Introduction
Gliomas are the most common primary tumors of the
central nervous system. The unique biological characteristics of
glioma cells, such as the invasiveness to surrounding tissues, the
difficulty of complete resection and the high probability of
recurrence in situ are important topics in present study.
The invasion of glioma cells into the surrounding tissue is a
complex process involving multiple steps, including the adherence
and migration of tumor cells and the degradation of the
extracellular matrix. Previous studies have demonstrated that
activating transcription factor 3 (ATF3) is highly expressed in
several malignant cancer tissues (1–3).
ATF3 can induce cells to enter the cell cycle from the stationary
phase, thus accelerating cell proliferation; this characteristic is
important in the processes of invasion and migration and is
significant for the prognosis for several types of tumor (4–6).
Maspin (SERPINB5) is a tumor suppressor gene that suppresses
angiogenesis, enhances the ability of cells to adhere and
suppresses cancer cell migration (7). Another important factor in glioma
invasion is matrix metalloproteinase 2 (MMP2), which has been
reported to destroy local tissue and enhance tumor angiogenesis,
thereby accelerating glioma invasion and migration (8). In the present study, we employed
immunohistochemical staining, western blot analysis and RT-qPCR to
assess the protein and mRNA expression levels of ATF3, maspin and
MMP2 in human brain glioma samples. We then conducted a series of
experiments using the human glioblastoma cell line, U373MG, in
which the cells were transfected with ATF3-siRNA or a control in
order to assess the cell proliferative capacity, cell cycle status
and apoptotic fraction, as well as the ability of the cells to
invade through fibronectin. We also used immunocytochemistry,
RT-qPCR and western blot anlaysis to assess the changes in the
protein and mRNA expression of ATF3, maspin and MMP2 in cultures of
U373MG cells in vitro. Finally, we determined the ability of
ATF3-siRNA to inhibit the growth of U373MG cells grown in
vivo as subcutaneous xenografts in nude mice. The objective was
to elucidate the role of ATF3, maspin and MMP2 in the development
of gliomas.
Materials and methods
Human tissues
Astrocytoma samples that were resected during
surgery from September 2008 to December 2009 at the First
Affiliated Hospital of the Medical College of Zhengzhou University
were collected. All patients provided signed informed consent and
the study was approved by the Research Ethics Committee of
Zhengzhou University.
Material from 100 glioma cases (58 males) was
examined. The age range was 18–66 years, with an average age of
42.3±3.1 years (SD). All pathological sections were analyzed by two
experienced pathologists. Cases were graded according to the WHO
classification criteria in 2007 (9) for central nervous system tumors: 15
cases were grade I (pilocytic astrocytoma), 32 cases were grade II
(diffuse astrocytoma), 30 cases were grade III (anaplastic
astrocytoma) and 23 cases were grade IV (glioblastoma multiforme).
Thirteen control brain tissue samples (8 males and 5 females) were
available from resection during surgery from patients with
craniocerebral trauma in the same hospital during the same time
period; the control samples were proven pathologically to be normal
brain tissues. From each tumor patient, two samples of central,
fresh tumor tissue without bleeding or necrosis were stored in
liquid nitrogen, and another sample was fixed with 10% formalin,
embedded in paraffin and cut into 5-μm-thick sections for
hematoxylin and eosin and immunohistochemical staining.
Main reagents
The rabbit polyclonal anti-human ATF3 (sc-188),
anti-maspin (sc-22762) and anti-MMP2 (sc-10736) antibodies, the
reference gene β-actin, goat anti-rabbit IgG antibody and the DAB
kit, as well as chemiluminescence reagents used for western blot
analysis were purchased from Santa Cruz Biotechnology, Inc. (Santa
Cruz, CA, USA). All PCR primers were designed and synthesized by
Shanghai Sangon Biological Engineering Technology & Services
Co., Ltd. (Shanghai, China). The PureLink® RNA Mini kit
(12183018A), the high-capacity cDNA reverse transcription kit
(4368813), Power SYBR®-Green PCR Master Mix (4367659),
the pSilencer2.1 U6 vector and Lipofectamine 2000 used for
transfection were purchased from Invitrogen (Life Technologies,
Carlsbad, CA, USA). The U373MG cells were obtained from the
American Type Culture Collection (ATCC® HTB-17; ATCC,
Rockefeller, MD, USA) in 2010; however, there is a possibility that
this glioblastoma cell line should be described as another
glioblastoma cell line, U-251 MG, as similarities have been
reported between these two cell lines (http://www.lgcstandards-atcc.org/support/faqs/cf245/U373%20MG%20ATCC%20HTB17-1055.aspx).
MTT tetrazolium substrate, propidium iodide (PI) for flow cytometry
and terminal deoxynucleotidyl transferase-mediated dUTP nick-end
labeling (TUNEL) reaction mixture were from Roche Diagnostics
(Laval, Quebec, Canada). The Transwell cell culture inserts were
obtained from Corning Inc. (Corning, NY, USA).
Immunohistochemistry
Sections of paraffin-embedded tissue were
deparaffinized in dimethylbenzene and hydrated using an alcohol
gradient. The sections were incubated in 0.3%
H2O2 in methanol for 30 min at room
temperature and in an ice bath for 5 min in 0.1% Triton X-100, and
blocked with 10% goat serum. The working dilutions of primary
antibodies against ATF3, maspin and MMP2 in the tissue sections
were 1:200. The development of color using
DAB-H2O2 was observed under a CX 31 Olympus
microscope (Olympus Optical Co., Ltd., Tokyo, Japan).
Counterstaining was performed with hematoxylin, and following
dehydration, the sections were coverslipped with neutral gum.
Normal goat serum was used as a substitute for the primary antibody
in the negative control group and tissue sections known to be
positive were used as positive controls. Obvious brownish yellow
particles observed in the nucleus or cytoplasm indicated positive
staining for ATF3, maspin and MMP2 proteins. We used the
semi-quantitative judgment method of Gatalica et al
(10) for the analysis of the
experimental results by the H-score (H=I × P) system. Five
high-power fields (×400, final magnification) were selected for
each section. The average positive rates were calculated and were
expressed as the means ± SD.
Measurement of mRNA levels by
RT-qPCR
Total RNA extraction, the reverse transcription of
mRNA into cDNA and fluorescence quantitative PCR (qPCR) were
performed according to the manufacturer’s instructions. We used
β-actin (ACTB) mRNA as an internal reference. The forward primer
for ATF3 was 5′-CCTCGGAAGTGAGTGCTTCT-3′ and the reverse primer was
5′-ATGGCAAACCTCAGCTCTTC-3′. The forward primer for maspin was
5′-AGACATTCTCGCTTCCCTGA-3′ and the reverse primer was
5′-AATTTTGACCCCTTATGGGC-3′. For MMP2, the forward primer was
5′-GCTATGGACTTGGGAGAA-3′ and the reverse primer was
5′-TGGAACGGAATGGAAAC-3′. The forward primer for β-actin was
5′-CACCACCATGTACCCTGGCA-3′ and the reverse primer was
5′-GCTGTCACCTTCACCGTTCC-3′. The reaction conditions for qPCR were
as follows: 95°C for 5 min and 40 cycles of 95°C for 15 sec, 60°C
for 1 min, and 72°C for 1 min. For each sample, 3 replicates were
assessed and a parallel reaction without primers was used as a
negative control. Relative mRNA expression levels were calculated
for each gene following normalization against β-actin, using the
ΔΔCt method, as previously described (11).
Measurement of protein levels by western
blot analysis
Tissues removed from storage at −80°C, or cells
collected from culture were extracted by grinding in RIPA lysis
buffer in a glass homogenizer and incubated on ice for 1 h. The
extracts were centrifuged for 10 min at 18,000 rcf and the
supernatants were collected and mixed with 2X sodium dodecyl
sulfate (SDS) loading buffer. Following denaturation for 5 min at
100°C and centrifugation for 10 min at 16,000 rcf, the lysates were
electrophoresed on 10% SDS-polyacrylamide gels. A semi-dry western
blot analysis transfer method was used. The membranes were blocked
with 5% non-fat milk powder for 1 h at room temperature, washed 3
times (5 min each) with TBS-T, incubated with primary antibodies
(the working dilutions were 1:1,000) overnight at 4°C, washed 3
times (5 min each) with TBS-T, incubated with secondary antibodies
for 2 h at 37°C and washed 3 times (5 min each) with TBS-T. The
presence of immunoreactive protein was detected using an ECL method
to expose X-ray film. Processed films were scanned or photographed,
and the integrated optical densities of the bands were analyzed
using a LabWorks 4.0 gel image analysis system. The ratios of the
integrated optical densities of the proteins to β-actin were
calculated.
Cell culture
The frozen human glioblastoma cell line, U373MG, was
inoculated onto a 25 cm2 culture flask after being
revived in a 37°C water bath. Low-glucose Dulbecco’s modified
Eagle’s medium (DMEM) with 10% heat-inactivated fetal calf serum
was added and the cells were placed in an incubator with 5%
CO2 and saturating oxygen at 37°C. The medium was
replaced every other day until the cells spread onto the bottom of
the flask. When the cell fusion state was about to occur, the
nutrient solution was discarded and the cells were digested by
0.25% pancreatin. When the cells retracted, the intercellular space
expanded and the cells became round, the nutrient solution was
added to terminate the digestion and the cells were transferred and
then centrifuged at 1,100 rcf for 5 min. The supernatant was
discarded and the low-sugar DMEM nutrient solution was added. The
cells were transferred to a new culture bottle and the bottle was
then placed in an incubator with a saturation humidity of 5%
CO2 at 37°C for cultivation. After 2–3 days, the cells
were passaged when the cell convergence degree was at 80–90%. The
third generation cells were used for the experiments.
Screening and identification of
ATF3-siRNA
Control and ATF3 hairpin loop siRNAs were cloned
using the expression vector, pSilencer2.1 U6. Gene-specific siRNA
hairpin loops targeting a 19-nucleotide sequence within human ATF3
were designed and synthesized by Shanghai Sangon Biological
Engineering Technology & Services Co., Ltd. Initially, 3
ATF3-siRNAs were tested to identify optimal ATF3-siRNA(s). As a
result, 2 of 3 siRNA sequences (5′-GAGCTGAGGTTTGCCATCC-3′ and
5′-GAGGCGACGAGAAAGAAAT-3′) were shown to be useful for ATF3
knockdown. An additional siRNA vector (5′-GCACCACGTGACGGAGCGT-3′)
was used as a negative control. The ATF3 and control siRNA
sequences contained the BamHI and HindIII restriction
sites included in the forward and reverse strands, respectively.
The synthesized oligonucleotides were annealed and inserted into
pSilencer2.1 U6 using the BamHI and HindIII sites.
The hairpin siRNA sequences were confirmed by nucleotide sequence
analysis. The U373MG cells were then transfected with pSilencer2.1
U6-ATF3 or control siRNAs using Lipofectamine 2000 according to the
manufacturer’s instructions. Three days following transfection,
cell lysates were harvested and processed for western blot analysis
to detect the protein expression of ATF3.
Cell groups
In the present study, the cells were divided into 5
experimental groups as follows: i) the control group: untreated
U373MG cells; ii) the control-siRNA group: the U373MG cells were
transfected with control siRNAs; iii) the ATF3-siRNA group: the
U373MG cells were transfected with pSilencer2.1 U6-ATF3; iv) the
cisplatin group: the U373MG cells were treated with cisplatin; and
v) the ATF3-siRNA + cisplatin group: the U373MG cells transfected
with pSilencer2.1 U6-ATF3 were treated with cisplatin.
In addition, a positive and negative control group
were used. In the positive control group, the cells were treated as
follows: the cells were fixed with 4% paraformaldehyde at room
temperature for 1 h followed by 2 washes (5 min each) with PBS. The
cells were then treated with 0.1% Triton X-100 for 5 min at room
temperature followed by 2 washes (5 min each) with PBS. This was
followed by the addition of 100 μl DNase I reagent (2,000 U
DNase I, 40 mM Tris-HCl pH 7.9, 10 mM NaCl, 6 mM MgCl2
and 10 mM CaCl2) and treatment for 30 min at room
temperature (37°C) followed by 3 washes (5 min each) with PBS.
TUNEL reagent (50 μl) was then added to each well and the
remaining steps were the same as those of the experimental
procedure in this study. The cells in the negative control group
was treated as follows: the cells were fixed with 4%
paraformaldehyde at room temperature for 1 h followed by 2 washes
(5 min each) with PBS. The cells were then treated with 0.1% Triton
X-100 for 5 min at room temperature followed by 2 washes (5 min
each) with PBS. This was followed by the addition of 50 μl
labeling buffer (5X 550 μl, with reagent containing
nucleotide mixture) without DNase I buffer to replace the TUNEL
reagent and the cells were incubated at 37°C in a dark, humid
environment for 60 min before being washed 3 times with PBS. DAPI
solution buffer was added and the following steps were the same as
those of the experimental procedure in this study. The addition of
DNase I in the positive control group made the cell sample produce
DNA double-strand breaks, thus leading to cell apoptosis to the
maximum degree, so that the final results presented the maximum
positive degree, so as to be used as the positive control. No TUNEL
reagent was added to the cells in the negative control group, and
thus no apoptotic cells could be tested, and the final result
presented complete negative, so as to be used as the negative
control. The cells in the control group (untreated group) were not
treated at all, so as to test the normal apoptotic rate of the
cells.
Analysis of cell proliferation
Cell numbers were estimated using an assay based on
the reduction of the formazan dye MTT by metabolically active
cells. Twenty-four hours post-transfection with the plasmids, the
cells were harvested, resuspended and seeded in 96-well cell
culture plate at a density of 5,000 cells/well. For each time
point, each group was set up in triplicate. An MTT assay was
undertaken at 24 h (day 1) and then cisplatin (sc-200896; Santa
Cruz Biotechnology, Inc.) at a concentration of 20 mg/l was added
to the appropriate wells (cisplatin group and the ATF3-siRNA +
cisplatin group) followed by further culture for an additional 24 h
(day 2), 48 h (day 3), 72 h (day 4) or 96 h (day 5). Cell number
was assessed by the addition of 20 μl (5 mg/ml) MTT solution
to each well of a plate followed by incubation for 4 h at 37°C with
5% CO2. The plates were then centrifuged at 2,000 rpm
for 5 min and the supernatants were discarded. The formazan
reaction product was extracted into 100 μl DMSO and the
absorbance at 490 nm was measured using a spectrophotometer.
Analysis of cell cycle phase by flow
cytometry
Forty eight hours following transfection, the cells
were seeded into 6-well plates, cultured for a further 48 h and
then rinsed with phosphate-buffered saline (PBS), digested with
trypsin-ethylenediaminetetraacetic acid (EDTA), harvested,
centrifuged at 450 rcf for 5 min and resuspended twice before
fixation by adding dropwise into to 95% ethanol precooled to −20°C
for storage. Prior to analysis, the cells were warmed, centrifuged
at 450 rcf for 5 min and resuspended twice, then stained with PI
(containing RNase A at 50 μg/ml) at room temperature in the
dark for 30 min. The DNA content was analyzed by flow cytometry
using the CellQuest program (Becton-Dickinson and Co., Franklin
Lakes, NJ, USA).
Analysis of cell apoptotic fraction by
TUNEL staining
Forty-eight hours following transfection, the cells
in each group were digested, counted and seeded in 96-well plates
at a density of 6×104 cells/ml followed by incubation at
37°C with 5% CO2 for 24 h. Cisplatin was added to the
appropriate wells at 20 mg/l followed by incubation at 37°C with 5%
CO2 for a further 24 h. The cells were then fixed with
4% paraformaldehyde at room temperature for 1 h followed by 2
washes (5 min each) with PBS. The cells were treated with 0.1%
Triton X-100 for 5 min at room temperature followed by 2 washes (5
min each) with PBS. TUNEL reagent (50 μl) was added to each
well and the cells were incubated at 37°C in a dark, humid
environment for 60 min before washing 3 times with PBS. DNA was
stained by the addition of 50 μl
4′,6-diamidino-2-phenylindole (DAPI) solution (0.01 mg/ml) to each
well, incubating at room temperature in the dark for 5 min followed
by washing 5 times with PBS. The cells were observed under a
fluorescence microscope (IX73-F22FL/PH; Olympus Optical Co., Ltd)
and images were captured using the OpenLab 4 imaging program
(Perkin Elmer Co., Ltd., Waltham, MA, USA).
Analysis of cell invasion
The ability of the glioblastoma cells to migrate
through fibronectin was assessed using a Transwell culture system.
Transfection was performed according to the protocol described
above. Cisplatin at 20 mg/l was added to the appropriate wells at
48 h after transfection. The cells were placed in an incubator at
37°C with 5% CO2 for 24 h. Subsequently, 10 μl of
fibronectin and 50 μl of Matrigel were coated on the
Transwell membranes. The cells in each group were digested and
counted. In total, 105 cells were placed into a 1.5 ml
microcentrifuge tube and centrifuged for 5 min at 450 rcf; the
supernatants were removed and 200 μl of medium without serum
were added, and the cells were resuspended before being added to
the wells in the Transwell plate. Endothelial cell medium was added
to the lower wells in the plate and cultured for 24 h in an
incubator at 37°C. The Transwell insert was removed from the
chamber, the cells remaining in the insert chamber were removed by
gentle wiping with a swab and the residues gently washed off with
PBS. A mixture of methanol and glacial acetic acid (3:1 v/v; 30
min) was used to fix cells that had moved through onto the lower
side of the Transwell insert and the cells were stained by
immersing the inserts in 0.1% Crystal violet dye (sc-207460; Santa
Cruz Biotechnology, Inc.) for 15 min. The total number of cells
that penetrated the Transwell was counted.
Growth of glioblastoma cells as tumor
xenografts in nude mice
These experiments were approved by the Institutional
Animal Care and Use Committee at the First Affiliated Hospital of
Zhengzhou University (Zhengzhou, China). BALB/c nude mice (n=40),
of the female gender, 4–5 weeks old, weighing 15–20 g, were
purchased from Hunan SJA Laboratory Animal Co., Ltd. (Changsha,
China). The nude mice were raised in isolation cages with
independent ventilation at 24–26°C and with free access to water
and food. All cages, bedding materials, drinking water and feed
were sterilized and surgical procedures were carried out
aseptically by experienced personnel.
The U373MG cells were cultured using standard
methods. The cells were digested with 0.2% trypsin for 3 min until
they reached tge log growth phase and digestion was terminated by
the addition of complete culture medium of low-glucose DMEM. The
cells were triturated to yield single cell suspensions and washed
twice with complete DMEM medium prior to resuspension in PBS. The
cells were adjusted to a density of 1×107 cells/ml and
0.2 ml was injected subcutaneously in the back of each nude mouse.
All procedures were undertaken in a laminar flow hood to minimize
infection.
The growth of the tumors was monitored regularly and
the size of each tumor was measured using a vernier caliper every 5
days. The longest (a) and shortest diameter (b) were measured and
the approximate volume of the tumor was calculated using the
following formula: transplanted tumor size (V) = longest diameter
(a) x square of the shortest diameter (b2)/2.
When the tumors attained an average volume of 100
mm3, the 40 nude mice were weighed and randomly
allocated into 5 groups receiving the following injections: i) the
vehicle control group: 0.6 ml saline; ii) the control-siRNA group:
0.5 ml irrelevant siRNA (5′-GAAGAAGGAGAAGACGGAG-3′) plus 0.1 ml
saline; iii) the ATF3-siRNA group: 0.5 ml ATF3-siRNA fragment (300
nM) plus 0.1 ml saline; iv) the cisplatin group: 0.1 ml cisplatin
solution (500 ng/ml) plus 0.5 ml saline; and v) the ATF3-siRNA +
cisplatin group: 0.5 ml ATF3-siRNA fragment and 0.1 ml cisplatin
solution. These treatments were administered 10 times by
peritumoral injection every other day. The tumor inhibition ratio
(%) = (average tumor weight in the control group - average tumor
weight in the treatment group)/average tumor weight in the control
group x100 was calculated for each treatment.
Statistical analyses
The data were processed with Statistical Product and
Service Solutions 18.0 software (IBM, Armonk, NY, USA). Two samples
of rank data were compared using the Mann-Whitney U test. Multiple
sample comparison of the rank data was carried out using the
Kruskal-Wallis H test. The expression of ATF3, maspin and MMP2 in
the patient glioma samples of variable grades was analyzed by
Spearman’s rank correlation analysis. The correlation of the
expression of ATF3, maspin and MMP2 with the pathological grade was
analyzed by Spearman’s rank correlation analysis. The mean values
of multiple samples were compared by one-way ANOVA. The
significance level was set at P<0.05.
Results
Immunohistochemical analysis of ATF3,
maspin and MMP2 in normal brain tissues and glioma tissues of each
histological grade
The presence of ATF3 and MMP2 proteins was mainly
manifested as brown particles in the tumor cytoplasm and marginally
in the nuclei. They were both irregularly distributed in the
lower-grade glioma tissue and diffusely distributed in the
higher-grade glioma tissue (Fig. 1A
and C). The protein expression of ATF3 and MMP2 in the glioma
tissues was evidently higher than that in the normal brain tissues
(P>0.05). Their expression in the glioma tissues increased with
the increasing glioma grade (I to IV) (Table I). Spearman’s rank correlation
analysis revealed that the protein expression of ATF3 and MMP2
positively correlated with the pathological grade of the glioma
(ϱ=0.735, P<0.01; ϱ =0.446, P<0.01, respectively).
 | Table IProtein expression of ATF3, maspin
and MMP2 in normal brain tissues and glioma tissues of each
grade. |
Table I
Protein expression of ATF3, maspin
and MMP2 in normal brain tissues and glioma tissues of each
grade.
| n | ATF3
n (%) | Maspin
n (%) | MMP2
n (%) |
|---|
| Normal brain
tissues | 13 | 2 (15.4) | 13 (100) | 1 (7.7) |
| Gliomas
tissues | 100 | 72 (72)a | 53 (53)a | 76 (76)a |
| WHO grading | | | | |
| I | 15 | 4 (26.7) | 12 (80)a | 4 (26.7) |
| II | 32 | 18 (56.3)a | 22 (78.1)a | 20 (62.5)a |
| III | 30 | 28 (93.3)a | 13 (43.3)a | 29 (90.6)a |
| IV | 23 | 22 (95.7)a | 6 (26.1)a | 23 (100)a |
Maspin protein expression was present both in the
nucleus and cytoplasm and it was highly expressed in the normal
brain tissues (Fig. 1B). The
protein expression of maspin decreased with the increasing glioma
grade (I to IV) (Table I) and
Spearman’s rank correlation analysis revealed that the protein
expression of maspin negatively correlated with the pathological
grade of the glioma (ϱ=−0.542, P<0.01).
Relative abundance of ATF3, maspin and
MMP2 mRNA expression in normal brain tissues and glioma tissues of
each histological grade
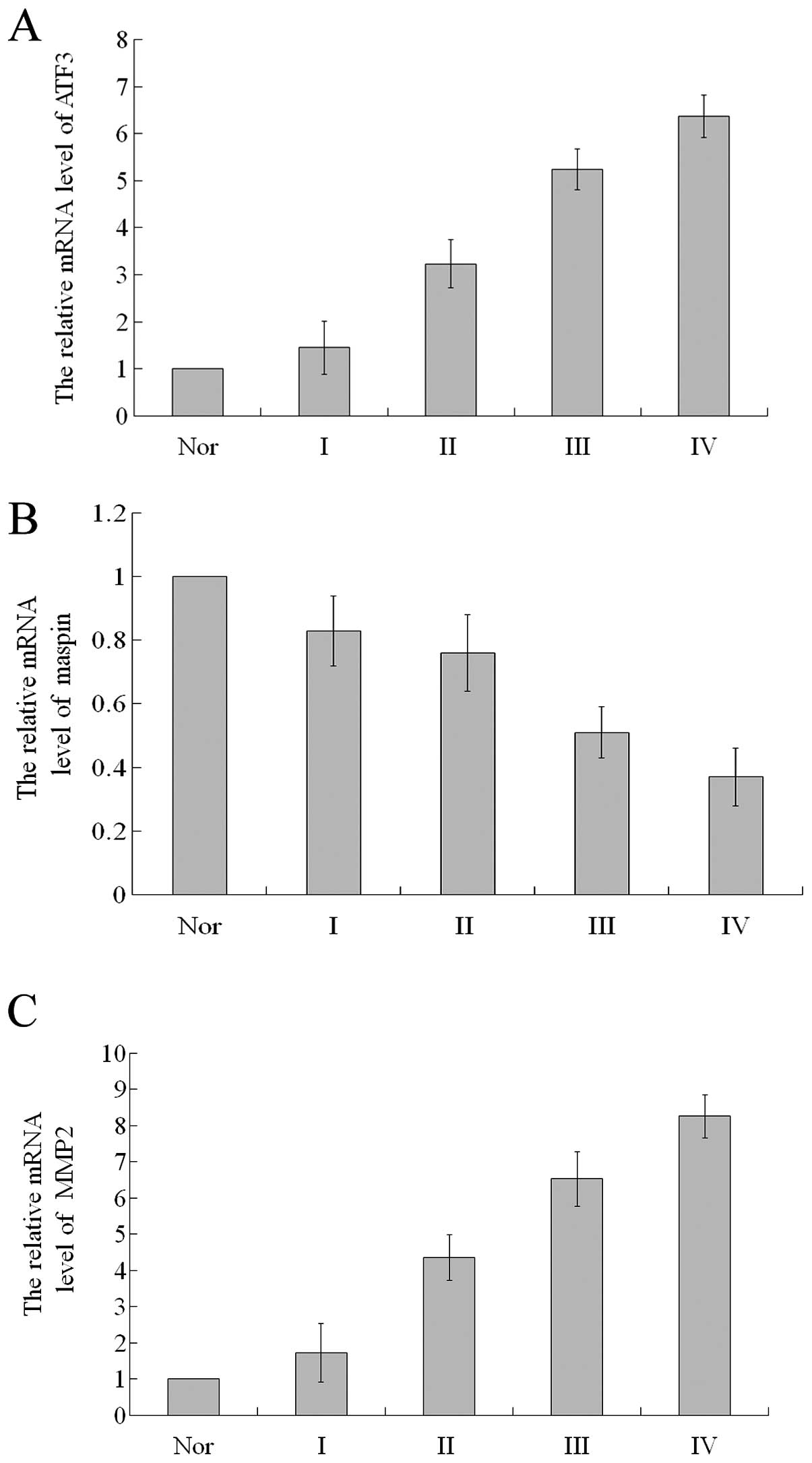
The relative mRNA expression of ATF3 in the glioma
tissues of grade II, III and IV was 3.23±0.51, 5.24±0.43 and
6.37±0.45, respectively, significantly higher compared to the
normal brain tissues (all P<0.05; Fig. 2A). The relative mRNA expression of
ATF3 in the glioma tissues of grade I did not differ significantly
from that in the normal brain tissues (P>0.05). The relative
mRNA expression of maspin in the glioma tissues of grade I, II, III
and IV was 0.83±0.11, 0.76±0.12, 0.51±0.08 and 0.37±0.09,
respectively, significantly lower compared to the normal brain
tissues (all P<0.05; Fig. 2B).
Similar to the mRNA expression of ATF3, in the glioma tissues of
grade I, the relative mRNA expression of MMP2 did not differ
significantly from that in the normal brain tissues (P>0.05;
Fig. 2C). In the glioma tissues
of grade II, III and IV, the relative mRNA expression of MMP2 was
4.36±0.63, 6.53±0.75 and 8.26±0.59, respectively, significanlty
higher compared to the normal brain tissues (all P<0.05;
Fig. 2C). Spearman’s rank
correlation analysis revealed that the mRNA expression of ATF3 and
MMP2 positively correlated with the pathological grade of glioma
(ϱ= 0.621, P<0.01 and ϱ=0.503, P<0.01, respectively), while
the mRNA expression of maspin negatively correlated with the
pathological grade of glioma (ϱ=−0.415, P<0.01).
Western blot analysis of ATF3, maspin and
MMP2 protein expression in normal brain tissues and glioma tissues
of each histological grade
The intensity of the β-actin bands was similar in
all the samples, indicating that the loading of each sample was
consistent, and thus that the results are reliable (Fig. 3A). ATF3 protein expression was
detected in both the normal brain tissues and glioma tissues of
each grade. In the glioma tissue of grade I, the relative protein
expression of ATF3 did not differ significantly from that in the
normal brain tissues (P>0.05). In the glioma tissue of grade II,
III and IV, the relative protein expression was 1.53±0.25,
2.57±0.34 and 3.45±0.41, respectively, significantly higher
compared to that in the normal brain tissues (P<0.05; Fig. 3B). In the glioma tissue of grade I
to IV, the expression of maspin was 2.04±0.32, 1.36±0.3, 0.73±0.25
and 0.42±0.21, respectively, signficantly lower compared to that in
the normal brain tissues (all P<0.05; Fig. 3B). In the glioma tissues of grade
I, the protein expression of MMP2 did not differ significantly from
that in the normal brain tissues (P>0.05). In the glioma tissues
of grade II to IV, the expression levels of MMP2 were 1.72±0.29,
4.15±0.45 and 5.82±0.53, respectively, significantly higher
compared to those in the normal brain tissues (P<0.05; Fig. 3B). Spearman’s rank correlation
analysis revealed that the protein expression of ATF3 and MMP2
positively correlated with the pathological grade of glioma (ϱ=
0.592, P<0.01 and ϱ=0.726, P<0.01, respectively), while the
protein expression of maspin negatively correlated with the
pathological grade of glioma (ϱ=−0.517, P<0.01).
Knockdown of ATF3 using ATF3-siRNA
inhibits the proliferative activity of U373MG cells in vitro
Compared with the control group (untreated cells)
and the control-siRNA group, the proliferative activity of the
cells in the ATF3-siRNA group (transfected with ATF3-siRNA), the
cisplatin group (treated with cisplatin) and the ATF3-siRNA +
cisplatin group (transfected with ATF3-siRNA and treated with
cisplatin) began to decrease by day 2 (P<0.05), and this
inhibitory effect occurred in time-dependent manner, i.e. the
inhibition intensity increased with time (Fig. 4). Following the addition of
cisplatin, the cell proliferative activity gradually decreased with
time (P<0.05); no significant difference was observed in the
cell proliferative activity between the cisplatin group and the
ATF3-siRNA + cisplatin group (P>0.05), which indicated that
inhibitory effects of transfection with ATF3-siRNA on the
proliferation of U373MG cells were equal to, or as effective to
those of treatment with a dose of a chemotherapeutic agent.
Knockdown of ATF3 using ATF3-siRNA
inhibits the cell cycle progression of U373MG cells in vitro
The percentage of cells in the S phase in the
ATF3-siRNA group and the cisplatin group was 29.42±1.13 and
28.79±0.95%, respectively, which was significantly lower compared
to that in the control (untreated) group (41.63±2.55%) and the
control-siRNA group (40.36±2.32%) (P<0.05; Fig. 5A). The percentage of cells in the
G0/G1 phase of the cell cycle in the
ATF3-siRNA group and the cisplatin group was 62.33±2.35 and
62.63±2.21%, respectively, which was significantly higher compared
to that in the untreated control group (55.84±2.23%) and the
control-siRNA group (56.27±2.13%; P<0.05). The proliferation
indices calculated for the ATF3-siRNA group and the cisplatin group
were significantly lower than those for the untreated control group
and the control-siRNA group (P<0.05; Fig. 5B). Compared with the ATF3-siRNA
group and the cisplatin group, the cells in the ATF3-siRNA +
cisplatin group exhibited a lower percentage of cells in the S
phase and a higher percentage of cells in the
G0/G1 phase, and had a significantly
decreased proliferation index (P<0.05). Following comparisons
between the ATF3-siRNA group and the cisplatin group, and between
the untreated control group and the control-siRNA group, no
significant differences were observed (P>0.05).
Knockdown of ATF3 using ATF3-siRNA
promotes the apoptosis of U373MG cells in vitro
Compared with the untreated control group and the
control-siRNA group, the proportion of apoptotic cells in the
ATF3-siRNA group, the cisplatin group and the ATF3 siRNA +
cisplatin group (25.84±3.107, 38.12±4.543 and 54.89±5.739%,
respectively) gradually increased (P<0.05) and intergroup
comparisons between these 3 groups revealed significant differences
(P<0.05). Apoptotic signals in the negative control group were
absent (0%), but were present in virtually all cells in the
positive control group (97.72±2.902%; Fig. 6).
Knockdown of ATF3 using ATF3-siRNA
suppresses the invasion ability of U373MG cells in vitro
As the initial counts of cells added into the
Transwell inserts were identical, changes in the invasion ability
of the cells were evaluated by comparing the number of cells that
penetrated the Transwell inserts to the initial number of cells
present. The percentage of migrating cells (the number of cells
that penetrated the Transwell inserts as a percentage of the
initial number of cells) in each group was as follows: 76±6.5% in
the untreated control group, 70±5.4% in the control-siRNA group,
43±3.8% in the ATF3-siRNA group, 26±4.2% in the cisplatin group and
25±3.2% in the ATF3 siRNA + cisplatin group. The difference between
the untreated control and the control-siRNA group was not
statistically significant (P>0.05). Compared to the untreated
control group and the control-siRNA group, the percentage of
migrating cells in the ATF3-siRNA group, the cisplatin group and
the ATF3 siRNA + cisplatin group was markedly decreased
(P<0.05). Compared with the ATF3-siRNA group, the percentage of
migrating cells in the cisplatin group and the ATF3 siRNA +
cisplatin group decreased even more substantially (P<0.05).
There was no significant difference observed between the number of
migrating cells in wells receiving ATF3 siRNA + cisplatin treatment
and wells treated only with cisplatin (P>0.05; Fig. 7).
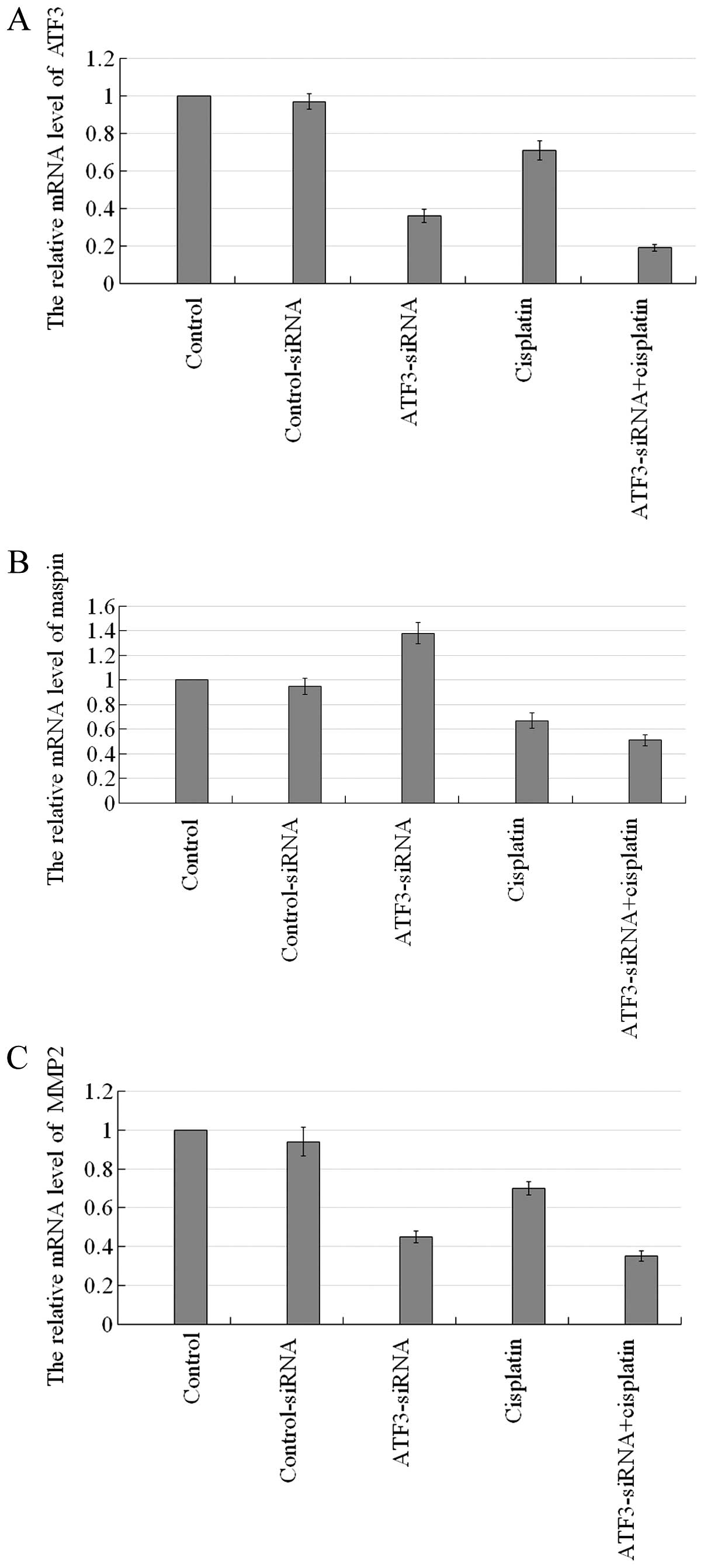
Knockdown of ATF3 using ATF3-siRNA
affects the mRNA levels of ATF3, maspin and MMP2 in U373MG cells in
vitro
A comparison between the untreated contorl group and
the control-siRNA group revealed no significant difference
(P>0.05), whereas a comparison between the other groups revealed
significant differences (P<0.05; Fig. 8). The mRNA levels of ATF3 and MMP2
in each experimental group were basically at the same level, while
they showed the highest expression level in the untreated control
group and the control-siRNA group, followed by, in decreasing
order, by the cisplatin group, the ATF3-siRNA group and the
ATF3-siRNA + cisplatin group. The relative mRNA expression level of
maspin was highest in the ATF3-siRNA group, followed by the
untreated control group and the control-siRNA group, the cisplatin
group, with the lowest expression being observed in the ATF3-siRNA
+ cisplatin group.
Knockdown of ATF3 using ATF3-siRNA
affects the protein levels of ATF3, maspin and MMP2 in U373MG cells
in vitro
A comparison between the untreated control group and
the control-siRNA group revealed no significant difference
(P>0.05), whereas a comparison between the other groups revealed
significant differences (P<0.05; Fig. 9). The protein expression levels of
ATF3 and MMP2 in each experimental group were basically at the same
level, with the highest expression level being observed the
untreated control group and the control-siRNA group, followed by,
in decreasing order, by the cisplatin group, the ATF3-siRNA group
and the ATF3-siRNA + cisplatin group. The relative protein
expression level of maspin was highest in the ATF3-siRNA group,
followed by the untreated control group and the control-siRNA
group, the cisplatin group, with the lowest expression being
observed in the ATF3-siRNA + cisplatin group.
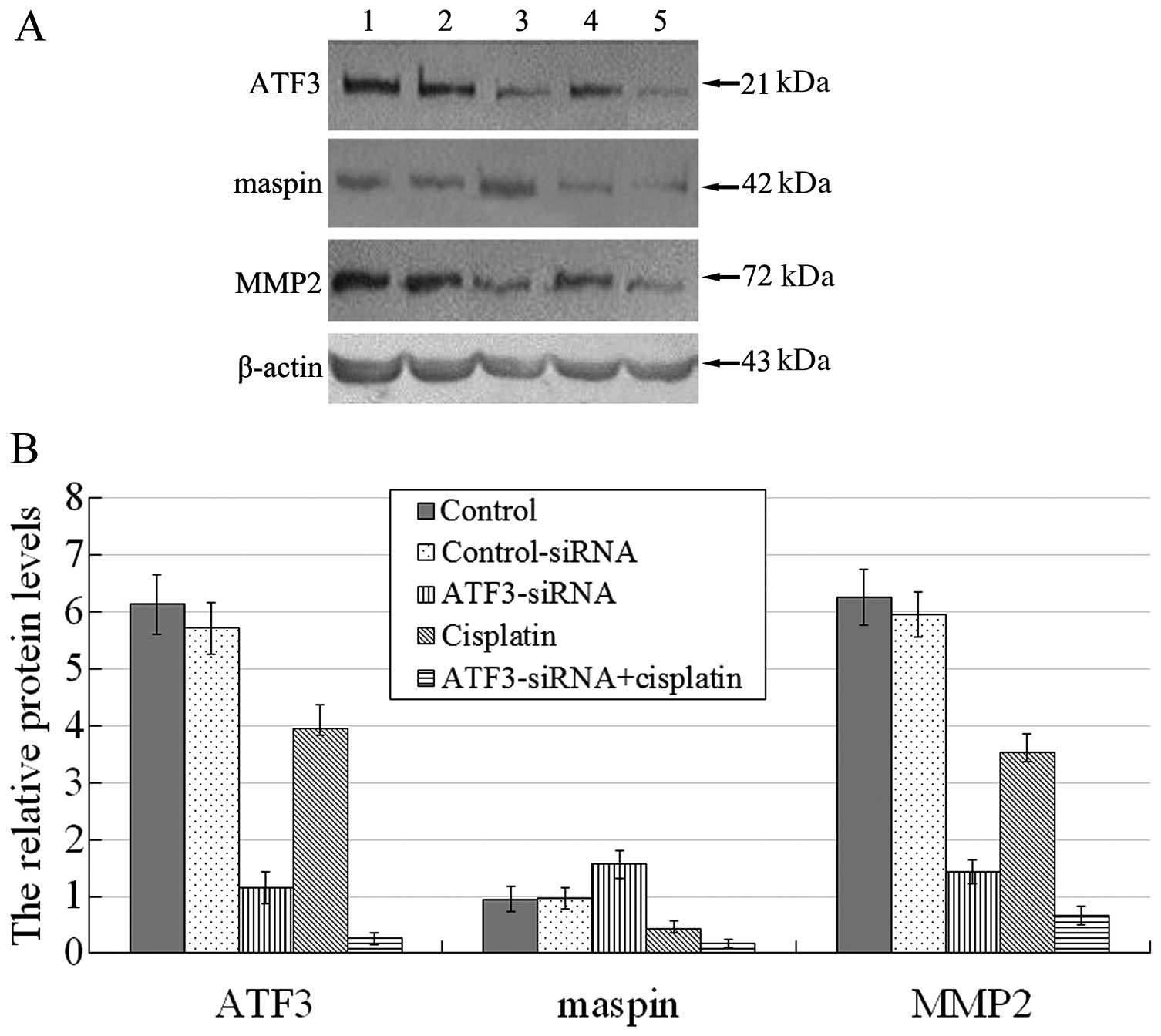 | Figure 9(A) Effects of transfection with
activating transcription factor 3 (ATF3)-siRNA and of cisplatin on
the protein levels of ATF3, maspin and matrix metalloproteinase 2
(MMP2). Lane 1, untreated control group; lane 2, control-siRNA;
lane 3, ATF3-siRNA group; lane 4, cisplatin group; lane 5,
ATF3-siRNA + cisplatin group. (B) Effects of transfection with ATF3
siRNA and of cisplatin on the relative protein levels of ATF3,
maspin and MMP2. ATF3 and MMP2 protein expression was consistent in
each experimental group and the expression of these 2 proteins
(relative to β-actin) in the control group and control-siRNA group
was the highest, followed by the cisplatin group; it was much lower
in the ATF3-siRNA group, and lowest in the ATF3-siRNA + cisplatin
group. By contrast, the relative protein expression of maspin was
highest in the ATF3-siRNA group, followed by the untreated control
group and control-siRNA group, much lower in the cisplatin group,
and lowest in the ATF3-siRNA + cisplatin group. |
Knockdown of ATF3 using ATF3-siRNA
inhibits the growth of U373MG xenograft tumors in vivo
Fifteen days after the subcutaneous injection of
U373MG cells, the 40 nude mice were randomly assigned into 5 groups
for treatment. Over the course of treatment several mice died: 2 in
the vehicle control group, 2 in the control-siRNA group, 3 in the
ATF3-siRNA group, 3 in the cisplatin group and 2 in the ATF3-siRNA
+ cisplatin group. The possible cause of death of the nude mice was
that the individual difference of the nude mice caused them to have
poor tolerance to the tumor load or they were allergic to the
injected preparations. A significant difference was observed in
tumor size between the ATF3-siRNA + cisplatin group and the other
groups by day 6 post-treatment (P<0.05), while inter-group
comparisons among other groups revealed no significant difference
(P>0.05; Fig. 10). Intergroup
comparisons among the vehicle control group, the control-siRNA
group and the ATF3-siRNA group revealed no significant difference
on day 11 post-treatment (P>0.05), while intergroup comparisons
among other groups revealed significant differences (P<0.05). A
comparison between the vehicle control group and the control-siRNA
revealed no significant difference (P>0.05) on day 16, while
intergroup comparisons among other groups revealed significant
differences (P<0.05). The statistical interpretations for day 21
were the same as those for day 16. Tumor growth curves indicated
that, compared with the vehicle control group and the control-siRNA
group, tumor growth in the ATF3-siRNA group was attenuated, with a
marked reduction in tumor size, although growth was slower and the
tumor size smaller in the cisplatin group, and growth was slowest
and tumor size smallest in the ATF3-siRNA + cisplatin group. The
tumor growth inhibition ratio calculated by tumor weight (values in
brackets) for day 21 with the vehicle control group as a negative
control were: vehicle control group, 0% (1.58±0.35 g);
control-siRNA group, 8.9±2.3% (1.44±0.31 g); ATF3-siRNA group,
41.1±3.7% (0.93±0.22 g); cisplatin group, 54.4±4.5% (0.72±0.17 g);
and ATF3-siRNA + cisplatin group, 75.3±5.5% (0.39±0.11 g). A
comparison between the vehicle control group and the control-siRNA
group revealed no significant difference (P>0.05), whereas
comparisons among other groups revealed significant differences
(P<0.05; Fig. 10B).
 | Figure 10(A) Growth curves for U373MG tumor
xenografts in vivo. From day 16 after treatment, compared to
the vehicle control group and the control-siRNA group, the growth
of U373MG cells in vivo was slower in the activating
transcription factor 3 (ATF3)-siRNA group and tumor volume was
significantly reduced (P<0.05). Growth in the cisplatin group
was much slower, and tumor volume was much smaller (P<0.05) and
tumor growth in the ATF3-siRNA + cisplatin group was the slowest,
with the smallest volume (P<0.05). (B) Tumor size on day 21. (a)
Vehicle control group, (b) control-siRNA group, (c) ATF3-siRNA
group, (d) cisplatin group, (e) ATF3-siRNA + cisplatin group.
Compared to the vehicle control group and the control-siRNA group,
tumor volume was significantly reduced (P<0.05) in the
ATF3-siRNA group. Tumor volume was much smaller (P<0.05) in the
cisplatin group, and smallest in the ATF3-siRNA + cisplatin group
(P<0.05). |
Discussion
ATF3 is a member of the ATF/CREB subfamily of the
basic region-leucine zipper family. ATF3 regulates the expression
of its target genes through complex mechanisms. It is an important
regulatory factor for transcription, apoptosis, cell division and
survival, and it plays an important role in controlling tumor
invasion and migration (4–6).
It has been reported that ATF3 is an oncogene and that it is
overexpressed in human cancer tissues; for example, the gene copy
number of ATF3 is significantly increased in breast cancers, which
may be due to the increase in the ATF3 gene on chromosome 1q
amplicon (the region with the largest increase in breast cancer)
(12). In contrast to
non-Hodgkin’s lymphoma and non-malignant tissue, a high level
expression of ATF3 has been observed in Reed-Sternberg cells of
patients with Hodgkin’s disease (2). Experimental results from the study
by Pelzer et al (1)
indicated that ATF3 was highly expressed in most prostate cancer
cell lines, and that the overexpression of ATF3 induced the
proliferation of prostate cancer cells and accelerated cell cycle
progression from the G1 to the S phase. In accordance
with these aforementioned experimental results, the present study
demonstrated that glial cells in normal brain tissue also had a
weak expression of ATF3, and that the expression of ATF3 was
upregulated in glioma tissues, and increased with the increasing
pathological grade of the glioma. ATF3 was also highly expressed in
the glioblastoma cell line, U373MG, suggesting that a high
expression level of ATF3 is closely related to the evolution of
glioma and its malignant progression.
There is evidence in the literature that ATF3 may
often promote the invasion and metastasis of cancer cell lines
in vitro and in vivo. For example, Ishiguro et
al (13) observed a high
expression level of ATF3 in a highly metastatic subline of B16
melanoma cells and demonstrated that low-migratory B16 cells were
changed into highly migratory cells by transfection with ATF3;
however, ATF3 was not expressed in the parental B16 cell line.
Bandyopadhyay et al (14)
found that the transcription of ATF3 in a prostate cancer model was
inhibited by the metastasis repressor, Drg-1; this suggests that
ATF3 promotes metastasis in prostate cancer.
Related studies have also confirmed that ATF3 may be
carcinogenic. For example, Ishiguro et al (15) abolished the expression of ATF3
using antisense oligonucleotides in vitro, and this reduced
the adhesion and invasion of HT29 colon cancer cells. The effects
of ATF3 antisense oligonucleotide intervention in mice inoculated
subcutaneously with HT29 cells were investigated; the results
revealed that the mice that received ATF3 antisense oligonucleotide
intervention therapay, compared with the controls, were less likely
to form tumors and had a longer average survival. A further
demonstration of the effects of the selective knockdown of ATF3
expression by RNA interference was shown in the study by Janz et
al (2) demonstrating that the
proliferation of Hodgkin’s lymphoma cells was inhibited, with a
reduced viability of the lymphoma cells, suggesting that ATF3 is
related to the proliferation of cancer cells.
Our study supports the aforementioned studies. Our
analysis of cell numbers following treatment of the U373MG cells
in vitro (MTT assay) also suggested that following
transfection with the plasmid expressing ATF3-siRNA, U373MG cell
proliferation was reduced, cell cycle progression was inhibited and
the growth inhibition rate of the U373MG cells gradually increased
progressively with time. Our cell cycle analysis by flow cytometry
revealed that, following transfection with ATF3-siRNA, the
proportion of cells in the S phase was significantly reduced, and
the percentage of cells arrested in the G0/G1
phase was significantly higher; the cell proliferation index was
significantly lower than that for the control group. Our TUNEL
staining results revealed that the proportion of U373MG cells
showing signs of apoptosis increased significantly following
transfection with ATF3-siRNA compared to the control group.
Additionally, the invasion ability of the U373MG cells in Transwell
culture was significantly weakened following transfection with
ATF3-siRNA. Taken together, our experiments all directly confirm
that transfection with ATF3-siRNA inhibits cell proliferation,
reduce cell viability, arrests cell cycle progression, promotes
apoptosis and impedes the migration of human glioblastoma cells,
thereby indirectly supporting that ATF3 plays a facilitating role
in the process of tumor invasion and metastasis.
Finally, our in vivo experiments confirmed
that the repeated peritumoral injection of ATF3-siRNA compared with
the vehicle-treated controls effectively inhibited the growth of
U373MG tumor xenografts in nude mice. Treatment of xenograft tumors
with ATF3-siRNA began to show effects at the 16th day of treatment;
tumor growth curves revealed that tumor growth in the ATF3-siRNA
group was attenuated and the tumor volume was significantly
reduced.
The inhibitory effect on tumors was remarkable and
similar effects were observed between ATF-siRNA and cisplatin
treatments. The growth curves of the xenograft tumors of the nude
mice indicated that tumor growth in the cisplatin group and the
ATF3-siRNA + cisplatin groups was attenuated in a time-dependent
manner; thus, the rate of increase in tumor volume decreased with
time. Our growth data for the U373MG cells in vitro (MTT
assay) indicated that cell growth was significantly suppressed in
the cisplatin group and the ATF3-siRNA + cisplatin group, and the
number of cells in both groups was significantly reduced compared
with the first day. The reason for this difference in results may
be that under in vitro conditions, human glioblastoma U373MG
cells were more sensitive to cisplatin and ATF3-siRNA +
cisplatin.
However, there is some evidence indicating the
contrary, suggesting that ATF3 inhibits cancer formation. For
example, the experimental results from the study by Lu et al
(16) demonstrated that ATF3
suppressed Rasstimulated tumorigenesis in vivo. Another
example is that the overexpression of ATF3 has been shown to reduce
the size of tumor xenografts of HCT-116 human colorectal cancer
cells placed subcutaneously in nude mice (17). The reason why these results differ
materially may be due to the different experimental conditions and
different experimental cell lines used. We confirm that, when faced
with different cell types and driving factors, ATF3 plays different
roles in the developmental process of cancer.
Maspin is a tumor suppressor gene discovered in 1994
(19). Maspin protein is
moderately to highly expressed in a number of normal tissues, and
plays an important role in inhibiting tumor growth, increasing cell
adherence, reducing cell movement and invasion, and suppressing
tumor angiogenesis; however, its expression is downregulated during
tumor progression (18). Its loss
is related to such factors as high malignancy, large tumor size, a
high histological grade, lymph node metastasis, local recurrence,
tumor development and a short survival time (19). The results of this study indicate
that maspin protein was present both in the nucleus and cytoplasm
and was highly expressed in the normal brain tissues. Its
expression decreased in glioma from grade I to IV and negatively
correlated with the pathological grade of the glioma. In U373MG
cells, maspin protein was only moderately expressed and the
expression rate was 47±6.4%. These results are consistent with the
experimental results from the studies of both Zhang and Zhang
(18) and Wang et al
(20).
The important role of MMP2 in tumor
neovascularization, cell infiltration and metastasis formation as a
tumor-enhancing gene has been well-established (21–25,32,33). The results of our study
demonstrated that the protein expression of MMP2 in glioma tissues
was substantially higher than that in normal brain tissues, and the
expression of MMP2 was also high in the U373MG cells. These results
are consistent with the published study by Herbst et al
(26).
In this study, we found that the ATF3 protein and
mRNA levels in the human glioma tissues positively correlated with
the expression of MMP2, and their expression was increased with the
increasing pathological grade of the glioma. ATF3 expression
negatively correlated with maspin expression, and the relative
protein and mRNA expression of maspin was reduced with the
increasing pathological grade of the glioma. In vitro, the
expression of ATF3 and MMP2 in the U373MG cells was also consistent
in each experimental group. Following transfection of the U373MG
cells with ATF3-siRNA, the protein and mRNA levels of MMP2
decreased significantly, while the maspin protein and mRNA levels
increased significantly; ATF3-siRNA downregulated the expression of
MMP2, but upregulated that of maspin. These effects correspond with
our experimental observations of human glioma tissues, that is, the
expression of ATF3 showed the same trend as that of MMP2, but an
opposite trend with maspin expression.
MMP2 is recognized as one of the strong
cancer-promoting genes which promotes the invasion and metastasis
of malignant glioma, and promotes tumor metastasis by degrading the
extracellular matrix (21). The
consistent association between ATF3 and MMP2 expression suggests
that ATF3 exerts a similar function with MMP2 in some ways, and
plays a role in promoting tumor metastasis. In contrast to MMP2,
maspin is one of the classic tumor suppressor genes (27–29), and the opposite changes observed
for ATF3 versus maspin expression suggest that the function of ATF3
opposes that of maspin, with ATF3 having a tumor-promoting role
that is opposite to that of maspin.
The correlations between ATF3, maspin and MMP2
expression suggest that that they are placed at the intersection of
molecular signaling pathways. One of the regulatory mechanisms of
maspin involves p53 signaling; the study by Zou et al
(30) demonstrated that infecting
breast and prostate cancer cell lines with wild-type p53 adenovirus
induced maspin expression. ATF3 and maspin both can bind p53 to
induce a series of responses, so they are likely to act on the p53
pathway in glioblastomas. Maspin can also lead to a cell stress
response through transcriptional regulation, and maspin
re-expression may also lead to the suppression of several genes
involved in the inflammatory response (27–30). ATF3 is not only an early stress
response gene, but is also a hub of the cellular adaptive-response
network for stress signals, and acts as the key regulation point
that can be induced by a variety of factors (31). Thus, it may be speculated that
ATF3 and maspin also act at the intersection of the stress response
pathway and participate in the regulation of pro-inflammatory
factors.
There are a number of sites on the MMP2 promoter
which can be bound by regulatory elements, including binding sites
for p53 and the cAMP response element binding protein (CREB)
(32,33). ATF3 is a member of the CREB
subfamily. In addition, MMP2 inhibits the inflammatory process, and
MMP2 gene expression regulates multiple genes at the
transcriptional and post-transcriptional level through the MAPK
pathway (32,33); the MAPK and p53 pathways are
closely related to ATF3 (1,34).
Thus, it can be speculated that the correlation in the expression
of MMP2 and ATF3 acts in a complex linkage to regulate signaling
pathways and the expression of inflammatory cytokines.
Contrary to our results, there are studies
describing that ATF3 inhibits the expression of MMP2. Stearns et
al (35) found that ATF3
suppressed MMP2 expression by directly binding with the MMP2
promoter, and Yan et al (36) demonstrated that ATF3 exerted its
inhibitory effect by interfering with the transcriptional
activation of MMP2 through p53. These inconsistent experimental
results may be related to different conditions and apparatus used
in different laboratories, and perhaps with MMP2 and ATF3 in
different tissues through different signal transduction
pathways.
In this study, the inhibitory effects of ATF3-siRNA
were observed in the human glioblastoma cell line, U373MG, in
vitro and in tumor xenografts in vivo. This indirectly
suggests that ATF3 exerts promoting effects on the development and
invasion process of glioblastoma, although the most relevant target
genes and the corresponding signaling pathways through which ATF3
promotes glioblastoma cells to invade, and factors regulating ATF3
in glioblastoma during the invasion process have not yet been fully
elucidated. Further studies are required to clarify this, in order
to identify new molecular targets and develop new treatment
strategies for the treatment of glioblastoma.
Acknowledgments
We acknowledge the valuable assistance of Dr Lv Xiao
Dong and Dr Zhang Jing for providing technical advice, reagents and
helpful discussion.
References
|
1
|
Pelzer AE, Bektic J, Haag P, Berger AP,
Pycha A, Schäfer G, Rogatsch H, Horninger W, Bartsch G and Klocker
H: The expression of transcription factor activating transcription
factor 3 in the human prostate and its regulation by androgen in
prostate cancer. J Urol. 175:1517–1522. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Janz M, Hummel M, Truss M, et al:
Classical Hodgkin lymphoma is characterized by high constitutive
expression of activating transcription factor 3 (ATF3), which
promotes viability of Hodgkin/Reed-Sternberg cells. Blood.
107:2536–2539. 2006. View Article : Google Scholar
|
|
3
|
Iyengar P, Combs TP, Shah SJ, et al:
Adipocyte-secreted factors synergistically promote mammary
tumorigenesis through induction of anti-apoptotic transcriptional
programs and protooncogene stabilization. Oncogene. 22:6408–6423.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hai T and Curran T: Cross-family
dimerization of transcription factors Fos/Jun and ATF/CREB alters
DNA binding specificity. Proc Natl Acad Sci USA. 88:3720–3724.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wolfgang CD, Chen BP, Martindale JL,
Holbrook NJ and Hai T: gadd153/Chop10, a potential target gene of
the transcriptional repressor ATF3. Mol Cell Biol. 17:6700–6707.
1997.PubMed/NCBI
|
|
6
|
Chen BPC, Liang G, Whelan J and Hai T:
ATF3 and ATF3 delta Zip. Transcriptional repression versus
activation by alternatively spliced isoforms. J Biol Chem.
269:15819–15826. 1994.PubMed/NCBI
|
|
7
|
Chen Z, Fan Z, McNeal JE, Nolley R,
Caldwell MC, Mahadevappa M, Zhang Z, Warrington JA and Stamey TA:
Hepsin and maspin are inversely expressed in laser capture
microdissectioned prostate cancer. J Urol. 169:1316–1319. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Forsyth PA, Wong H, Laing TD, et al:
Gelatinase-A (MMP-2), gelatinase-B (MMP-9) and membrane type matrix
metallopro-teinase-1 (MT1-MMP) are involved in different aspects of
the pathophysiology of malignant gliomas. Br J Cancer.
79:1828–1835. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Louis David N, Ohgaki Hiroko, Wiestler
Otmar D, Cavenee Webster K, et al: Astrocytic tumours. Who
Classification of Tumours of the Central Nervous System. World
Health Organization; Geneva: pp. 13–45. 2007
|
|
10
|
Gatalica Z, Lele SM, Rampy BA and Norris
BA: The expression of Fhit protein is related inversely to disease
progression in patients with breast carcinoma. Cancer.
88:1378–1383. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
12
|
Yin X, Dewille JW and Hai T: A potential
dichotomous role of ATF3, an adaptive-response gene, in cancer
development. Oncogene. 27:2118–2127. 2008. View Article : Google Scholar
|
|
13
|
Ishiguro T, Nakajima M, Naito M, Muto T
and Tsuruo T: Identification of genes differentially expressed in
B16 murine melanoma sublines with different metastatic potentials.
Cancer Res. 56:875–879. 1996.PubMed/NCBI
|
|
14
|
Bandyopadhyay S, Wang Y, Zhan R, et al:
The tumor metastasis suppressor gene Drg-1 down-regulates the
expression of activating transcription factor 3 in prostate cancer.
Cancer Res. 66:11983–11990. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ishiguro T, Nagawa H, Naito M and Tsuruo
T: Inhibitory effect of ATF3 antisense oligonucleotide on ectopic
growth of HT29 human colon cancer cells. Jpn J Cancer Res.
91:833–836. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lu D, Wolfgang CD and Hai T: Activating
transcription factor 3, a stress-inducible gene, suppresses
Rasstimulated tumorigenesis. J Biol Chem. 281:10473–10481. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bottone FG Jr, Moon Y, Kim JS,
Alston-Mills B, Ishibashi M and Eling TE: The anti-invasive
activity of cyclooxygenase inhibitors is regulated by the
transcription factor ATF3 (activating transcription factor 3). Mol
Cancer Ther. 4:693–703. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang W and Zhang M: Tissue microarray
analysis of maspin expression and its reverse correlation with
mutant p53 in various tumors. Int J Oncol. 20:1145–1150.
2002.PubMed/NCBI
|
|
19
|
Zou Z, Anisowicz A, Hendrix MJ, Thor A,
Neveu M, Sheng S, Rafidi K, Seftor E and Sager R: Maspin, a serpin
with tumor-suppressing activity in human mammary epithelial cells.
Science. 263:526–529. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang DL, Wang YF, Shi GS and Huang H:
Correlation of hTERT expression to maspin and bFGF expression and
their significance in glioma. Ai Zheng. 26:601–606. 2007.In
Chinese. PubMed/NCBI
|
|
21
|
Song H, Li Y, Lee J, Schwartz AL and Bu G:
Low-density lipoprotein receptor-related protein 1 promotes cancer
cell migration and invasion by inducing the expression of matrix
metalloproteinases 2 and 9. Cancer Res. 69:879–886. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kodera T, Nakagawa T, Kubota T, Kabuto M,
Sato K and Kobayashi H: The expression and activation of matrix
metallo-proteinase-2 in rat brain after implantation of C6 rat
glioma cells. J Neurooncol. 46:105–114. 2000. View Article : Google Scholar
|
|
23
|
Musso O, Théret N, Campion JP, Turlin B,
Milani S, Grappone C and Clément B: In situ detection of matrix
metalloproteinase-2 (MMP2) and the metalloproteinase inhibitor
TIMP2 transcripts in human primaryhepatocellular carcinoma and in
liver metastasis. J Hepatol. 26:593–605. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mendes O, Kim HT and Stoica G: Expression
of MMP2, MMP9 and MMP3 in breast cancer brain metastasis in a rat
model. Clin Exp Metastasis. 22:237–246. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Danilewicz M, Sikorska B and
Wagrowska-Danilewicz M: Prognostic significance of the
immunoexpression of matrix metalloproteinaseMMP2 and its inhibitor
TIMP2 in laryngeal cancer. Med Sci Monit. 9:MT42–MT47.
2003.PubMed/NCBI
|
|
26
|
Herbst RS, Onn A and Sandler A:
Angiogenesis and lung cancer: Prognostic and therapeutic
implications. J Clin Oncol. 23:3243–3256. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bodenstine TM, Seftor RE, Khalkhali-Ellis
Z, Seftor EA, Pemberton PA and Hendrix MJ: Maspin: Molecular
mechanisms and therapeutic implications. Cancer Metastasis Rev.
31:529–551. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sheng S: The promise and challenge toward
the clinical application of maspin in cancer. Front Biosci.
9:2733–2745. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ngamkitidechakul C, Warejcka DJ, Burke JM,
O’Brien WJ and Twining SS: Sufficiency of the reactive site loop of
maspin for induction of cell-matrix adhesion and inhibition of cell
invasion. Conversion of ovalbumin to a maspin-like molecule. J Biol
Chem. 278:31796–31806. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zou Z, Zhang W, Young D, Gleave MG, Rennie
P, Connell T, Connelly R, Moul J, Srivastava S and Sesterhenn I:
Maspin expression profile in human prostate cancer (CaP) and in
vitro induction of Maspin expression by androgen ablation. Clin
Cancer Res. 8:1172–1177. 2002.PubMed/NCBI
|
|
31
|
Hai T, Wolford CC and Chang YS: ATF3, a
hub of the cellular adaptive-response network, in the pathogenesis
of diseases: Is modulation of inflammation a unifying component?
Gene Expr. 15:1–11. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
McCawley LJ and Matrisian LM: Matrix
metalloproteinases: Multifunctional contributors to tumor
progression. Mol Med Today. 6:149–156. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Fang J, Shing Y, Wiederschain D, Yan L,
Butterfield C, Jackson G, Harper J, Tamvakopoulos G and Moses MA:
Matrix metal-loproteinase-2 is required for the switch to the
angiogenic phenotype in a tumor model. Proc Natl Acad Sci USA.
97:3884–3889. 2000. View Article : Google Scholar
|
|
34
|
Rosenberger CM, Clark AE, Treuting PM,
Johnson CD and Aderem A: ATF3 regulates MCMV infection in mice by
modulating IFN-gamma expression in natural killer cells. Proc Natl
Acad Sci USA. 105:2544–2549. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Stearns ME, Kim G, Garcia F and Wang M:
Interleukin-10 induced activating transcription factor 3
transcriptional suppression of matrix metalloproteinase-2 gene
expression in human prostate CPTX-1532 Cells. Mol Cancer Res.
2:403–416. 2004.PubMed/NCBI
|
|
36
|
Yan C, Wang H and Boyd DD: ATF3 represses
72-kDa type IV collagenase (MMP-2) expression by antagonizing
p53-dependent trans-activation of the collagenase promoter. J Biol
Chem. 277:10804–10812. 2002. View Article : Google Scholar : PubMed/NCBI
|