Introduction
The number of patients with diabetes is markedly
increasing worldwide. Diabetes leads to vascular changes and
dysfunction, as well as complications that result in increased
morbidity and mortality (1).
Among diabetic vascular complications, nephropathy contributes to
the development of cardiovascular disease and is the leading cause
of end-stage renal disease in developed countries (2,3).
Therefore, the prevention of renal insufficiency may improve the
prognosis of patients with diabetes.
Numerous factors contribute to the development of
diabetic nephropathy. Hyperglycemia alters both extracellular and
intracellular metabolism, leading to effects such as glomerular
hyperfiltration (4), oxidative
stress (5), the accumulation of
advanced glycation end products (AGEs) (6), the activation of protein kinase C
(7), abnormal polyol metabolism
(8), the overexpression of
transforming growth factor-β (TGF-β) (9) and inflammation. These effects have
been recognized as classical characteristics of the pathogenesis of
diabetic nephropathy. Additionally, intracellular stress associated
with renal hypoxia (10,11), reactive oxygen species (ROS)
production by mitochondria (12–15) and endoplasmic reticulum (ER)
stress (16–18) have recently been proposed as key
mechanisms underlying the pathogenesis of diabetic nephropathy.
Thus, the maintenance of cellular homeostasis against stress
conditions may be a new therapeutic target for the treatment of
diabetic nephropathy.
Autophagy is a tightly regulated process in which
endogenous cellular proteins aggregate and damaged organelles are
degraded by the lysosomal pathway. This process functions to
maintain intracellular homeostasis and cell integrity. It has
recently been highlighted as it can be stimulated by multiple types
of cellular stressors, including starvation, hypoxia and ER stress.
Emerging evidence also indicates that autophagy plays a critical
role in several organs, particularly in highly metabolic organs,
and that its alteration is involved in the pathogenesis of
metabolic disease, immunity and autoimmunity, inflammation,
development, aging and cancer (19,20). It has been suggested that
autophagy is enhanced and plays a protective role during kidney
disease (21). Its renoprotective
role in several animal models, including those used for aging and
acute kidney injury, has also been demonstrated (22–24).
One possible mechanism through which autophagy
protects cells is that it may eliminate damaged mitochondria.
Autophagy involves the sequestration of proteins and cellular
organelles into autophagosomes, which directs them to lysosomes
(25). The formation of
autophagosomes is dependent on the induction of several genes,
including microtubule-associated protein 1 light chain 3 (LC3),
Beclin1 and autophagy-related genes (Atgs) (26). Nonetheless, autophagy may also
represent a form of programmed cell death known as autophagic cell
death or type II programmed cell death, and altered autophagy is
associated with the loss of renal tubular epithelial cell mass in
diabetes. The role of autophagy in diabetic nephropathy remains a
largely undetermined; thus, its underlying mechanisms are presently
unclear.
Glucagon-like peptide-1 (GLP-1) is a gut incretin
hormone and is currently considered an attractive agent for the
treatment of type 2 diabetes. It has been shown to exert various
beneficial effects on pancreatic β-cells, such as the enhancement
of glucose-dependent insulin secretion (27), the acceleration of β-cell
proliferation and the inhibition of β-cell apoptosis (28). In the gut and hypothalamus, GLP-1
has been shown to inhibit motility, gastric emptying (29) and the central regulation of
feeding (30), resulting in the
loss of body weight (27). It has
previously been demonstrated that GLP-1 receptor (GLP-1R) is
produced not only in the pancreas, gut and hypothalamus, but also
in the kidneys (31). Liraglutide
is a human incretin-GLP-1 analogue with a high degree of homology
to the native hormone. It shares 97% sequence identity with native
human GLP-1. A recent study demonstrated that liraglutide inhibited
cytokine-induced apoptosis in primary rat islet cells in a
dose-dependent manner and reduced free fatty acid-induced apoptosis
by approximately 50% (32).
In the present study, we evaluated the effects of
high glucose concentrations on the induction of autophagy in the
human renal tubular epithelial cell line, HK-2 cells, as well as in
the kidneys of diabetic rats. We also investigated the ability of
GLP-1 to protect HK-2 cells and diabetic rat kidneys against
apoptosis induced by high glucose levels by targeting autophagy. As
shown by our results, autophagy plays a potential role in
nephropathy, which affects cell viability through the GLP-1
receptor.
Materials and methods
Cell culture
The human renal tubular epithelial cell line (HK-2)
was purchased from the American Type Culture Collection (ATCC,
Manassas, VA, USA) and was cultured in Dulbecco’s modified Eagle’s
medium (DMEM) supplemented with 5.5 mM D-glucose [normal glucose
(N)], 10% fetal bovine serum (FBS), epidermal growth factor, 100
U/ml penicillin, and 100 μg/ml streptomycin (all from
HyClone, Logan, UT, USA) in a 5% CO2 incubator at
37°C.
3-(4,5-Dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
(MTS) assay
The HK-2 cells were plated on 96-well plates at a
density of 1.2×105 cells/cm2 in medium
containing either 5.5, 16.7, 25 or 40 mM glucose [high glucose
(HG)] and incubated for up to 72 h. Liraglutide (LG) was
administrated at various concentrations (1, 10 and 100 nM) or HG
with LG with glucagon-like peptide-1 receptor (GLP-1R) antagonist
{exendin-(9-39) [EX-(9-39)]; 1,000 nM}. At 24, 48 and 72 h, cell
viability was assessed by the ability of metabolically active cells
to reduce the tetrazolium salt to formazan compounds using MTS
reagent (Promega, Madison, WI, USA). The absorbance of the samples
was measured using a microplate reader at a 450-nm wavelength after
3 h of incubation with MTS solution (0.19 mg/ml). The results are
expressed as the means ± standard error of the mean (SEM) and are
representative of 3 independent experiments.
Trypan blue exclusion assay
The cells were plated on 12-well plates at a density
of 1.2×105 cells/cm2 in medium containing
either 5.5 or 40 mM glucose, collected and stained with trypan blue
(Invitrogen, Carlsbad, CA, USA). The total number of total cells,
as well as the number of trypan blue-stained cells were counted
using a hemocytometer (Cat. no. 02-671-6; Hausser Scientific,
Horsham, PA, USA).
Western blot analysis
The cells were harvested and lysed in RIPA buffer
(150 mM sodium chloride, 1.0% Triton X-100, 0.5% sodium
deoxycholate, 0.1% sodium dodecyl sulfate, 50 mM Tris, pH 8.0) with
protease and phosphatase inhibitors, and centrifuged at 12,000 rpm
for 20 min at 4°C. Protein samples were then mixed with loading
buffer and boiled at 95–100°C for 5 min. Total protein extracts
were subjected to 12% SDS-PAGE. The separated proteins were
transferred ono polyvinylidene fluoride membranes (Bio-Rad,
Hercules, CA, USA) by electrotransfer. The blots were subsequently
blocked with 5% (v/v) skimmed milk (Nacalai Tesque, Kyoto, Japan)
and incubated with GLP-1R rabbit antibody (ab39072, 1:1,000; Abcam,
Cambridge, MA, USA), LC3 rabbit antibody (12741S, 1:1,000) and
Beclin1 rabbit antibody (3495S, 1:1,000) (both from Cell Signaling
Technology, Danvers, MA, USA) for 12 h at 4°C, or with
glyceraldehyde 3-phosphate dehydrogenase (GAPDH) antibody (G8795,
1:1,000; Sigma-Aldrich; St. Louis, MO, USA) for 1 h at room
temperature. The membranes were incubated with horseradish
peroxidase-linked, goat anti-rabbit (sc-2054) or anti-mouse IgG
(sc-2055, 1:5,000; both from Santa Cruz Biotechnology, Inc., Santa
Cruz, CA, USA) at room temperature for 2 h. The blots were then
visualized using a western blotting detection system (ECL plus; GE
Healthcare, Princeton, NJ, USA).
Reverse transcription-quantitative
(real-time) polymerase chain reaction (RT-qPCR)
Total RNA was extracted from each sample using the
Total RNA kit I (Omega Bio-Tek, Norcross, GA, USA). cDNA was
synthesized from the individual samples of 1 μg of total RNA
using the PrimeScript® RT reagent kit (Takara Bio, Inc.,
Tokyo, Japan). Following the addition of each set of primers (final
concentration, 0.4 μmol/l) and template DNA to the master
mix, quantitative (real-time) PCR was performed using a LightCycler
(Roche Diagnostics, Tokyo, Japan) and SYBR Premix Ex Taq (Takara
Bio, Inc.). The PCR protocol was as follows: initial denaturation
(95°C for 30 sec) followed by 40 cycles of denaturation (95°C for
30 sec, 60°C for 30 sec and 72°C for 30 sec) and annealing and
extension (72°C for 1 min). The specific oligonucleotide primers
were designed by Takara Bio Inc., and the primer sequences were as
follows: GLP-1R sense, TCAAGGTCAACGGCTTATTAGTGAA and
antisense, CCCAAGTGATGCAAGCAGAG; GAPDH sense, GCAC
CGTCAAGGCTGAGAAC and antisense, TGGTGAAGACGCCAGTGGA. To visualize
gene expression, individual DNA fragments were electrophoresed on a
2% (w/v) agarose gel (Sigma-Aldrich) and treated with ethidium
bromide. cDNA of the human pancreas (Takara Bio, Inc.) was used as
a positive control.
Electron microscopy
The HK-2 cells were plated in 0.01% poly-L-lysine
(Sigma-Aldrich)-coated glass slides at a density of
1.5×104 cells/slide (area, 1.8 cm2), cultured
for 1 day, and finally treated for 72 h in medium containing either
a 5.5 or 40 mM glucose concentration. The cells were fixed in 2.5%
glutaraldehyde (Electron Microscopy Sciences, Hatfield, PA, USA)
for 2 h and dissolved in 0.1 M phosphate buffer (PB; pH 7.4). The
HK-2 cells were then post-fixed for 1 h in 1% osmium tetroxide in
PB and then stained with 70% ethanol containing 1% uranyl acetate
(Sigma-Aldrich). The HK-2 cells were then dehydrated in a graded
alcohol series and embedded in epon (Sigma-Aldrich). Ultrathin
sections (with silver to gray interference) were cut using a
diamond knife (Diatome, Biel, Switzerland), mounted on
Formvar-coated single-slot grids and then counterstained with 3%
uranyl acetate and then with 0.2% lead citrate (Sigma-Aldrich). The
sections were visualized under a Philips CM100 transmission
electron microscope (Philips Electron Optics, Hillsboro, OR,
USA).
Animals
The animal care, handling and in vivo studies
complied with the guidelines provided by the Animal Care Committee
of the Fourth Military Medical University. Male Sprague-Dawley rats
(4 weeks old) were divided into the following groups: i)
non-diabetic group (n=10); ii) diabetic group (n=10); iii) diabetic
group treated with liraglutide (n=10); and iv) diabetic group
treated with liraglutide and the GLP-1R antagonist, exendin-(9-39)
(synthesized by Sigma-Aldrich; n=10). At the age of 5 weeks, the
mice in the diabetic groups were administered intravenous
injections of streptozotocin (MP Biomedicals, Santa Ana, CA, USA)
at 60 mg/kg body weight in citrate buffer (pH 4.5). Only the rats
with blood glucose concentrations >16.7 mM at 3 and 7 days
following the injection of streptozotocin were included in the
diabetic groups. The non-diabetic groups were administered
injections of citrate buffer alone.
The groups treated with liraglutide were
administered liraglutide (Novo Nordisk, Copenhagen, Denmark)
subcutaneously at the dose of 0.3 mg/kg/12 h for 5 weeks as
previously described (33).
Twice-daily dosing was used as the pharmacokinetic half-life of
liraglutide is only approximately 4 h in rats. The mice in the
group treated with liraglutide and the GLP-1R antagonist,
exendin-(9-39), were subcutaneously administered liraglutide at the
dose of 0.3 mg/kg/12 h and exendin-(9-39) at the dose of 25
nmol/kg/12 h for 5 weeks, beginning at 1 week after the
streptozotocin or citrate buffer injections. The placebo groups
were administered water alone using the same schedule as for the
liraglutide treatment groups. All rats were allowed free access to
standard food and tap water. All rats were euthanized at 5 weeks
after the induction of diabetes in the diabetic groups, and the
kidneys were weighed and fixed in 10% (v/v) formalin or frozen in
liquid nitrogen.
Assays of metabolic variables
Serum creatinine and blood urea nitrogen levels were
measured using a BioMajesty JCA-BM12 analyzer (Hitachi, Tokyo,
Japan). Body weight was monitored weekly from 4 weeks of age. Food
intake was calculated as an average over a period of 3 days.
Immunohistochemistry
Hematoxylin and eosin (H&E) and immunoperoxidase
staining were performed as previously described (34). The kidneys were fixed in 10%
formaldehyde, and embedded in paraffin. Paraffin sections were cut
at 3 μm and deparaffinized for staining. Briefly, for
immunoperoxidase staining, the primary antibodies used were GLP-1R
rabbit antibody (1:200; Abcam) and LC3 rabbit antibody (1:200; Cell
Signaling Technology), and were applied for 12 h at 4°C. Secondary
antibodies were biotin-labeled anti-rabbit IgG (Santa Cruz
Biotechnology, Inc.), which were applied for 60 min at room
temperature. The sections were counterstained with hematoxylin
before being examined under a light microscope (Olympus, Tokyo,
Japan).
Statistical analysis
Data are presented as means ± SEM. Statistical
analysis was assessed by one-way ANOVA followed by the least
significant difference (LSD) t-test or Tamhane’s T2 for multiple
comparisons. A P-value <0.05 was considered to indicate a
statistically significant difference.
Results
Chronic exposure to high glucose
concentrations decreases cell proliferation and induces
apoptosis
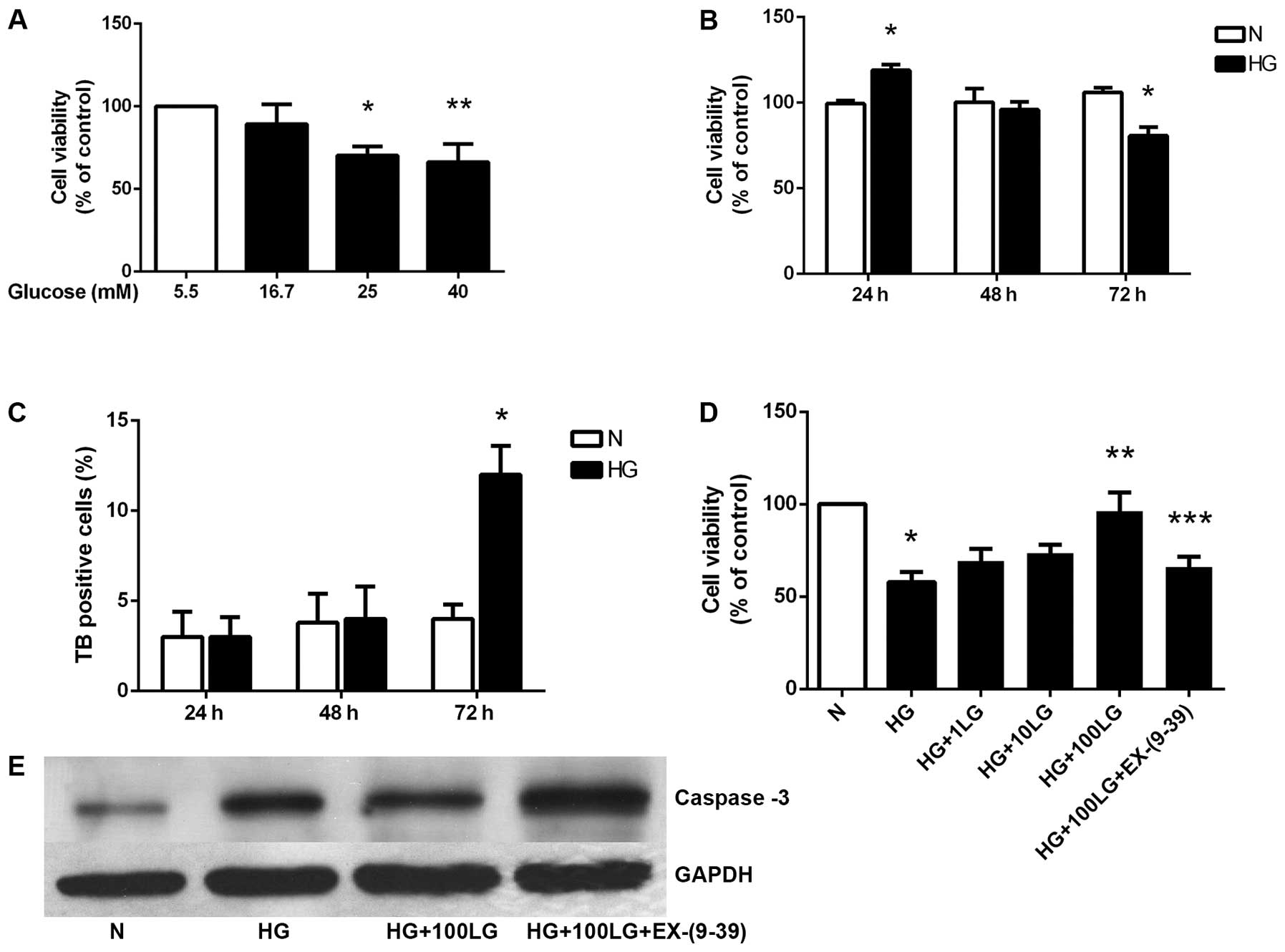
DMEM containing 5.5 mM glucose has been used as a
standard culture medium for HK-2 human renal tubular epithelial
cells. In order to examine the cytotoxicity resulting from exposure
to high glucose concentrations, cell viability was first measured
by MTS assay, which is dependent on the metabolic activity of
viable cells. The HK-2 cells were cultured in medium containing
5.5, 16.7, 25 or 40 mM glucose for 72 h. Cell viability
significantly decreased in a glucose concentration-dependent manner
(Fig. 1A). Of note, the HK-2
cells cultured in 40 mM glucose for 24 h at a density of
1.2×105 cells/cm2 showed increased viability.
After 72 h of culture in 40 mM glucose, however, there was a
significant decrease in viability as compared to the cells
incubated in medium with 5.5 mM glucose (Fig. 1B).
The decrease in MTS values observed with 40 mM
glucose may be due to reduced proliferation or increased cell
death. When cell death was assessed by the trypan blue exclusion
assay, increased numbers of dead cells were observed at 72 h in the
cells cultured with 40 mM glucose (Fig. 1C).
Effects of liraglutide on HK-2 cell
viability are mediated through GLP-1R
The HK-2 cells cultured in a high concentration of
glucose for 72 h showed a significantly decreased cell viability.
Liraglutide significantly enhanced cell viability in a
dose-dependent manner. Additionally, the effects of liraglutide
were significantly blocked by exendin-(9-39), a GLP-1R antagonist
(Fig. 1D). The loss of viability
that occurred in the cells at 72 h cultured in the presence of 5.5
or 40 mM glucose was associated with the activation of the common
executioner caspase, caspase-3 at the protein level. Liraglutide
downregulated caspase-3 expression, and exendin-(9-39) partly
blocked this effect (Fig.
1E).
Effect of liraglutide on GLP-1R
expression in HK-2 cells
The HK-2 cells stimulated with a high concentration
of glucose for 72 h showed significantly decreased levels of GLP-1R
expression. Liraglutide significantly enhanced GLP-1R mRNA
expression in a dose-dependent manner. Additionally, the effects of
liraglutide were significantly blocked by exendin-(9-39). The
protein expression of GLP-1R was altered in a similar manner
(Fig. 2).
Chronic exposure to high glucose
concentrations alters cellular ultrastructure
Changes in the ultrastructure of HK-2 cells induced
by high glucose were examined under a transmission electron
microscope. No changes in cellular nuclei or membranes were
observed in the HK-2 cells cultured in the presence of high glucose
at 24 h (Fig. 3A). A large number
of free-standing membrane structures and double-membrane vacuoles
was observed in the cytoplasm after 48 h, which resembled
pre-autophagosomal structures. Typical apoptotic changes, with
chromatin condensation and nuclear fragmentation, were observed in
the cells exposed to high glucose concentrations, and the
accumulation of glycogen significantly increased after 48 h.
Nuclear fragmentation and disappearance and the formation of a
large number of vacuoles indicated cytoplasmic vacuolization
(Fig. 3B).
High glucose concentrations promote
autophagy in HK-2 cells
The promotion of autophagy by exposure of the HK-2
cells to high glucose was confirmed by electron microscopy, showing
an accumulation of autophagosomes (Fig. 3C and D, black arrows).
The HK-2 cells stimulated with a high concentration
of glucose for 72 h exhibited an increased expression of the
autophagic markers, LC3-II and Beclin1. Liraglutide significantly
attenuated the increase in LC3-II and Beclin1 gene expression in a
dose-dependent manner. Additionally, the effects of liraglutide
were significantly blocked by the GLP-1R antagonist, exendin-(9-39)
(Fig. 4).
Metabolic variables
Body and organ weights are presented in Table I. The
body weights of the rats in the diabetic groups at 5 weeks after
the initiation of liraglutide treatment were significantly lower
than those of the rats in the non-diabetic group. The weight of the
kidneys in the diabetic rats, normalized to the body weight of the
rats in the non-diabetic groups, was significantly higher than that
of the rats in the non-diabetic group. No significant differences
were observed among the different diabetic groups.
Food intake was significantly increased in the
diabetic groups, but was decreased in the diabetic groups at 5
weeks after the initiation of liraglutide treatment compared with
the diabetic groups not receiving liraglutide. Serum creatinine and
blood urea nitrogen levels, which are markers of renal injury,
progressively increased in the diabetic groups during the
experiment. Liraglutide treatment significantly reduced serum
creatinine levels, but not the blood urea nitrogen levels, compared
to the values in the diabetic groups not treated with liraglutide
at 5 weeks.
Kidney morphology
The level of glomerular hypertrophy was
significantly greater in the diabetic groups (Fig. 5B) than in the non-diabetic group
(Fig. 5A). By contrast,
liraglutide treatment inhibited glomerular hypertrophy in the rats
in the diabetic group (Fig. 5C).
The renal interstitium showed a significantly greater level of
tubular hypertrophy and vacuolar degeneration in the diabetic
groups than in the non-diabetic group. Liraglutide treatment
improved this situation; however, its effects were blocked by
exendin-(9-39) (Fig. 5D).
High glucose concentrations promote
autophagy in the kidneys of diabetic rats
Immunohistochemistry revealed that the levels of the
autophagic marker, LC3-II, were significantly upregulated in the
kidneys of the diabetic rats (Fig.
6B) compared to those of the rats in non-diabetic group
(Fig. 6A). Similar to the results
from the in vitro experiments, liraglutide significantly
decreased the expression of LC3-II (Fig. 6C). Additionally, the effects of
liraglutide were partially blocked by exendin-(9-39) (Fig. 6D).
Discussion
The balance between the survival and death of renal
tubular epithelial cells plays a crucial role in the pathogenesis
of diabetic nephropathy. In the present study, we characterized
high glucose-induced toxicity in renal tubular epithelial cells
both in vivo and in vitro by means of several
complementary assays, including cell viability, cell death assays
and the assessment of the changes in the ultrastructure of HK-2
cells. The viability of renal tubular epithelial cells was markedly
reduced and the number of dead cells, including those undergoing
apoptosis, was significantly increased when the cells were treated
for a prolonged period of time with high glucose
concentrations.
The exposure of renal tubular epithelial cells to
high glucose concentrations for 72 h in vitro resulted in
the downregulation of GLP-1R expression. Treatment with liraglutide
increased cell viability, while exendin-(9-39) reversed this
effect. These results support the concept that the changes in cell
viabiltiy are caused by the reversible effects of glucotoxicity.
These findings are consistent with the hypothesis that the
unresponsiveness to GLP-1 is partly due to the changes in its
receptor, suggesting that liraglutide has a renoprotective function
that at least partially involves an increase in GLP-1R
expression.
Importantly, we demonstrated that the chronic
exposure of renal tubular epithelial cells and the kidneys of
nephritic rats to elevated glucose levels induced autophagy.
Autophagy is a cellular pathway involved in protein and organelle
degradation. It occurs at a basal level in the majority of cells
and is important in cellular homeostasis and responses to human
disease (35). Autophagy can be
induced by a variety of conditions, including nutrient deprivation
and growth-factor depletion and hypoxia. However, little is known
of the mechanisms underlying glucose-induced autophagy in renal
tubular epithelial cells. Electron microscopy, the gold standard
for monitoring the formation of autophagosomes, was used to
demonstrate an elevated autophagic activity in HK-2 cells, where
abundant vacuolization, a widely known morphologic indicator for
autophagic cell death, was observed after 72-h of incubation in
mediujm containing a high glucose concentration.
The pro-autophagic effect of high glucose was
associated with the expression of LC3-II. The formation of
autophagosomes includes the formation of pre-autophagosomal
membranes (phagophore or isolation membrane), which is initiated by
a class III phosphoinositide 3-kinase (PI3K) complex that includes
Beclin1, and the elongation of the isolation membrane, which is
stimulated by two ubiquitin-like conjugation systems (Atg12-Atg5
and LC3-phosphatidyletha nolamine) (36,37). It has been demonstrated that the
activation of Beclin1 is consistently associated with the induction
of autophagy in cancer cells (38). LC3, which is associated with the
control of autophagosome elongation, is the second essential
ubiquitin-like protein. Cytosolic LC3-I is recruited to the
membrane and interacts with phosphatidylethanolamine and is
converted to LC3-II. Thus, LC3-II has been used as a marker of
autophagy (39). In the present
study, autophagy was induced by high glucose concentrations, as
directly observed by the accumulation of autophagic vacuoles in the
cytoplasm, as well as by the increase in the content of the
autophagic markers, LC3-II and Beclin1. The prominent role of
autophagy in the response of renal tubular epithelial cells to
prolonged exposure to high glucose concentrations suggests that
autophagy may be an effective therapeutic target for the treatment
of diabetic nephropathy.
The addition of liraglutide to the high-glucose
medium significantly increased the viability of the HK-2 cells that
were exposed to high glucose alone. Liraglutide is a human incretin
and GLP-1 analogue with high homology to the native hormone. The
intestinal absorption of glucose stimulates its secretion, which
increases insulin and decreases glucagon secretion (27). GLP-1 is associated with enhanced
satiety, reduced food intake and weight loss. GLP-1 preserves
β-cell morphology and function, and reduces cellular apoptosis
(40). In the present study, we
also found that liraglutide inhibited the apoptosis of HK-2 cells,
which was accompanied by a significant decrease in autophagy, and
that exendin-(9-39), a GLP-1R antagonist, blocked the
cytoprotective effects of liraglutide. The same trends in cell
viability, GLP-1R expression and autophagy suggested the potential
connections in this pathological procedure of high glucose-induced
toxicity in renal tubular epithelial cells.
The question of whether autophagy is the driver of
cell death or a pro-survival process in response to certain stress
conditions remains controversial. Although autophagy was initially
described as a cytoprotective mechanism under conditions of
nutrient deprivation, several lines of evidence have indicated a
role for autophagy in promoting cell death (41–43), similar to the in vivo and
in vitro results obseved in the present study. This
indicates that liraglutide may also prevent injury in renal tubular
epithelial cells by reducing the autophagy induced by exposure to
high glucose and may be a promising therapeutic agent. Future
studies are required to determine the precise mechanisms underlying
the differential effects of liraglutatide on apoptosis and
autophagy, and to optimize its cytoprotective effects.
Acknowledgments
This study was supported by the grants from the
National Science Foundation of China, Grant No. 30870948 and
81102006. The authors thank all the staff at the Department of
Endocrinology, The First Affiliated Hospital of The Fourth Military
Medical University, Xi’an, P.R. China for their assistance with
this project. The authors declare no potential conflicts of
interest with respect to the authorship and/or publication of this
article.
References
|
1
|
Chen L, Magliano DJ and Zimmet PZ: The
worldwide epidemiology of type 2 diabetes mellitus - present and
future perspectives. Nat Rev Endocrinol. 8:228–236. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
de Zeeuw D, Parving HH and Henning RH:
Microalbuminuria as an early marker for cardiovascular disease. J
Am Soc Nephrol. 17:2100–2105. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Go AS, Chertow GM, Fan D, McCulloch CE and
Hsu CY: Chronic kidney disease and the risks of death,
cardiovascular events, and hospitalization. N Engl J Med.
351:1296–1305. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Magee GM, Bilous RW, Cardwell CR, Hunter
SJ, Kee F and Fogarty DG: Is hyperfiltration associated with the
future risk of developing diabetic nephropathy? A meta-analysis.
Diabetologia. 52:691–697. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Baynes JW: Role of oxidative stress in
development of complications in diabetes. Diabetes. 40:405–412.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Brownlee M, Cerami A and Vlassara H:
Advanced glycosylation end products in tissue and the biochemical
basis of diabetic complications. N Engl J Med. 318:1315–1321. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Koya D, Jirousek MR, Lin YW, Ishii H,
Kuboki K and King GL: Characterization of protein kinase C beta
isoform activation on the gene expression of transforming growth
factor-beta, extracellular matrix components, and prostanoids in
the glomeruli of diabetic rats. J Clin Invest. 100:115–126. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dunlop M: Aldose reductase and the role of
the polyol pathway in diabetic nephropathy. Kidney Int Suppl.
77:S3–S12. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ziyadeh FN, Sharma K, Ericksen M and Wolf
G: Stimulation of collagen gene expression and protein synthesis in
murine mesangial cells by high glucose is mediated by autocrine
activation of transforming growth factor-beta. J Clin Invest.
93:536–542. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Singh DK, Winocour P and Farrington K:
Mechanisms of disease: the hypoxic tubular hypothesis of diabetic
nephropathy. Nat Clin Pract Nephrol. 4:216–226. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Miyata T and de Strihou Cv: Diabetic
nephropathy: a disorder of oxygen metabolism? Nat Rev Nephrol.
6:83–95. 2010. View Article : Google Scholar
|
|
12
|
Giacco F and Brownlee M: Oxidative stress
and diabetic complications. Circ Res. 107:1058–1070. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kashihara N, Haruna Y, Kondeti VK and
Kanwar YS: Oxidative stress in diabetic nephropathy. Curr Med Chem.
17:4256–4269. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hakim FA and Pflueger A: Role of oxidative
stress in diabetic kidney disease. Med Sci Monit. 16:pp. RA37–RA48.
2010, PubMed/NCBI
|
|
15
|
Ha H, Hwang IA, Park JH and Lee HB: Role
of reactive oxygen species in the pathogenesis of diabetic
nephropathy. Diabetes Res Clin Pract. 82(Suppl 1): S42–S45. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wu J, Zhang R, Torreggiani M, et al:
Induction of diabetes in aged C57B6 mice results in severe
nephropathy: an association with oxidative stress, endoplasmic
reticulum stress, and inflammation. Am J Pathol. 176:2163–2176.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Qi W, Mu J, Luo ZF, et al: Attenuation of
diabetic nephropathy in diabetes rats induced by streptozotocin by
regulating the endoplasmic reticulum stress inflammatory response.
Metabolism. 60:594–603. 2011. View Article : Google Scholar
|
|
18
|
Cybulsky AV, Takano T, Papillon J, Kitzler
TM and Bijian K: Endoplasmic reticulum stress in glomerular
epithelial cell injury. Am J Physiol Renal Physiol. 301:F496–F508.
2011. View Article : Google Scholar
|
|
19
|
Masini M, Bugliani M, Lupi R, et al:
Autophagy in human type 2 diabetes pancreatic beta cells.
Diabetologia. 52:1083–1086. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ebato C, Uchida T, Arakawa M, et al:
Autophagy is important in islet homeostasis and compensatory
increase of beta cell mass in response to high-fat diet. Cell
Metab. 8:325–332. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hartleben B, Gödel M, Meyer-Schwesinger C,
et al: Autophagy influences glomerular disease susceptibility and
maintains podocyte homeostasis in aging mice. J Clin Invest.
120:1084–1096. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kimura T, Takabatake Y, Takahashi A, et
al: Autophagy protects the proximal tubule from degeneration and
acute ischemic injury. J Am Soc Nephrol. 22:902–913. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jiang M, Liu K, Luo J and Dong Z:
Autophagy is a renoprotective mechanism during in vitro hypoxia and
in vivo ischemia-reper-fusion injury. Am J Pathol. 176:1181–1192.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kume S, Uzu T, Horiike K, et al: Calorie
restriction enhances cell adaptation to hypoxia through
Sirt1-dependent mitochondrial autophagy in mouse aged kidney. J
Clin Invest. 120:1043–1055. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ohsumi Y and Mizushima N: Two
ubiquitin-like conjugation systems essential for autophagy. Semin
Cell Dev Biol. 15:231–236. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xie Z and Klionsky DJ: Autophagosome
formation: core machinery and adaptations. Nat Cell Biol.
9:1102–1109. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Baggio LL and Drucker DJ: Biology of
incretins: GLP-1 and GIP. Gastroenterology. 132:2131–2157. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Farilla L, Hui H, Bertolotto C, et al:
Glucagon-like peptide-1 promotes islet cell growth and inhibits
apoptosis in Zucker diabetic rats. Endocrinology. 143:4397–4408.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nauck MA, Niedereichholz U, Ettler R, et
al: Glucagon-like peptide 1 inhibition of gastric emptying
outweighs its insulino-tropic effects in healthy humans. Am J
Physiol. 273:pp. E981–E988. 1997, PubMed/NCBI
|
|
30
|
Turton MD, O’Shea D, Gunn I, et al: A role
for glucagon-like peptide-1 in the central regulation of feeding.
Nature. 379:69–72. 1996. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bullock BP, Heller RS and Habener JF:
Tissue distribution of messenger ribonucleic acid encoding the rat
glucagon-like peptide-1 receptor. Endocrinology. 137:2968–2978.
1996.PubMed/NCBI
|
|
32
|
Wajcberg E and Amarah A: Liraglutide in
the management of type 2 diabetes. Drug Des Devel Ther. 4:279–290.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hayes MR, Kanoski SE, Alhadeff AL and
Grill HJ: Comparative effects of the long-acting GLP-1 receptor
ligands, liraglutide and exendin-4, on food intake and body weight
suppression in rats. Obesity (Silver Spring). 19:1342–1349. 2011.
View Article : Google Scholar
|
|
34
|
Sugimoto H, Shikata K, Hirata K, et al:
Increased expression of intercellular adhesion molecule-1 (ICAM-1)
in diabetic rat glomeruli: glomerular hyperfiltration is a
potential mechanism of ICAM-1 upregulation. Diabetes. 46:2075–2081.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Martinet W and De Meyer GR: Autophagy in
atherosclerosis: a cell survival and death phenomenon with
therapeutic potential. Circ Res. 104:304–317. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang H, Kong X, Kang J, et al: Oxidative
stress induces parallel autophagy and mitochondria dysfunction in
human glioma U251 cells. Toxicol Sci. 110:376–388. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kiyono K, Suzuki HI, Matsuyama H, et al:
Autophagy is activated by TGF-beta and potentiates
TGF-beta-mediated growth inhibition in human hepatocellular
carcinoma cells. Cancer Res. 69:8844–8852. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kawakami T, Inagi R, Takano H, et al:
Endoplasmic reticulum stress induces autophagy in renal proximal
tubular cells. Nephrol Dial Transplant. 24:2665–2672. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Marsh BJ, Soden C, Alarcón C, et al:
Regulated autophagy controls hormone content in secretory-deficient
pancreatic endocrine beta-cells. Mol Endocrinol. 21:2255–2269.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Law BY, Wang M, Ma DL, et al: Alisol B, a
novel inhibitor of the sarcoplasmic/endoplasmic reticulum Ca(2+)
ATPase pump, induces autophagy, endoplasmic reticulum stress, and
apoptosis. Mol Cancer Ther. 9:718–730. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhu J, Wang KZ and Chu CT: After the
banquet: mitochondrial biogenesis, mitophagy, and cell survival.
Autophagy. 9:1663–1676. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Petersen M, Hofius D and Andersen SU:
Signaling unmasked: Autophagy and catalase promote programmed cell
death. Autophagy. 10:520–521. 2014. View Article : Google Scholar : PubMed/NCBI
|