Introduction
Hepatic fibrosis is the final consequence of many
chronic liver injuries that later develop in cirrhosis and
hepatocellular carcinoma (HCC), which are leading causes of
morbidity and mortality worldwide (1). Regardless of the cause, hepatic
fibrosis is always characterized by the abnormal accumulation of
extracellular matrix (ECM). Accumulating evidence has showed that
in liver injury, hepatic stellate cells (HSCs) undergo a phenotypic
transformation from quiescent, non-proliferating, retinoid-storing
cells to a proliferating, matrix-producing phenotype similar to
myofibroblasts (MFBs) (2–4). Thus, the factors that regulate the
activation, proliferation and functions of HSCs represent important
antifibrotic targets (5).
Clinical reports suggest that advanced hepatic fibrosis is
potentially reversible (6);
however, therapeutic options are limited. Therefore, there is an
urgent need for novel effective agents capable of inhibiting the
function of HSCs.
The Janus kinases (JAKs) are a family of
intracellular tyrosine kinases that play essential roles in the
signaling of numerous cytokines that have been implicated in the
pathogenesis of many diseases. The family of JAKs comprises four
members in mammals: JAK1, JAK2, JAK3 and tyrosine kinase 2 (Tyk2)
(7). After the engagement of
cytokine receptors constitutively bound to JAK, JAK is activated by
a conformational change and phosphorylated. This in turn
phosphorylates the cytokine receptors, resulting in the
phosphorylation of signal transducer and activator of transcription
(STAT) that subsequently translocates into the nucleus, in order to
regulate gene expression (8). The
JAK/STAT pathway mediates a plethora of cellular functions
including defense against pathogens, differentiation,
proliferation, apoptosis, metabolism and cellular transformation
(7,9). Some JAK1/2 inhibitors have been
demonstrated to exert protective effects in fibrotic diseases, such
as bone marrow fibrosis and myelofibrosis (10). However, the therapeutic effect of
JAK inhibition in hepatic fibrosis has not been investigated to
date, to the best of our knowledge. The JAK inhibitor SHR0302
(C18H22N8O2S·H2SO4,
MW, 512), binds to JAK1 with a stronger affinity than to other JAKs
(selectivity for JAK1 is >10 times for JAK2, 77 times for JAK3
and 420 times for Tyk2). This study examined the effects of the JAK
inhibitor SHR0302 on the activation, proliferation, migration,
collagen production and apoptosis of HSCs and the underlying
mechanisms responsible for these effects. Our findings demonstrated
the protective effects of SHR0302 on hepatic fibrosis by inhibiting
HSC functions.
Materials and methods
Chemicals and reagents
SHR0302 was obtained from Jiangsu Hengrui Medicine
Co., Ltd. (Jiangsu, China). MTT was purchased from Sigma Chemical
Co. (St. Louis, MO, USA). The cell apoptosis Annexin V/PI detection
kit was obtained from Shanghai Bestbio (Shanghai, China).
Transforming growth factor β1 (TGF-β1) was obtained from Peprotech
EC, Ltd. (London, UK). Phosphorylated (p-)STAT3 (9145), STAT3
(4904), p-Akt (4058) and Akt (4691) primary antibodies were
purchased from Cell Signaling Technology (Beverly, MA, USA).
p-JAK1, JAK, cleaved caspase-3 (sc-98785), α-smooth muscle actin
(α-SMA; sc-53015), collagen I (sc-59772) and collagen III
(sc-8780-R) primary antibodies were purchased from Santa Cruz
Biotechnology (Santa Cruz, CA, USA). Bcl-2 (TA803003), Bax
(TA346891) and β-actin (TA-09) primary antibodies were purchased
from ZSGB-Bio (Beijing, China). Dulbecco's modified Eagle's medium
(DMEM) and fetal bovine serum (FBS) were obtained from Gibco
(Carlsbad, CA, USA). Other chemicals used in the experiment were of
analytical grade and obtained from commercial sources.
Cell culture
The HSC-T6 cell line was obtained from the Institute
of Liver Disease at the Shanghai University of Traditional Chinese
Medicine (Shanghai, China). The HSCs were cultured at 37°C in an
atmosphere of 5% CO2 in DMEM containing 10% FBS, 2
mmol/l L-glutamine, 100 U/ml of penicillin and 100 µg/ml of
streptomycin.
Cell proliferation assay
The MTT assay was used to evaluate the proliferation
of HSCs. Briefly, the HSCs were plated at a density of
5×104 cells/ml in 96-well culture plates. The confluent
cells were growth-arrested in DMEM containing 0.5% FBS for 24 h.
Subsequently, the cells were incubated with SHR0302 at various
concentrations (10−9, 10−8, 10−7,
10−6 and 10−5 mol/l) for 48 h.
Following treatment, MTT solution (5.0 mg/ml in PBS) was added
(20.0 µl/well), and the plates were incubated at 37°C in 5%
CO2 for a further 4 h. The MTT-formazan product was
dissolved in 150 µl dimethyl sulfoxide (DMSO)/well. After 10
min, the plates were read on a BioTek Elx808 microplate reader
(Winooski, VT, USA) at 570 nm.
Migration assay
To examine the effects of SHR0302 on the migration
of HSCs, a wound-healing assay was performed. The HSCs were seeded
in 6-well plates at 80–90% confluency. The cell monolayer was then
wounded with a 200 µl-pipette. After washing with PBS three
times to remove cell debris, the cells were incubated with 5 ng/ml
TGF-β1 and SHR0302 at various concentrations (10−9,
10−8, 10−7, 10−6 and
10−5 mol/l) for 24 h, and then images were captured
under a microscope (IX71; Olympus, Tokyo, Japan) at 0 and 24 h
after treatment. Cell migration was determined by measuring the
movement of cells into the scraped area and quantitative analysis
showing the percentage wound closure relative to different
conditioned media from the HSCs was performed using Adobe Photoshop
Elements 6.0 software.
Flow cytometric analysis
The HSCs were plated in 6-well plates and treated
with SHR0302 (10−9, 10−8, 10−7,
10−6 and 10−5 mol/l) for 48 h. After being
harvested by trypsinization, the cells were washed twice with cold
PBS, and 400 µl 1X binding buffer was added to each sample
tube at a density of 1.0×l06 cells/ml. The sample
solutions were then transferred to a 5 ml culture tube and
incubated with 5 µl FITC-conjugated Annexin V for 15 min at
2–8°C in the dark, and 10 µl PI for 5 min at 2–8°C in the
dark. The cells were collected and apoptosis was examined using a
flow cytometer (FC 500; Beckman Coulter, Inc., Brea, CA, USA).
Western blot analysis
Proteins were then extracted from the cells in RIPA
lysis buffer [50 mmol/l Tris-HCl, pH 7.4, 150 mmol/l NaCl, 10
mmol/l phenylmethylsulfonyl fluoride (PMSF), 1 mmol/l
ethylenediaminetetraacetic acid (EDTA), 0.1% sodium dodecyl sulfate
(SDS), 1% Triton X-100, 1% sodium deoxycholate]. The protein
concentration was determined using the Bradford assay. A protein
sample was mixed with the 5X sample buffer (4:1) (Bio-Rad
Laboratories, Inc., Hercules, CA, USA) and heated in boiling water
for 10 min. The proteins were resolved by SDS-PAGE, transferred to
a polyvinylidene fluoride (PVDF) membrane (Millipore, Bedford, MA,
USA). After blocking with 5% non-fat milk (blocking solution) at
room temperature, the membrane was incubated with α-SMA, collagen
I, collagen III, cleaved caspase-3, Bcl-2, Bax, p-JAK1, JAK1,
p-STAT3, STAT3, p-Akt, Akt and β-actin primary antibodies (1:1,000)
overnight at 4°C. After washing the blot in TBST three times,
horseradish peroxidase (HRP)-conjugated secondary antibodies were
applied for 2 h at room temperature. After extensive washing in
TBST, immunodetection was visualized by enhanced chemiluminescence
(Pierce, Rockford, IL, USA) using hydrogen peroxide and luminol as
substrates. Autoradiographs were scanned using ImageQuant LAS 4000
mini (GE Healthcare Bio-Sciences AB, Uppsala, Sweden). The density
of the specific bands was quantified using ImageJ software
(National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
Data are expressed as the means ± SD and statistical
analysis was performed using one-way ANOVA. All statistical
analysis was performed with the statistical package SPSS 13.0
(SPSS, Inc., Chicago, IL, USA). P<0.05 was considered to
indicate a statistically significant difference.
Results
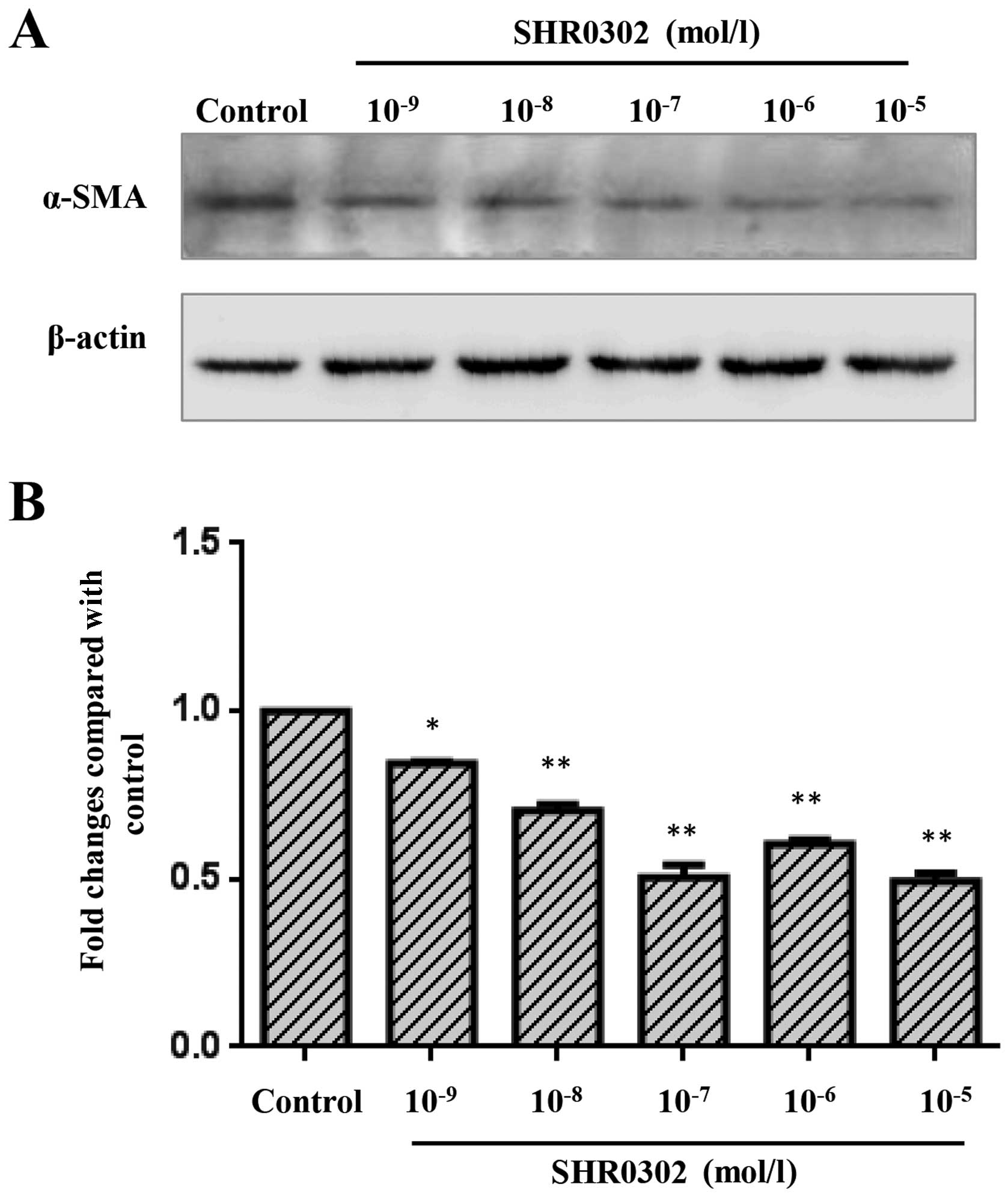
SHR0302 suppresses the activation of
HSCs
Since α-SMA is an marker of activated HSCs (11), we investigated the activity of
HSCs by examining the expression of α-SMA. As illustrated in
Fig. 1, SHR0302 at various
concentrations markedly inhibited the expression of α-SMA compared
with the untreated group. These results suggest that SHR0302
inhibits the activation of HSCs by suppressing the expression of
α-SMA.
SHR0302 inhibits the proliferation of
HSCs
The effect of SHR0302 on the proliferation of HSCs
was detected using the MTT assay. HSCs were incubated with SHR0302
at various concentrations (10−9, 10−8,
10−7, 10−6 and 10−5 mol/l). As
shown in Table I, the inhibitory
rates of SHR0302 (10−9, 10−8,
10−7, 10−6 and 10−5 mol/l) in HSCs
were 6.376, 6.650, 14.551, 19.143 and 48.640%, respectively,
following treatment for 48 h. The MTT experiment confirmed that
HSCs were sensitive to SHR0302, that SHR0302 displayed an
inhibitory effect on the proliferation of HSCs, and that inhibition
ocurred in a concentration-dependent manner.
 | Table IEffect of SHR0302 on the
proliferation of HSCs (means ± SD, n=8). |
Table I
Effect of SHR0302 on the
proliferation of HSCs (means ± SD, n=8).
| Group | Concentration
(mol/l) | 48 h
|
|---|
|
A570nm | Inhibitory rate
(%) |
|---|
| Control | – | 0.782±0.060 | – |
| SHR0302 |
10−9 | 0.733±0.055 | 6.376 |
|
10−8 | 0.730±0.056 | 6.650 |
|
10−7 | 0.669±0.080a | 14.551 |
|
10−6 | 0.632±0.025a | 19.143 |
|
10−5 | 0.402±0.046a | 48.640 |
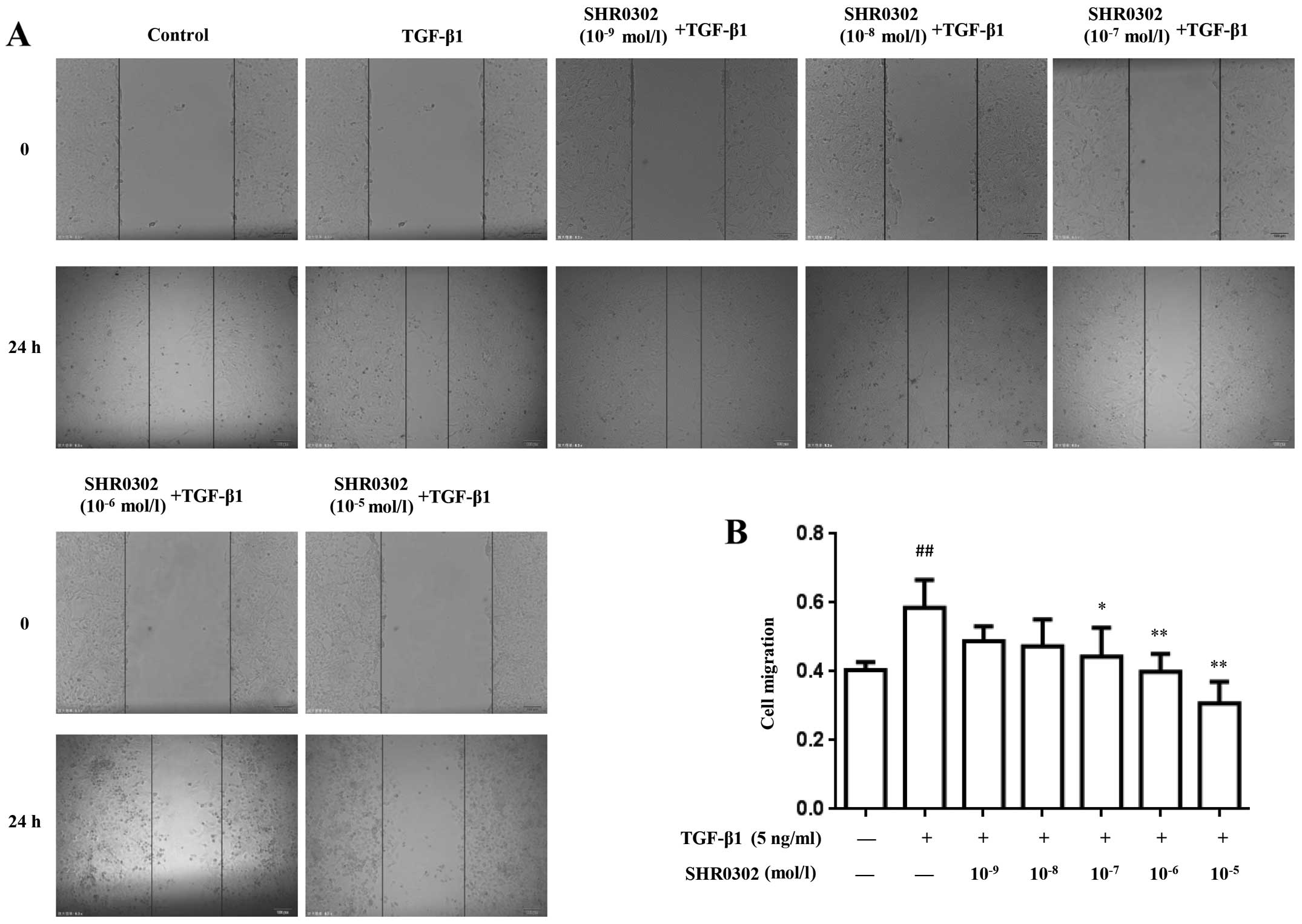
SHR0302 inhibits the migration of
HSCs
To determine the effect of SHR0302 on the migration
of HSCs, wound-healing assays was performed after exposing cells to
different concentrations of SHR0302 in the presence or absence of
TGF-β1. As shown in Fig. 2,
TGF-β1 markedly enhanced cell migration during the wound-healing
process compared with the control group. By contrast, SHR0302
effectively attenuated the migration ability of HSCs at
concentrations of 10−7, 10−6 and
10−5 mol/l, which shows that SHR0302 directly inhibited
the migration of HSCs.
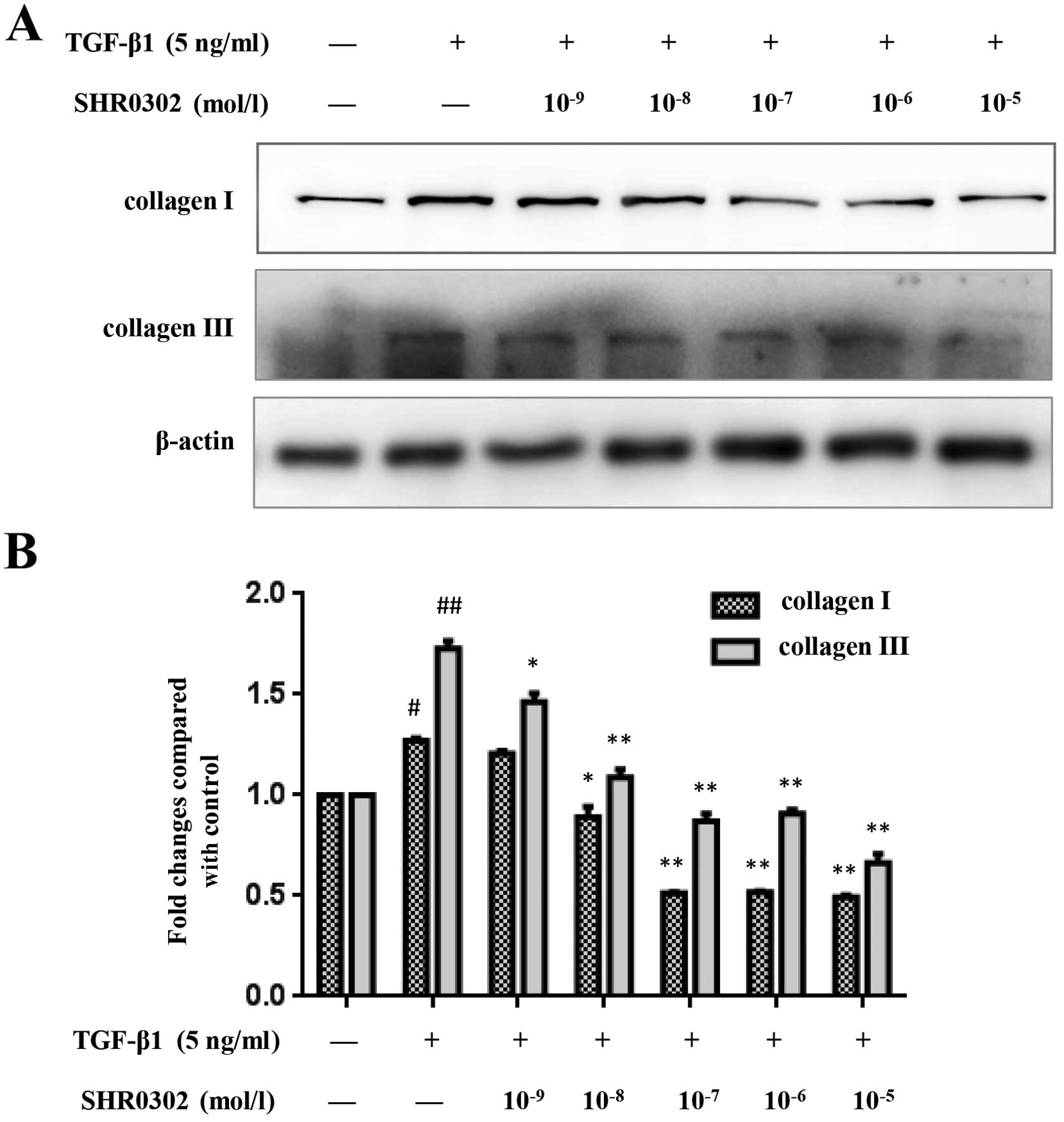
SHR0302 decreases the expression of
collagen I and collagen III in HSCs
To determine the effect of JAK inhibitor SHR0302 on
collagen I and collagen III synthesis in HSCs, western blot
analysis was performed. TGF-β1 strongly promotes HSCs to synthesize
ECM, such as collagen and fibronetin (12,13). The control groups showed low
levels of collagen I and collagen III whereas treatment with TGF-β1
significantly increased the expression of collagen I and collagen
III in HSCs. SHR0302 treatment at concentrations of
10−8, 10−7, 10−6 and
10−5 mol/l markedly reduced the production of both
collagen I and collagen III in HSCs (Fig. 3). Our data therefore demonstrated
that SHR0302 reduces collagen deposition.
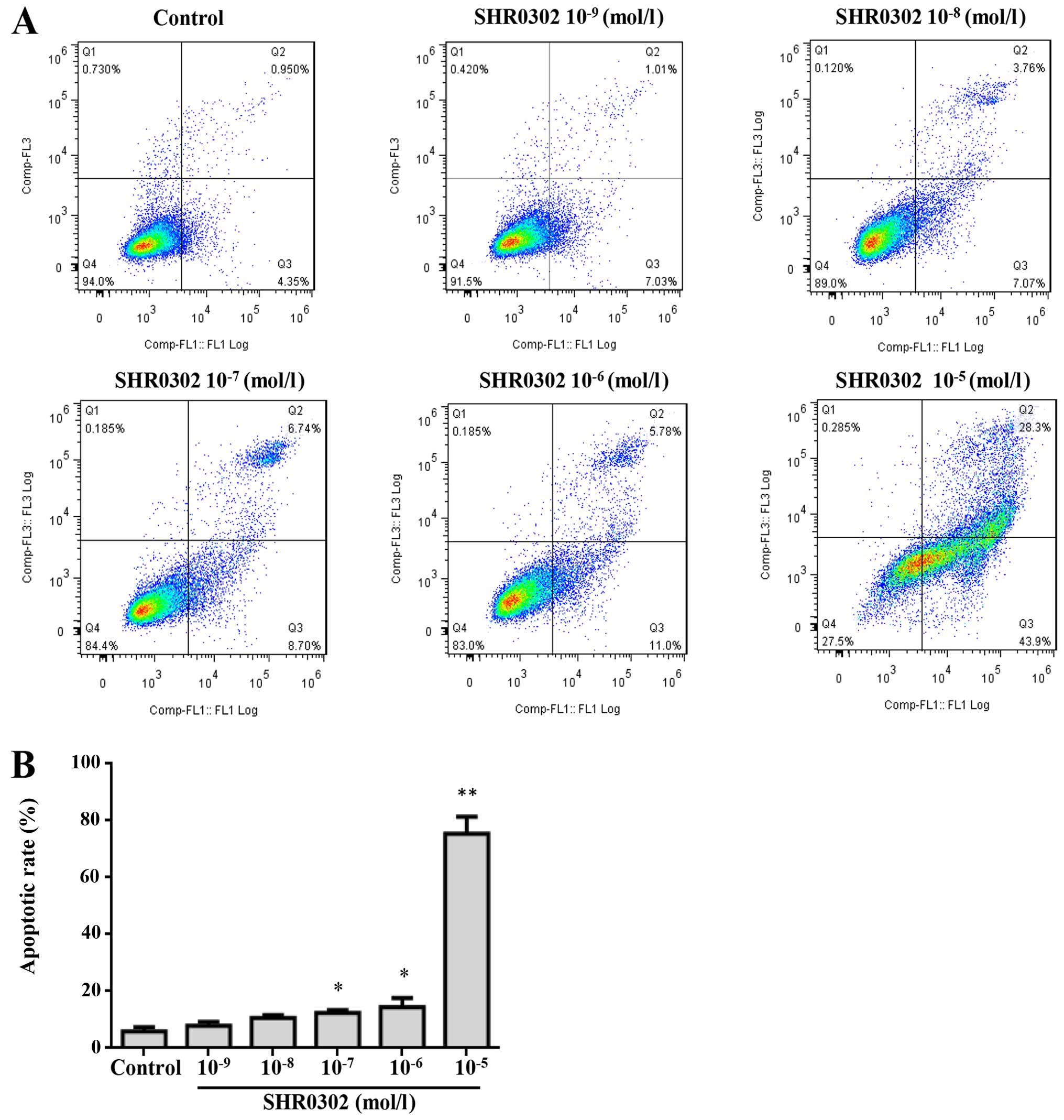
SHR0302 induces the apoptosis of
HSCs
To determine whether SHR0302 induced the apoptosis
of HSCs, the apoptosis rates were detected by Annexin V/PI
staining. As shown in Fig. 4, the
percentage of apoptotic cells increased from 6.76% in the control
group to 75.45% in the SHR0302 10−5 mol/l treatment
group of HSCs after 48 h. These results showed that SHR0302
significantly induced the apoptosis of HSCs.
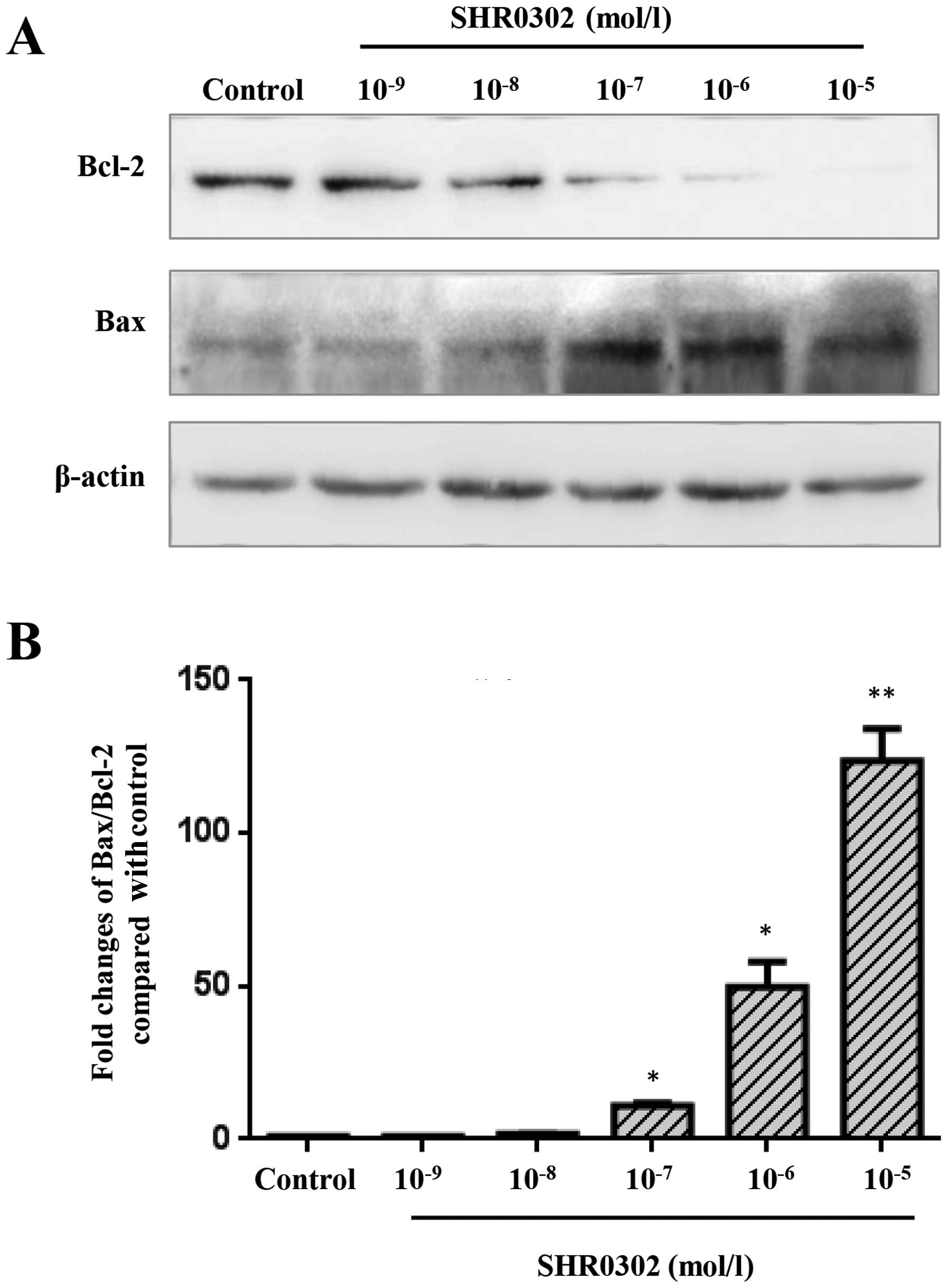
SHR0302 modulates the expression of Bcl-2
and Bax in HSCs
To identify a possible mechanism by which SHR0302
induced the apoptosis of HSCs, western blot analysis was performed
(Fig. 5A). The results of western
blot analysis revealed the decreased expression of Bcl-2 in the
SHR0302-treated HSCs compared with the control group. SHR0302 at
various concentrations markedly inhibited Bcl-2 expression.
However, the expression of Bax was elevated at various
concentrations of SHR0302-treated cells compared with the control
group. As shown in Fig. 5B, the
Bax to Bcl-2 ratio was significantly increased in the
SHR0302-treated cells compared with the control group. The results
indicated that SHR0302 induced the apoptosis of HSCs mainly by
regulating the expression of Bcl-2 family proteins.
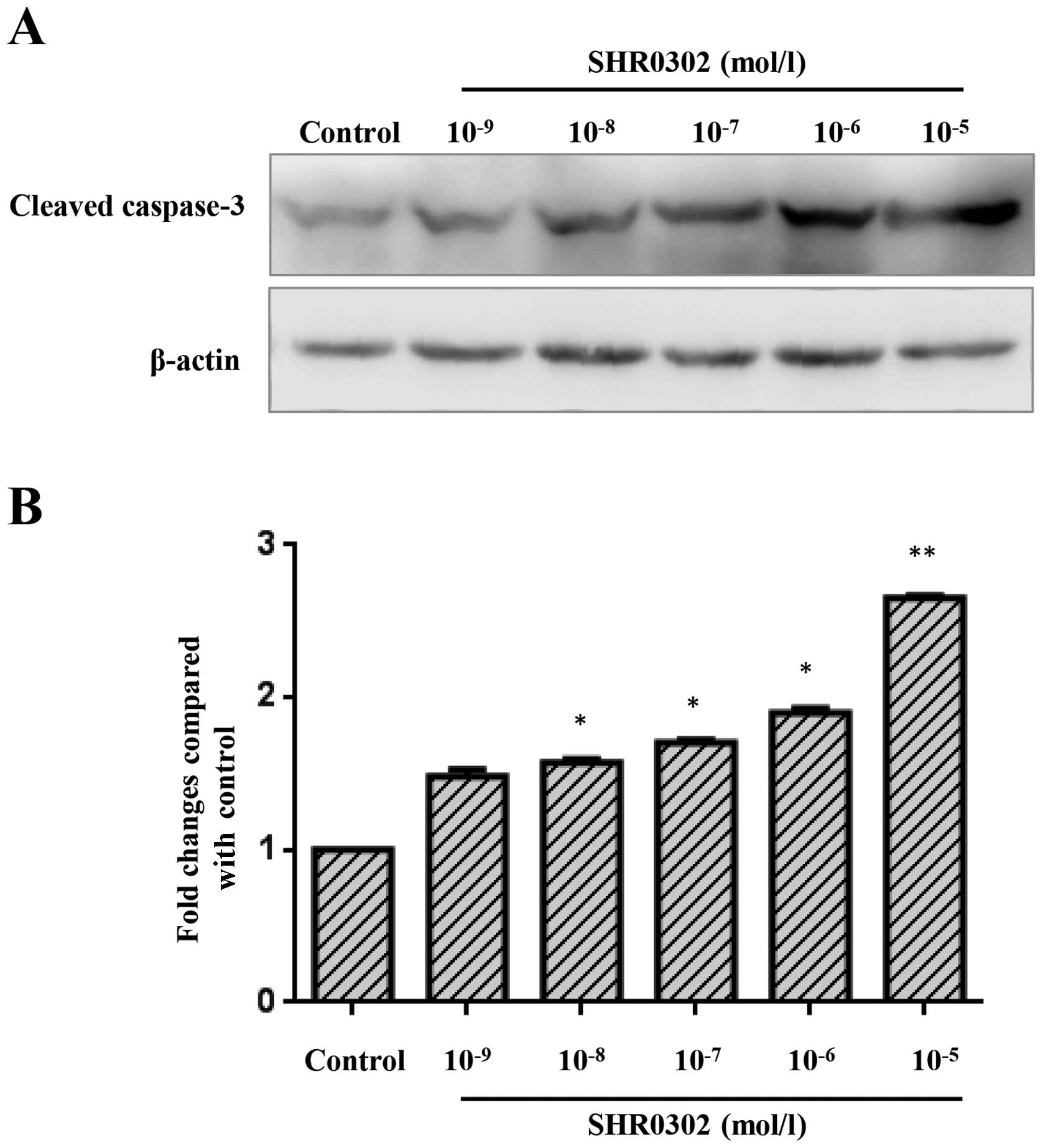
SHR0302 increases caspase-3 activation in
HSCs
To determine whether SHR0302 affects the activation
of caspase-3 in HSCs, the activation of caspase-3 was quantified
through western blot analysis. As shown in Fig. 6, a low level of cleaved caspase-3
was observed in the control group. Treatment with various
concentrations of SHR0302 markedly increased the levels of cleaved
caspase-3 in HSCs.
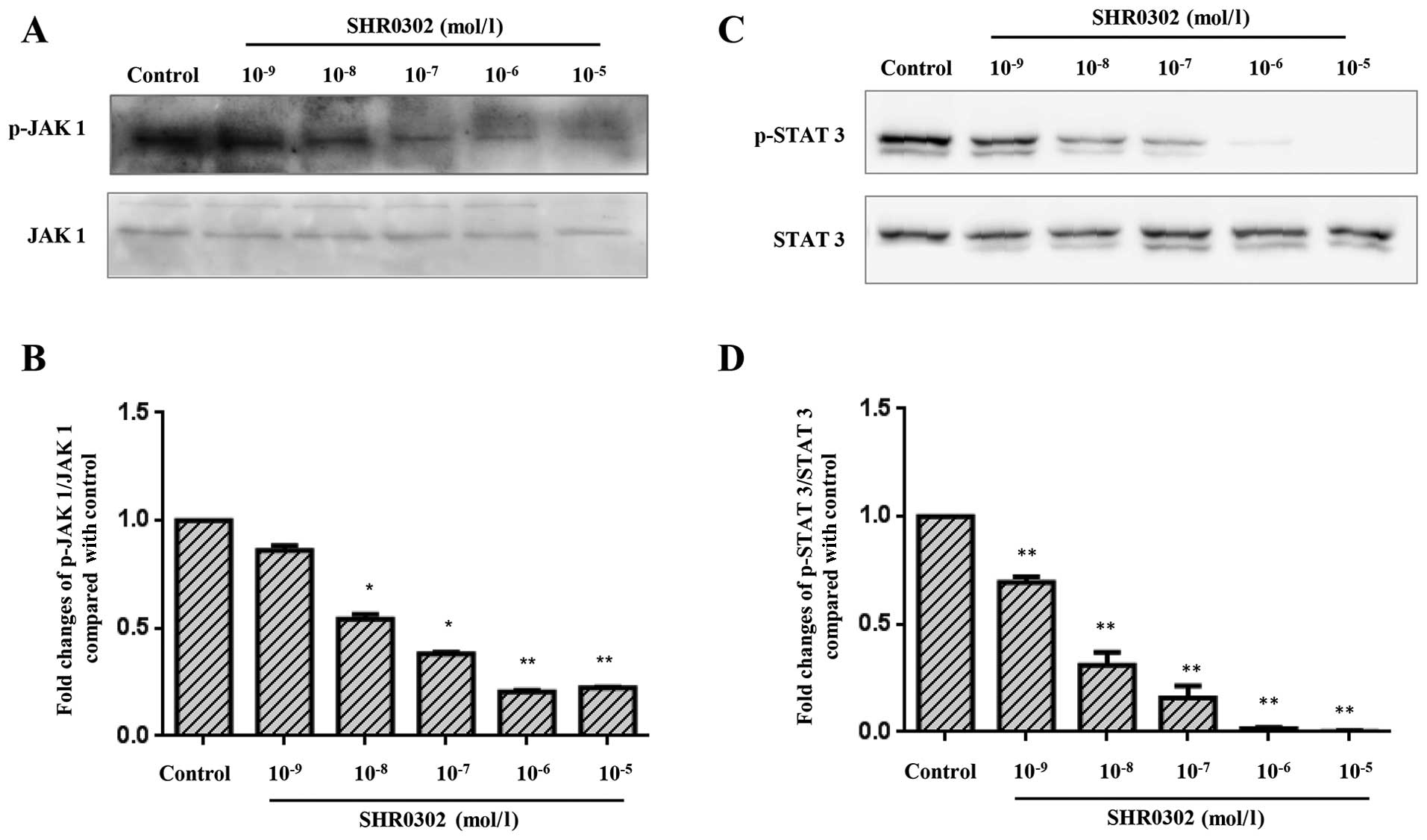
SHR0302 suppresses the JAK1/STAT 3
signaling pathway in HSCs
The pathogenesis of hepatic fibrosis is closely
associated with the activation of STAT3 (14). SHR0302 resulted in dose-dependent
decreases in JAK1 and STAT3 phosphorylation (Fig. 7). Also, similar concentrations of
SHR0302 exerted effects on the activation, proliferation, collagen
deposition and apoptosis of HSCs. These findings suggest that the
mechanism responsible for the effects of SHR0302 on HSC function
occurs through the inhibition of JAK1/STAT3 signaling pathways.
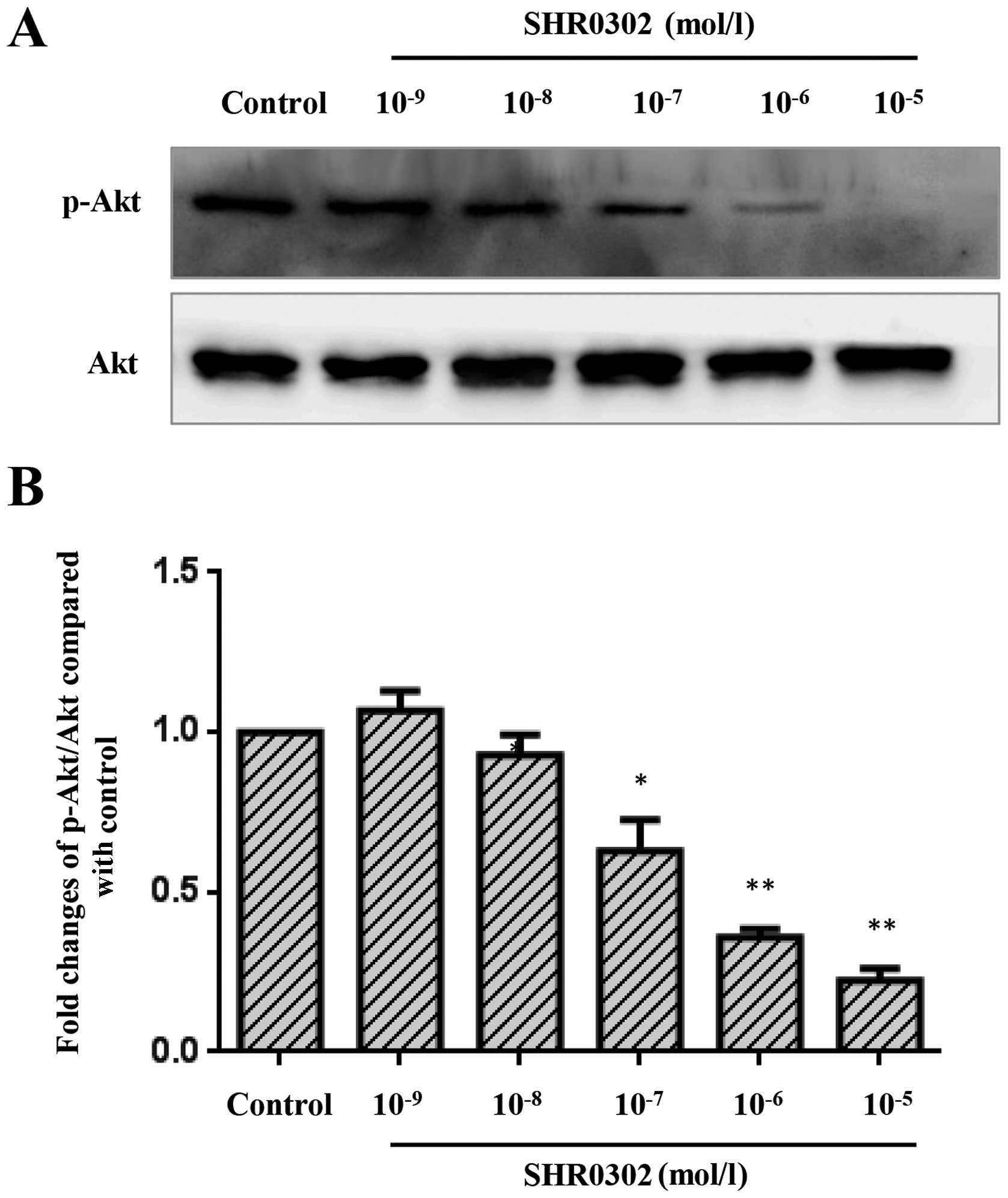
SHR0302 suppresses the Akt signaling
pathway in HSCs
The Akt signaling pathway is a major regulator of
crucial cell functions such as cell growth, survival and
proliferation. The inhibition of Akt activation may induce the
apoptosis of HSCs and suppress collagen synthesis (15,16) in hepatic fibrosis. Thus, we
examined whether SHR0302 has an effect on the Akt signaling
pathway. The results (Fig. 8)
showed that SHR0302 significantly suppressed the Akt signaling
pathway at concentrations of 10−5, 10−6 and
10−7 mol/l.
Discussion
Hepatic fibrosis is the pathological consequence of
chronic liver diseases, resulting from the progressive accumulation
of ECM, which is mainly enriched in types I and III fibrillar
collagens. In the advanced stages, fibrosis leads to cirrhosis, a
condition characterized by abnormal liver architecture, failing
liver function and portal hypertension as well as a high
susceptibility to infection and to developing HCC (17,18). To date, the most effective therapy
for the treatment of hepatic fibrosis involves removal of the
causative agent. During liver injury, HSCs undergo
transdifferentiation from the lipocyte phenotype to MFBs; during
this process, lipid droplets decrease in size and number whereas
proliferation, migration rate and ECM production are increased
(19,20), and this appears to be the dominant
driving force in fibrosis. These studies explored the rationale for
choosing HSCs as a target for pharmacological, molecular and other
novel therapeutics, for potential application in the treatment of
hepatic fibrosis. Our data demonstrated that the abrogation of the
JAK1/STAT3 signaling pathway, induced by JAK inhibitor SHR0302 in
HSCs, SHR0302 (10−9–10−5 mol/l) exerted an
inhibitory effect on the activation, proliferation and migration of
HSCs. Additionally, the expression of collagen I and collagen III
was decreased after treatment with SHR0302. Furthermore, SHR0302
induced the apoptosis of HSCs, which may occur through the
regulation of caspase-3 and the Bcl-2 family.
Once HSCs are activated, they become
α-SMA-expressing MFBs and migrate to the site of hepatic injury.
Once there, they proliferate and express various signal
transduction proteins, producing both pro-inflammatory cytokines
and a great deal of collagen-rich ECM (21,22). In fact, the majority of
antifibrotic treatments currently under evaluation are aimed at
inhibiting the activation and proliferation of HSCs as well as ECM
synthesis (23). For this reason,
in hepatic fibrosis, α-SMA is considered as an indicator of
activated HSCs, and contributes to the proliferation and migration
of HSCs as well as collagen deposition. In previous studies, the
activation of renal interstitial fibroblasts was accompanied by the
phosphorylation of STAT3; the inhibition of the STAT3 pathway
inhibited the expression of α-SMA and fibronectin in a dose- and
time-dependent manner (24), and
suppressed collagen expression in renal fibrosis (25). Furthermore, a JAK inhibitor
through STAT3 signaling reduced the leptin-mediated increase in the
levels of α-SMA and the activity of HSCs in hepatic fibrosis
(26). The MTT assay in our study
demonstrated that the JAK inhibitor SHR0302 significantly decreased
the proliferation of HSCs. Furthermore, western blot analysis
demonstrated that following treatment with SHR0302, the expression
of α-SMA was decreased in HSCs which suggests that the activation
of HSCs was suppressed.
Cell migration is a vital step in the development
and aggravation of several diseases, from organ fibrosis to cancer
(27). The inhibition of
interleukin (IL)-6-induced JAK/STAT3 phosphorylation strongly
reduced the proliferation and migration of glioblastoma cells
(28), and the inhibition of
STAT3 expression delayed cell migration in keloid fibroblasts
(29). The migration of HSCs may
contribute to their accumulation at sites of liver injury.
Following activation, cultured HSCs migrate in response to many
stimuli. In the present study, wound-healing assays were designed
to determine the migratory ability of HSCs following treatment with
SHR0302. The results showed that SHR0302 significantly inhibited
the migration of HSCs and provided new insights into the potential
use of SHR0302 for controlling the development of hepatic
fibrosis.
The most striking biological consequence of the
activation of HSCs is the marked increase in collagen deposition.
In the human body, collagen represents about one third of the total
protein, but only 5 to 10% of the protein in a normal liver. In
cirrhosis, however, it amounts to 50% or more (30). The principal ECM protein products
in the progression of liver fibrogenesis are predominantly collagen
types I and III (31,32). The blockade of STAT3 signaling
inhibits collagen I in hepatic fibrosis (26,33). During the development of fibrosis,
HSCs respond to TGF-β by moving to a MFB phenotype, which in turn
produces a higher deposition of ECM proteins including collagen I
and collagen III (13,34). The results of western blot
analysis revealed that TGF-β1 induced the activation of HSCs which
expressed high levels of collagen I and collagen III. Following
treatment with SHR0302, type I and III collagen expression was
suppressed, suggesting that SHR0302 inhibits the expression of
collagen, and further decreased the deposition of ECM.
Apoptosis is a physiological process of programmed
cell death that plays a vital role in maintaining tissue
homeostasis (35). The apoptosis
of activated HSCs is a key factor in the regression of liver
fibrosis; emerging experimental and clinical evidence indicates
that even cirrhosis is potentially reversible (5,6).
The key to this is the discovery that reversion of fibrosis is
accompanied by the clearance of HSCs by apoptosis. Furthermore,
proof-of-concept studies in rodents have demonstrated that the
experimental augmentation of the apoptosis of HSCs promotes the
regression of fibrosis (36,37). Our flow cytometry assays revealed
that SHR0302 significantly induces the apoptosis of HSCs. To
further elucidate the possible pro-apoptotic mechanisms induced by
SHR0302, the expression of the Bcl-2 family proteins and caspase-3,
which are acknowledged apoptosis-related regulators, was examined.
Activated HSCs are resistant to many pro-apoptotic stimuli, and the
main survival signal is the overexpression of Bcl-2 family members
(38,39). The previous study showed evidence
that freshly isolated HSCs possessed high levels of the
pro-apoptotic molecule Bax and undetectable expression of Bcl-2, a
potent inhibitor of apoptotic cell death (39). Activated HSCs/MFBs had a complete
reversal in the Bcl-2/Bax ratio, and Bcl-2-silenced cells were
susceptible to apoptosis. The inhibition of STAT3 is associated
with decreased Bcl-2 expression and increased Bax expression in
many diseases (40–42). And in our study, western blot
analysis revealed the decreased expression of Bcl-2 and elevated
levels of Bax in the HSCs treated with SHR0302 compared with the
control group. These results suggest that SHR0302 modulates the
expression of Bcl-2 family proteins. Caspases are known for playing
an important role in the execution of apoptosis, which frequently
activates death proteases, catalyzing the specific cleavage of many
pivotal cellular proteins (43).
Caspase-3 is a frequently activated death protease; cleaved
caspase-3 expression was much less apparent in the cirrhotic livers
of wild-type mice compared with those from mice receiving drug
treatment (44,45). JAK inhibitor-treated HSCs are
positive for active caspase-3, which indicates the presence of a
higher apoptotic rate (46).
Western blot analysis in this study demonstrated that SHR0302
enhanced the expression of cleaved caspase-3 in HSCs. In agreement
with the data regarding the Bcl-2 family of proteins, our study
indicated that SHR0302 may induce apoptosis through mechanisms
which modulate the Bcl-2 family proteins and caspase-3
activity.
Following the binding of cytokines to their cognate
receptor, JAKs phosphorylate STAT to modulate gene expression
(47). The JAK/STAT pathway is
activated in response to cytokines, growth factors and hormones,
mediating a plethora of cellular functions including defense
against pathogens, differentiation, proliferation, apoptosis,
metabolism and cellular transformation (9). STAT3 plays a key role in many
cellular processes such as cell growth and apoptosis, and mediates
the expression of a variety of genes in response to stimuli as
discussed above; these broad ranging activities make JAK/STAT3 an
attractive therapeutic target. Herein, we demonstrated that SHR0302
suppressed the activation of the JAK1/STAT3 signaling pathway, and
may be involved in the proliferation, migration, and apoptosis of
HSCs as well as HSC collagen production.
Akt signal transduction regulates ECM deposition,
HSC activation and is implicated in the development and progression
of hepatic fibrosis (15,16). Previous findings have demonstrated
that there are complex interactions between the Akt signaling
pathway and the JAK signaling pathway. In study of MCF-7 human
breast cancer cells and mouse embryonic fibroblasts, it was found
that the activity of STAT3 regulates Akt gene expression (48). In hepatic diseases, the
JAK-dependent Akt signaling pathway plays a important role in
disease progression (49,50). Thus, our results showing that
SHR0302 inhibits the Akt signaling pathway is consistent with the
findings of other studies of JAK inhibitors suggesting that they
potentially inhibit the Akt signaling pathway (51,52).
The effects of JAK inhibitors have been demonstrated
in rheumatoid arthritis, colon cancer, lymphoblastic leukemia,
myelofibrosis and so on (53–56). For example, ruxolitinib, a JAK1
and JAK2 inhibitor, in clinical trials, alleviated the burdensome
manifestations of myelofibrosis, namely splenomegaly and core
symptoms (57). The JAK inhibitor
AG490 inhibits the leptin-stimulated mRNA expression of JAK1, JAK2
and α1(I) collagen in HSCs (58).
Thus, we hypothesized that the JAK inhibitor SHR0302 suppresses
hepatic fibrosis, and herein, we proved that the selective JAK
inhibitor SHR0302 suppresses certain functions in HSCs.
In conclusion, our hypothesis will require further
examination. However, taken together, these findings suggest that
the blockade of the JAK/STAT3 signaling pathways significantly
decreased cell function and suppressed the Akt signaling pathway in
HSCs. JAKs represent an attractive target for the development of
novel targeted therapies in various clinical settings including
hematologic malignancies, autoimmune disease and organ
transplantation. Our study has shown that SHR0302 blocks the
downstream STAT3 signaling pathway by abrogating JAK1 activity, and
thereby inhibits the activation, proliferation and migration of
HSCs as well as collagen synthesis by HSCs. SHR0302 also induces
the apoptosis of HSCs. Currently, no optimal antifibrotic drugs
available for the clinical treatment of hepatic fibrosis. These
results indicate that the JAK inhibitor SHR0302 may have the
potential to alleviate hepatic fibrosis.
Acknowledgments
The present study was supported by grants from the
National Natural Science Foundation of China (nos. 81300332,
81173075 and 81330081), the Specialized Research Fund for the
Doctoral Program of Higher Education in China (no. 20113420120002),
the Natural Science Foundation of the Higher Education Institutions
of Anhui Province (nos. KJ2012A153 and KJ2014A119) and Anhui
Provincial Natural Science Foundation (no. 1308085QH130).
References
|
1
|
Leon DA and McCambridge J: Liver cirrhosis
mortality rates in Britain from 1950 to 2002: an analysis of
routine data. Lancet. 367:52–56. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Priya S and Sudhakaran PR: Cell survival,
activation and apoptosis of hepatic stellate cells: modulation by
extracellular matrix proteins. Hepatol Res. 38:1221–1232.
2008.PubMed/NCBI
|
|
3
|
Wang P, Liu T, Cong M, Wu X, Bai Y, Yin C,
An W, Wang B, Jia J and You H: Expression of extracellular matrix
genes in cultured hepatic oval cells: an origin of hepatic stellate
cells through transforming growth factor beta? Liver Int.
29:575–584. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Moles A, Tarrats N, Morales A, Domínguez
M, Bataller R, Caballería J, García-Ruiz C, Fernández-Checa JC and
Marí M: Acidic sphingomyelinase controls hepatic stellate cell
activation and in vivo liver fibrogenesis. Am J Pathol.
177:1214–1224. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fallowfield JA: Therapeutic targets in
liver fibrosis. Am J Physiol Gastrointest Liver Physiol.
300:G709–G715. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Povero D, Busletta C, Novo E, di Bonzo LV,
Cannito S, Paternostro C and Parola M: Liver fibrosis: a dynamic
and potentially reversible process. Histol Histopathol.
25:1075–1091. 2010.PubMed/NCBI
|
|
7
|
Seavey MM and Dobrzanski P: The many faces
of Janus kinase. Biochem Pharmacol. 83:1136–1145. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tanaka Y: Recent progress and perspective
in JAK inhibitors for rheumatoid arthritis: from bench to bedside.
J Biochem. 158:173–179. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kisseleva T, Bhattacharya S, Braunstein J
and Schindler CW: Signaling through the JAK/STAT pathway, recent
advances and future challenges. Gene. 285:1–24. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pardanani A, Vannucchi AM, Passamonti F,
Cervantes F, Barbui T and Tefferi A: JAK inhibitor therapy for
myelofibrosis: critical assessment of value and limitations.
Leukemia. 25:218–225. 2011. View Article : Google Scholar
|
|
11
|
Johnson SJ, Hines JE and Burt AD:
Phenotypic modulation of perisinusoidal cells following acute liver
injury: a quantitative analysis. Int J Exp Pathol. 73:765–772.
1992.PubMed/NCBI
|
|
12
|
Cheng K, Yang N and Mahato RI: TGF-beta1
gene silencing for treating liver fibrosis. Mol Pharm. 6:772–779.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kanzler S, Lohse AW, Keil A, Henninger J,
Dienes HP, Schirmacher P, Rose-John S, zum Büschenfelde KH and
Blessing M: TGF-beta1 in liver fibrosis: an inducible transgenic
mouse model to study liver fibrogenesis. Am J Physiol.
276:G1059–G1068. 1999.PubMed/NCBI
|
|
14
|
Mair M, Blaas L, Österreicher CH, Casanova
E and Eferl R: JAK-STAT signaling in hepatic fibrosis. Front Biosci
(Landmark Ed). 16:2794–2811. 2011. View
Article : Google Scholar
|
|
15
|
Paik YH, Kim JK, Lee JI, Kang SH, Kim DY,
An SH, Lee SJ, Lee DK, Han KH, Chon CY, et al: Celecoxib induces
hepatic stellate cell apoptosis through inhibition of Akt
activation and suppresses hepatic fibrosis in rats. Gut.
58:1517–1527. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Son G, Hines IN, Lindquist J, Schrum LW
and Rippe RA: Inhibition of phosphatidylinositol 3-kinase signaling
in hepatic stellate cells blocks the progression of hepatic
fibrosis. Hepatology. 50:1512–1523. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Schuppan D and Kim YO: Evolving therapies
for liver fibrosis. J Clin Invest. 123:1887–1901. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mallat A and Lotersztajn S: Cellular
mechanisms of tissue fibrosis. 5. Novel insights into liver
fibrosis. Am J Physiol Cell Physiol. 305:C789–C799. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Senoo H, Yoshikawa K, Morii M, Miura M,
Imai K and Mezaki Y: Hepatic stellate cell (vitamin A-storing cell)
and its relative-past, present and future. Cell Biol Int.
34:1247–1272. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hernandez-Gea V and Friedman SL:
Pathogenesis of liver fibrosis. Annu Rev Pathol. 6:425–456. 2011.
View Article : Google Scholar
|
|
21
|
Choi JH, Hwang YP, Choi CY, Chung YC and
Jeong HG: Anti-fibrotic effects of the anthocyanins isolated from
the purple-fleshed sweet potato on hepatic fibrosis induced by
dimethylnitrosamine administration in rats. Food Chem Toxicol.
48:3137–3143. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Clément S, Pascarella S, Conzelmann S,
Gonelle-Gispert C, Guilloux K and Negro F: The hepatitis C virus
core protein indirectly induces alpha-smooth muscle actin
expression in hepatic stellate cells via interleukin-8. J Hepatol.
52:635–643. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wu J and Zern MA: Hepatic stellate cells:
a target for the treatment of liver fibrosis. J Gastroenterol.
35:665–672. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pang M, Ma L, Gong R, Tolbert E, Mao H,
Ponnusamy M, Chin YE, Yan H, Dworkin LD and Zhuang S: A novel STAT3
inhibitor, S3I-201, attenuates renal interstitial fibroblast
activation and interstitial fibrosis in obstructive nephropathy.
Kidney Int. 78:257–268. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yu Y, Wang Y, Niu Y, Fu L, Chin YE and Yu
C: Leukemia inhibitory factor attenuates renal fibrosis through
Stat3-miR-29c. Am J Physiol Renal Physiol. 309:F595–F603. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang W, Niu M, Yan K, Zhai X, Zhou Q,
Zhang L and Zhou Y: Stat3 pathway correlates with the roles of
leptin in mouse liver fibrosis and sterol regulatory element
binding protein-1c expression of rat hepatic stellate cells. Int J
Biochem Cell Biol. 45:736–744. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ridley AJ, Schwartz MA, Burridge K, Firtel
RA, Ginsberg MH, Borisy G, Parsons JT and Horwitz AR: Cell
migration: integrating signals from front to back. Science.
302:1704–1709. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Michaud-Levesque J, Bousquet-Gagnon N and
Béliveau R: Quercetin abrogates IL-6/STAT3 signaling and inhibits
glioblastoma cell line growth and migration. Exp Cell Res.
318:925–935. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lim CP, Phan TT, Lim IJ and Cao X: Stat3
contributes to keloid pathogenesis via promoting collagen
production, cell proliferation and migration. Oncogene.
25:5416–5425. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Schuppan D: Structure of the extracellular
matrix in normal and fibrotic liver: collagens and glycoproteins.
Semin Liver Dis. 10:1–10. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Rojkind M and Martinez-Palomo A: Increase
in type I and type III collagens in human alcoholic liver
cirrhosis. Proc Natl Acad Sci USA. 73:539–543. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Grimaud JA, Druguet M, Peyrol S, Chevalier
O, Herbage D and El Badrawy N: Collagen immunotyping in human
liver: light and electron microscope study. J Histochem Cytochem.
28:1145–1156. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lakner AM, Moore CC, Gulledge AA and
Schrum LW: Daily genetic profiling indicates JAK/STAT signaling
promotes early hepatic stellate cell transdifferentiation. World J
Gastroenterol. 16:5047–5056. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hellerbrand C, Stefanovic B, Giordano F,
Burchardt ER and Brenner DA: The role of TGFbeta1 in initiating
hepatic stellate cell activation in vivo. J Hepatol. 30:77–87.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Thompson HJ, Strange R and Schedin PJ:
Apoptosis in the genesis and prevention of cancer. Cancer Epidemiol
Biomarkers Prev. 1:597–602. 1992.PubMed/NCBI
|
|
36
|
Bataller R and Brenner DA: Hepatic
stellate cells as a target for the treatment of liver fibrosis.
Semin Liver Dis. 21:437–451. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhao W, Su W, Kuang P, Zhang L, Liu J, Yin
Z and Wang X: The role of hepatic stellate cells in the regulation
of T-cell function and the promotion of hepatocellular carcinoma.
Int J Oncol. 41:457–464. 2012.PubMed/NCBI
|
|
38
|
Kawada N: Human hepatic stellate cells are
resistant to apoptosis: implications for human fibrogenic liver
disease. Gut. 55:1073–1074. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Novo E, Marra F, Zamara E, Valfrè di Bonzo
L, Monitillo L, Cannito S, Petrai I, Mazzocca A, Bonacchi A, De
Franco RS, et al: Overexpression of Bcl-2 by activated human
hepatic stellate cells: resistance to apoptosis as a mechanism of
progressive hepatic fibrogenesis in humans. Gut. 55:1174–1182.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Nielsen M, Kaestel CG, Eriksen KW,
Woetmann A, Stokkedal T, Kaltoft K, Geisler C, Röpke C and Odum N:
Inhibition of constitutively activated Stat3 correlates with
altered Bcl-2/Bax expression and induction of apoptosis in mycosis
fungoides tumor cells. Leukemia. 13:735–738. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lee SY, Kwok SK, Son HJ, Ryu JG, Kim EK,
Oh HJ, Cho ML, Ju JH, Park SH and Kim HY: IL-17-mediated Bcl-2
expression regulates survival of fibroblast-like synoviocytes in
rheumatoid arthritis through STAT3 activation. Arthritis Res Ther.
15:R312013. View
Article : Google Scholar : PubMed/NCBI
|
|
42
|
Moodley YP, Misso NL, Scaffidi AK,
Fogel-Petrovic M, McAnulty RJ, Laurent GJ, Thompson PJ and Knight
DA: Inverse effects of interleukin-6 on apoptosis of fibroblasts
from pulmonary fibrosis and normal lungs. Am J Respir Cell Mol
Biol. 29:490–498. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Galluzzi L, Kepp O and Kroemer G:
Caspase-3 and prostaglandins signal for tumor regrowth in cancer
therapy. Oncogene. 31:2805–2808. 2012. View Article : Google Scholar
|
|
44
|
Yang B, El Nahas AM, Thomas GL, Haylor JL,
Watson PF, Wagner B and Johnson TS: Caspase-3 and apoptosis in
experimental chronic renal scarring. Kidney Int. 60:1765–1776.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhou TB, Qin YH, Zhou C, Lei FY, Zhao YJ,
Chen J, Su LN and Huang WF: Less expression of prohibitin is
associated with increased caspase-3 expression and cell apoptosis
in renal interstitial fibrosis rats. Nephrology (Carlton).
17:189–196. 2012. View Article : Google Scholar
|
|
46
|
Jiang JX, Mikami K, Venugopal S, Li Y and
Török NJ: Apoptotic body engulfment by hepatic stellate cells
promotes their survival by the JAK/STAT and Akt/NF-kappaB-dependent
pathways. J Hepatol. 51:139–148. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
O'Sullivan LA, Liongue C, Lewis RS,
Stephenson SE and Ward AC: Cytokine receptor signaling through the
Jak-Stat-Socs pathway in disease. Mol Immunol. 44:2497–2506. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Xu Q, Briggs J, Park S, Niu G, Kortylewski
M, Zhang S, Gritsko T, Turkson J, Kay H, Semenza GL, et al:
Targeting Stat3 blocks both HIF-1 and VEGF expression induced by
multiple oncogenic growth signaling pathways. Oncogene.
24:5552–5560. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Saxena NK, Sharma D, Ding X, Lin S, Marra
F, Merlin D and Anania FA: Concomitant activation of the JAK/STAT,
PI3K/AKT, and ERK signaling is involved in leptin-mediated
promotion of invasion and migration of hepatocellular carcinoma
cells. Cancer Res. 67:2497–2507. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Niu L, Wang X, Li J, Huang Y, Yang Z, Chen
F, Ni H, Jin Y, Lu X and Cao Q: Leptin stimulates alpha1(I)
collagen expression in human hepatic stellate cells via the
phosphatidylinositol 3-kinase/Akt signalling pathway. Liver Int.
27:1265–1272. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Monaghan KA, Khong T, Burns CJ and Spencer
A: The novel JAK inhibitor CYT387 suppresses multiple signalling
pathways, prevents proliferation and induces apoptosis in
phenotypically diverse myeloma cells. Leukemia. 25:1891–1899. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Gross ER, Hsu AK and Gross GJ: The
JAK/STAT pathway is essential for opioid-induced cardioprotection:
JAK2 as a mediator of STAT3, Akt, and GSK-3 beta. Am J Physiol
Heart Circ Physiol. 291:H827–H834. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Tanaka Y and Yamaoka K: JAK inhibitor
tofacitinib for treating rheumatoid arthritis: from basic to
clinical. Mod Rheumatol. 23:415–424. 2013. View Article : Google Scholar
|
|
54
|
An HJ, Choi EK, Kim JS, Hong SW, Moon JH,
Shin JS, Ha SH, Kim KP, Hong YS, Lee JL, et al: INCB018424 induces
apoptotic cell death through the suppression of pJAK1 in human
colon cancer cells. Neoplasma. 61:56–62. 2014. View Article : Google Scholar
|
|
55
|
Suryani S, Bracken LS, Harvey RC, Sia KC,
Carol H, Chen IM, Evans K, Dietrich PA, Roberts KG, Kurmasheva RT,
et al: Evaluation of the in vitro and in vivo efficacy of the JAK
inhibitor AZD1480 against JAK-mutated acute lymphoblastic leukemia.
Mol Cancer Ther. 14:364–374. 2015. View Article : Google Scholar :
|
|
56
|
Swaim SJ: Ruxolitinib for the treatment of
primary myelofibrosis. Am J Health Syst Pharm. 71:453–462. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Ostojic A, Vrhovac R and Verstovsek S:
Ruxolitinib for the treatment of myelofibrosis: its clinical
potential. Ther Clin Risk Manag. 8:95–103. 2012.PubMed/NCBI
|
|
58
|
Cao Q, Mak KM and Lieber CS: Leptin
enhances alpha1(I) collagen gene expression in LX-2 human hepatic
stellate cells through JAK-mediated
H2O2-dependent MAPK pathways. J Cell Biochem.
97:188–197. 2006. View Article : Google Scholar
|