Introduction
Diabetic nephropathy (DN) is a major microvascular
complication of diabetes mellitus (DM) that invariably leads to
end-stage renal disease (ESRD). Although DN was traditionally
considered a primarily glomerular disease, accumulating evidence
indicates that renal tubules play an important role in the
pathogenesis of DN (1–3). However, the mechanisms through which
the deregulation of renal tubules contributes to the development of
DN remain largely unknown.
Transforming growth factor (TGF)-β1 initiates
intracellular signaling by binding and activating transmembrane
type I and II serine/threonine kinase receptors, which in turn
activate the downstream mediators, Smad2 and Smad3. Activated Smad2
and Smad3 undergo phosphorylation and heteroligomerize with Smad4
to form the Smad complex, which then translocates to the nucleus to
regulate the transcription of TGF-β1 target genes (4,5).
Under normal physiological conditions, TGF-β1/Smad signaling is
tightly controlled by a negative regulatory mechanism. Ski-related
novel protein N (SnoN) is a Smad transcriptional co-repressor
(6,7) that negatively regulates TGF-β1/Smad
signaling by binding and repressing Smad complexes to activate gene
transcription (8,9). Thus, the abundance and activity of
SnoN in a given circumstance may determine the final response of
cells to TGF-β1 stimulation. TGF-β1 is a profibrotic cytokine that
has been shown to play a key role in the pathophysiology of DN, in
both experimental models of DN and in patients (4). High glucose activates TGF-β1
signaling, which in turn stimulates tubule epithelial cells to
overproduce extracellular matrix (10). A large body of evidence has
indicated that TGF-β1 plays a critical role in the development of
tubulointerstitial fibrosis in DN (4,10–12). However, the molecular mechanisms
underlying the role of TGF-β1 in the pathogenesis of DN remain
elusive.
In our previous study, we demonstrated that SnoN
protein was downregulated progressively in the kidneys of diabetic
rats and primary proximal tubule epithelial cells under
high-glucose conditions (13).
Moreover, we demonstrated that SnoN inhibited high-glucose-induced
epithelial-mesenchymal transition (EMT) in renal tubule cells
(13). These data suggest that
the loss of SnoN may lead to tubulointerstitial damage in DN
through uncontrolled TGF-β1/Smad signaling. However, the regulatory
mechanisms responsible for the decrease in SnoN expression under
high-glucose conditions remain to be elucidated. Of note, Smad
ubiquitination regulatory factor-2 (Smurf2), an E3 ubiquitin
ligase, has been shown to be increased in renal fibrosis induced by
obstructive injury and to promote the downregulation of SnoN
(14,15). Considering that TGF-β1 signaling
is commonly upregulated in obstructive nephropathy and DN, we
hypothesized that the upregulation of Smurf2 through TGF-β1/Smad
signaling may contribute to the downregulation of SnoN in DN.
In this study, we examined the expression of SnoN in
human renal proximal tubule epithelial cells (hRPTECs) cultured in
high-glucose (30 mmol/l D-glucose) medium in the presence or
absence of either the proteasome inhibitor, MG132, or the TGF-β
type I receptor inhibitor, SB-431542. We further determined the
protein levels of SnoN following the silencing of Smurf2 by small
interfering RNA (siRNA) in the hRPTECs under high-glucose
conditions.
Materials and methods
Cell culture
The hRPTECs were purchased from ScienCell Research
Laboratories (San Diego, CA, USA). The cells were cultured in
fibronectin-coated flasks using epithelial cell medium (ScienCell
Research Laboratories) supplemented with growth additives, 2% fetal
bovine serum (FBS) and penicillin/streptomycin. The cells were
cultured at 37°C in a humidified incubator in the presence of 5%
CO2. Actively proliferating hRPTECs at the third passage
were used in the subsequent experiments.
Cell treatments
When the cells reached 60–80% confluence, the
culture medium was replaced with serum-free epithelial cell medium
and the cells were starved for 16 h to synchronize cell growth. The
cells were incubated in media containing various concentrations of
glucose: normal glucose (5.5 mmol/l D-glucose), high glucose (30
mmol/l D-glucose) or in medium of a high osmolarity (5.5 mmol/l
D-glucose + 24.5 mmol/l D-mannitol). The cells were incubated for
2, 12, 24, 48 and 72 h, and collected at various time points for
RNA and protein extraction. D-glucose and D-mannitol were purchased
from Sigma-Aldrich (St. Louis, MO, USA).
For treatment with MG132 (proteasome inhibitor),
following starvation for 16 h, the cells were treated with 1.0
µmol/l MG132 (Selleck Chemicals, Houston, TX, USA) for 48 h
in media containing 5.5 mmol/l D-glucose or 30 mmol/l D-glucose.
For treatment with SB-431542 (TGF-β type I receptor inhibitor),
following starvation for 16 h, the cells were treated with various
concentrations of SB-431542 (Sigma-Aldrich) or the vehicle (0.1%
DMSO) for 48 h in medium containing 30 mmol/l D-glucose.
Gene silencing by siRNA
To facilitate the optimization of lipid-mediated
transfection for RNA interference (RNAi) experiments, BLOCK-iT™
Alexa Fluor® Red Fluorescent Control (Invitrogen,
Carlsbad, CA, USA), an Alexa Fluor® 555-labeled,
double-stranded RNA (dsRNA) duplex were transfected into the
hRPTECs according to the manufacturer's instructions. The
transfection efficiency was roughly determined by calculating the
ratio of red fluorescence cells among the total cells.
Subsequently, the hRPTECs were transiently transfected with
negative control siRNA (Cat. no. 12935200), Smurf2 siRNA-1
(ID#VHS41440), or Smurf2 siRNA-2 (ID#VHS41441; Invitrogen) using
Lipofectamine RNAiMAX reagent according to the manufacturer's
instructions (Invitrogen). Briefly, the cells were plated into
6-well plates in medium without antibiotics and grown to 50–60%
confluence at the time of transfection. The siRNAs and
Lipofectamine RNAiMAX reagent were diluted in Opti-MEM I reduced
serum medium (Invitrogen), separately. The diluted siRNA was then
added to the diluted Lipofectamine RNAiMAX reagent and incubated at
room temperature for 5 min. The final concentration of siRNA in the
medium was 20 nM. The cells were incubated in serum-free medium
with siRNA-lipid complex for 6 h at 37°C. The medium was then
replaced with medium with or without 30 mmol/l D-glucose, and the
cells were incubated for an additional 48 h. The control cells
refer to the untransfected cells cultured for 48 h cultured under
high-glucose conditions. The mock-transfected cells refer to the
cells cultured for 48 h in media containing high glucose and
diluted transfection reagent.
Western blot analysis
Total protein from the cultured cells was extracted
using RIPA lysis buffer containing phosphatase inhibitors and
protease inhibitors (Roche, Mannheim, Germany). Protein
concentrations were determined using the bicinchoninic acid (BCA)
assay. Equal amounts of proteins were separated by 10% sodium
dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and
then transferred onto polyvinylidene fluoride (PVDF) membranes
(Millipore Corp., Bedford, MA, USA). The membranes were blocked in
5% non-fat milk for 1 h and incubated with primary antibody at a
dilution recommended by the manufacturer overnight at 4°C. The
primary antibodies used were as follows: anti-Smad2 (ab40855),
anti-phospho-Smad2 (ab53100; both from Abcam, Cambridge, MA, USA),
anti-Smurf2 (sc-25511), anti-SnoN (sc-9141), anti-SnoN (sc-9595)
and anti-β-actin (sc-130656; all from Santa Cruz Biotechnology,
Inc., Santa Cruz, CA, USA). After washing with TBST (10 mM Tris, pH
8.0, 150 mM NaCl, and 0.1% Tween-20), the membranes were incubated
for 1 h with horseradish peroxidase-conjugated secondary antibody
(Cell Signaling Technology, Inc., Boston, MA, USA) at a dilution of
1,3000. The membranes were processed using an enhanced
chemiluminescence kit (Millipore Corp.) and the images were
captured using the ChemiDoc XRS+ imaging system (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). β-actin was used as a
loading control. Quantitative analysis of the western blot analysis
data was performed by measuring the intensity of the band signals
using Image Lab 3.0 software (Bio-Rad Laboratories, Inc.).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was isolated with TRIzol reagent
(Invitrogen) according to the manufacturer's instructions. RNA
concentration and quality were determined by spectrophotometry.
Complementary (c)DNA was synthesized from 1 µg total RNA
using oligo(dT) primers and the Reverse Transcription system
(Promega Corp., Madison, WI, USA) at 42°C for 60 min as recommended
by the manufacturer. qPCR with 1 µl cDNA was performed using
a TaqMan Gene Expression assay for each mRNA and TaqMan Gene
Expression Master Mix (Applied Biosystems, Foster City, CA, USA) in
a total of 20 µl/reaction. The PCR conditions were as
follows: 95°C for 10 min, 40 cycles at 95°C for 15 sec, and 60°C
for 60 sec for amplification. All reactions were performed in
triplicate. The results were analyzed using the 2−ΔΔCt
method.
Co-immunoprecipitation
The hRPTECs were harvested after the cells were
cultured in medium containing 30 mmol/l D-glucose for 24 h. The
cells were lysed and centrifuged at 12,000 × g for 20 min at 4°C.
The supernatants were collected for immunoprecipitation. After
preclearing with normal host IgG (Santa Cruz Biotechnology, Inc.),
the lysates were immunoprecipitated overnight at 4°C with
anti-Smurf2 antibody (1/100 µg total protein), followed by
precipitation with 20 µl of protein A/G Plus-Agarose (Santa
Cruz Biotechnology, Inc.) for 4 h at 4°C. After 3 washes, the
precipitated complexes were separated by 10% SDS-PAGE which was
followed by western blot analysis.
Statistical analysis
All data are expressed as the means ± SD.
Statistical analysis of the data was performed using SPSS 17.0
software (SPSS, Inc., Chicago, IL, USA). Comparisons among the
experimental groups were performed using one-way analysis of
variance (ANOVA) followed by Scheffe's test. A value of P<0.05
was considered to indicate a statistically significant
difference.
Results
High-glucose conditions induce the
downregulation of SnoN in hRPTECs at the post-transcriptional
level
To determine whether high-glucose conditions
decrease SnoN protein levels, we cultured the hRPTECs in media
containing normal glucose (5.5 mmol/l D-glucose), high glucose (30
mmol/l D-glucose) or in medium of a high osmolarity (5.5 mmol/l
D-glucose + 24.5 mmol/l D-mannitol) for different periods of time
and we measured the SnoN protein levels by western blot analysis.
Compared with the cells cultured under normal-glucose conditions,
in the cells cultured under high-glucose conditions, SnoN protein
expression was significantly downregulated after 24 h (Fig. 1A and B). This may not be due to
the hypertonic pressure of 30 mmol/l D-glucose, as 24.5 mmol/l
mannitol, which has an equal osmotic pressure as 30 mmol/l
D-glucose, did not apparently alter the abundance of SnoN protein
(Fig. 1A and B). To determine
whether the decrease in SnoN protein expression resulted from the
downregulation of SnoN mRNA expression, we measured the SnoN mRNA
levels by RT-qPCR. Surprisingly, the SnoN mRNA levels were
significantly upregulated in the hRPTECs cultured under
high-glucose conditions from 12–72 h (Fig. 1C) in contrast to those of the
cells grown in medium containing normal glucose and of high
osmolarity. These results indicated that SnoN expression was
reduced under high-glucose conditions in the hRPTECs at the
post-transcriptional level.
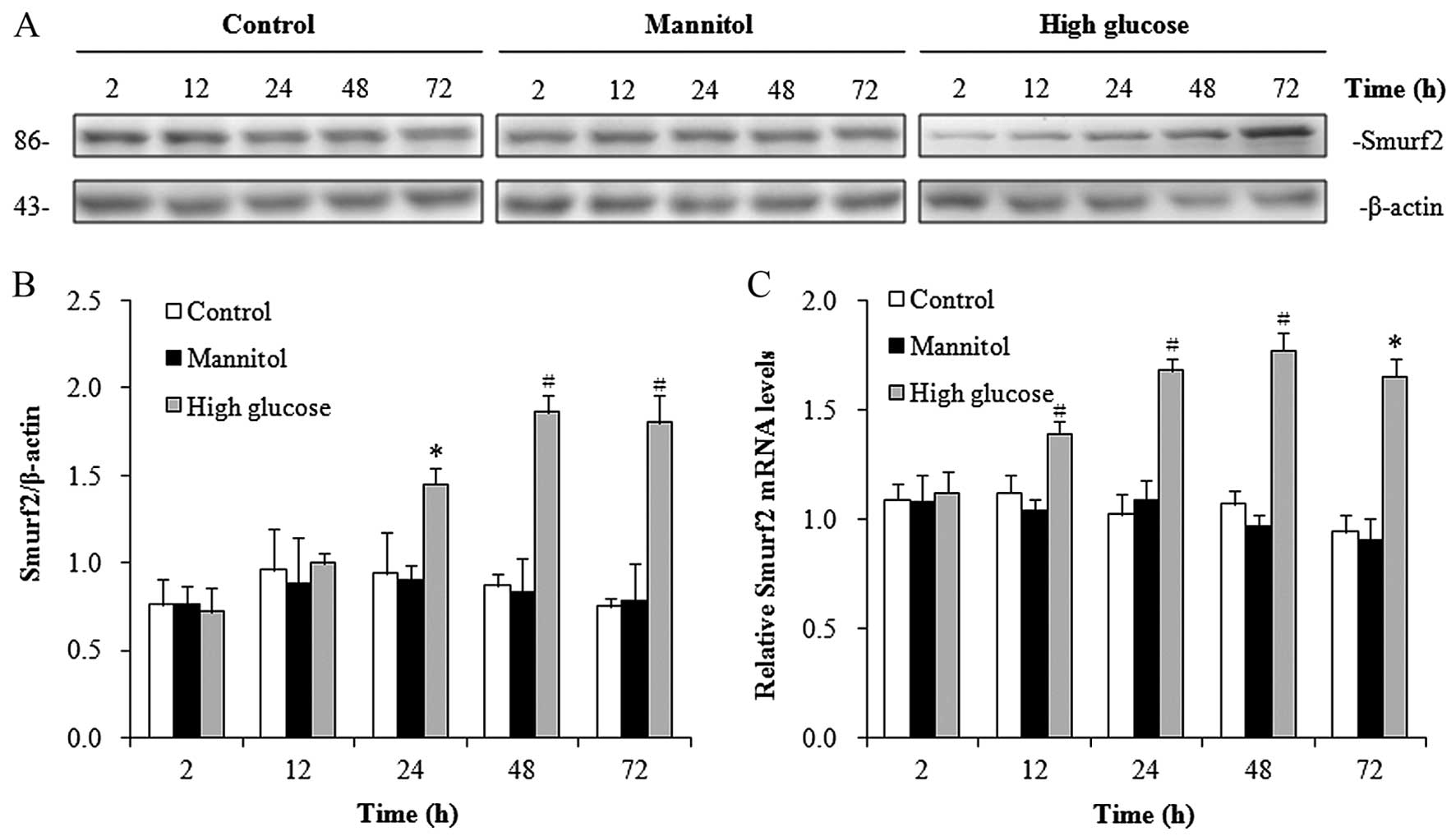
Smurf2 expression is induced by
high-glucose conditions in hRPTECs
Previous studies have shown that the E3 ubiquitin
ligase, Smurf2, targets SnoN for proteasome-mediated degradation
(14,15). In this study, to determine whether
Smurf2 contributes to the downregulation of SnoN under high-glucose
conditions, we measured the protein expression levels of Smurf2 in
the hRPTECs under high-glucose conditions. Western blot analysis
revealed that Smurf2 expression was significantly induced from 24 h
in the hRPTECs cultured under high-glucose conditions in a
time-dependent manner (Fig. 2A and
B). RT-qPCR demonstrated the increased mRNA expression of
Smurf2 in the hRPTECs cultured under high-glucose conditions from
12 h (Fig. 2C). Similar to the
results observed for SnoN, the hypertonic culture environment did
not affect the neither protein expression of Smurf2 (Fig. 2A and B) nor its mRNA expression
(Fig. 2C) in the hRPTECs. These
results demonstrated that the high-glucose conditions enhanced the
expression of Smurf2.
Knockdown of Smurf2 by siRNA stabilizes
SnoN expression in hRPTECs under high-glucose conditions
Our above-mentioned observations suggest that the
upregulation of Smurf2 may contribute to the downregulation of SnoN
under high-glucose conditions. To further examine this hypothesis,
we first used an RNA interference (RNAi) approach to knockdown
Smurf2 expression in the hRPTECs under high-glucose conditions. To
ensure the specificity of Smurf2 inhibition, two different
double-stranded Smurf2 siRNAs and a control siRNA were separately
transiently transfected into the hRPTECs. The transfection
efficiency was observed by calculating the ratio of red fluorescent
cells among the total cells. Approximately 70–80% of the hRPTECs
transfected with siRNA expressed a red fluorescence signal in the
nuclei, indicating efficient transfection (Fig. 3A). The Smurf2 mRNA levels were
reduced by 62% following transfection with Smurf2 siRNA-1 and by
45% following transfection with Smurf2 siRNA-2 compared with the
cells transfected with the control siRNA (Fig. 3B). Similarly, the Smurf2 protein
levels were reduced by 50% following transfection with Smurf2
siRNA-1 and by 34% following transfection with Smurf2 siRNA-2
compared with the cells transfected with the control siRNA
(Fig. 3C).
Subsequently, we examined the SnoN protein levels
following the knockdown of Smurf2 expression in the hRPTECs under
high-glucose conditions. The Smurf2 protein levels were increased
while the SnoN protein levels were decreased when the cells were
cultured in high-glucose medium (Fig.
4A and B). Following the knockdown of Smurf2 expression, the
SnoN protein levels were increased by 1.5-fold in the cells
cultured in high-glucose medium, to similar levels observed under
normal glucose conditions (Fig.
4B). These results suggest that high-glucose conditions
downregulate SnoN expression by upregulating Smurf2 in the
hRPTECs.
Smurf2 physically interacts with SnoN
under high-glucose conditions
To confirm the involvement of the
ubiquitin-proteasomal degradation pathway in the downregulation of
SnoN expression under high-glucose conditions, we used the
proteasome specific inhibitor, MG132, to block the
ubiquitin-proteasomal pathway in the hRPTECs. As shown in Fig. 5A, treatment with MG132 prevented
the degradation of SnoN protein in the hRPTECs under high-glucose
conditions. We then examined the potential physical interaction
between Smurf2 and SnoN in the hRPTECs by protein
co-immunoprecipitation. SnoN was detected in the immunocomplexes
that were precipitated with Smurf2-specific antibody under
high-glucose conditions, while it was undetectable when applied
with control IgG or under normal glucose conditions (Fig. 5B). These results demonstrated that
Smurf2 physically interacted with SnoN under high-glucose
conditions and suggested that SnoN protein was subjected to
proteasome-mediated degradation by Smurf2 in hRPTECs under
high-glucose conditions.
Pharmacological inhibition of TGF-β1
signaling with SB-431542 inhibits high-glucose-induced Smad2
activation and Smurf2 expression
It has been well documented that TGF-β1 plays a key
role in the pathophysiology of DN and that high glucose activates
TGF-β1 signaling (4,10). Thus, to determine whether the
upregulation of Smurf2 results from enhanced TGF-β1 signaling in
the hRPTECs under high-glucose conditions, we treated the hRPTECs
under high-glucose conditions with the TGF-β type I receptor
specific inhibitor, SB-431542, and examined the expression of
Smurf2. The phosphorylation of Smad2, a downstream target of TGF-β1
signaling, was stimulated by high glucose and prevented by
SB-431542 (Fig. 6A), supporting
the conclusion that high glucose activates TGF-β1 signaling
(4). Intriguingly, the
high-glucose induced increase in the protein levels of Smurf2 was
significantly inhibited by SB-431542 in a dose-dependent manner
(Fig. 6B). These results
demonstrated that SB-431542 blocked the high-glucose-induced
activation of Smad2 and inhibited the protein expression of the
Smurf2.
Discussion
In this study, we found that high-glucose conditions
decreased the protein level, while increasing the mRNA level of
SnoN in the hRPTECs, and concurrently upregulated Smurf2. Moreover,
Smurf2 physically interacted with SnoN under high-glucose
conditions and the knockdown of Smurf2 by siRNA or MG132 abolished
the downregulation of SnoN under high-glucose conditions.
Furthermore, the pharmacological inhibition of TGF-β1 signaling
with SB-431542 inhibited the high-glucose-induced Smad2 activation
and Smurf2 expression. Our findings suggest that the downregulation
of SnoN expression in hRPTECs under high-glucose conditions is
mediated by an increase in Smurf2 expression through TGF-β1/Smad
signaling.
SnoN is a negative regulator of TGF-β signaling as a
consequence of binding to Smad proteins (8,9).
SnoN disrupts the formation of a functional complex between Smad4
and activated Smad2/3, thereby blocking the Smad complexes from
activating the transcription of TGF-β1 target genes (16,17). Furthermore, SnoN may recruit other
transcriptional co-repressors, such as the nuclear hormone receptor
co-repressor (N-CoR), and prevent the binding of Smads to the
transcriptional co-activator, p300/CREB-binding protein (CBP)
(6,8,17).
These mechanisms may operate together to suppress TGF-β1/Smad
signaling through SnoN. On the other hand, Yang et al
(18) demonstrated that the
threshold for the TGF-β1 response was reduced significantly by the
complete depletion of SnoN and that a minimal amount of TGF-β1 was
sufficient to trigger a full-scale TGF-β1 response under chronic
disease conditions. It is widely accepted that hyperactive TGF-β1
signaling plays a crucial role in the genesis and progression of
diabetic renal injuries (11,12,19). Thus, it is possible that the
downregulation of SnoN protein expressino under high-glucose
conditions may lead to hyperactive TGF-β1/Smad signaling and
promote the pathogenesis of DN.
However, the mechanisms that regulate the expression
of SnoN are not yet fully understood. SnoN transcription is
strongly induced by TGF-β itself (6). In this study, we demonstrated that
the mRNA expression levels of SnoN were significantly increased in
the hRPTECs under high-glucose conditions, through an unknown
mechanism. This upregulation of SnoN transcription may function as
a negative feedback mechanism to inhibit TGF-β1 signaling. Although
SnoN mRNA levels were significantly increased, the SnoN protein
levels were decreased under these conditions, suggesting that SnoN
is mainly regulated at the post-transcriptional level.
Intracellular protein degradation is a tightly
regulated process that is essential to sustain normal cellular
functions and is mainly controlled by the ubiquitin-proteasome
system, which contains an E1 ubiquitin-activating enzyme, an E2
ubiq-uitin-conjugating enzyme, and a substrate specific E3
ubiquitin ligase. The E3 ubiquitin ligase defines the substrate
selectivity and the subsequent degradation by the 26S proteasome
(20,21). As previously demonstrated, the
deregulation of the ubiquitin-proteasome system disrupts normal
cellular homeostasis and leads to the development of a number of
human diseases, including Liddle syndrome, ischemic acute renal
failure and obstructive nephropathy (14,15,20). Smurf2, homologous to the E6-AP
carboxyl terminus (HECT) domain-containing E3 ubiquitin ligase,
targets SnoN for ubiquitin-mediated degradation by the proteasomes
(22). In the present study, we
demonstrated that SnoN reduction in the hRPTECs, cultured under
high-glucose conditions, was mediated by enhanced
proteasome-dependent degradation. Firstly, SnoN protein levels were
decreased in spite of an increase in its mRNA expression. Secondly,
Smurf2 mRNA and protein levels were induced in the hRPTECs cultured
in high-glucose medium. Thirdly, the knockdown of Smurf2 expression
by siRNA stabilized the SnoN protein levels in the hRPTECs.
Finally, Smurf2 physically interacted with SnoN, and treatment with
MG132 restored SnoN expression. Thus, Smurf2 targeting of SnoN for
degradation by the ubiquitin-proteasome system is one of the
mechanisms responsible for the downregulation of SnoN in hRPTECs
under high-glucose conditions. However, other E3 ubiquitin ligases,
such as the anaphase-promoting complex and Arkadia, have also been
shown to target SnoN for degradation in response to TGF-β1
(23,24). Whether they are also involved in
the proteasome-dependent degradation of SnoN under high-glucose
conditions remains to be determined.
The mRNA expression of Smurf2 is rapidly induced by
TGF-β itself (25,26). Furthermore, Smurf2 induction by
TGF-β1 requires Smad signaling rather than the p38
mitogen-activated protein kinase (MARK), phosphoinositide 3-kinase
(PI3K), c-Jun N-terminal kinase (JNK), or MEK signaling pathways
(26). SB-431542 is a potent and
specific inhibitor of the TGF-β type I receptor (27,28). Inman et al (28) reported that SB-431542 inhibited
the TGF-β-induced phosphorylation of Smad2 and blocked the
activation of TGF-β signaling. In this study, we found that
SB-431542 suppressed the expression of phosphorylated (p-)Smad2 and
Smurf2 in a dose-dependent manner. These observations suggest that
the upregulation of Smurf2 by TGF-β/Smad signaling contributes to
the degradation of SnoN under high-glucose conditions.
In conclusion, in this study, we demonstrated that
the down-regulation of SnoN expression in hRPTECs under
high-glucose conditions is mediated by an incresae in Smurf2
expression through the TGF-β1/Smad signaling pathway. Our findings
suggest that a decrease in SnoN expression may eliminate the
negative regulatory mechanism for TGF-β1 signaling and amplify
profibrotic TGF-β1 signaling, leading to a vicious cycle between
Smurf2 expression and TGF-β1 action. The preservation of SnoN by a
proteasome inhibitor or by the knockdown of Smurf2, may be an
effective approach for confining pathological TGF-β1 activity.
Acknowledgments
The present study was supported by grants from the
National Natural Science Foundation of China (no. 81300607) and the
Beijing Municipal Science and Technology Commission Fund (no.
D131100004713001).
References
|
1
|
Tang SC and Lai KN: The pathogenic role of
the renal proximal tubular cell in diabetic nephropathy. Nephrol
Dial Transplant. 27:3049–3056. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Phillips AO and Steadman R: Diabetic
nephropathy: the central role of renal proximal tubular cells in
tubulointerstitial injury. Histol Histopathol. 17:247–252.
2002.PubMed/NCBI
|
|
3
|
Vallon V and Thomson SC: Renal function in
diabetic disease models: the tubular system in the pathophysiology
of the diabetic kidney. Annu Rev Physiol. 74:351–375. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Loeffler I and Wolf G: Transforming growth
factor-β and the progression of renal disease. Nephrol Dial
Transplant. 29(Suppl 1): i37–i45. 2014. View Article : Google Scholar
|
|
5
|
Massagué J and Gomis RR: The logic of
TGFbeta signaling. FEBS Lett. 580:2811–2820. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Stroschein SL, Wang W, Zhou S, Zhou Q and
Luo K: Negative feedback regulation ofTGF-beta signaling by the
SnoN oncoprotein. Science. 286:771–774. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wotton D and Massagué J: Smad
transcriptional corepressors in TGF beta family signaling. Curr Top
Microbiol Immunol. 254:145–164. 2001.PubMed/NCBI
|
|
8
|
Luo K: Ski and SnoN: Negative regulators
of TGF-beta signaling. Curr Opin Genet Dev. 14:65–70. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Deheuninck J and Luo K: Ski and SnoN,
potent negative regulators of TGF-beta signaling. Cell Res.
19:47–57. 2009. View Article : Google Scholar
|
|
10
|
Chen S, Jim B and Ziyadeh FN: Diabetic
nephropathy and transforming growth factor-beta:transforming our
view of glomerulosclerosis and fibrosis build-up. Semin Nephrol.
23:532–543. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hills CE and Squires PE: The role of TGF-β
and epithelial-to mesenchymal transition in diabetic nephropathy.
Cytokine Growth Factor Rev. 22:131–139. 2011.PubMed/NCBI
|
|
12
|
Lan HY: Transforming growth factor-β/Smad
signalling in diabetic nephropathy. Clin Exp Pharmacol Physiol.
39:731–738. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu R, Wang Y, Xiao Y, Shi M, Zhang G and
Guo B: SnoN as a key regulator of the high glucose-induced
epithelial-mesenchymal transition in cells of the proximal tubule.
Kidney Blood Press Res. 35:517–528. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tan R, Zhang J, Tan X, Zhang X, Yang J and
Liu Y: Downregulation of SnoN expression in obstructive nephropathy
is mediated by an enhanced ubiquitin-dependent degradation. J Am
Soc Nephrol. 17:2781–2791. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fukasawa H, Yamamoto T, Togawa A, Ohashi
N, Fujigaki Y, Oda T, Uchida C, Kitagawa K, Hattori T, Suzuki S, et
al: Ubiquitin-dependent degradation of SnoN and Ski is increased in
renal fibrosis induced by obstructive injury. Kidney Int.
69:1733–1740. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu X, Sun Y, Weinberg RA and Lodish HF:
Ski/Sno and TGF-beta signaling. Cytokine Growth Factor Rev. 12:1–8.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wu JW, Krawitz AR, Chai J, Li W, Zhang F,
Luo K and Shi Y: Structural mechanism of Smad4 recognition by the
nuclear oncoprotein Ski: insights on Ski-mediated repression of
TGF-beta signaling. Cell. 111:357–367. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yang J, Zhang X, Li Y and Liu Y:
Downregulation of Smad transcriptional corepressors SnoN and Ski in
the fibrotic kidney: an amplification mechanism for TGF-beta1
signaling. J Am Soc Nephrol. 14:3167–3177. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ziyadeh FN: Mediators of diabetic renal
disease: The case for TGF-β as the major mediator. J Am Soc
Nephrol. 15(Suppl 1): S55–S57. 2004. View Article : Google Scholar
|
|
20
|
Debigaré R and Price SR: Proteolysis, the
ubiquitin-proteasome system, and renal diseases. Am J Physiol Renal
Physiol. 285:F1–F8. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mani A and Gelmann EP: The
ubiquitin-proteasome pathway and its role in cancer. J Clin Oncol.
23:4776–4789. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bonni S, Wang HR, Causing CG, Kavsak P,
Stroschein SL, Luo K and Wrana JL: TGF-beta induces assembly of a
Smad2-Smurf2 ubiquitin ligase complex that targets SnoN for
degradation. Nat Cell Biol. 3:587–595. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wan Y, Liu X and Kirschner MW: The
anaphase-promoting complex mediates TGF-beta signaling by targeting
SnoN for destruction. Mol Cell. 8:1027–1039. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nagano Y, Mavrakis KJ, Lee KL, Fujii T,
Koinuma D, Sase H, Yuki K, Isogaya K, Saitoh M, Imamura T, et al:
Arkadia induces degradation of SnoN and c-Ski to enhance
transforming growth factor-beta signaling. J Biol Chem.
282:20492–20501. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ohashi N, Yamamoto T, Uchida C, Togawa A,
Fukasawa H, Fujigaki Y, Suzuki S, Kitagawa K, Hattori T, Oda T, et
al: Transcriptional induction of Smurf2 ubiquitin ligase by
TGF-beta. FEBS Lett. 579:2557–2563. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tan R, He W, Lin X, Kiss LP and Liu Y:
Smad ubiquitination regulatory factor-2 in the fibrotic kidney:
Regulation, target specificity, and functional implication. Am J
Physiol Renal Physiol. 294:F1076–F1083. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Callahan JF, Burgess JL, Fornwald JA,
Gaster LM, Harling JD, Harrington FP, Heer J, Kwon C, Lehr R,
Mathur A, et al: Identification of novel inhibitors of the
transforming growth factor beta1 (TGF-β1) type 1 receptor (ALK5). J
Med Chem. 45:999–1001. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Inman GJ, Nicolás FJ, Callahan JF, Harling
JD, Gaster LM, Reith AD, Laping NJ and Hill CS: SB-431542 is a
potent and specific inhibitor of transforming growth factor-beta
superfamily type I activin receptor-like kinase (ALK) receptors
ALK4, ALK5, and ALK7. Mol Pharmacol. 62:65–74. 2002. View Article : Google Scholar : PubMed/NCBI
|