cancer. 90:59–67. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wei MC, Zong WX, Cheng EH, Lindsten T,
Panoutsakopoulou V, Ross AJ, et al: Proapoptotic BAX and BAK: a
requisite gateway to mitochondrial dysfunction and death. Science.
292:727–730. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Green DR and Reed JC: Mitochondria and
apoptosis. Science. 281:1309–1312. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Marsden VS, O’Connor L, O’Reilly LA, Silke
J, Metcalf D, Ekert PG, et al: Apoptosis initiated by
Bcl-2-regulated caspase activation independently of the cytochrome
c/Apaf-1/caspase-9 apoptosome. Nature. 419:634–637. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tsujimoto Y: Cell death regulation by the
Bcl-2 protein family in the mitochondria. J Cell Physiol.
195:158–167. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kowaltowski AJ, Vercesi AE and Fiskum G:
Bcl-2 prevents mitochondrial permeability transition and cytochrome
c release via maintenance of reduced pyridine nucleotides. Cell
Death Differ. 7:903–910. 2000. View Article : Google Scholar
|
|
28
|
Reed JC: Proapoptotic multidomain
Bcl-2/Bax-family proteins: mechanisms, physiological roles, and
therapeutic opportunities. Cell Death Differ. 13:1378–1386. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Schuler M, Bossy-Wetzel E, Goldstein JC,
Fitzgerald P and Green DR: p53 induces apoptosis by caspase
activation through mitochondrial cytochrome c release. J Biol Chem.
275:7337–7342. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Miyashita T and Reed JC: Tumor suppressor
p53 is a direct transcriptional activator of the human bax gene.
Cell. 80:293–299. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ahmad N, Cheng P and Mukhtar H: Cell cycle
dysregulation by green tea polyphenol epigallocatechin-3-gallate.
Biochem Biophys Res Commun. 275:328–334. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Weinert TA and Hartwell LH: The RAD9 gene
controls the cell cycle response to DNA damage in Saccharomyces
cerevisiae. Science. 241:317–322. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Smerdon MJ, Kastan MB and Lieberman MW:
Distribution of repair-incorporated nucleotides and nucleosome
rearrangement in the chromatin of normal and xeroderma pigmentosum
human fibroblasts. Biochemistry. 18:3732–3739. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kastan MB, Canman CE and Christopher J:
p53, cell cycle control and apoptosis: implications for cancer.
Cancer Metastasis Rev. 14:3–15. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chow HH, Cai Y, Hakim IA, Crowell JA,
Shahi F, Brooks CA, Dorr RT, Hara Y and Alberts DS:
Pharmacokinetics and safety of green tea polyphenols after
multiple-dose administration of epigallocatechin gallate and
polyphenon E in healthy individuals. Clin Cancer Res. 9:3312–3319.
2003.PubMed/NCBI
|
|
36
|
Chow HH, Cai Y, Alberts DS, et al: Phase I
pharmacokinetic study of tea polyphenols following single-dose
administration of epigallocatechin gallate and polyphenon E. Cancer
Epidemiol Biomarkers Prev. 10:53–58. 2001.PubMed/NCBI
|
|
37
|
Li QS, Wang CY, Han GZ, Zou LL, Li L and
Li N: Application of LC-MS/MS method in pharmacokinetic study of
multi-components from tea polyphenols in rats. Chin J New Drugs.
20:817–823. 2011.
|