Introduction
Interest in the therapeutic application of bone
marrow (BM)-derived mesenchymal stem cells (MSCs) has increased in
recent years, arising from the possibility that MSCs not only
support hematopoiesis (1,2), but may also ameliorate
graft-versus-host disease (GVHD) following allogeneic hematopoietic
stem cell (HSC) transplantation (3). Allogeneic HSC transplantation is
currently the only curative option for a number of hematological
malignancies, although large numbers of MSCs are required to
achieve clinical efficacy (4),
and the effects of MSCs on tumor initiation, survival, progression
and metastasis remain poorly understood. It is considered that MSCs
suppress tumor growth. Otsu et al demonstrated that the
direct inoculation of MSCs into subcutaneous melanomas induced
apoptosis and abrogated tumor growth by inhibiting angiogenesis
(5). Khakoo et al
demonstrated that systemically injected MSCs reduced tumor growth
in a model of Kaposi’s sarcoma through the inhibition of Akt
(6), Zhu et al reported
that human MSCs inhibited the proliferation of K562 cells by the
secretion of Dickkopf-related protein 1 (DKK-1) (7). Wang et al recently reported
that MSCs inhibit the proliferation of hepatic stellate cells
through the inhibition of Toll-like receptor 4 (TLR4) signaling
(8), and Menge et al
reported that MSCs inhibit endothelial cell proliferation and
angiogenesis through the modulation of the VE-cadherin/β-catenin
signaling pathway (9). However,
MSCs have also been reported to promote tumor growth. Galiè et
al reported that MSCs co-implanted with cancer cells in
syngeneic animals accelerated the appearance of tumors (10), possibly by promoting the
angiogenic switch. MSCs have also been shown to increase the
metastatic potential of breast cancer cell lines without altering
primary tumor progression (11).
Clearly, these data present a confusing picture of the contribution
of MSCs to tumor formation, indicating that much study lies ahead
in this field.
The aim of this study was to evaluate the
therapeutic potential application of MSCs in allogeneic bone marrow
transplantation (BMT) in hemotological malignanciess. First, we
observed that in cell culture, C57BL/6 (B6) mouse MSCs inhibited
the proliferation of leukemia and lymphoma cells, leading to cell
cycle arrest and promoting apoptosis. In addition, in model of
allogeneic BMT, transplanted MSCs inhibited the development of
tumors induced by an injection of A20 B lymphoma cells. Our
findings suggest that the clinical application of MSCs may
contribute to the effectiveness of HSC transplantation in
hematological malignancies.
Materials and methods
Mice
BALB/c (H-2d) and C57BL/6
(H-2b) (commonly known as B6 mice) mice (6–8 weeks old)
were obtained from the Shanghai SLAC Laboratory Animal Co., Ltd.
(Shanghai, China) and housed in plastic cages under specific
pathogen-free conditions at the Institute for Animal Experiments,
the Second Military Medical University (Shanghai, China). Chow and
water were available at all times. The mice used in the experiments
were gender- and age-matched. All animal experiments were performed
following the approval of the Animal Care and Use Committee of the
Changhai Hospital, Second Military Medical University (Shanghai,
China).
Preparation of MSCs
The B6 mice were sacrificed by cervical dislocation,
and the femurs and tibias were removed and cleaned of all
connective tissue. BM cells were collected by flushing the femurs
and tibias with medium using a 26-gauge needle (Shandong Weigao
Group Medical Polymer Co., Ltd., Shandong, China), filtered, and
washed twice by centrifugation at 1,500 rpm for 6 min. The cells
were cultivated in 21-cm2 plates (BD Biosciences,
Franklin lakes, NJ, USA) at 106 cells/cm2 in
Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, Grand Island, NY,
USA), supplemented with 10% FCS (Gibco), 100 IU/ml penicillin, 100
µg/ml streptomycin (Sigma, St. Louis, MO, USA) and 2 mM
L-glutamine (Sigma) at 37°C in 5% CO2. After 48 h, the
non-adherent cells were removed and the medium was replenished
every 3 days. At 70–80% confluence, the adherent cells were
incubated for 2–3 min at 37°C with a 0.05% trypsin solution
containing 0.02% EDTA (PAA). Trypsin was neutralized by the
addition of fresh complete medium, and the cells were then
harvested, and re-plated at a ratio of 1:2. MSCs were identified by
detecting the expression of CD29, CD44, CD90.2, stem cell antigen-1
(Sca-1), fms-related tyrosine kinase 1 (Flk-1), c-kit and major
histocompatibility complex (MHC)-I, and by the absence of the
expression of CD34, CD45, MHC-II, determined by flow cytometry
(using a flow cytometer, BD Biosciences). All antibodies used
[PE-CD29 (12-0291), PE-CD44 (12-0441), APC-CD90.2 (17-0902),
PE-Sca-1 (15-5981), PE-Flk-1 (12-5821), FITC-c-Kit (11-1171),
PE-MHC-I (12-5999), Biotin-CD34 (13-0341), FITC-CD45 (11-0451),
PE-MHC-II (12-5322)] were purchased from eBioscience, Inc. (San
Diego, CA, USA). MSCs at passages 3–10 were used in the
experiments.
Cell culture
The A20 B lymphoma cell line (H-2d), FBL3
erythroleukemia cell line (H-2b), P388 acute lymphocytic
leukemia cell line (H-2k) (all from the Cell Resource
Center of the Shanghai Institute of Life Science of the Chinese
Academy of Sciences) were kept in liquid nitrogen. The cells were
maintained in RPMI-1640 medium (Gibco) containing 10%
heat-inactivated newborn calf serum (NCS) (purchased from Gibco),
100 U/ml penicillin and 100 µg/ml streptomycin in a
humidified atmosphere of 5% CO2 at 37°C.
Co-culture of MSCs and leukemia and
lymphoma cell lines
The mouse MSCs were harvested and pre-treated with
25 µg/ml mitomycin C (MMC) (Sigma) at 37°C for 20 min, then
washed twice with PBS and resuspended in DMEM supplemented with 10%
FCS, 2 Mm glutamine, 100 U/ml penicillin and 100 µg/ml
streptomycin, and plated into a 96-well plate at 2×103,
5×103, 2×104 and 5×104 cells/well
in 0.2 ml medium. After 10 h, the MSCs had adhered to the plates,
and the medium was removed. The A20, FBL3, P388 cancer cell lines
were added to the wells to achieve an MSC:cancer cell ratio of
1:10, 1:4, 1:1, or 1:0.4 in 0.2 ml medium, and co-cultured for 48
h. The cancer cell lines were cultured alone or with the MSCs for
24, 48 and 72 h. The non-adherent cells (leukemia and lymphoma
cells) were collected to assess cell proliferation and the cell
cycle.
To determine whether the MSCs inhibit A20 cell
proliferation through the Akt protein kinase pathway, an Akt
inhibitor (Cat. no. 124005; Calbiochem, La Jolla, CA, USA) was
added to the A20 cells co-cultured with the MSCs at a ratio of 1:1
at a final concentration of 5 µM. After 48 h, the cells were
collected for further analysis.
Assessment of cell proliferation
The floating A20 cells cocultured with the MSCs were
isolated from the MSCs which were attached to the Transwell
membrane (Corning, Inc., Corning, NY, USA). Cell viability was
assessed using a Cell Counting kit-8 (CCK-8) (Dojindo Laboratories,
Kumamoto, Japan). A total of 6 wells was included in each sample
and MSCs were used as controls. CCK-8 was added 4 h before the end
of the culture time. Wells without cells were set as blanks. The
absorbance at 450 nm was measured using a Universal Microplate
Spectrophotometer (Thermo Fisher Scientific, Inc., Waltham, MA,
USA). The relative proliferation rate (%) of the cancer cells was
calculated as follows: (OD450 of cancer cell lines with
MSCs - OD450 of MSC control)/OD450 of cancer
cell lines ×100.
Assessment of cell cycle progression and
early apoptosis
To determine the effect of MSCs on cancer cell
apoptosis and cell cycle distribution, the MSCs were seeded into
6-well plates at 1×105 cells/well. After 10 h, the MSCs
had adhered to the plates, the medium was removed followed by the
addition of 1×105/well of cancer cells After 72 h, the
floating cancer cells were harvested. The apoptosis of the cancer
cells was analyzed by Annexin V/propidium iodide (PI) staining
(eBio-science, Inc.) according to the manufacturer’s instructions.
For cell cycle analysis, 1 million cancer cells were fixed with 70%
cold ethanol at 4°C overnight, washed with PBS twice, and 10
µg (1 mg/ml) RNase and 0.3 ml (50 µg/ml) PI (Sigma)
were then added to the cell suspension for 30 min. Cell
fluorescence was assessed using a BD FACSCalibur Flow Cytometer and
analyzed using CellQuest software (BD Biosciences). Data were
analyzed using ModFit software.
In order to determine the expression of cell cycle-
and apoptosis-related genes, the mRNA expression of p21 and
caspase-3 was analyzed by reverse transcription-quantitative
PCR (RT-qPCR). Total RNA was extracted using TRIzol reagent
(Invitrogen Life Technologies, Carlsbad, CA, USA), and the
concentration and purity of the RNA were estimated by optical
density measurements. For PCR, the cDNA samples were standardized
based on the mRNA expression of β-actin. Total RNA (500 ng) was
reverse transcribed and amplified using the Takara PrimeScript One
Step RT-PCR kit [Takara Biotechnology (Dalian) Co., Ltd., Liaoning,
China]. RT-PCR was performed using the following primers for 43
cycles at 95°C for 2 min, at 95°C for 13 sec, and at 58°C for 1
min: p21 forward, 5′-CCCGAGAACGGTGGAACT-3′ and reverse,
5′-AGAGGGCAGGCAGCGTAT-3′; caspase-3 forward,
5′-ATGTCATCTCGCTCTGGT-3′ and reverse, 5′-TCTG TTTCTTTGCGTGGA-3′;
and β-actin forward, 5′-GCCA TGTACGTAGCCATCCA-3′ and
reverse, 5′-AACCGCT CATTGCCGATAGT-3′. Quantitative (real-time) PCR
(qPCR) was performed using an ABI PRISM Sequence Detection System
7500 (Applied Biosystems, Foster City, CA, USA) with the
QuantiTect™ SYBR-Green PCR kit (Qiagen, Hilden, Germany).
Triplicate wells were averaged and the relative quantities of
p21 and caspase-3 were then calculated using the
comparative Ct method. The mRNA expression levels were normalized
using the β-actin control 1/2ΔCT value. The relative
gene expression was calculated in triplicate as follows: the amp
lication of (cancer cells co-cultured with MSCs)/(cancer cells
alone).
Measurement of extracellular cytokine and
intracellular interleukin (IL)-10 levels by ELISA
The A20 cells were cultured alone or with MSCs at a
ratio of 1:1. After 24, 48 or 72 h the supernatants were harvested,
and the concentrations of IL-10, transforming growth factor
(TGF)-β, tumor necrosis factor (TNF)-α, and interferon (IFN)-γ were
measured by ELISA (eBioscience, Inc.) according to the
manufacturer’s instructions. Intracellular IL-10 staining was
performed on the cells after 48 h of co-culture using the
anti-mouse IL-10 staining kit according to the directions for use
(eBioscience, Inc.). The cells were collected, washed with PBS and
resuspended in PBS. Mouse FITC-CD19 antibody (11-0193) was added,
followed by the addition of fixation and penetrating solutions.
Thereafter, mouse PE-IL-10 antibody (12-7101) was added. After 20
min, the cells were washed with penetrating solution and
resuspended in PBSC. The numers of
CD19+IL-10+ cells were assessed using a BD
FACSCalibur Flow Cytometer and analyzed using CellQuest software
(BD Biosciences).
Allogeneic BMT in mice injected with A20
B lymphoma cells
The BABL/c recipients were conditioned with a lethal
7-Gy (60Co, 80c Gy/min) dose of total body irradiation,
6 h before transplantation. The mice were divided into 5 groups
according to the intravenous infusion of cell types as follows: i)
the control group (n=10), no cells but only PBS was administered;
ii) the BM group (n=10), 1×107 donor BM cells were
injected; iii) the BM-MSC group (n=10), 1×107 donor BM
cells and 5×105 MSCs were injected; iv) the A20 group
(n=17), 1×107 donor BM cells and 1×104 A20
cells were injected; and v) the MSC-A20 group (n=17),
1×107 donor BM cells, 1×104 A20 cells and
5×105 MSCs were injected. The survival and appearance of
the mice were monitored daily, and body weight was measured every
other day. Kaplan-Meier survival curves were established for each
group. Mice suffering from advanced-stage disease were sacrificed
for histological examination, and this event was considered as a
death in the survival curve.
In order to assess the homing of the allogenic MSCs,
we injected the above-mentioned mice with MSCs stained with a
fluorescence marker. The MSCs were resuspended in DMEM supplemented
with 10% FBS and washed twice with PBS. Fluorescence labeling was
performed by incubating 107 MSCs in 1 ml Diluent C
supplemented with freshly prepared 16 µM PKH67 membrane
linker (Sigma) for 5 min at room temperature, and the staining
reaction was then terminated by the addition of an equal volume of
serum. Donor BM cells (1×107) were injected
intravenously with or without 5×105 murine MSCs. The
mice were sacrificed 24 h or 7 days following transplantation for
the frozen section examination of the tissues of the heart, lungs,
liver, spleen, small intestine and kidneys. The 8 µm
cryosections were then observed under a fluorescence microscope
(Leica, Wetzlar, Germany).
Analysis of T cell subsets
On days 7 and 14 following transplantation, 100
µl peripheral blood was collected from the retro-orbital
vein of each mouse under ether anesthesia in a heparinized tube,
and lymphocyte subset analysis was performed using
fluorochrome-conjugated anti-mouse monoclonal antibodies (mAbs):
murine PE-cy5.5-CD3e, FITC-CD4, PE-CD8, APC-CD25 and PE-MHC-I
(H-2Kb) (eBioscience, Inc.), followed by 30 min of
incubation and then by erythrocyte lysis using BD FACS Lysing
Solution (BD Biosciences) according to manufacturer’s instructions.
The samples were washed twice in PBS and the pellet was resuspended
in 200 µl of PBS. Approximately 20,000 events/sample were
acquired on a BD FACSCalibur Flow Cytometer and analyzed using
CellQuest software (BD Biosciences).
Histological examination
Four weeks after allogeneic BMT, the remaining mice
were sacrificed. The livers, small intestine, lungs and spleen were
obtained in order to evaluate histological changes. The tissue
samples were fixed in 4% formaldehyde solution for several days and
embedded in paraffin, and 5-µm sections were stained with
hematoxylin and eosin (H&E) for histological examination.
Statistical analysis
Unless otherwise stated, the experiments were
repeated at least 3 times. Statistical analysis was performed using
Excel (Microsoft) and SPSS 11.0 statistical analysis software
(SPSS, Inc., Chicago, IL, USA). All experimental quantitative data
are expressed as the means ± standard deviation. A Student’s t-test
or covariance analysis were performed for statistical analysis.
Percentages were compared using the Chi-square test (Fisher’s exact
test). Kaplan-Meier survival curves were established for each
group. A value of P<0.05 was considered to indicate a
statistically significant difference.
Results
MSCs inhibit the proliferation of
leukemia and lymphoma cells
We investigated the effects of the MSCs on the
prolif-erative activity of leukemia and lymphoma cells of different
lineages. The A20 B lymphoma cells (H-2d), the FBL3
erythroleukemia cells (H-2b) and the P388 acute
lymphocytic leukemia cells (H-2k) were cultivated with
the MSCs for 48 h. When the B6 MSCs were added to the culture in
concentrations equivalent to 1 MSC to 1 or 0.4 leukemia cells, the
proliferation of the leukemia and lymphoma cells was inhibited;
however when the leukemia and lymphoma cells were in excess, the
proliferation was not inhibited (Fig.
1A). Furthermore, the anti-proliferative effects of the MSCs
increased with increasing co-culture times (Fig. 1B), but these effects were lost
when the MSCs and leukemia/lymphoma cells were separated by a
permeable membrane (Fig. 1C),
indicating that the MSCs inhibited the proliferation of the
leukemia and lymphoma cells in a contact-dependent manner. In
addition, although it has previously been reported that human MSCs
suppress tumor development by inhibiting target-cell Akt activity
(6), we did not find that Akt
inactivation affected the proliferation of the A20 cells
co-cultured with MSCs (Fig. 1C),
suggesting that the inhibition of lymphoma cell proliferation by
mouse MSCs may not involve the inhibition of Akt.
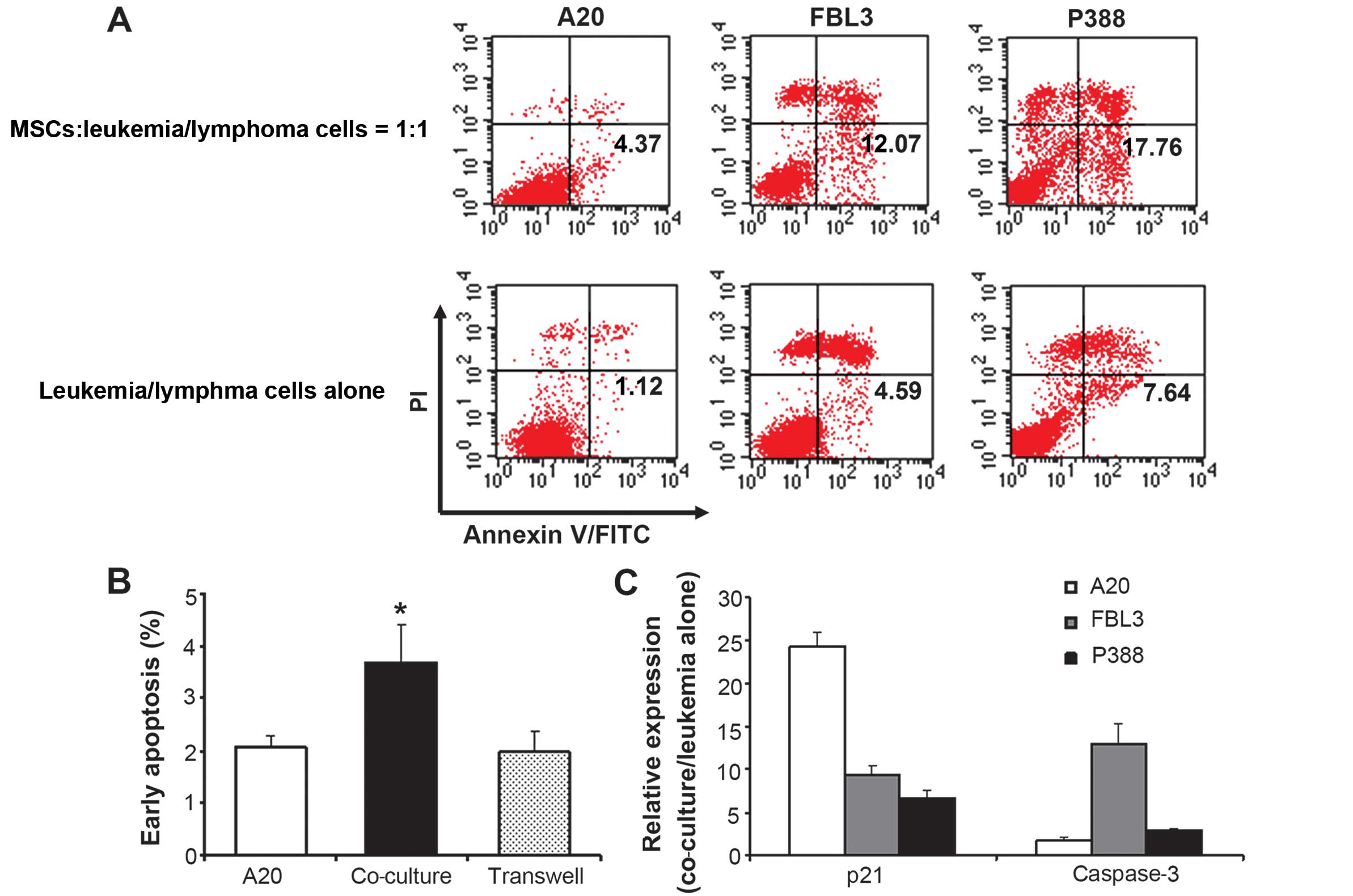
MSCs induce early apoptosis and cell
cycle arrest in leukemia and lymphoma cells in a contact-dependent
manner
When the A20 B lymphoma cells, the FBL3
erythroleukemia cells and the P388 acute lymphocytic leukemia cells
were co-cultured with the MSCs for 72 h, the proportion of
apoptotic cells, as measured by Annexin V and PI staining, was
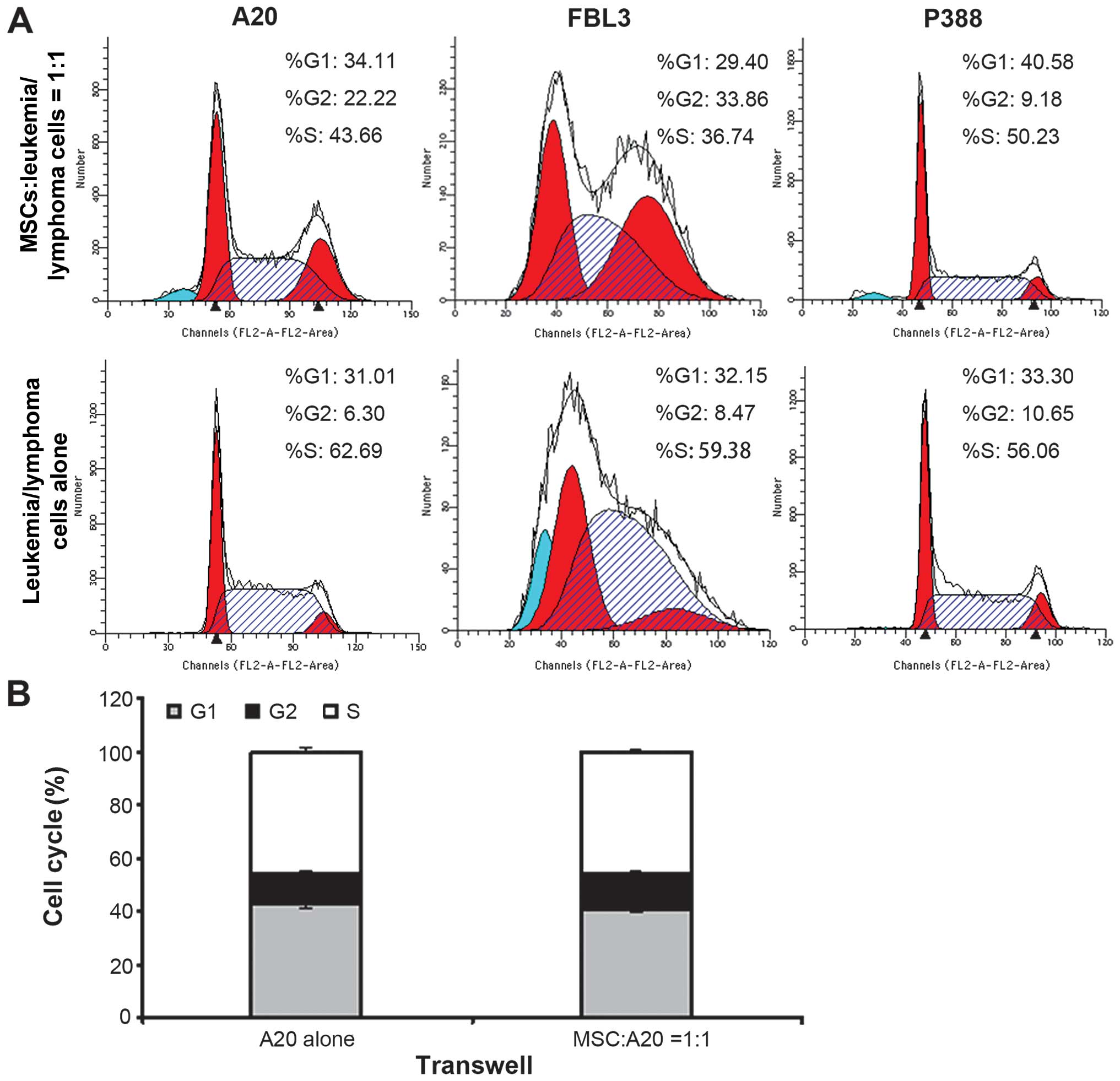
significantly increased (Fig. 2A
and B). The proportion of cells in the G0/G1
phase was also significantly higher in the cells incubated with the
MSCs than in those incubated alone, while the proportion of cells
in the S phase was significantly decreased following co-culture
with the MSCs for 72 h (Fig. 3A
and Table I). When the MSCs were
physically separated from the A20 cells using a Transwell system,
they no longer influenced the early apoptotic rate of the A20 cells
(Fig. 2B) or the cell cycle
(Fig. 3B), suggesting that MSCs
influence the cell cycle and apoptosis of lymphoma cells in a
contact-dependent manner.
 | Table IChanges in the cell cycle in leukemia
and lymphoma cells detected by flow cytometry. |
Table I
Changes in the cell cycle in leukemia
and lymphoma cells detected by flow cytometry.
| Groups | Cell cycle phase
|
|---|
|
G0/G1 (%) | S (%) | G2/M
(%) |
|---|
| A20 cells
alone | 31.08±1.21 | 62.93±1.20 | 5.99±0.27 |
| MSCs:A20 = 1:1 | 35.86±1.26b | 45.77±1.56b | 18.36±2.74 |
| FΒL3 cells
alone | 27.00±3.68 | 63.38±14.47 | 9.62±7.84 |
| MSCs:FBL3 =
1:1 | 32.69±1.26a | 38.40±7.25a | 28.91±4.31 |
| P388 cells
alone | 33.14±0.43 | 56.27±0.55 | 10.59±0.11 |
| MSCs:P388 =
1:1 | 39.65±1.85b | 51.03±2.10b | 9.32±0.33 |
Furthermore, we assessed the mRNA levels of the cell
cycle negative regulator, p21, and the apoptosis-associated
protease, caspase-3. qPCR revealed that incubation with the
MSCs induced a significant upregulation in the mRNA levels of
p21 and caspase-3 in the leukemia and lymphoma cells
when compared with the cancer cells cultured alone (Fig. 2C).
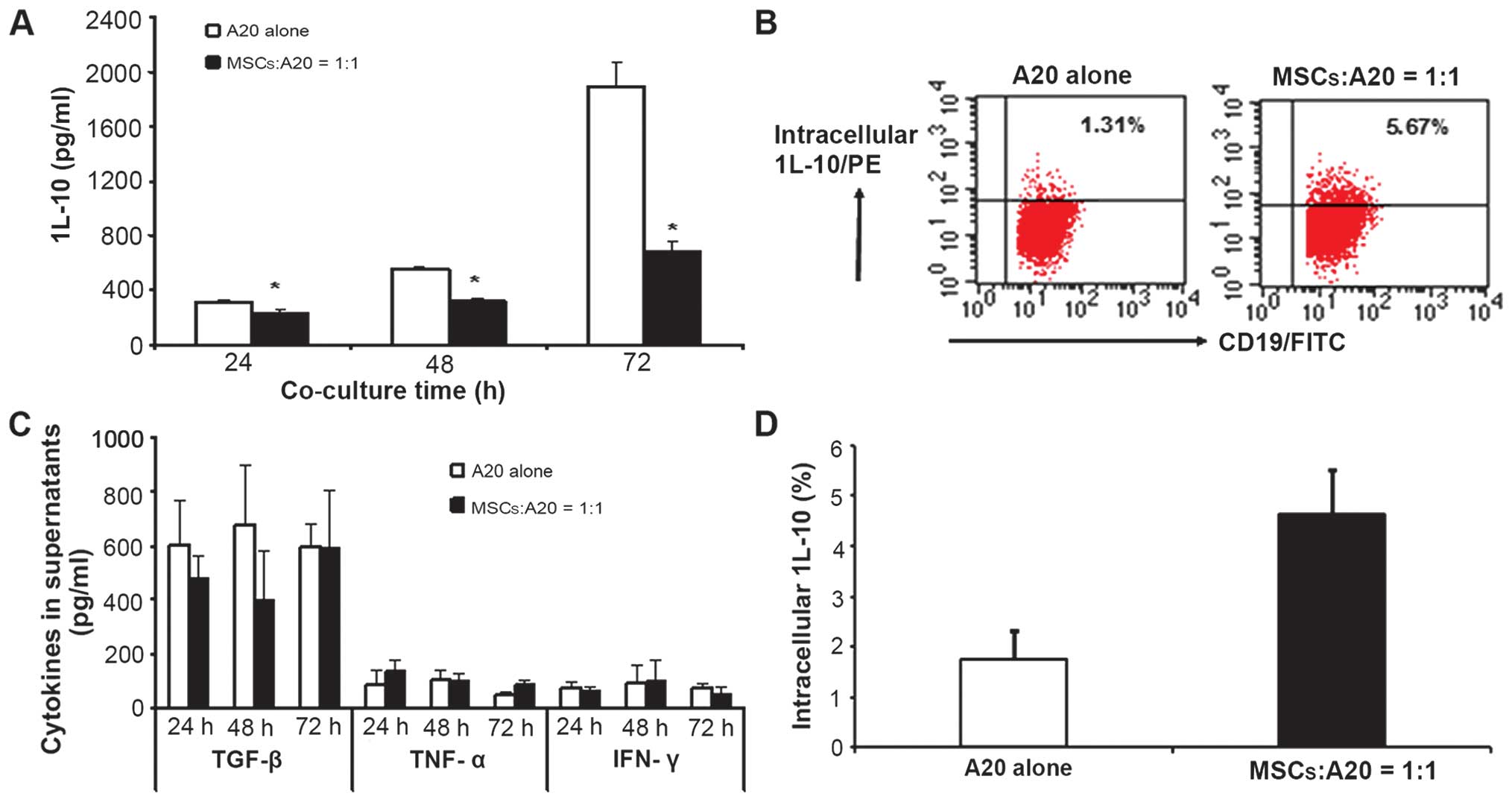
MSCs inhibit the secretion of cytokines
from A20 cells
As cytokines play an important role in the
regulation of adaptive and innate immune responses to tumors
(12–14), we measured the contents of
cytokines in the A20 cells incubated in the presence or absence of
MSCs. The A20 cells were found to express low levels of TNF-α and
IFN-γ, moderate levels of TGF-β, and high levels of IL-10 (Fig. 4A and B). When co-cultured with the
MSCs at a ratio of 1:1, the levels of IL-10 in the supernatant were
significantly decreased, and the inhibitory effects of the MSCs on
IL-10 secretion became more prominent with time (Fig. 4A). However, the levels of TGF-β,
TNF-α, and IFN-γ in the supernatant were not affected by the
presence of MSCs (Fig. 4C).
To determine whether the decrease in the content of
IL-10 in the co-culture supernatant was related to IL-10 secretion,
we measured the intracellular IL-10 levels in the A20 cells. When
the A20 cells were co-cultured with the MSCs for 48 h, the fraction
of cells in which intracellular IL-10 could be detected increased
from 1.74±0.59% to 4.64±0.89%, suggesting that MSCs influence the
capacity of lymphoma cells to release IL-10 (Fig. 4B and D).
MSCs inhibit A20 lymphoma cell growth in
the mouse model of allogeneic BMT
MSCs have been reported to both promote (15) and abrogate (5) tumor growth in vivo. In this
study, in order to investigate the effect of MSCs in a model of
allogeneic BMT, we implanted A20 B lymphoma cells into mice.
Lethally irradiated BABL/c female mice were either injected with
PBS (n=10), grafted with 1×107 B6 BM cells (n=10),
1×107 donor BM cells and 5×105 MSCs (n=10);
1×107 donor BM cells and 1×104 A20 cells
(n=17); or 1×107 donor BM cells, 1×104 A20
cells and 5×105 MSCs (n=17). Ninety percent of the
grafted mice in the BM group and 100% in the BM-MSC group survived
for >28 days, while the ungrafted animals died before day 21
(Fig. 5B). When 1×104
A20 cells were injected, the mice in the BM group exhibited a
characteristic infiltration of leukemia/lymphoma cells presenting
as paralysis and splenohepatomegalia. The mice administered the A20
and MSCs did not exhibit paralysis, but exhibited mild
splenohepatomegalia. The mice injected with A20 cells also
exhibited a hunched posture, dull fur and slight diarrhea, and the
histological examination revealed necrosis, defluxion, vacuolar
degeneration of the small intestinal mucosa, and atrophy and
collapse in the alveolae. These symptoms were less severe in the
mice that also received MSCs (Fig.
6), and the mean body weight was significantly higher in the
mice that received MSCs in addition to A20 cells from 8 days after
irradiation (Fig. 5A). Excluding
those mice that died of complications associated with irradiation,
the mice administered the A20 cells and MSCs survived longer than
the mice administered only A20 cells (Fig. 5C).
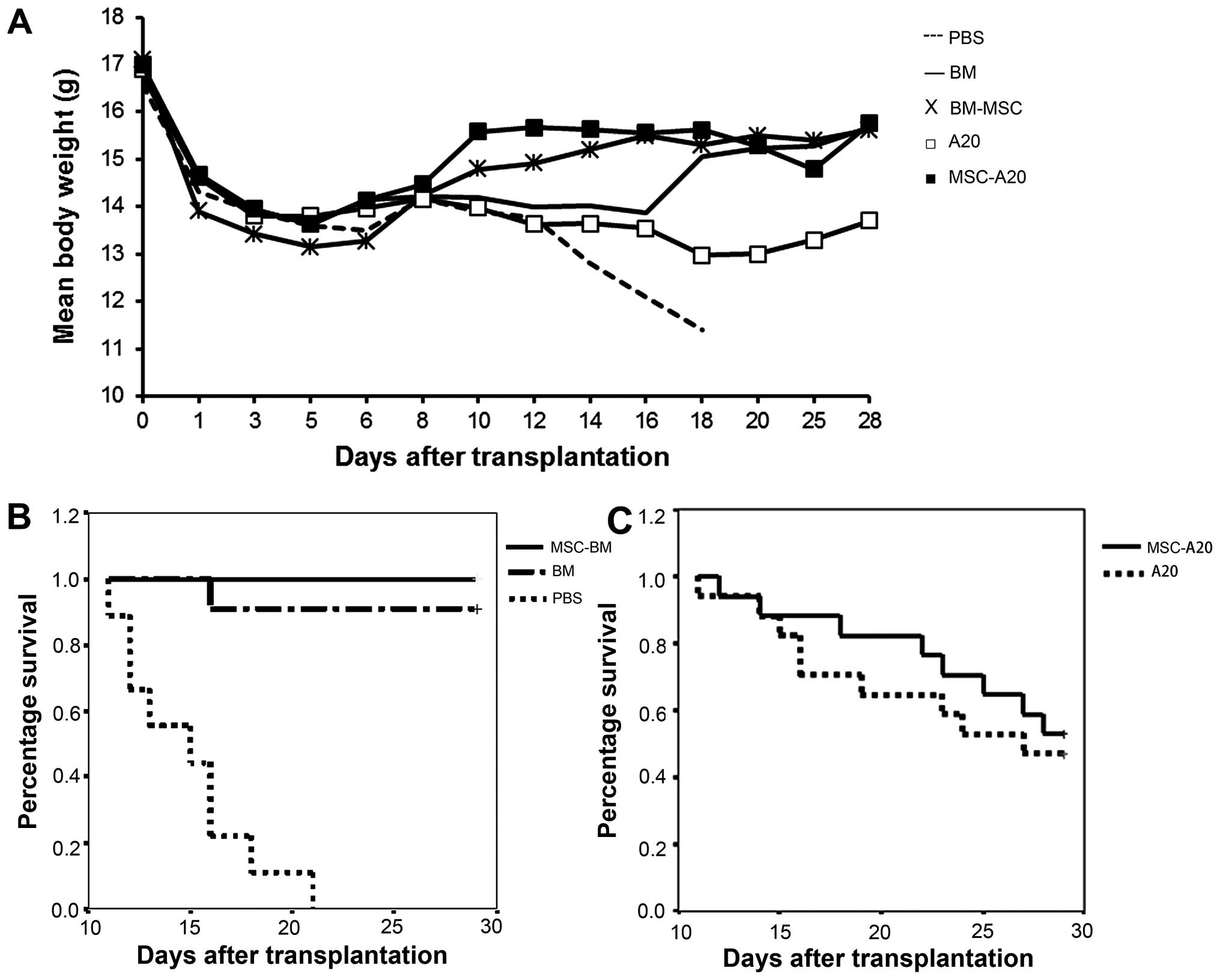 | Figure 5Mesenchymal stem cells (MSCs)
increase the weight and extend the survival of mice in a model of
allogeneic bone marrow transplantation (BMT). (A) Mice were weighed
following BMT, and the mean wild-type curves ± SEM were established
for the mice receiving PBS (–, n=10), bone marrow (BM) cells alone
(−, n=10), BM cells supplemented with MSCs (×, n=10), BM cells plus
A20 cells (◻, n=17), or BM cells with A20 cells plus MSCs (■,
n=17). (B and C) Results are represented as a Kaplan-Meier survival
curve. (B) There was significant difference between the PBS group
and the other 2 groups (BM and BM-MSC group) (P<0.01). (C)
Lethally irradiated BABL/c mice were transplanted with
1×107 BM cells and 1×104 A20 cells with or
without 5×105 MSCs. The survival rate in the 2 groups
was not significant, but the the time of death of the mice injected
with MSCs and A20 cells was delayed. |
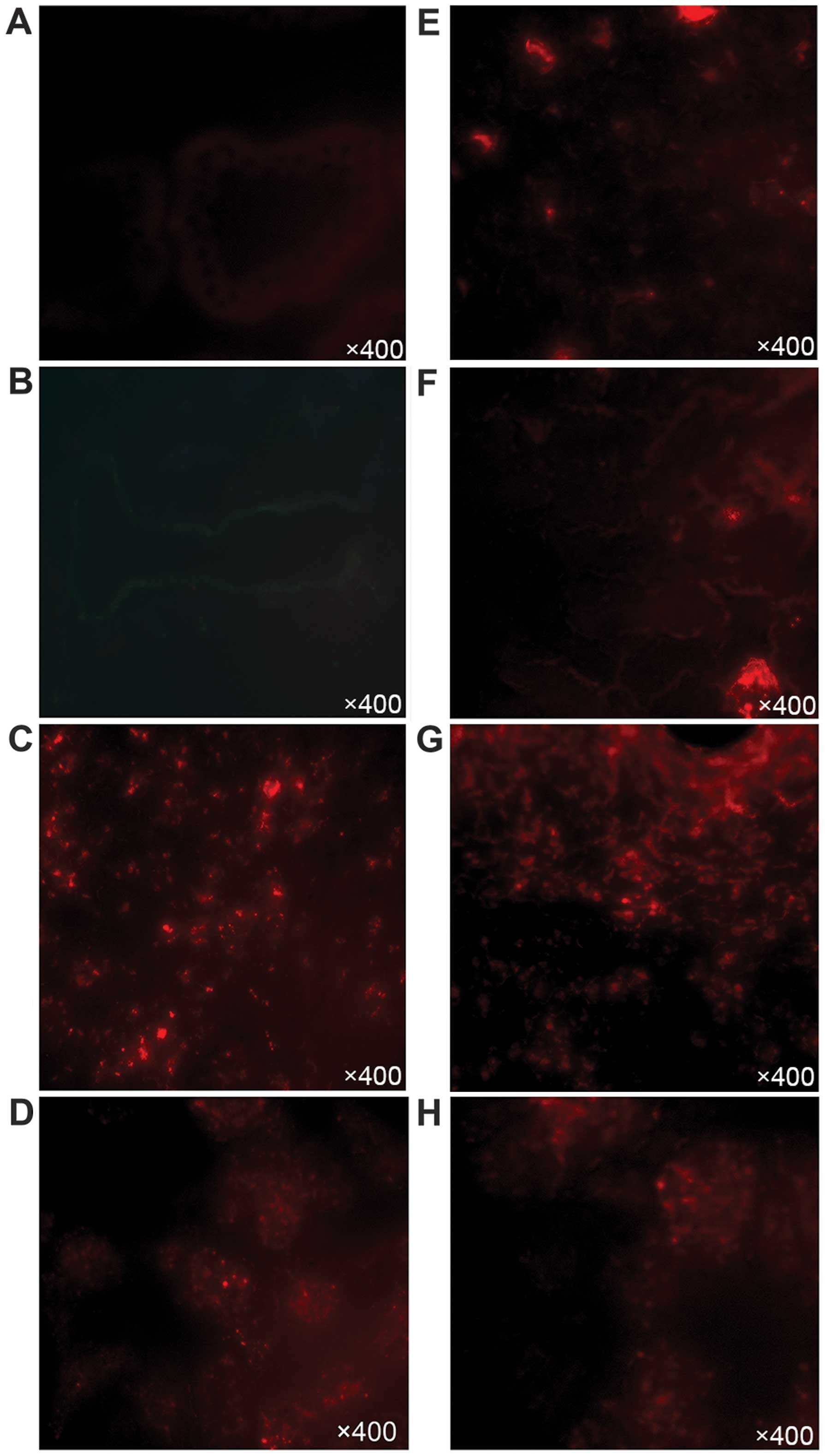
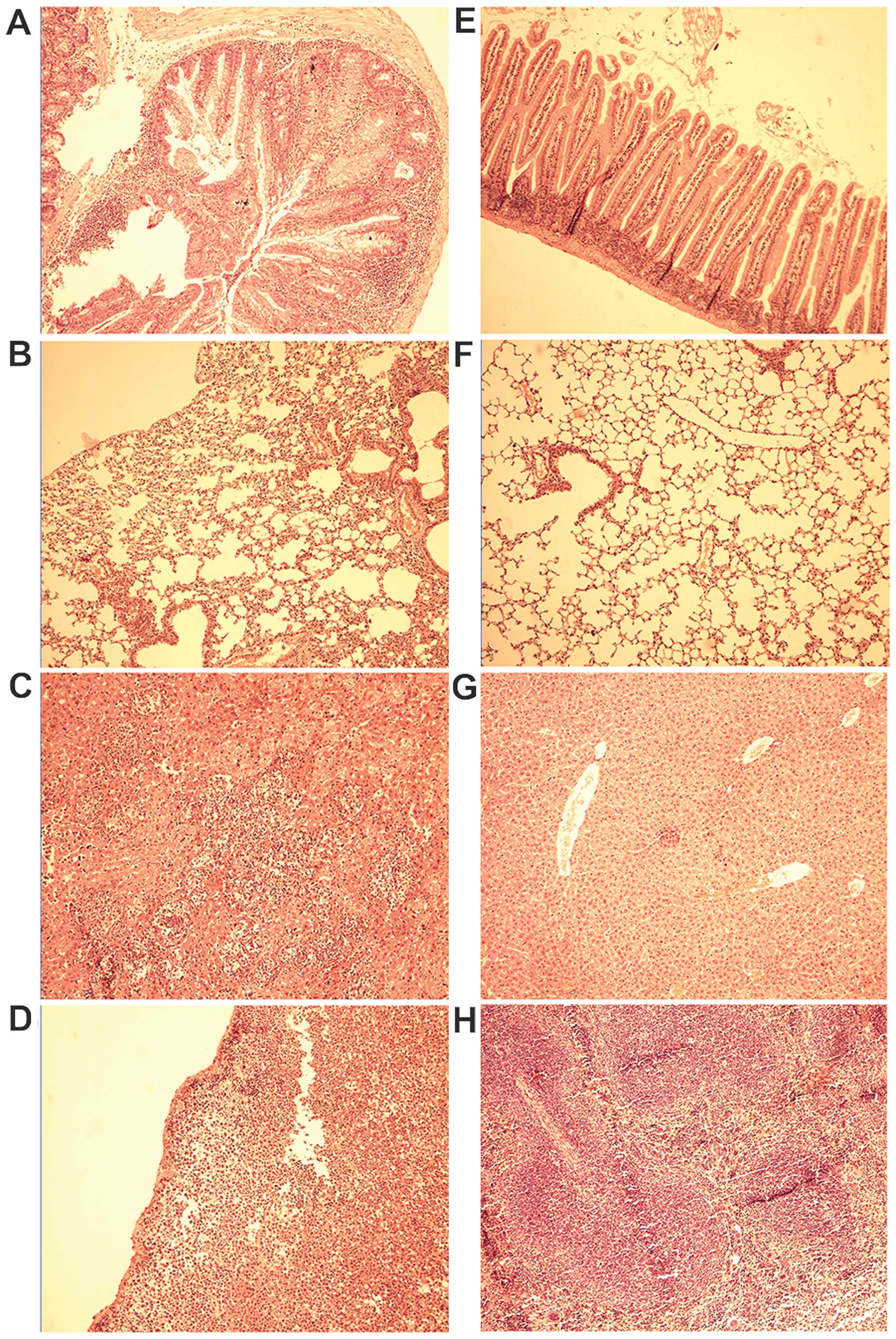 | Figure 6Histological analysis of the mice
injected with A20 cells alone and mesenchymal stem cell (MSC) plus
A20 cells on day 28 following transplantation. Mice in the (A-D)
A20 group and (E-H) MSC-A20 group were sacrificed, and the small
intestine, lungs, liver, and spleen were removed, fixed,
paraffin-embedded, and sectioned. Paraffin-embedded 5-µm
tissue sections were stained with hematoxylin and eosin. (A) Small
intestinal mucosa presented necrosis, defluxion and vacuolar
degeneration. (B) Collapse of pulmonary alveoli. (C) Large tumor
nodule infiltrating the liver, crushing normal liver tissues. (D)
A20 cells infiltrated the spleen. (E-F) Normal small intestine and
lung structures. (G-H) Normal liver and spleen structures.
Objective lens: magnification, ×10. |
To investigate the effect of MSCs on the
proliferation of A20 cells in vivo, we compared the
incidence of lymphoma between the mice injected with MCCs and those
that were not, and found that the injection of MSCs significantly
decreased the incidence of lymphoma in the mice injected with A20
cells (P<0.05, Table II).
| Table IITumor incidence in the mice injected
with A20 cells alone and MSCs plus A20 cells on day 28 following
BMT. |
Table II
Tumor incidence in the mice injected
with A20 cells alone and MSCs plus A20 cells on day 28 following
BMT.
| Groups | Tumor | No tumor | Total | Incidence (%) |
|---|
| A20 | 16 | 1 | 17 | 94.1 |
| MSC-A20 | 10 | 7 | 17 | 58.5a |
| Total | 26 | 8 | 34 | 76.5 |
Tracing the PKH26-labeled MSCs in the
allogeneic BMT setting
Since our results revealed that the B6 MSCs
inhibited the proliferation of the A20 cells, increased early
apoptosis and led to cell cycle arrest in vitro, we wished
to determine whether intravenously injected mouse MSCs can home to
A20 lymphoma-infiltrated organs (such as the spleen and liver). On
day 0, 1×106 MSCs were injected with 1×107 BM
cells and 1×104 A20 cells into the tail vein of the
irradiated BABL/c mice. To track the distribution of the MSCs
within the mouse over time, we labeled the MSCs with PKH26 (a
fluorescent membrane linker) to allow for the easy identification
in histopathological sections. We have previously established that
PKH26-labeling efficiency is almost 100% and does not affect the
growth of MSCs in vitro (data not shown), and a similar
approach has been previously demonstrated to effectively track the
in vivo location of MSCs without alterating cellular
functions (16).
Twenty-four-hours post-grafting, the labeled MSCs
were observed to be diffusely distributed in the spleen and kidneys
(Fig. 7C and D), while no MSCs
were observed in the heart, liver, small intestine (Fig. 7A) or lungs (Fig. 7B). On day 7, we could still
observe numerous MSCs within the spleen and kidneys (Fig. 7G and H), which appeared to
maintain the elongated, fibroblast-like appearance that they adopt
in culture. Furthermore, on day 7, the donor MSCs were present in
the liver and lungs of the grafted mice. This indicated that
following their infusion, the MSCs mainly migrated to the tumor
sites (spleen and liver) and the damaged tissues (lungs and
kidneys). We were, however, surprised not to find any MSCs in the
easily injured small intestine 24 h or 7 days post-grafting.
Effect of MSCs on peripheral blood T cell
subsets in the model of allogeneic BMT
When blood was taken from the mice on day 7
post-grafting, we found that the percentage of
CD3+CD8+ T cells was 32.5±7.29% higher in the
peripheral blood of the mice administered MSCs and A20 cells in
comparison to those administered only A20 cells, while the
percentages of CD3+CD4+ T cells and
CD4+CD25+ T cells were significantly
decreased (Table III). However
no significant differences were observed in the T cell subsets on
day 14 following transplantation (Table III).
| Table IIIDistribution of T cell subsets in the
peripheral blood on days 7 and 14 following BMT. |
Table III
Distribution of T cell subsets in the
peripheral blood on days 7 and 14 following BMT.
| Cell culture | Time since BMT
(days) |
CD3+CD4+ (%) |
CD3+CD8+ (%) |
CD4+CD25+ (%) |
|---|
| A20 | 7 | 78.41±3.98 | 4.90±1.74 | 4.73±1.67 |
| 14 | 4.61±0.81 | 34.66±14.51 | 0.12±0.11 |
| MSC-A20 | 7 | 50.85±11.85a | 37.40±9.03b | 1.88±0.43a |
| 14 | 4.82±1.57 | 30.86±8.90 | 0.27±0.17 |
Discussion
MSCs have been reported to suppress immune responses
in some contexts, and to enhance them in others [reviewed in
(17)]. While MSCs show great
promise in the treatment of autoimmune diseases, such as GVHD
(3), Crohn’s disease (18) and multiple sclerosis (19), MSCs can also contribute negatively
in diseases, such as cancer, and the effect of MSCs on the
proliferation of hematopoietic cells has not been well documented
(20,21). In this study, we found that MSCs
from B6 mice inhibited the proliferation of leukemia and lymphoma
cells in a dose- and time-dependent manner in vitro. This
finding is consistent with that of a previous study demonstrating
that MSCs exhibit a similar anti-proliferative activity in cancer
cells of hematopoietic and non-hematopoietic origin (22).
The mechanisms through which MSCs inhibit the
proliferation of leukemia/lymphoma cells have not been well
characterized. Otsu et al found that in a Matrigel
angiogenesis assay, high numbers of MSCs increased the production
of reactive oxygen species, inhibiting capillary growth, and
abrogating tumor growth (5).
Similar to the findings of the study by Lu et al (20), we demonstrated that MSCs
upregulated p21 and caspase-3 mRNA expression in
leukemia and lymphoma cells, increasing the fraction of cells
undergoing early apoptosis, and leading to cell cycle arrest at the
G0/G1 phase, thus decreasing the fraction of
leukemia and lymphoma cells in the S phase. In contrast to the
results reported in the study by Khakoo et al, who found
that human MSCs inhibited the proliferation of Kaposi’s sarcoma
cells by inhibiting the in vitro activation of the Akt
protein kinase (6), we did not
find that the inhibitory effects of MSCs were affected by the
addition of an Akt inhibitor, indicating that the effects of MSCs
on lymphoma cells are not medaited through the Akt pathway.
BALB/c-derived B lymphoma A20 cells express high
levels of IL-10 and this cytokine may contribute to their immune
evasion by affecting various arms of the immune system, including
Treg cells and dendritic cells (DCs) (23). In this study, we found that A20
cells secreted high levels of IL-10 and moderate levels of TGF-β,
but very low levels of TNF-α and IFN-γ. The levels of IL-10 in the
supernatant of A20 cell cultures were significantly decreased by
co-culture with MSCs in a time-dependent manner. Furthermore, when
co-cultured with the MSCs, the fraction of A20 cells expressing
intracellular IL-10 significantly increased, suggesting that MSCs
inhibited the secretion of IL-10 by A20 cells.
We found that both the survival rates and body
weights of the mice increased significantly following
transplantation of the MSCs, and the symptoms were significantly
ameliorated in the mice were injected with the MSCs. Previous
studies on the use of MSCs in murine models of tumors have focused
on the NOD-SCID or athymic nude mouse model (20,24) or subcutaneous model (15,25). Ramasamy et al reported that
MSCs formed a cancer stem cell niche in which the potential of
cancer cells to proliferate is preserved, sustaining the malignant
process (22). However, in their
study, the experiments were performed using immunodeficient nude
mice, and do not reflect the situation of autologous tumor
development. Djouad et al reported that the subcutaneous
injection of B16 melanoma cells led to tumor growth in allogeneic
recipients only when MSCs were co-injected, which were related with
the immunosuppressive properties in mixed lymphocyte reaction
(15). However, these mouse tumor
models do not represent the leukemia/lymphoma environment well. By
contrast, in this study, we observed the amelioration of aGVHD in
mice in a model of BMT, and found the incidence of lymphoma in the
liver/spleen to be lower in the mice administered MSCs. Thus,
consistent with the results of the study by Baron et al, who
reported in a clinical study that the co-transplantation of MSCs
prevented death from GVHD without abrogating graft-versus-tumor
effects after HLA-mismatched allogeneic transplantation following
non-myeloablative conditioning (26), our results support the hypothesis
that allogeneic BM-derived MSCs control aGVHD without increasing
the incidence of lymphoma. To the best of our knowledge, this study
represents the first report aiming to evaluate the effects of MSCs
on the development of lymphoma/leukemia cells in an allogeneic BMT
model of minimal residual leukemia. Although the A20 lymphoma model
used in this study does not fully capitulate the physiological
circumstances of leukemia/lymphoma cell proliferation in humans, it
provides some insight as to whether the systemic injection of
allogeneic BM-derived MSCs in a BMT setting may influence the
growth of malignant cells.
We also observed the abrogation of T cell subsets in
the peripheral blood of mice. The percentage of
CD3+CD8+ T cells increased and the fraction
of CD3+CD4+ T cells and
CD4+CD5+ T cells decreased in the mice
administered MSCs 7 days following transplantation. However, these
differences had disappeared by day 14. This perhaps reflects the
reduction in the numbers of A20 cells in vivo at an early
stage. We also detected large numbers of MSCs, not only in the
target organs of GVHD, but also in the spleen, and furthermore
detected some MSCs in the livers of the recipients 7 days after
grafting. We also found that the inhibition of A20 cell
proliferation by MSCs in vitro was contact-dependent,
suggesting that cell-cell contact may be necessary for MSCs to
inhibit the leukemia/lymphoma cell cycle and cell proliferation. It
has also been recently reported that the capacity of MSCs to
inhibit hepatic stellate or endothelial cell proliferation is also
contact-dependent (8,9). The present study in combination with
previously reported results suggests that the involvement of a
communication microenvironment between MSCs and leukemia cells is
required. We also thus speculate that it is the MSCs present in the
spleen and liver that directly inhibit the proliferation of A20
cells, which are also primarily present in the spleen and liver of
the recipient mice.
In conclusion, we found that the inhibition of the
proliferation of leukemia and lymphoma cells by MSCs in
vitro is dependent on cell-cell contact and that MSCs inhibit
the release of IL-10 from lymphoma cells. Our findings also
indicate that when administered intravenously in a model of
allogeneic BMT, MSCs can reduce GVHD without increasing tumor
growth or tumor incidence. However, the mechanisms through which
MSCs interact with the malignant cells remain unknown, and thus
further studies on MSCs are warranted. Although the safety and
clinical efficacy of MSC infusion are under evaluation in humans
for the support of BMT (26–28), the benefit of using MSCs in the
clinical setting still needs to be explored in prospective,
controlled studies.
References
|
1
|
Sorrentino A, Ferracin M, Castelli G,
Biffoni M, Tomaselli G, Baiocchi M, Fatica A, Negrini M, Peschle C
and Valtieri M: Isolation and characterization of CD146+
multipotent mesenchymal stromal cells. Exp Hematol. 36:1035–1046.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Le Blanc K, Samuelsson H, Gustafsson B,
Remberger M, Sundberg B, Arvidson J, Ljungman P, Lönnies H, Nava S
and Ringdén O: Transplantation of mesenchymal stem cells to enhance
engraftment of hematopoietic stem cells. Leukemia. 21:1733–1738.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Herrmann RP and Sturm MJ: Adult human
mesenchymal stromal cells and the treatment of graft versus host
disease. Stem Cells Cloning. 7:45–52. 2014.PubMed/NCBI
|
|
4
|
Shlomchik WD: Graft-versus-host disease.
Nat Rev Immunol. 7:340–352. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Otsu K, Das S, Houser SD, Quadri SK,
Bhattacharya S and Bhattacharya J: Concentration-dependent
inhibition of angiogenesis by mesenchymal stem cells. Blood.
113:4197–4205. 2009. View Article : Google Scholar :
|
|
6
|
Khakoo AY, Pati S, Anderson SA, et al:
Human mesenchymal stem cells exert potent antitumorigenic effects
in a model of Kaposi’s sarcoma. J Exp Med. 203:1235–1247. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhu Y, Sun Z, Han Q, et al: Human
mesenchymal stem cells inhibit cancer cell proliferation by
secreting DKK-1. Leukemia. 23:925–933. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang PP, Xie DY, Liang XJ, Peng L, Zhang
GL, Ye YN, Xie C and Gao ZL: HGF and direct mesenchymal stem cells
contact synergize to inhibit hepatic stellate cells activation
through TLR4/NF-kB pathway. PLoS One. 7:e434082012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Menge T, Gerber M, Wataha K, Reid W, Guha
S, Cox CS Jr, Dash P, Reitz MS Jr, Khakoo AY and Pati S: Human
mesenchymal stem cells inhibit endothelial proliferation and
angiogenesis via cell-cell contact through modulation of the
VE-Cadherin/β-catenin signaling pathway. Stem Cells Dev.
22:148–157. 2013. View Article : Google Scholar :
|
|
10
|
Galiè M, Konstantinidou G, Peroni D, et
al: Mesenchymal stem cells share molecular signature with
mesenchymal tumor cells and favor early tumor growth in syngeneic
mice. Oncogene. 27:2542–2551. 2008. View Article : Google Scholar
|
|
11
|
Karnoub AE, Dash AB, Vo AP, Sullivan A,
Brooks MW, Bell GW, Richardson AL, Polyak K, Tubo R and Weinberg
RA: Mesenchymal stem cells within tumour stroma promote breast
cancer metastasis. Nature. 449:557–563. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cirone M, Lucania G, Aleandri S, Borgia G,
Trivedi P, Cuomo L, Frati L and Faggioni A: Suppression of
dendritic cell differentiation through cytokines released by
Primary Effusion Lymphoma cells. Immunol Lett. 120:37–41. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang L, Zhao Y, Qian J, Sun L, Lu Y, Li H,
Li Y, Yang J, Cai Z and Yi Q: Toll-like receptor-4 signaling in
mantle cell lymphoma: effects on tumor growth and immune evasion.
Cancer. 119:782–791. 2013. View Article : Google Scholar
|
|
14
|
Shen L, Chiang AK, Liu WP, Li GD, Liang RH
and Srivastava G: Expression of HLA class I, beta(2)-microglobulin,
TAP1 and IL-10 in Epstein-Barr virus-associated nasal
NK/T-celllymphoma: Implications for tumor immune escape mechanism.
Int J Cancer. 92:692–696. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Djouad F, Plence P, Bony C, Tropel P,
Apparailly F, Sany J, Noël D and Jorgensen C: Immunosuppressive
effect of mesen-chymal stem cells favors tumor growth in allogeneic
animals. Blood. 102:3837–3844. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen WJ, Huang JW, Niu CC, Chen LH, Yuan
LJ, Lai PL, Yang CY and Lin SS: Use of fluorescence labeled
mesenchymal stem cells in pluronic F127 and porous hydroxyapatite
as a bone substitute for posterolateral spinal fusion. J Orthop
Res. 27:1631–1636. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang Y, Chen X, Cao W and Shi Y:
Plasticity of mesenchymal stem cells in immunomodulation:
Pathological and therapeutic implications. Nat Immunol.
15:1009–1016. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dalal J, Gandy K and Domen J: Role of
mesenchymal stem cell therapy in Crohn’s disease. Pediatr Res.
71:445–451. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cohen JA: Mesenchymal stem cell
transplantation in multiple sclerosis. J Neurol Sci. 333:43–49.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lu YR, Yuan Y, Wang XJ, et al: The growth
inhibitory effect of mesenchymal stem cells on tumor cells in vitro
and in vivo. Cancer Biol Ther. 7:245–251. 2008. View Article : Google Scholar
|
|
21
|
Ilmer M, Vykoukal J, Recio Boiles A,
Coleman M and Alt E: Two sides of the same coin: Stem cells in
cancer and regenerative medicine. FASEB J. 28:2748–2761. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ramasamy R, Lam EW, Soeiro I, Tisato V,
Bonnet D and Dazzi F: Mesenchymal stem cells inhibit proliferation
and apoptosis of tumor cells: Impact on in vivo tumor growth.
Leukemia. 21:304–310. 2007. View Article : Google Scholar
|
|
23
|
Elpek KG, Lacelle C, Singh NP, Yolcu ES
and Shirwan H: CD4+CD25+ T regulatory cells
dominate multiple immune evasion mechanisms in early but not late
phases of tumor development in a B cell lymphoma model. J Immunol.
178:6840–6848. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhu W, Xu W, Jiang R, Qian H, Chen M, Hu
J, Cao W, Han C and Chen Y: Mesenchymal stem cells derived from
bone marrow favor tumor cell growth in vivo. Exp Mol Pathol.
80:267–274. 2006. View Article : Google Scholar
|
|
25
|
Djouad F, Bony C, Apparailly F,
Louis-Plence P, Jorgensen C and Noël D: Earlier onset of syngeneic
tumors in the presence of mesenchymal stem cells. Transplantation.
82:1060–1066. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Baron F, Lechanteur C, Willems E, et al:
Cotransplantation of mesenchymal stem cells might prevent death
from graft-versus- host disease (GVHD) without abrogating
graft-versus-tumor effects after HLA-mismatched allogeneic
transplantation following nonmyeloablative conditioning. Biol Blood
Marrow Transplant. 16:838–847. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Le Blanc K, Rasmusson I, Sundberg B,
Götherström C, Hassan M, Uzunel M and Ringdén O: Treatment of
severe acute graft-versus-host disease with third party
haploidentical mesenchymal stem cells. Lancet. 363:1439–1441. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ning H, Yang F, Jiang M, et al: The
correlation between cotrans-plantation of mesenchymal stem cells
and higher recurrence rate in hematologic malignancy patients:
Outcome of a pilot clinical study. Leukemia. 22:593–599. 2008.
View Article : Google Scholar : PubMed/NCBI
|