Introduction
Pregnancy is both a complex and unique physiological
process that involves many events, including successful
implantation, decidualization, placentation and parturition
(1). The placenta is an organ
whcih grows in the uterus and connects the fetus to the uterine
wall of the mother; it allows not only for the uptake of nutrients,
but also for the exchange of gases and the transport of metabolic
waste (2,3). Invasion of extravillous trophoblasts
into the uterine decidua and myometrium occurs within the first 20
weeks of gestation. This is of great importance to maternal
circulation at the maternal-fetal interface during pregnancy
(4,5). Inadequate trophoblast invasion into
the uterine spiral arteries and impaired placentation affect fetal
growth and lead to serious pregnancy complications, such as
intrauterine growth restriction (IUGR), preeclampsia (PE), preterm
birth and spontaneous abortion (6,7).
Additionally, excessive trophoblast apoptosis can also be observed
in the placentas of patients who have been affected by pregnancy
complications (5,8). However, the precise mechanisms
remain unclear.
Pleckstrin homology-like domain, family A, member 2
(PHLDA2; also known as BRW1C, BWR1C, HLDA2, IPL and TSSC3), a
maternally expressed imprinted gene (9), is negatively correlated with size at
birth and has been linked to fetal growth restriction in a number
of studies (10–13). In the placentae of babies affected
by IUGR (10,14,15) and with low birth weight (LBW)
(12), upregulated expression of
PHLDA2 has been observed. In a mouse model, it was noted that
knockdown of PHLDA2 resulted in a significant increase in placental
size during gestation (16).
Previous research has also shown that downregulated PHLDA2
expression in one-cell zygotes using siRNA resulted in accelerated
blastocyst development (17).
Therefore, high expression of PHLDA2 may have a close correlation
with abnormal placental function and pregnancy complications.
However, the effects of PHLDA2 on trophoblast invasion and cell
apoptosis, which play a critical role in the onset and development
of pregnancy complications, have yet to be fully clarified.
In the present study, we isolated primary
trophoblasts and detected cytokeratin 18 (CK18), vimentin and human
placental lactogen (hPL) expression. The human placental
choriocarcinoma cell line JEG-3 and primary trophoblasts were
infected with lentiviruses, and subsequently we examined the impact
of PHLDA2 overexpression on cell proliferation, apoptosis,
mitochondrial function, migration and invasion.
Materials and methods
Tissue collection
Human villous tissues were obtained from women who
underwent artificial abortion within 7 weeks of pregnancy. Written
informed consent was signed by the patients who were recruited to
the study. Ethics approval was provided by The China Medical
University Ethics Committee (Shenyang, China).
Primary culture of primary trophoblasts
and cell culture
The human placental villous tissues were first
rinsed in PBS and minced into 1–2 mm3 fragments, placed
in a 35-mm culture dish and subsequently subjected to sequential
10-min treatments with 0.125% trypsin at 37°C. The supernatant was
collected, and then Dulbecco's Modified Eagle's medium (DMEM)
(Gibco, Grand Island, NY, USA) containing fetal bovine serum (FBS)
(HyClone, Logan, UT, USA) was added in order to terminate the
digestion. The cell suspension was filtered through a 70-µm
mesh nylon strainer to remove undigested tissue fragments. The
filtrate was centrifuged, washed with PBS and cultured in minimum
essential medium (MEM) for 48 h. The adherent cells were primary
trophoblasts.
Human placental choriocarcinoma JEG-3 cells were
purchased from the Chinese Academy of Sciences Cell Bank (Shanghai,
China). The cells were cultured in DMEM supplemented with 10% FBS
and incubated in a humidified atmosphere with 5% CO2 at
37°C.
Treatment groups
Primary trophoblasts and JEG-3 cells that were
transfected with lentivirus containing control vector or lentivirus
overexpressing PHLDA2 were termed Vector or PHLDA2. Primary
trophoblasts and JEG-3 cells, which were not transfected with
lentiviruses, were used as controls in our experiments.
Immunofluorescence staining
In the present study, the primary trophoblast slides
were first fixed with 4% paraformaldehyde for 15 min and then
permeabilized with 0.1% Triton X-100 for 30 min at room
temperature. Goat serum (purchased from Solarbio, Beijing, China)
was added in order to block non-specific binding sites, followed by
incubation with CK18 antibody (dilution 1:50; #sc-6259; Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA), vimentin antibody
(dilution 1:100; #WL0274; Wanleibio, Shenyang, China) and hPL
antibody (dilution 1:100; #ab11396; Abcam, Cambridge, MA, USA) at
4°C overnight. The slides were then washed three times with PBS for
5 min and incubated with Cy3-labeled goat anti-rabbit secondary
antibody (#A0516) or Cy3-labeled goat anti-mouse secondary antibody
(#A0521) (Beyotime Institute of Biotechnology, Haimen, China) for 1
h at room temperature. Subsequently, 4′,6-diamidino-2-phenylindole
(DAPI) was added in order to stain nuclei, and they were then
examined by fluorescence microscopy (BX53; purchased from Olympus,
Tokyo, Japan).
Generation of overexpressing cell
lines
Lentivirus overexpressing PHLDA2 or containing
control vector was purchased from HanBio (Shanghai, China). JEG-3
cells and primary trophoblasts were cultured in 6-well plates.
After reaching 70% confluence, medium containing lentiviruses and
polybrene (8 µg/ml; Hanbio) was added at a multiplicity of
infection (MOI) of 10 and mixed with the cells. Polybrene was used
to improve infection efficiency. After incubation for 24 h,
supernatants in the wells were replaced by DMEM containing FBS and
cultured for 24 and 48 h for subsequent analyses.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was isolated from the cells in each group
with an RNAsimple Total RNA kit (Tiangen Biotech Co., Ltd.,
Beijing, China) and reverse transcription was carried out. RT-qPCR
was performed using SYBR-Green Master Mix (Solarbio) on an
Exicycler™ 96 quantitative fluorescence analyzer (Bioneer
Corporation, Daejeon, Korea). The reaction conditions were as
follows: 95°C for 10 min; 40 cycles of 95°C for 10 sec, 60°C for 20
sec and 72°C for 30 sec. Gene expression levels were calculated
using the 2−ΔΔCt method, as previously described
(18). β-actin was used as the
normalization internal control and sequences of primers are shown
in Table I.
 | Table IGenes and primer sequences. |
Table I
Genes and primer sequences.
| Gene | Primer
sequence | Accession no. | Temp (°C) |
|---|
| PHLDA2 | F:
CCATCCTCAAGGTGGACTGC | NM_003311.3 | 59.9 |
| R:
TTCCTGGCGGCTGCGAAAGT | | 67.5 |
| β-actin | F:
CCATCGTCCACCGCAAAT | NM_001101.3 | 59.5 |
| R:
GCTGTCACCTTCACCGTTC | | 55.6 |
Cell Counting Kit-8 (CCK-8) cell
proliferation assay
Cell proliferation was detected by CCK-8 (Beyotime
Institute of Biotechnology) according to the manufacturer's
instructions. In brief, cells were seeded in 96-well plates at a
density of 2×103 cells/well. CCK-8 reagent was added at
0, 24, 48 and 72 h. The plates were incubated for an additional 1 h
at 37°C. The absorbance at 450 nm was measured with a microplate
reader (ELX-800; BioTek, Winooski, VT, USA).
Western blot analysis
After the cells were lysed using NP-40 lysis buffer
(Beyotime Institute of Biotechnology), total proteins were
extracted from the supernatants of cell lysates. In addition, cells
were collected and ice-cold PBS was used to resuspend the pelleted
cell. Mitochondrial and cytosolic proteins were isolated with a
Cell Mitochondria Isolation kit (Beyotime Institute of
Biotechnology). Subsequently, 1 ml mitochondrial lysis buffer
containing phenylmethanesulfonyl fluoride (PMSF) was added to
incubate with the isolated mitochondria, and the mitochondrial
proteins were obtained. Protein concentrations were determined
using a BCA protein assay kit (Beyotime Institute of
Biotechnology). Equal amounts of proteins (40 µg) were
separated on sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE) and then transferred to PVDF membranes
(Millipore Corp., Bedford, MA, USA). The membranes were incubated
overnight at 4°C with PHLDA2 antibody (dilution 1:400; #bs-6884R;
BIOSS, Beijing, China), cleaved-caspase-3 antibody (dilution
1:1,000; #WL0146), Bax antibody (dilution 1:1,000; #WL0101), Bcl-2
antibody (dilution 1:1,000; #WL0104) and cytochrome c
antibody (dilution 1:1,000; #WL0483; Wanleibio). We also used COX
IV antibody (#WL0933; Wanleibio) and β-actin (#WL0001; Wanleibio).
COX IV was used as a loading control for mitochondrial proteins and
β-actin for cytosolic proteins. After being washed with TTBS
buffer, the membranes were incubated with HRP-conjugated goat
anti-rabbit IgG secondary antibody (dilution 1:5,000; #A0208;
Beyotime Institute of Biotechnology) for 45 min at 37°C. Band
intensities were determined using Gel-Pro Analyzer software.
Analysis of apoptosis by flow
cytometry
Cells were trypsinized, counted and seeded.
Subsequently, cells were harvested and stained with Annexin
V-FITC/propidium iodide (PI) (KeyGen, Nanjing, China) according to
the manufacturer's instructions. Briefly, cells were washed twice
with PBS and resuspended in binding buffer. Cells were subsequently
incubated with 5 µl Annexin V-FITC and 5 µl PI for 15
min at room temperature in the dark and analyzed by flow cytometry
(BD Accuri C6; Becton-Dickinson, Franklin Lakes, NJ, USA).
Hoechst staining
After 24 h infection, cells from each group were
cultured on the slides in 12-well plates at a density of
1×105 cells/slide. When cells reached 80% confluence
(cultured for approximately 24 h), the supernatant was discarded
and they were washed twice with PBS. Cells were then fixed for 20
min at room temperature. Hoechst solution (Beyotime Institute of
Biotechnology) was added, and cells were observed and photographed
under a microscope (AE31; Motic, Xiamen, China).
Measurement of reactive oxygen species
(ROS) using 2′7′-dichlorofluorescin diacetate (DCFH-DA)
Intracellular ROS levels were measured using a
Reactive Oxygen Species Assay kit (Beyotime Institute of
Biotechnology) and flow cytometry (C6; Becton-Dickinson). In brief,
cells in each group were plated in a T25 culture flask at a density
of 5×105 cells/flask. When they reached 70% confluence,
the supernatant was discarded and 2 ml of diluted DCFH-DA (10
µM final concentration) solution was added to each well and
then incubated at 37°C for 20 min and washed with serum-free medium
to remove extracellular DCFH-DA. The cells were harvested, washed,
resuspended in PBS and then detected by flow cytometry.
Mitochondrial membrane potential
assay
Mitochondrial membrane potential (also known as ΔΨm)
was studied using MitoTracker Red CMXRos (Invitrogen Life
Technologies, Grand Island, NY, USA) according to the
manufacturer's instructions. In brief, cells were harvested and
labeled with MitoTracker (a dilution of 1:200) at 37°C for 20 min.
Cells were harvested, washed twice with PBS and resuspended in PBS.
Mitochondrial membrane potential was analyzed by flow cytometry
(FACSCalibur; Becton-Dickinson).
Wound healing assay
Cells in each group were cultured in 6-well plates.
Subsequently, the supernatant was aspired and a wound was created
with a sterile 200-µl pipette tip. The cells were then
washed twice with serum-free medium (Gibco) to remove cell debris
and photographed immediately. After 24 h of incubation,
photographic images were acquired under an inverted microscope
(AE31; Motic) and the migration distance was calculated. The cell
migration rates were calculated using the following formula: cell
migration rate (%) = (1 − the distance following healing/the
distance prior to healing) × 100%. Each experiment was repeated
three times.
Transwell assay
A Transwell chamber (Corning Life Sciences,
Tewksbury, MA, USA) pre-coated with Matrigel gel (Becton-Dickinson)
was placed into a 24-well plate. After incubation for 2 h at 37°C,
the Matrigel gel solidified. Cells in each group were digested with
trypsin, resuspended in serum-free medium, and then added to the
upper chamber (2×104 cells/well). Subsequently, 800
µl medium containing 20% FBS was added to the lower chamber.
After incubation for 24 h at 37°C, the cells which had not invaded
were removed with a cotton swab. The invading cells in the lower
chamber were fixed with paraformaldehyde at room temperature for 20
min and stained with hematoxylin staining solution for 5 min. Five
fields were randomly selected and the number of invading cells was
counted under an inverted microscope (AE31; Motic).
Statistical analysis
All experiments were performed three times. Data are
presented as the means ± SD (n=3 replicates for each analysis). All
analyses were performed using GraphPad Prism 5.0 software (GraphPad
Software, Inc., San Diego, CA, USA). One-way ANOVA followed by the
Bonferroni post hoc test was performed. A P-value <0.05 was
considered to indicate a statistically significant difference.
Results
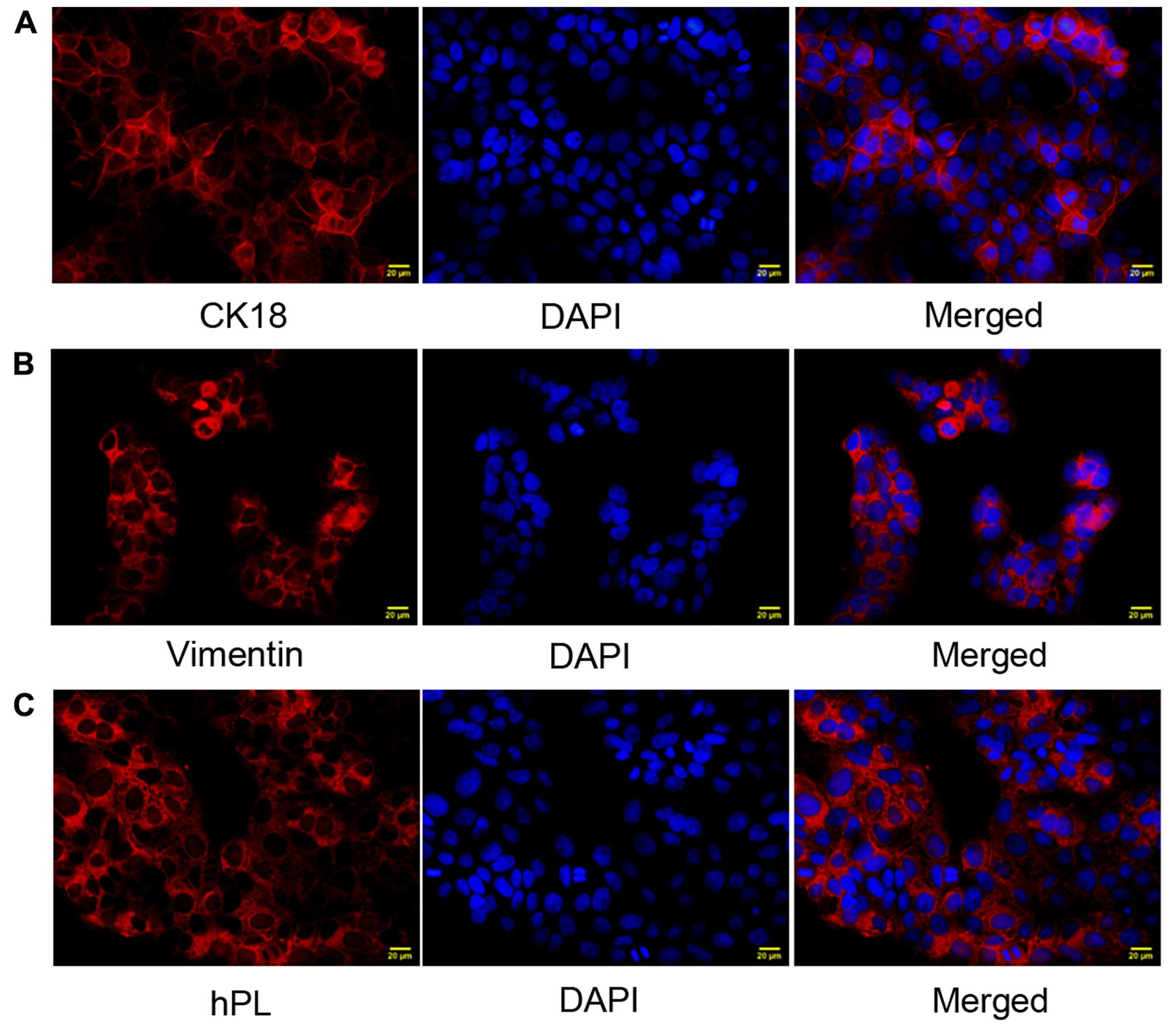
Isolation and characterization of primary
trophoblasts
We examined the expression of CK18, vimentin and hPL
in the isolated cells. Immunofluorescence staining demonstrated
that CK18 (Fig. 1A), vimentin
(Fig. 1B) and hPL (Fig. 1C) were highly expressed,
suggesting that we successfully isolated and characterized primary
trophoblasts.
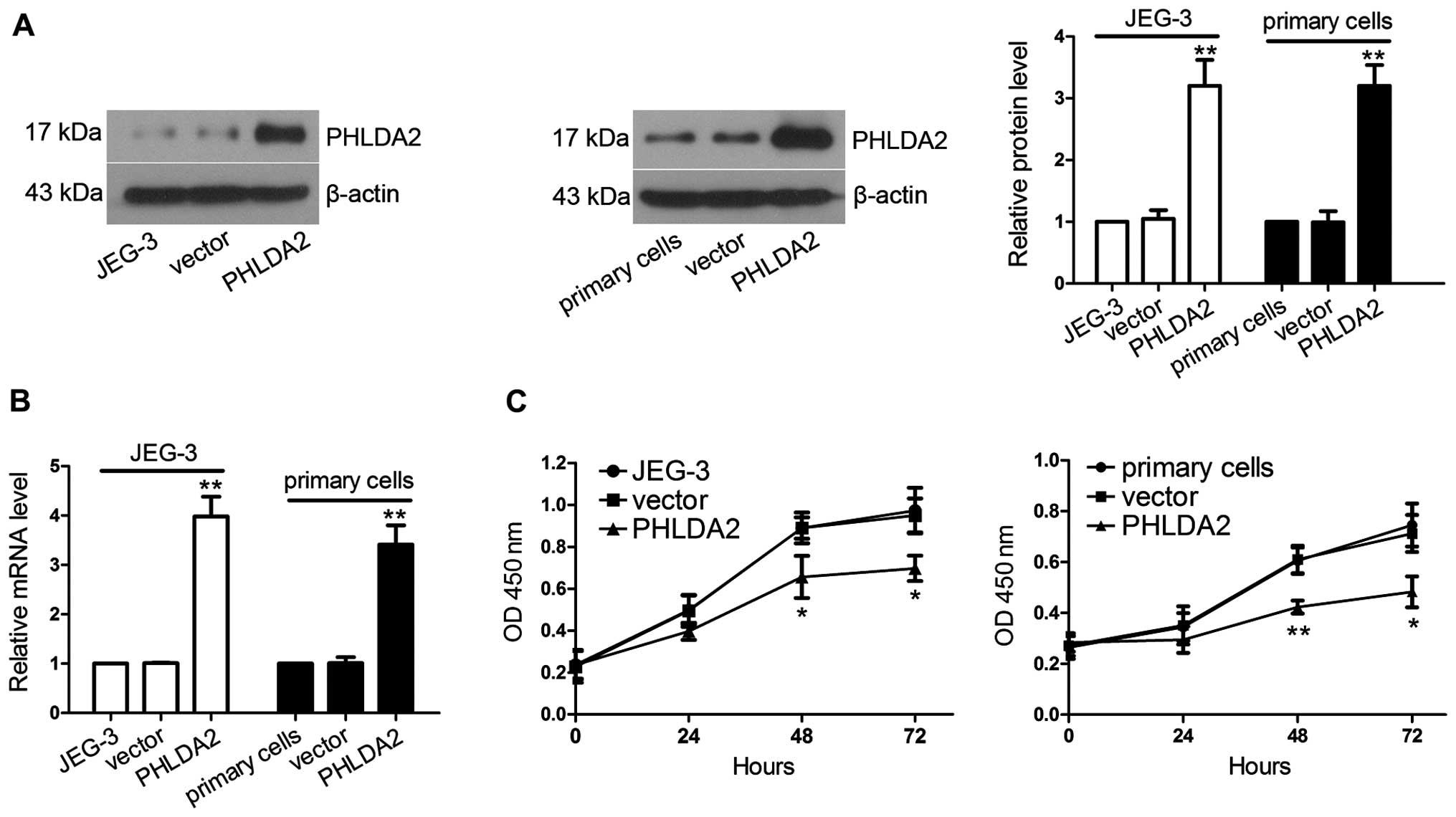
Establishment of cell lines
overexpressing PHLDA2
JEG-3 cells and primary trophoblasts were infected
with lentivirus. RT-qPCR and western blot analysis were used to
identify the increased expression of PHLDA2 in lentivirus-infected
JEG-3 cells and lentivirus-infected primary trophoblasts at 48-h
post-infection. As shown in Fig. 2A
and B, PHLDA2 mRNA and protein expression levels in the JEG-3
cells infected with PHLDA2-overexpressing lentivirus were 3.94-fold
(P<0.01) and 3.05-fold (P<0.01) higher than those in the
JEG-3 cells infected with the lentivirus containing control vector.
PHLDA2 mRNA and protein expression levels in the primary cells
infected with PHLDA2-overexpressing lentivirus were increased to
3.38-fold (P<0.01) and 3.23-fold (P<0.01), respectively, when
compared with levels in the primary cells infected with the
vector-containing lentivirus.
PHLDA2 overexpression inhibits cell
proliferation
To evaluate the effect of PHLDA2 overexpression in
regulating the proliferation of JEG-3 cells and primary
trophoblasts, a CCK-8 cell proliferation assay was used at 24-h
post-infection. As shown in Fig.
2C, the proliferative capabilities at 48 h (JEG-3, P<0.05;
primary cells, P<0.01) and 72 h (JEG-3, P<0.05; primary
cells, P<0.05) were severely impaired in the JEG-3 and primary
cells infected with the lentivurus overexpressing PHLDA2.
PHLDA2 overexpression promotes
apoptosis
The ability of PHLDA2 to induce apoptosis was
detected by Hoechst staining, flow cytometry and western blot
analysis at 24 h post-infection. After the cells were stained with
Annexin V-FITC/PI, a significant increase in the PHLDA2-induced
apoptotic rate was observed by flow cytometric analysis in the
JEG-3 cells infected with PHLDA2-overexpressing
lentivirus(25.37±4.11%, P<0.01) (Fig. 3A) and also the primary cells
infected with PHLDA2-overexpressing lentivirus (36.14±3.48%,
P<0.01) compared with those in the control vector group (JEG-3,
3.50±1.15%; primary cells, 12.78±2.59%), respectively. The
condensed and fragmented nuclei were regarded as apoptotic cells.
As observed from the photomicrographs, PHLDA2 overexpression
markedly induced chromatin condensation (Fig. 3B). As shown in Fig. 3C, during the apoptotic process,
PHLDA2 overexpression in JEG-3 cells and also in primary cells
sharply increased cleaved-caspase-3 (JEG-3, 2.25-fold, P<0.01;
primary cells, 2.00-fold, P<0.01) and pro-apoptotic protein Bax
expression (JEG-3, 2.98-fold, P<0.01; primary cells, 2.54-fold,
P<0.01), respectively, compared with the vector-infected cells.
Anti-apoptotic protein Bcl-2 levels were decreased 0.33-fold
(JEG-3, P<0.01) and 0.48-fold (primary cells, P<0.01).
PHLDA2 overexpression induces ROS
production and also mitochondrial injury
To examine the impact of PHLDA2 overexpression on
ROS accumulation, mitochondrial injury and cytochrome c
release, we measured mitochondrial membrane potential and ROS
content using flow cytometric analysis at 24 h post-infection.
Mitochondrial ROS was measured using DCFH-DA staining. The JEG-3
cells infected with PHLDA2-overexpressing lentivurus (Fig. 4A) (P<0.01) and the primary
cells infected with PHLDA2-overexpressing lentivirus (P<0.01)
demonstrated significantly increased ROS levels as compared with
the groups infected with the vector-containing lentivirus. PHLDA2
overexpression induced significant loss of mitochondiral membrane
potential (JEG-3, P<0.01; primary cells, P<0.01) (Fig. 4B). Additionally, western blot
analysis detected that cytochrome c in the cytosol (JEG-3,
1.81-fold, P<0.01; primary cells, 1.81-fold, P<0.01)
(Fig. 4C) was upregulated,
whereas cytochrome c in the mitochondria was downregulated
(JEG-3, 0.46-fold, P<0.01; primary cells, 0.47-fold,
P<0.01).
PHLDA2 overexpression inhibits cell
migration and invasion
To evaluate the role of PHLDA2 in the regulation of
trophoblast migration, we carried out a wound healing assay at 24 h
post-infection. The wound healing assay revealed that the migration
rates of the cells in the JEG-3 group infected with
PHLDA2-overexpressing lentivirus (32.30±3.93%, P<0.05) (Fig. 5A) and the primary cells infected
with PHLDA2-overexpressing lentivirus (15.19±3.16%, P<0.05) were
significantly decreased compared with those treated with the
control vector-containing lentivirus (JEG-3, 48.72±4.73%; primary
cells, 32.59±8.07%). Subsequently, we assessed the effect of PHLDA2
on cell invasion with a Transwell assay. The results indicated that
the number of invading cells in the JEG-3 group treated with
lentivirus overexpressing PHLDA2 (14.00±2.45 cells/well, P<0.01)
(Fig. 5B) and the primary cells
infected with PHLDA2-overexpressing lentivirus (13.00±1.87
cells/well, P<0.05) were significantly lower than those in
groups infected with the control vector-containing lentivirus
(JEG-3, 38.80±4.66 cells/well; primary cells, 24.60±2.97
cells/well).
Discussion
PHLDA2 is a maternally expressed and paternally
imprinted gene (19) and is
associated with fetal growth restriction (13). Our present study is the first, to
the best of our knowledge, to demonstrate the impact of PHLDA2 on
trophoblast function. We obtained primary trophoblasts, and CK18
(20), vimentin and hPL (21) were used as markers to characterize
and identify trophoblasts. We detected high expression levels of
CK18, vimentin and hPL in primary trophoblasts with
immunofluorescence staining. Subsequently, primary trophoblasts and
JEG-3 cells were infected with lentiviruses and PHLDA2
overexpression cell lines were established. We noted that PHLDA2
inhibited cell proliferation, migration and invasion, and induced
cell apoptosis.
Bcl-2 family members act as regulators of cell
apoptosis (22) and have been
divided into three groups: antiapoptotic proteins (Bcl-2, Bcl-xl
and Mcl-1), proapoptotic proteins (Bax and Bak) and BH3-only
proteins (Bad, Bik and Bid) (23). Once the cells receive apoptosis
signals, proapoptotic proteins Bax and Bak are activated
(oligomerization of Bax and Bak), inserted into the outer
mitochondrial membrane and then trigger the release of cytochrome
c into the cytosol. Subsequently, cytochrome c
activates caspases and induces cell apoptosis (24,25). The role of antiapoptotic members
is to inhibit their proapoptotic partners (26). A previous study has reported that
PHLDA2 inhibits tumor growth and induces tumor cell apoptosis in
vivo. Knockdown of PHLDA2 promotes human osteosarcoma SaOS-2
cell proliferation and decreases the apoptotic rate in vitro
and in vivo (27).
However, the roles of PHLDA2 in the proliferation and apoptosis of
trophoblasts have not yet been reported. In the present study,
JEG-3 cells and primary trophoblasts were infected with
lentiviruses. We then evaluated the impact of PHLDA2 overexpression
on cell proliferation capability and apoptosis. The results showed
that PHLDA2 mRNA and protein expression levels were remarkably
upregulated in both sets of cells infected with the
PHLDA2-overexpressing lentivirus. In accordance with previous
research (27), the
overexpression of PHLDA2 significantly suppressed cell
proliferation and induced apoptosis, and the activation of
caspase-3 and pro-apoptotic protein Bax, as well as the inhibition
of antiapoptotic protein Bcl-2.
ROS, such as superoxide anions, hydroxyl radicals
and hydrogen peroxide, are the most common metabolic products of
organism (28). Low levels of ROS
serve as 'redox messengers' in the regulation of multiple signaling
pathways and cellular processes, whereas excess ROS lead to the
inhibition of protein functions and the promotion of cell apoptosis
(29). When ROS reach a threshold
level, they cause a decrease in mitochondrial membrane potential,
and also cause the release of cytochrome c from the
mitochondria into the cytosol and the release of caspase-activating
proteins (29,30). PHLDA2 has been shown to induce
apoptosis of osteosarcoma tumor-initiating cells by activating
caspase-3, releasing cytochrome c and decreasing the
mitochondrial membrane potential (31). The results of the present study
demonstrated that PHLDA2 significantly promoted the release of
cytochrome c, and induced loss of mitochondrial membrane
potential and ROS accumulation in trophoblasts. Our study suggests
that PHLDA2 overexpression induces trophoblast apoptosis via the
mitochondrial pathway and the accumulation of ROS.
The invasion of trophoblasts into the maternal
endometrium plays an important role in successful implantation and
placentation during pregnancy (32–36). Various researchers have
demonstrated that shallow trophoblast invasion and inadequate
uterine remodeling of spiral arteries are ubiquitous in the
placenta of patients with PE in previous studies (37,38). Additionally, the expression of
PHLDA2 is elevated in the placenta of infants who are affected by
IUGR (10). PHLDA2 is a regulator
of embryonic development. It has previously been suggested that
PHLDA2 overexpression results in placental abnormalities, including
IUGR and PE, whereas low expression of PHLDA2 causes hypertrophic
placenta or placentomegaly (39).
Based on these statements, we hypothesized that PHLDA2 was
associated with trophoblast invasion. Therefore, in the present
study, trophoblasts were infected with a lentivirus overexpressing
PHLDA2 in order to examine the functions of PHLDA2. As expected,
our results demonstrated that PHLDA2 overexpression inhibited
trophoblast migration and invasion.
Our results indicate that PHLDA2 overexpression
inhibited trophoblast proliferation and induced cell apoptosis
through the involvement of the mitochondrial pathway. Moreover, we
suggest that PHLDA2 inhibits trophoblast migration and invasion.
These results suggest that PHLDA2 overexpression causes inadequate
trophoblast invasion. Further studies are warranted to validate our
results. Our study provides evidence for investigating the possible
role of PHLDA2 in the occurence and progression of
pregnancy-associated complications.
References
|
1
|
Cha J, Sun X and Dey SK: Mechanisms of
implantation: strategies for successful pregnancy. Nat Med.
18:1754–1767. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Garnica AD and Chan WY: The role of the
placenta in fetal nutrition and growth. J Am Coll Nutr. 15:206–222.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nagashima T, Li Q, Clementi C, Lydon JP,
DeMayo FJ and Matzuk MM: BMPR2 is required for postimplantation
uterine function and pregnancy maintenance. J Clin Invest.
123:2539–2550. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Romero R, Kusanovic JP, Chaiworapongsa T
and Hassan SS: Placental bed disorders in preterm labor, preterm
PROM, spontaneous abortion and abruptio placentae. Best Pract Res
Clin Obstet Gynaecol. 25:313–327. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Whitley GS, Dash PR, Ayling LJ, Prefumo F,
Thilaganathan B and Cartwright JE: Increased apoptosis in first
trimester extravillous trophoblasts from pregnancies at higher risk
of developing preeclampsia. Am J Pathol. 170:1903–1909. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Valenzuela FJ, Vera J, Venegas C, Pino F
and Lagunas C: Circadian system and melatonin hormone: risk factors
for complications during pregnancy. Obstet Gynecol Int.
2015(825802)2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Khong Y and Brosens I: Defective deep
placentation. Best Pract Res Clin Obstet Gynaecol. 25:301–311.
2011. View Article : Google Scholar
|
|
8
|
Huang Y, Dong F, Du Q, Zhang H, Luo X,
Song X, Zhao X, Zhang W and Tong D: Swainsonine induces apoptosis
through mitochondrial pathway and caspase activation in goat
trophoblasts. Int J Biol Sci. 10:789–797. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Qian N, Frank D, O'Keefe D, Dao D, Zhao L,
Yuan L, Wang Q, Keating M, Walsh C and Tycko B: The IPL gene on
chromosome 11p15.5 is imprinted in humans and mice and is similar
to TDAG51, implicated in Fas expression and apoptosis. Hum Mol
Genet. 6:2021–2029. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
McMinn J, Wei M, Schupf N, Cusmai J,
Johnson EB, Smith AC, Weksberg R, Thaker HM and Tycko B: Unbalanced
placental expression of imprinted genes in human intrauterine
growth restriction. Placenta. 27:540–549. 2006. View Article : Google Scholar
|
|
11
|
Jensen AB, Tunster SJ and John RM: The
significance of elevated placental PHLDA2 in human growth
restricted pregnancies. Placenta. 35:528–532. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Apostolidou S, Abu-Amero S, O'Donoghue K,
Frost J, Olafsdottir O, Chavele KM, Whittaker JC, Loughna P,
Stanier P and Moore GE: Elevated placental expression of the
imprinted PHLDA2 gene is associated with low birth weight. J Mol
Med Berl. 85:379–387. 2007. View Article : Google Scholar
|
|
13
|
Tunster SJ, Van De Pette M and John RM:
Isolating the role of elevated Phlda2 in asymmetric late fetal
growth restriction in mice. Dis Model Mech. 7:1185–1191. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kumar N, Leverence J, Bick D and Sampath
V: Ontogeny of growth-regulating genes in the placenta. Placenta.
33:94–99. 2012. View Article : Google Scholar
|
|
15
|
Diplas AI, Lambertini L, Lee MJ, Sperling
R, Lee YL, Wetmur J and Chen J: Differential expression of
imprinted genes in normal and IUGR human placentas. Epigenetics.
4:235–240. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Frank D, Fortino W, Clark L, Musalo R,
Wang W, Saxena A, Li CM, Reik W, Ludwig T and Tycko B: Placental
overgrowth in mice lacking the imprinted gene Ipl. Proc Natl Acad
Sci USA. 99:7490–7495. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Driver AM, Huang W, Kropp J, Peñagaricano
F and Khatib H: Knockdown of CDKN1C (p57(kip2)) and PHLDA2 results
in developmental changes in bovine pre-implantation embryos. PLoS
One. 8:e694902013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
19
|
Demetriou C, Abu-Amero S, Thomas AC,
Ishida M, Aggarwal R, Al-Olabi L, Leon LJ, Stafford JL, Syngelaki
A, Peebles D, et al: Paternally expressed, imprinted insulin-like
growth factor-2 in chorionic villi correlates significantly with
birth weight. PLoS One. 9:e854542014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang L, Zhang W, Shao C, Zhang J, Men K,
Shao Z, Yan Y and Xu D: Establishment and characterization of a
spontaneously immortalized trophoblast cell line (HPT-8) and its
hepatitis B virus-expressing clone. Hum Reprod. 26:2146–2156. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Aboagye-Mathiesen G, Laugesen J,
Zdravkovic M and Ebbesen P: Isolation and characterization of human
placental trophoblast subpopulations from first-trimester chorionic
villi. Clin Diagn Lab Immunol. 3:14–22. 1996.PubMed/NCBI
|
|
22
|
Liu Z, Lu H, Jiang Z, Pastuszyn A and Hu
CA: Apolipoprotein l6, a novel proapoptotic Bcl-2 homology 3-only
protein, induces mitochondria-mediated apoptosis in cancer cells.
Mol Cancer Res. 3:21–31. 2005.PubMed/NCBI
|
|
23
|
Brinkmann K and Kashkar H: Targeting the
mitochondrial apoptotic pathway: a preferred approach in
hematologic malignancies? Cell Death Dis. 5:e10982014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Green DR and Kroemer G: The
pathophysiology of mitochondrial cell death. Science. 305:626–629.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Adams JM and Cory S: The Bcl-2 apoptotic
switch in cancer development and therapy. Oncogene. 26:1324–1337.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hardwick JM and Soane L: Multiple
functions of BCL-2 family proteins. Cold Spring Harb Perspect Biol.
5(5)2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dai H, Huang Y, Li Y, Meng G, Wang Y and
Guo QN: TSSC3 overexpression associates with growth inhibition,
apoptosis induction and enhances chemotherapeutic effects in human
osteosarcoma. Carcinogenesis. 33:30–40. 2012. View Article : Google Scholar
|
|
28
|
Xu Q, Zhang B, Li XM and Gao X:
Traditional Chinese medicine formula Qing Huo Yi Hao as superoxide
anion scavenger in high glucose-treated endothelial cells. Acta
Pharmacol Sin. 33:496–502. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Circu ML and Aw TY: Reactive oxygen
species, cellular redox systems, and apoptosis. Free Radic Biol
Med. 48:749–762. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zorov DB, Juhaszova M and Sollott SJ:
Mitochondrial ROS-induced ROS release: an update and review.
Biochim Biophys Acta. 1757:509–517. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Huang Y, Dai H and Guo QN: TSSC3
overexpression reduces stemness and induces apoptosis of
osteosarcoma tumor-initiating cells. Apoptosis. 17:749–761. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Pijnenborg R, Robertson WB, Brosens I and
Dixon G: Review article: trophoblast invasion and the establishment
of haemochorial placentation in man and laboratory animals.
Placenta. 2:71–91. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Aplin JD: Implantation, trophoblast
differentiation and haemochorial placentation: mechanistic evidence
in vivo and in vitro. J Cell Sci. 99:681–692. 1991.PubMed/NCBI
|
|
34
|
Bischof P, Meisser A and Campana A:
Biochemistry and molecular biology of trophoblast invasion. Ann N Y
Acad Sci. 943:157–162. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tapia-Pizarro A, Argandoña F, Palomino WA
and Devoto L: Human chorionic gonadotropin (hCG) modulation of
TIMP1 secretion by human endometrial stromal cells facilitates
extravillous trophoblast invasion in vitro. Hum Reprod.
28:2215–2227. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yang Q, Chen SP, Zhang XP, Wang H, Zhu C
and Lin HY: Smurf2 participates in human trophoblast cell invasion
by inhibiting TGF-beta type I receptor. J Histochem Cytochem.
57:605–612. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lim KH, Zhou Y, Janatpour M, McMaster M,
Bass K, Chun SH and Fisher SJ: Human cytotrophoblast
differentiation/invasion is abnormal in pre-eclampsia. Am J Pathol.
151:1809–1818. 1997.PubMed/NCBI
|
|
38
|
George EM and Granger JP: Recent insights
into the pathophysiology of preeclampsia. Expert Rev Obstet
Gynecol. 5:557–566. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Salas M, John R, Saxena A, Barton S, Frank
D, Fitzpatrick G, Higgins MJ and Tycko B: Placental growth
retardation due to loss of imprinting of Phlda2. Mech Dev.
121:1199–1210. 2004. View Article : Google Scholar : PubMed/NCBI
|